
Abstract
Key message
A major novel QTL was identified in a recombinant inbred line population derived from a cross of ‘Wyandot’ × PI 567301B for Fusarium graminearum, a seed and seedling pathogen of soybean.
Abstract
Fusarium graminearum is now recognized as a primary pathogen of soybean, causing root, seed rot and seedling damping-off in North America. In a preliminary screen, ‘Wyandot’ and PI 567301B were identified with medium and high levels of partial resistance to F. graminearum, respectively. The objective of this study was to characterise resistance towards F. graminearum using 184 recombinant inbred lines (RILs) derived from a cross of ‘Wyandot’ × PI 567301B. The parents and the RILs of the mapping population were evaluated for resistance towards F. graminearum using the rolled towel assay in a randomized incomplete block design. A genetic map was constructed from 2545 SNP markers and 2 SSR markers by composite interval mapping. One major and one minor QTL were identified on chromosomes 8 and 6, respectively, which explained 38.5 and 8.1 % of the phenotypic variance. The major QTL on chromosome 8 was mapped to a 300 kb size genomic region of the Williams 82 sequence. Annotation of this region indicates that there are 39 genes including the Rhg4 locus for soybean cyst nematode (SCN) resistance. Based on previous screens, PI 567301B is susceptible to SCN. Fine mapping of this locus will assist in cloning these candidate genes as well as identifying DNA markers flanking the QTL that can be used in marker-assisted breeding to develop cultivars with high levels of resistance to F. graminearum.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The incidence of seedling diseases in soybean [Glycine max (L.) Merr] caused by both oomycete and true fungi has increased substantially as the number of acres and the number of fields in continuous soybean has increased (Broders et al. 2007; Ellis et al. 2011). Phytophthora sojae and Pythium spp. have long been known in Ohio for association with seed and seedling diseases causing pre- and post-emergence damping-off in soybean and are responsible for severe production losses in the state (Schmitthenner 1985). In addition to these, another residue-borne pathogen, Fusarium graminearum [telemorph: Gibberella zeae (Schwein) Petch] has also been identified as a primary pathogen of soybean causing seed rot and seedling damping-off in North America (Broders et al. 2007; Díaz Arias et al. 2013a; Xue et al. 2007).
Symptoms of seed rot and seedling damping-off caused by F. graminearum appear first as water soaked lesions followed by light brown or pink discoloration around the inoculation point. At later stages seed, seedling, and root rots develop (Broders et al. 2007; Ellis et al. 2011; Xue et al. 2007). In South America, infections can also occur at pod filling and symptoms include spreading of discoloration vertically on the stem, interveinal chlorosis of leaves leading to plant wilting, pod blight and death (Martinelli et al. 2004; Pioli et al. 2004).
F. graminearum is a homothallic, ascomycete fungus and its sexual reproduction involves the development of perithecia that produce ascospores. This fungus produces macroconidia as asexual spores but lacks microconidia (Leslie and Summerell 2006). The macroconidia are slender and elongated, containing 3–5 cells separated by septa and play an important role in the dissemination of the pathogen (Harris 2005; Leslie and Summerell 2006). In addition to F. graminearum, many closely related species were also found to cause disease, hence, the fungi that contribute to these infections are now known as the Fusarium graminearum species complex (FGSC) (O’Donnell et al. 2004; Starkey et al. 2007; Taylor et al. 2000; Wang et al. 2011). In the United States, F. graminearum sensu stricto is the predominant species causing head blight (Gale et al. 2007). Fusarium graminearum has been more extensively studied as a pathogen of wheat and barley, which caused estimated yield losses of close to $3 billion during the scab epidemics in the US in the 1990s (Windels 2000). More importantly, Fusarium isolates with wide differences in aggressiveness have been detected, but no biological races have been reported to date in the Fusarium head blight––wheat system (review by Buerstmayr et al. 2009). This residue-borne pathogen also causes Gibberella ear and stalk rot of corn. In addition to causing yield loss, they produce mycotoxins, secondary metabolites, such as deoxynivalenol (DON), nivalenol (NIV) and zearalenone (ZEN). Type B trichothecenes (DON and NIV) are produced by every species in the Fusarium graminearum species complex (O’Donnell et al. 2008), are toxic to humans, animals, plants, insects and other microorganisms and are also important in fungal pathogenesis and antagonism (Mule et al. 2005).
Numerous quantitative trait loci (QTL) for resistance towards F. graminearum have been identified in both wheat and corn. Interestingly, different QTL have been identified for different types of diseases caused by the same pathogen, and for toxin production (Somers et al. 2003; Tamburic-Ilincic et al. 2009). For instance, QTL controlling seedling blight, head blight and DON accumulation, caused by F. graminearum, were different in wheat (Somers et al. 2003; Tamburic-Ilincic et al. 2009). In corn, F. graminearum can infect ears through the silk channel or through wounds made by birds and insects damaging the husk tissue and grain (Chungu et al. 1996; Reid and Hamilton 1996; Reid et al. 1992a, b). Previous studies identified quantitative resistance loci for silk or kernel resistance; the former prevents the fungus from growing down from silk to kernel and the latter prevents spread of the fungus from kernel to kernel when silk resistance is ineffective (Ali et al. 2005; Chungu et al. 1996; Reid et al. 1992a, b). Ali et al. (2005) detected 11 QTL for silk resistance and 18 QTL for kernel resistance in corn of which only two were the same for both silk and kernel resistance. Similarly, Martin et al. (2011) identified six major effects QTL that significantly enhanced ear rot resistance, including four QTL that significantly lowered DON accumulation and five QTL that contributed to lower zearalenone levels. Quantitative resistance has also been identified to Gibberella stalk rot in corn (Pè et al. 1993; Yang et al. 2010) including a single resistance gene, Rfg1 (Yang et al. 2004).
