
Abstract
Quantitative trait loci (QTLs) for resistance to rice blast offer a potential source of durable disease resistance in rice. However, few QTLs have been validated in progeny testing, on account of their small phenotypic effects. To understand the genetic basis for QTL-mediated resistance to blast, we dissected a resistance QTL, qBR4-2, using advanced backcross progeny derived from a chromosome segment substitution line in which a 30- to 34-Mb region of chromosome 4 from the resistant cultivar Owarihatamochi was substituted into the genetic background of the highly susceptible Aichiasahi. The analysis resolved qBR4-2 into three loci, designated qBR4-2a, qBR4-2b, and qBR4-2c. The sequences of qBR4-2a and qBR4-2b, which lie 181 kb apart from each other and measure, 113 and 32 kb, respectively, appear to encode proteins with a putative nucleotide-binding site (NBS) and leucine-rich repeats (LRRs). Sequence analysis of the donor allele of qBR4-2a, the region with the largest effect among the three, revealed sequence variations in the NBS-LRR region. The effect of qBR4-2c was smallest among the three, but its combination with the donor alleles of qBR4-2a and qBR4-2b significantly enhanced blast resistance. qBR4-2 comprises three tightly linked QTLs that control blast resistance in a complex manner, and thus gene pyramiding or haplotype selection is the recommended strategy for improving QTL-mediated resistance to blast disease through the use of this chromosomal region.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rice blast, a destructive disease of rice, is caused by the fungal pathogen Magnaporthe oryzae (Ou 1985). To date, more than 80 genes for blast resistance have been recorded, of which 60 are genetically mapped (Gramene database: http://www.gramene.org/). Most are race specific and are characterized by a hypersensitive reaction (Greenberg and Yao 2004). However, genes for race-specific resistance are rapidly been overcome by the pathogen (Bonman et al. 1992; Kiyosawa 1982) and so cannot support sustainable crop production.
In contrast to race-specific resistance, resistance controlled by quantitative trait loci (QTLs) is characterized by a susceptible infection type, usually without race specificity or gene-for-gene interaction (Ezuka 1972; Parlevliet 1979). In general, cultivars carrying resistance QTLs have maintained their resistance for a long time, possibly because of decreased selection pressure against the pathogen. Therefore, the discovery of QTLs in resistant cultivars is crucial to our understanding of the genetic control of QTL-mediated blast resistance (Huang et al. 2011; Jia and Liu 2011; Liu et al. 2011; Shi et al. 2010).
Japanese upland rice cultivars are potential donors of QTL-mediated resistance (Abe et al. 1976). Their resistance is controlled by multiple genes (Fukuoka and Okuno 2001; Higashi and Kushibuchi 1978; Kato et al. 2002; Miyamoto et al. 2001). For example, the resistance QTLs identified in cultivar Owarihatamochi have been detected in three regions, of which pi21, on chromosome 4, explains 45.7% of the phenotypic variation (Fukuoka and Okuno 2001). Although extensive efforts have been made to introduce blast resistance from upland cultivars into elite irrigated cultivars, substantial QTL-mediated resistance has not been introduced, on account of poor eating quality and low yield potential caused by linkage drag (Higashi 1995; Morimoto 1980; Saka 2006). Recently, pi21 has been cloned by map-based cloning, allowing us to solve the long-term problem of linkage drag (Fukuoka et al. 2009). This example is a clear demonstration of the value of validation and fine mapping of QTLs for blast resistance.
The other two QTLs in Owarihatamochi, on chromosomes 4 (qBR4-2) and 12 (qBR12-1), explain 29.4 and 13.7%, respectively, of the phenotypic variation (Fukuoka and Okuno 2001). As pi21 confers moderate resistance, the resistance of elite cultivars carrying pi21 can be enhanced by combination with other resistance QTLs. To increase the set of genes for QTL-mediated resistance, extensive genetic studies have investigated QTLs with different magnitudes of effects from several cultivars (Fukuoka and Okuno 2001; Fukuoka et al. 2009; Huang et al. 2011; Jia and Liu 2011; Kato et al. 2002; Liu et al. 2011; Miyamoto et al. 2001; Nguyen et al. 2006; Shi et al. 2010; Suh et al. 2009; Terashima et al. 2008; Wang et al. 1994; Xu et al. 2008; Zenbayashi et al. 2002; Zenbayashi-Sawata et al. 2007). Although the elimination of undesirable characters closely linked to loci of interest is a key factor in the successful transfer of genes to commercial cultivars (Fukuoka et al. 2009), most QTLs have not been delimited in advanced progeny lines. Our aim here was to analyze qBR4-2, with the second largest effect after pi21, in order to map it precisely by fine genetic analysis using lines derived from a chromosome segment substitution line (CSSL) carrying this QTL.