F. graminearum was found to be highly aggressive in causing severe root rot in soybean compared to other Fusarium species (Broders et al. 2007; Díaz Arias et al. 2013a; Ellis et al. 2011; Zhang et al. 2010). In Ohio, F. graminearum was readily isolated from soybeans with symptoms of seedling blight collected in the field (Broders et al. 2007). F. graminearum was also the most frequently recovered species of Fusarium in fields in Iowa (Díaz Arias et al. 2013b) and in Eastern Ontario (Zhang et al. 2010). Since corn and wheat are often grown in rotation with soybean in the north central region of the USA, and because of the increased reports of this species contributing to seedling blights and root rots, it is important to examine potential risk of this pathogen and to establish disease management strategies.
A combined total of more than 250 soybean genotypes were screened with F. graminearum in three previous studies that readily identified soybean genotypes with resistance (Acharya 2014; Ellis et al. 2012; Zhang et al. 2010). However, genetic analysis of this resistance has thus far been limited to only one study (Ellis et al. 2012). They reported five QTL conferring resistance to F. graminearum, of which resistance alleles were contributed from ‘Conrad’ for four loci and from ‘Sloan’ for the other (Ellis et al. 2012). Among the 200 cultivars and plant introductions screened for resistance by Acharya (2014), plant introduction (PI) 567301B has high levels of resistance and ‘Wyandot’ has moderate level of resistance towards F. graminearum. A recombinant inbred line (RIL) population derived from a cross of Wyandot × PI 567301B had been developed and used to map QTL associated with resistances to soybean aphid (Aphis glycines) and powdery mildew (Microsphaera diffusa) (Jun et al. 2012a, b). A high density genetic map of this mapping population was also constructed recently (Lee et al. 2015). Therefore, the objective of this study was to identify QTL for resistance to F. graminearum using this same Wyandot × PI 567301B-derived RIL population.
Materials and methods
Plant materials
A subset of 184 F 7:10 RILs derived from the cross of Wyandot × PI 567301B was used to map QTL associated with resistance towards F. graminearum. A high-density linkage map was completed on the full population of 357 F 7:10 RILs of the same population (Lee et al. 2015). The cross was made in 2006 at Ohio Agricultural Research and Development Center (OARDC), Wooster, OH and advanced by single seed descent to develop RILs used in this study. Wyandot (OARDC–OSU) is a maturity group (MGII) soybean cultivar and PI 567301B is a MG IV accession originally collected from Gansu, China. DNA was extracted from young leaf tissue from each RIL as previously described (Lee et al. 2015).
Inoculum production
Inoculations were carried out with F. graminearum isolate Fay 11, originally collected from soybean and previously shown to be highly aggressive when compared to several other isolates (Ellis et al. 2011). Macroconidia of this isolate were stored at −80 °C in 10 % glycerol in cryovials (Corning, New York) until used. Macroconidia were transferred to Petri plates with mungbean agar and grown for 10 days under fluorescent light with a 12 h light/dark cycle. Plates were then flooded with 5–10 ml sterile distilled water, macroconidia were dislodged with a glass rod and the resulting suspension was filtered through 4–6 layers of cheese cloth. The final macroconidia suspension was measured using a hemacytometer (Bright-Line Hemacytometer, Hausser Scientific, Horsham, PA) and sterile distilled water was added for a final concentration of 2.5 × 104 macroconidia ml−1. Inoculation of seed with this concentration of macroconidia had the greatest disease severity in rolled towel assays (Ellis et al. 2011) and the same concentration was used to map QTL for resistance to F. graminearum in a different population (Ellis et al. 2012).
Phenotypic evaluation
A rolled towel assay was used as described by Ellis et al. (2011) with slight modifications. Fifteen seeds from each RIL and checks were placed on a germination paper moistened with deionized water and inoculated with a 100 µl suspension of 2.5 × 104 macroconidia ml−1. A second moistened towel was placed over the inoculated seeds, rolled up and placed on the top of a wire mesh in a 25 l bucket. The buckets were covered and stored in the dark at room temperature (~23–25 °C). Ten days after inoculation (dai), lesion length and total plant length were measured for each seedling of each genotype. A disease severity index (DSI) was calculated by dividing lesion length by total length of the seedlings and multiplying by 100. Disease severity was also assessed by a score using a 1–5 scale where, 1 = germination and healthy seedlings with no visible signs of colonization; 2 = germination and colonization of the root with 1–19 % of the root with lesions; 3 = germination and colonization of the root with 20–74 % of the root with lesions; 4 = germination and complete colonization of the root with 75 % or more of the seedling rot with lesions and 5 = no germination and complete colonization of the seed.
The 184 F 7:10 RILs were divided into two groups of 92 RILs each and evaluated separately. Each group of 92 RILs was evaluated in an incomplete block which consisted of 40–48 RILs and, the two parents of the mapping population, ‘Conrad’, ‘Williams’ and ‘Sloan’ as checks in each individual assay. It took 2–3 assays to complete one full rep, and this was repeated two more times so that each RIL was evaluated three times.
The mean DSI and disease score of each RIL was calculated from 15 seedlings in each rolled towel in each replication of each experiment. Since the mean DSI and disease score were very similar, only DSI data was used for the following statistical analyses. DSI data from each replication were analysed to obtain the best linear unbiased predictor (BLUP) (Stroup 1989) using proc mixed procedure of SAS version 9.3 (SAS Institute Inc., Cary, NC, USA). The model applied in the analysis was Y ijklm = μ + G i + R(G)ij + S(RG)ijk + C l + L(C)lm + ε ijklm where, μ is overall mean, G i is the effect of ith group, R(G) ij is the effect of jth replication in ith group, S(RG)ijk is the effect of kth set in jth replication in ith group, C l is the effect of lth class of entry (l = 1, 2, 3, 4, 5 and 6 for Conrad, Sloan, Williams, Wyandot, PI 567301B and RILs, respectively), L(C)lm is the effect of mth genotype within class for recombinant inbred lines only (genotypic variance, \(\sigma_{\text{g}}^{2}\)) and ε ijklm is the experimental error (σ 2). Class of entry was treated as a fixed effect; all other terms were treated as random effects. The heritability was calculated as: \(\sigma_{\text{g}}^{2} /(\sigma_{\text{g}}^{2} + \sigma^{2} /r)\), where r is the number of replications per RIL.
Genotypic assay
Single nucleotide polymorphism (SNP) genotyping was performed using the Illumina Infinium BARCSoySNP6k BeadChip and genotypes were scored as described in Lee et al. (2015), where a high-density genetic map with 2545 SNPs based on 357 RILs of Wyandot × PI 567301B population was reported. For the present study, the genetic map was generated using JoinMap4 (Van Ooigen 2006) based on only the subset of 184 RILs used in the phenotypic assay. Two SSR markers (Sat_157 and Sat_162) on chromosome 8 were also added to increase marker density in the genomic region of the F. graminearum resistance QTL identified in this study. Polymerase chain reactions (PCR) for the SSR markers were performed as previously described (Ellis et al. 2012) using genomic DNA (50 ng) as a template in a volume of 12.5 µl and amplicons were visualized under ultraviolet light by electrophoresis through 4 % high resolution agarose gels (Amresco, Solon, OH) and prestained with GelRed (Biotium Inc., Hayward, CA in 1× RapidRun buffer (USB, Cleveland, OH). For QTL identification, composite interval mapping (CIM) was conducted using MapQTL® 5 (Van Ooigen 2004). For CIM, the significance threshold was determined by a 1000-permutation test at α = 0.05 (Churchill and Doerge 1994).
Results
Phenotypic evaluation
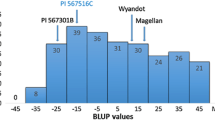
The mean DSI was significantly different among the two parents and the checks indicating that PI 567301B had a high level of resistance and Wyandot had a moderate level of resistance towards F. graminearum (Table 1). The mean DSI and root rot scores of parents and checks were significantly different from one another (P < 0.0001; Fisher’s protected least significant difference test) (Table 1). The mean DSI of individual RILs ranged from 0.8 to 63.7 %, and the overall mean DSI of all the RILs was 32.6 %. Both Wyandot and PI 567301B had BLUP values less than Conrad, a moderately resistant cultivar, indicating that these parents had a higher level of resistance to F. graminearum than Conrad and that this was a moderately resistant by resistant cross (Fig. 1).

Frequency distribution of BLUP values for disease severity index of F 7:10 recombinant inbred lines (RILs) population derived from the cross of ‘Wyandot’ × PI 567301B. Estimates of two parents and checks are indicated by arrows. A lower BLUP value means higher level of resistance to F. graminearum
The BLUP values calculated for DSI were normally distributed in this population (Fig. 1). Lower BLUP values indicate higher level of resistance to F. graminearum. Using the mixed model analysis, the BLUP values were estimated −17.3 and 9.6 for PI 567301B and Wyandot, respectively. The BLUP values of the checks Conrad, Williams, and Sloan were estimated to be 17.0, 40.4, and 59.9, respectively. Six RILs had BLUP values lower than PI 567301B and 38 RILs had BLUP values higher than Wyandot. The broad-sense heritability for the mean DSI was 0.79.
Genetic linkage map
Genotyping with the BARCSoy6K BeadChip array and the two SSR markers included on Gm08 resulted in integrating a total of 2547 markers in 20 linkage groups, corresponding to the 20 chromosomes of soybean. Total length of this genetic linkage map was 2,342 cM and marker order was comparable with the genetic map of 357 RILs described in Lee et al. (2015). The average marker interval ranged from 0.9 to 1.6 cM by chromosome with the overall average marker distance of 1.2 cM. Based on physical positions of the SNPs, each linkage map covered 94.4–99.8 % of each chromosome.
QTL identification
Two QTL conferring resistance to F. graminearum were identified in the Wyandot × PI 567301B population by CIM, one major QTL on chromosome 8 and one minor QTL on chromosome 6 (Table 2). Both QTL were highly significant with respective LOD scores of 23.9 and 6.3 compared to the genome-wide LOD threshold of 3.4 calculated by a 1000-permutation test. The major QTL on chromosome 8 was flanked by markers Sat_157 and ss715602786 and accounted for 38.5 % of the phenotypic variance (Table 2; Fig. 2a). A minor QTL, which explained 8.1 % of the phenotypic variance, was closely linked to marker ss715593768 (Glyma.Wm82.a2, Gm06 at 17672411 bp) on chromosome 6 (Table 2; Fig. 2b). The resistance alleles for both QTL were contributed by PI 567301B (Table 2).


Graphical presentation of quantitative trait loci (QTL) for resistance to Fusarium graminearum identified on chromosome a 8 and b 6 in the ‘Wyandot’ × PI 567301B population by genome-wide LOD threshold 3.4 (a hatched line in LOD plots). The 1- and 2-LOD intervals are presented by a black bar and solid lines between the chromosome and the LOD plot for each QTL. The enlarged image was presented side by side to highlight approximate 20 cM region around LOD peak of each QTL
Based on the current annotation of the ‘Williams 82’ sequence, Glyma.Wm82.a2.v1, in Soybase (www.soybase.org, accessed June 2014), the flanking markers on chromosome 8 encompassed a region of 305 kb in which 39 genes were annotated, many of which have a high potential as candidate genes for resistance to F. graminearum. Among these genes are those with functions related to a serine hydroxymethyl transferase (SHMT), a leucine-rich repeat, rapid alkalinzation factor (RALF); heat shock transcription factor, hydroxymethylglutaryl-CoA tranferase involved in chalcone and stilbene synthesis, two subtilisin-like proteases; a gene coding for lectins, and amino acid synthesis with aspartokinase-homoserine dehydrogenase as well as those with unknown function.
The minor QTL identified on chromosome 6 was flanked by markers ss715593740 and 715593784 (829-kb interval) and has 47 genes (www.soybase.org, accessed on August, 2014). Among these genes are at least four transcription factors, one leucine-rich repeat, several genes involved with initiation of translation and DNA repair as well as four genes with unknown functions.
Discussion
We characterized the resistance towards F. graminearum in PI 567301B and identified a major QTL using a population with over 2,500 molecular markers. The major QTL detected on chromosome 8 is a novel QTL associated with resistance to F. graminearum and, due to the marker density, was narrowed to a 305-kb region on chromosome 8 using a RIL population derived from a cross of Wyandot × PI 567301B. The QTL which contributed a major effect (38.5 % of phenotypic variance) for resistance, potentially overlaps the Rhg4 locus (www.soybase.org, accessed on July, 2014) located on chromosome 8 in the Soybean Consensus map 4.0 (Ariagada et al. 2012; Ferreira et al. 2011; Guo et al. 2006; Mahalingam and Skorupska 1995; Meksem et al. 2001; Vuong et al. 2011; Webb et al. 1995; Wu et al. 2009). Significant association of two molecular markers with resistance to three different isolates of P. sojae was also identified in this region on chromosome 8 (Wang et al. 2012). Interestingly, one of the minor resistance QTL for F. graminearum from the cultivar Conrad mapped near 102 cM (BARC-051847-11270) on chromosome 8 (Ellis et al. 2012), which is approximately, 56 cM (28 Mb) distant from the QTL identified in the present study.
Large effect QTL are atypical for race non-specific pathogens, although F. graminearum is now believed to behave as a hemibiotroph during infection of wheat heads (Brown et al. 2010; Trail 2009). As such, several major QTL have been identified for resistance to F. graminearum in wheat. The QTL Fhb1 for resistance to spread of infection within wheat spikes explains up to 41.6 % of the variation and has been verified in numerous populations (Bai et al. 1999; Anderson et al. 2001; Buerstmayr et al. 2003; Pumphrey et al. 2007). Interestingly, wheat genotypes carrying Fhb1 are able to detoxify DON (Lemmens et al. 2005). The DON detoxification reaction is carried out by uridine diphosphate (UDP)-glycosyltransferase. Subsequently, a gene putatively encoding UDP-glycosyltransferase, which was upregulated following inoculation in Fhb1 lines, was proposed as a candidate for Fhb1 (Schweiger et al. 2013). A third major QTL in wheat, Qfhs.ifa-5A, acts primarily through a reduction in the initial rate of infection and explained up to 24 % of the variation (Buerstmayr et al. 2003). A lipid transferase protein which is highly upregulated in a QTL-specific manner following inoculation represents candidate gene for this QTL (Schweiger et al. 2013). One QTL qRfg1 for resistance towards F. graminearum infection of corn stalks contributed to reducing disease severity and accounted for over 30 % of variation (Yang et al. 2010). This QTL was fine mapped and has been proposed to encode a transcription regulator (Ye et al. 2013). A transcriptome analysis of maize near isogenic lines with or without qRfg1 indicated that secondary metabolic pathways for biosynthesis of phenylpropanoids, biosynthesis of plant hormones, and biosynthesis of terpenoids and steroids may play important roles in resistance towards F. graminearum in corn roots and stalks (Ye et al. 2013).
Based on the Williams 82 annotated sequence (Glyma.Wm82.a2.v1), the interval to which the large effect QTL on chromosome 8 is located in the present study includes a nine-member family of genes putatively encoding hydroxymethylglutaryl-CoA, which is a key enzyme in flavonoid biosynthesis pathway and may indicate that secondary metabolite biosynthesis is also important in the defense response to F. graminearum in soybean. In addition, there are three annotated genes putatively encoding rapid alkanization factor (RALF) which can initiate a signal transduction pathway (Pearce et al. 2010). RALF peptides are present throughout the plant kingdom and have been shown to cause a loss of root growth inhibition in tomato seedlings (Pearce et al. 2010). Two genes encoding subtilisin-like proteases are also located within the region, which have a predicted function in recognition of cell wall components during the plant development and in the plant defense responses (Nelsen et al. 2004). Another candidate for a defense response is a gene encoding a brassinosteroid-regulated protein, which regulates the gene expression and promotes elongation in soybean (Zurek and Clouse 1994). These will all make excellent candidate genes for future studies that focus on expression and functional analyses to explore the mechanism of this resistance response to F. graminearum.
This QTL also contains a gene putatively encoding a serine hydromethyl transferase (SHMT), which was reported to function in photorespiratory pathway influencing resistance to biotic and abiotic stresses in Arabidopsis (Moreno et al. 2005). A study by Liu et al. (2012) confirmed that SHMT is the Rhg4 gene using mutation analysis, gene silencing, and transgenic complementation techniques and also cloned the gene from soybean cultivar ‘Forrest’. Interestingly, Forrest was found to be susceptible to the same isolate of F. graminearum used in this study with a disease severity index of 70.7 % and mean disease score of 3.9 (Acharya 2014). PI 567301B is reportedly moderately susceptible and susceptible to SCN races 3 and 4, respectively (Germplasm Resources Information Network, www.ars-grin.gov, accessed July 2014). This indicates that while resistance to F. graminearum is potentially linked to Rhg4, the SHMT allele contributing to SCN resistance is unlikely to be involved in the resistance response to F. graminearum.
The minor QTL on chromosome 6 is also first identified for resistance to F. graminearum in the present study but has been reported for resistance to multiple biotic stresses in several previous studies. This QTL mapped close to the resistance QTL for sudden death syndrome (SDS) which is caused by Fusarium virguliforme (Kazi et al. 2008), Sclerotinia sclerotiorum (Huynh et al. 2010), P. sojae (Li et al. 2010), Phomopsis seed decay (Sun et al. 2013), soybean aphid (Jun et al. 2013) and corn earworm (Terry et al. 2000). Among the 47 genes within the genomic region defined by the markers flanking this QTL are four transcriptional factors, which are also good candidate genes for functional analysis for resistance to F. graminearum.
Fusarium proliferatum was also recently reported to be highly pathogenic to soybean (Díaz Arias et al. 2011). Therefore, future investigation is needed into the potential resistance from PI 567301B to other pathogenic species of Fusarium such as F. proliferatum, as it has been observed in wheat (Toth et al. 2008) and corn (Mesterházy 1982). Resistance towards F. virguliforme in PI 567301B was evaluated previously in a greenhouse assay and this accession was reported to be susceptible (Hartman et al. 1997). The occurrence of F. graminearum isolates containing 3 ADON (more aggressive compared to 15 ADON) and NIV chemotypes were reported recently in North America (Foroud et al. 2012; Gale et al. 2011; Starkey et al. 2007). Hence, future investigation into interactions between the chemotype of F. graminearum and resistance responses in these and other soybean genotypes is warranted. Albeit, a limited survey of F. graminearum chemotypes collected primarily from Iowa found the 15-ADON genotype (Ellis and Munkvold 2014).
To date, F. graminearum has only been reported as a root, seed and seedling pathogen in North America, however, the observation of pod blight in Brazil (Martinelli et al. 2004) and pod and seed infection in Argentina (Barros et al. 2012; Garcia et al. 2012) signals the possibility that infections in these later growth stages may occur in the areas where wheat, corn and soybean are grown in rotation and in fields with high inoculum levels and favorable environmental conditions. However, more studies are needed to determine if the same QTL will confer resistance to infections that occur at later soybean growth stages.
PI 567301B contains resistance alleles conferring resistance to at least three different pathogens and pests of soybean including F. graminearum, soybean aphid (Jun et al. 2012a), and soybean powdery mildew (Jun et al. 2012b) but is susceptible to P. sojae (Lohnes et al. 1996), F. virguliforme (Hartman et al. 1997), Phakopsora pachyrhizi (Miles et al. 2006), Phialophora gregata (Bachman et al. 1997), soybean cyst nematode (Noel, GRIN database), and moderately susceptible to Macrophomina phaseolina (Mengistu et al. 2013). Marker-assisted breeding to prevent or limit the introgression of susceptible alleles into elite germplasm will greatly enhance the rapid development of high-yielding adapted cultivars which are resistant to targeted pests and pathogens.
Author contribution statement
Mr. B. Acharaya conducted phenotypic assays and co-wrote the manuscript, Dr. S. Lee constructed the genetic map and conducted the QTL analysis and co-wrote the manuscript. Dr. MAR Mian, conceived and oversaw development of the population, mapping design, and contributed to the analysis and map construction; Dr. T. Jun, development of population and earlier map construction, and Dr. L. K. McHale contributed to the writing and provided key information regarding the importance of these genomic locations. Dr. A. P. Michel, provided funding for the development of the populations, contributed to the writing. Dr. A. E. Dorrance (and United Soybean Board and Ohio Soybean Council) provided funding for the phenotyping study and analysis, conceived of the screening for resistance, oversaw phenotyping experiments, extensive guidance to BA and SL for completion of the study, and co-wrote the manuscript.
References
Acharya B (2014) Evaluation of soybean germplasm for additional sources of resistance and characterization of resistance towards Fusarium graminearum. M.S. Thesis. The Ohio State University
Ali ML, Taylor JH, Jie L, Sun G, Williams M, Kasha KJ, Reid LM, Pauls KP (2005) Molecular mapping of QTLs for resistance to Gibberella ear rot, in corn, caused by Fusarium graminearum. Genome 48:521–533
Anderson JA, Stack RW, Liu S, Waldron BL, Field AD, Coyne C, Moreno-Sevilla B, Mitchell Fetch J, Song QJ, Cregan PB, Frohberg RC (2001) DNA markers for Fusarium head blight resistance QTLs in two wheat populations. Theor Appl Genet 102:1164–1168
Ariagada O, Mora F, Dellarosa J, Ferreira M, Cervigni G, Schuster I (2012) Bayesian mapping of quantitative trait loci (QTL) controlling soybean cyst nematode resistant. Euphytica 186:907–917
Bachman MS, Nickell CD, Stephens PA, Nickell AD (1997) Brown stem rot resistance in soybean germplasm from central China. Plant Dis 81:953–956
Bai G, Kolb FL, Shaner G, Domier LL (1999) Amplified fragment length polymorphism markers linked to a major quantitative trait locus controlling scab resistance in wheat. Phytopathology 89:343–348
Barros G, Alaniz Zanon MS, Abod A, Oviedo MS, Ramirez ML, Reynoso MM, Torres A, Chulze S (2012) Natural deoxynivalenol occurrence and genotype and chemotype determination of a field population of the Fusarium graminearum complex associated with soybean in Argentina. Food Addit Contam 29:293–303
Broders KD, Lipps PE, Paul PA, Dorrance AE (2007) Evaluation of Fusarium graminearum associated with corn and soybean seed and seedling in Ohio. Plant Dis 91:1155–1160
Brown NA, Urban M, van de Meene AM, Hammond-Kosack KE (2010) The infection biology of Fusarium graminearum: defining the pathways of spikelet to spikelet colonization in wheat ears. Fungal Biol 114:555–571
Buerstmayr H, Lemmens M, Hartl L, Doldi L, Steiner B, Stierschneider M, Ruckenbauer P (2003) Molecular mapping of QTLs for Fusarium head blight resistance in spring wheat. II. Resistance to fungal penetration and spread. Theor Appl Genet 107:503–508
Buerstmayr H, Ban T, Anderson JA (2009) QTL mapping and marker-assisted selection for Fusarium head blight resistance in wheat: a review. Plant Breed 128:1–26
Chungu C, Mather DE, Reid LM, Hamilton RI (1996) Inheritance of kernel resistance to Fusarium graminearum in maize. J Hered 87:382–385
Churchill DA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Díaz Arias MM, Munkvold GP, Ellis ML, Leandro LFS (2011) First report of Fusarium proliferatum causing root rot on soybean (Glycine max) in the United States. http://dx.doi.org.proxy.lib.ohio-state.edu/10.1094/PDIS-04-11-0346
Díaz Arias MM, Leandro LF, Munkvold GP (2013a) Aggressiveness of Fusarium species and impact of root infection on growth and yield of soybeans. Phytopathology 103:822–832
Díaz Arias MM, Munkvold GP, Ellis ML, Leandro LFS (2013b) Distribution and frequency of Fusarium species associated with soybean roots in Iowa. Plant Dis 97:1557–1562
Ellis ML, Munkvold GP (2014) Trichothecene genotype of Fusarium graminearum isolates from soybean (Glycine max) seedling and root diseases in the United States. Plant Dis 98:1012
Ellis ML, Broders KD, Paul PA, Dorrance AE (2011) Infection of soybean seed by Fusarium graminearum and effect of seed treatments on disease under controlled conditions. Plant Dis 95:401–407
Ellis ML, Wang H, Paul PA, St Martin SK, McHale LK, Dorrance AE (2012) Identification of soybean genotypes resistant to Fusarium graminearum and genetic mapping of resistance quantitative trait loci in the cultivar Conrad. Crop Sci 52:2224–2233
Ferreira M, Cervigni G, Ferreira A, Schuster I, Santana F, Pereira W, Barros E, Moreira M (2011) QTL for resistance to soybean cyst nematode races 3, 9, and 14 in cultivar Hartwig. Pesqui Agropecu Bras 46:420–428
Foroud NA, McCormick SP, MacMillan T, Badea A, Kendra DF, Ellis BE, Eudes F (2012) Greenhouse studies reveal increased aggressiveness of emergent Canadian Fusarium graminearum chemotypes in wheat. Plant Dis 96:1271–1279
Gale LR, Ward TJ, Balmas V, Kistler HC (2007) Population subdivision of Fusarium graminearum sensu stricto in the upper Midwestern United States. Phytopathology 97:1434–1439
Gale LR, Harrison SA, Ward TJ, O’Donnell K, Milus EA, Gale SW, Kistler HC (2011) Nivalenol-type populations of Fusarium graminearum and F. asiaticum are prevalent on wheat in southern Louisiana. Phytopathology 101:124–134
Garcia D, Barros G, Chulze S, Ramos AJ, Sanchis V, Marín S (2012) Impact of cycling temperatures on Fusarium verticillioides and Fusarium graminearum growth and mycotoxins production in soybean. J Sci Food Agric 92:2952–2959
Guo B, Sleper DA, Arelli PR, Shannon JG, Nguyen HT (2006) Identification of QTLs associated with resistance to soybean cyst nematode races 2, 3 and 5 in soybean PI 90763. Theor Appl Genet 103:1167–1173
Harris SD (2005) Morphogenesis in germinating Fusarium graminearum macroconidia. Mycologia 97:880–887
Hartman GL, Huang YH, Nelson RL, Noel GR (1997) Germplasm evaluation of Glycine max for resistance to Fusarium solani, the causal organism of sudden death syndrome. Plant Dis 81:515–518
Huynh T, Bastien M, Iquira E, Turcotte P, Belzile F (2010) Identification of QTLs associated with partial resistance to white mold in soybean using field-based inoculation. Crop Sci 50:969–979
Jun T-H, Mian MA, Michel AP (2012a) Genetic mapping revealed two loci for soybean aphid resistance in PI 567301B. Theor Appl Genet 124:13–22
Jun T-H, Mian MA, Kang ST, Michel AP (2012b) Genetic mapping of the powdery mildew resistance gene in soybean PI 567301B. Theor Appl Genet 125:1159–1168
Jun T-H, Mian MAR, Michel AP (2013) Genetic mapping of three quantitative trait loci for soybean aphid resistance in PI 567324. Heredity 111:16–22
Kazi S, Shultz J, Afzal J, Johnson J, Njiti V, Lightfoot D (2008) Separate loci underlie resistance to root infection and leaf scorch during soybean sudden death syndrome. Theor Appl Genet 116:967–977
Lee S, Freewalt KR, McHale LK, Song Q, Jun T-H, Michel AP, Dorrance AE, Mian MAR (2015) A high resolution genetic linkage map of soybean based on 357 recombinant inbred lines genotyped with BARCSoySNP6k. Mol Breed. doi:10.1007/s11032-015-0209-5
Lemmens M, Scholz U, Berthiller F, Dall’Asta C, Koutnik A, Schuhmacher R, Adam G, Buerstmayr H, Mesterházy A, Krska R, Ruckenbauer P (2005) The ability to detoxify the mycotoxin deoxynivalenol colocalizes with a major quantitative trait locus for Fusarium head blight resistance in wheat. Mol Plant Microbe Interact 18:1318–1324
Leslie JF, Summerell BA (2006) The Fusarium laboratory manual. Blackwell Publishing, Ames
Li X, Han Y, Teng W, Zhang S, Yu K, Poysa V, Anderson T, Ding J, Li W (2010) Pyramided QTL underlying tolerance to Phytophthora root rot in mega-environments from soybean cultivars Conrad and Hefeng 25. Theor Appl Genet 121:651–658
Liu S, Kandoth PK, Warren SD, Yeckel G, Heinz R, Alden J, Yang C, Jamai A, El-Mellouki T, Juvale PS, Hill J, Baum TJ, Cianzio S, Whitham SA, Korkin D, Mitchum MG, Meksem K (2012) A soybean cyst nematode resistance genes points to a new mechanism of plant resistance to pathogens. Nature 492:256–262
Lohnes DG, Nickell CD, Schmitthenner AF (1996) Origin of soybean alleles for Phytophthora resistance in China. Crop Sci 36:1689–1692
Mahalingam R, Skorupska HT (1995) DNA markers for resistance to Heterodera glycines I. Race 3 in soybean cultivar Peking. Breed Sci 45:435–443
Martin M, Miedaner T, Dhillon BS, Ufermann U, Kessel B, Ouzunova M, Schipprack W, Melchinger AE (2011) Colocalization of QTL for Gibberella ear rot resistance and low mycotoxin contamination in early European maize. Crop Sci 51:1935–1945
Martinelli JA, Bocchese CAC, Xie W, O’Donnell K, Kistler HC (2004) Soybean pod blight and root rot caused by lineages of the Fusarium graminearum and the production of mycotoxins. Fitopatol Bras 29:492–498
Meksem K, Pantazopoulos P, Njiti VN, Hyten LD, Arelli PR, Lightfoot DA (2001) Forrest resistance to the soybean cyst nematode is bigenic: saturation mapping of the Rhg1 and Rhg4 loci. Theor Appl Genet 103:710–717
Mengistu A, Bond J, Nelson R, Rupe J, Shannon G, Arelli P, Wrather A (2013) Identification of soybean accessions resistant to Macrophomina phaseolina by field screening and laboratory validation. Plant Health Prog. doi:10.1094/PHP-2013-0318-01-RS
Mesterházy Á (1982) Resistance of corn to Fusarium ear rot and its relation to seedling resistance. Phytopathology 103:218–231
Miles MR, Frederick RD, Hartman GL (2006) Evaluation of soybean germplasm for resistance to Phakopsora pachyrhizi. Plant Health Prog. doi:10.1094/PHP-2006-0104-01-RS
Moreno JI, Martin R, Castresana C (2005) Arabidopsis SHMT1, a serine hydroxylmethyltransferase that functions in the photorespiratory pathway influences resistance to biotic and abiotic stress. Plant J 41:451–463
Mule G, González-Jaén MT, Hornok L, Nicholson P, Waalwijk C (2005) Advances in molecular diagnosis of toxigenic Fusarium species: a review. Food Addit Contam 22:316–323
Nelsen NS, Li Z, Warner AL, Matthews BF, Knap HT (2004) Genomic polymorphism indentifies a subtilisin-like portease near the Rhg4 locus in soybean. Crop Sci 44:265–273
O’Donnell K, Ward TJ, Geiser DM, Kistler HC, Aoki T (2004) Genealogical concordance between the mating type locus and seven other nuclear genes supports formal recognition of nine phylogenetically distinct species within the Fusarium graminearum clade. Fungal Genet Biol 41:600–623
O’Donnell K, Ward TJ, Aberra D, Kistler HC, Aoki T, Orwig N, Kimura M, Bjornstad A, Klemsdal SS (2008) Multilocus genotyping and molecular phylogenetics resolve a novel head blight pathogen within the Fusarium graminearum species complex from Ethiopia. Fungal Genet Biol 45:1514–1522
Otto CD, Kianian SF, Elias EM, Stack RW, Joppa LR (2002) Genetic dissection of a major Fusarium head blight QTL in tetraploid wheat. Plant Molec Biol 48:625–632
Pè ME, Gianfranceschi L, Taramino G, Tarchini R, Angelini P, Dani M, Binelli G (1993) Mapping quantitative trait loci (QTLs) for resistance to Gibberella zeae infection in maize. Mol Gen Genet 241:11–16
Pearce G, Yamaguchi Y, Munske G, Ryan CA (2010) Structure-activity studies of RALF, rapid alkalinization factor, reveal an essential—YISY—motif. Peptides 31:1973–1977
Pioli RN, Mozzoni L, Morandi EN (2004) First report of pathogenic association between Fusarium graminearum and soybean. Plant Dis 88:220
Pumphrey MO, Bernardo R, Anderson JA (2007) Validating the Fhb1 QTL for Fusarium head blight resistance in near isogenic wheat lines developed from breeding populations. Crop Sci 47:200–206
Reid LM, Hamilton RI (1996) Effects of inoculation position, timing, macroconidial concentration, and irrigation on resistance of maize to Fusarium graminearum infection through kernels. Can J Plant Pathol 18:279–285
Reid LM, Mather DE, Hamilton RI, Bolton AT (1992a) Diallel analysis of resistance in maize to Fusarium graminearum infection via the silk. Can J Plant Sci 72:915–923
Reid LM, Mather DE, Hamilton RI, Bolton AT (1992b) Genotypic differences in the resistance of maize silk to Fusarium graminearum. Can J Plant Pathol 14:211–214
Schmitthenner AF (1985) Problems and progress control of Phytophthora root rot of soybean. Plant Dis 69:362–368
Schweiger W, Steiner B, Ametz C, Siegwart G, Wiesenberger G, Berthiller F, Lemmens M, Jia H, Adam G, Muehlbauer GJ, Kreil DP, Buerstmayr H (2013) Transcriptomic characterization of two major Fusarium resistance quantitative loci (QTLs), Fhb1 and Qfhs.ifa-5A, identifies novel candidate genes. Mol Plant Pathol 14:772–785
Somers DJ, Fedak G, Savard M (2003) Molecular mapping of novel genes controlling Fusarium head blight resistance and deoxynivalenol accumulation in spring wheat. Genome 46:555–564
Starkey DE, Ward TJ, Aoki T, Gale LH, Kistler HC, Geiser DM, Suga H, Tóth B, Varga J, O’Donnell K (2007) Global molecular surveillance reveals novel Fusarium head blight species and trichothecene toxin diversity. Fungal Genet Biol 44:1191–1204
Stroup WW (1989) Why mixed models? In: Application of mixed models in agriculture and related disciplines. Southern Coop Serv Bull, Vol 343. Louisiana Agricultural Experiment Station, Baton Rouge, pp 1–8
Sun S, Kim MY, Van K, Lee YW, Li B, Lee SH (2013) QTLs for resistance to Phomopsis seed decay are associated with days to maturity in soybean (Glycine max). Theor Appl Genet 126:2029–2038
Tamburic-Ilincic L, Somers D, Fedak G, Schaafsma A (2009) Different quantitative trait loci for Fusarium resistance in wheat seedlings and adult stage in the Wuhan/Nyubai wheat population. Euphytica 165:453–458
Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, Hibbett DS, Fisher MC (2000) Phylogenetic species recognition and species concept in fungi. Fungal Genet Biol 31:21–32
Terry LI, Chase K, Jarvik T, Orf JH, Mansur LM, Lark KG (2000) Soybean quantitative trait loci for resistance to insects. Crop Sci 40:375–382
Toth B, Kaszonyi G, Bartok T, Varga J, Mesterhazy A (2008) Common resistance of wheat to members of the Fusarium graminearum species complex and F. culmorum. Plant Breed 127:1–8
Trail F (2009) For blighted waves of grain: Fusarium graminearum in the postgenomics era. Plant Physiol 149:103–110
Van Ooigen JW (2004) MapQTL 5, Software for the mapping of quantitative trait loci in experimental populations. Kyazma B. V, Wageningen
Van Ooigen JW (2006) JoinMap 4, Software for the calculation of genetic linkage maps in experimental populations. Kyazma B. V, Wageningen
Vuong T, Sleper D, Shannon J, Wu X, Nguyen H (2011) Confirmation of quantitative trait loci for resistance to multiple-HG types of soybean cyst nematode (Heterodera glycines Ichinohe). Euphytica 181:101–113
Wang H, St. Martin S, Dorrance A (2012) Comparison of phenotypic methods and yield contributions of quantitative trait loci for partial resistance to Phyophthora sojae in soybean. Crop Sci 52:609–622
Wang JH, Ndoye M, Zhang JB, Li HP, Liao YC (2011) Population structure and genetic diversity of the Fusarium graminearum species complex. Toxins 3:1020–1037
Webb DM, Baltazar BM, Rao-Arelli AP, Schupp J, Clayton K, Keim P, Beavis WD (1995) Genetic mapping of soybean cyst nematode race-3 resistance loci in the soybean PI 437654. Theor Appl Genet 91:574–581
Windels CE (2000) Economic and social impacts of Fusarium head blight: changing farms and rural communities in the Northern Great Plains. Phytopathology 90:17–21
Wu X, Blake S, Sleper D, Shannon J, Cregan P, Nguyen H (2009) QTL, additive and epistatic effects for SCN resistance in PI 437654. Theor Appl Genet 118:1093–1105
Xue AG, Cober E, Voldeng HD, Babcook C, Clear RM (2007) Evaluation of the pathogenicity of Fusarium graminearum and Fusarium pseudograminearum on soybean seedlings under controlled conditions. Can J Plant Pathol 29:35–40
Yang DE, Zhang CL, Zhang DS, Jin DM, Weng ML, Chen SJ, Nguyen H, Wang B (2004) Genetic analysis and molecular mapping of maize (Zea mays L.) stalk rot resistance gene Rfg1. Theor Appl Genet 108:706–711
Yang Q, Yin Guangming Y, Guo Y, Zhang D, Chen S, Xu M (2010) A major QTL for resistance to Gibberella stalk rot in maize. Theor Appl Genet 121:673–687
Ye J, Guo Y, Zhang D, Zhang N, Wang C, Xu M (2013) Cytological and molecular characterization of quantitative trait locus qRfg1, which confers resistance to Gibberella stalk rot in maize. Mol Plant-Microbe Interact 26:1417–1428
Zhang JZ, Xue AG, Zhang HJ, Nagasawa AE, Tambong JT (2010) Response of soybean cultivars to root rot caused by Fusarium species. Can J Plant Sci 90:767–776
Zurek DM, Clouse SD (1994) Molecular cloning and characterization of a brassinosteroid-related gene from elongating soybean (Glycine max L.) epicotyls. Plant Physiol 104:161–170
Acknowledgments
This study was funded in part by United Soybean Board and Ohio Soybean Council through soybean producer’s check-off dollars. We would like to thank Tim Mendiola for his efforts in developing this population; Andreas Cruz, Anna Dixon, Jaqueline Huzar Novakowiski, Austin Pelyak and Thomas Fitz Gibbon for technical assistance. For SNP genotyping, we acknowledge Vanessa Rashbrook at DNA Technologies, University of California, Davis. We also thank Drs. Perry Cregan and Qijian Song at the Soybean Genomics and Improvement Laboratory, USDA–ARS, Beltsville, MD, for developing the BARCSoySNP6k BeadChip array and for the updated physical positions of the SNPs. In addition, we wish to acknowledge support from the Molecular Cellular Imaging Center at Ohio Agricultural Research and Development Center and the Ohio Bioproducts Innovation Center (OBIC). Salaries and research support for this project was provided by State and Federal Funds appropriated to the Ohio Agricultural Research and Development Center, The Ohio State University, and USDA–ARS. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by The Ohio State University, or the United States Department of Agriculture.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H. T. Nguyen.
B. Acharya and S. Lee have contributed equally to first author.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Acharya, B., Lee, S., Rouf Mian, M.A. et al. Identification and mapping of quantitative trait loci (QTL) conferring resistance to Fusarium graminearum from soybean PI 567301B. Theor Appl Genet 128, 827–838 (2015). https://doi.org/10.1007/s00122-015-2473-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-015-2473-5