Materials and methods
Plant materials
Their highly homogeneous genetic background makes CSSLs suitable for identifying QTLs under complicated genetic control in several crop species, including rice (Ali et al. 2010; Ebitani et al. 2005; Eshed and Zamir 1995; Hirabayashi et al. 2010; Howell et al. 1996; Kubo et al. 2002; Xu et al. 2010; Yoshimura et al. 2010). Using three rounds of backcrossing and marker-assisted selection, we developed a CSSL in which the 30- to 34-Mb region of chromosome 4 between the DNA marker loci RM317 and C1016 from the resistant upland rice cultivar Owarihatamochi was substituted into the genetic background of the susceptible lowland cultivar Aichiasahi (Fig. 1). Owarihatamochi has a high level of resistance controlled by multiple QTLs, whereas Aichiasahi which carries the race-specific genes Pia and Pi19(t) (Koide et al. 2011) is highly susceptible to blast under field condition. The rest of the genome was homogeneous for Aichiasahi, as confirmed by the DNA markers used in our previous study (Fukuoka and Okuno 2001).

Development of the plant materials used in the present study
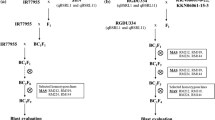
The CSSL was further crossed with Aichiasahi to select recombinants within the substituted region in order to develop inbred lines having recombination within and around qBR4-2. First, 36 plants with independent recombination events were selected from 780 plants in the BC4F2 population. The position and size of the Owarihatamochi region in the recombinants were analyzed using an additional 7 restriction fragment length polymorphism (RFLP) and 14 simple sequence repeat (SSR) markers in the target region. The RFLP markers were obtained from the Nipponbare/Kasalath map (Kurata et al. 1994) and the SSR markers from previous studies (Akagi et al. 1996; IRGSP 2005; McCouch et al. 2002). DNA marker analysis followed our previous procedures (Fukuoka and Okuno 2001). Homozygous plants were selected from the progeny of recombinants, and self-pollinated progeny lines, called sub-CSSLs, were used for phenotyping. Sub-CSSLs were classified into eight genotypes B to I (Fig. 2). To dissect qBR4-2, we crossed a sub-CSSL with Aichiasahi to select recombinants within the 2.5-Mb region between the DNA marker loci RM5503 and RM348. The DNA markers used are listed in Supplemental Table S1. These sub-CSSLs were classified into 12 genotypes J to U on the basis of DNA marker profiles (Fig. 3). We used a total of 30 sub-CSSLs selected from 3,103 individuals for mapping. As we detected three QTLs in the qBR4-2 region, we crossed sub-CSSLs carrying one or two of the QTLs and selected lines having each combination of pairs or all three by means of marker-assisted selection in order to validate the effects of the combined QTLs.

Genetic linkage map and graphical genotypes of sub-CSSLs around qBR4-2. Black bars indicate chromosome regions derived from the resistant Owarihatamochi; white bars indicate those derived from the susceptible Aichiasahi. The locations of qBR4-2a/qBR4-2b and qBR4-2c, indicated at the bottom, are based on the phenotypic data tabulated on the right
Assessment of blast resistance
In order to determine the map position of qBR4-2, we evaluated the blast resistance of homozygous progeny of the sub-CSSLs. We evaluated the resistance in an experimental field at the Aichi Agricultural Research Center (AARC), Mountainous Region Institute (Toyota, Aichi), or at the National Institute of Agrobiological Sciences (NIAS; Tsukuba, Ibaraki), where the disease pressure from blast fungus is particularly high and its progress is well monitored. The predominant fungal races are 007.0 in the former and 037.3 in the latter, and produced susceptible lesions on the plants.
Plants from 50 seeds per line were grown in 3 replications. The lesion area of 60- to 70-day-old plants was scored against a published reference scale (http://www.gene.affrc.go.jp/pdf/manual/micro-18.pdf, page 13). The susceptible recurrent parent Aichiasahi was grown on either side of each line.
Data analysis
We constructed a genetic linkage map around the qBR4-2 region using 192 F2 plants of the cross between the CSSL and Aichiasahi using Mapmaker software (Lander et al. 1987). The PROC GLM program of the Statistical Analysis Systems package (SAS Institute Inc.) was used to test differences in the phenotypic values among genotypes.
Construction of bacterial artificial chromosome library, sequencing, and gene prediction
Megabase-size rice DNA was prepared from young leaves of Owarihatamochi as described (Zhang et al. 1995). A bacterial artificial chromosome (BAC) library was constructed by ligation of the megabase DNA with the pIndigoBAC vector (Epicenter) and transformation of BACs into E. coli DH10B cells (Invitrogen) (Osoegawa et al. 1998). The library consisted of 20,380 clones with an average insert size of 100 kb. The clones containing the qBR4-2a locus were screened using DNA markers ID03-34 and ID03-35, and two positive clones (Owa28H01 and Owa28A18) were shotgun sequenced (Fleischmann et al. 1995). Putative coding sequences (CDSs) were predicted by the Rice Genome Automated Annotation System (RiceGAAS, http://ricegaas.dna.affrc.go.jp/; Sakata et al. 2002).
Results
Blast resistance in CSSL and sub-CSSLs
In the primary genetic analysis, we evaluated the CSSL and 36 sub-CSSLs representing nine genotypes (A to I) for blast resistance for 3 years in the field at the AARC (Fig. 2). The mean lesion area ranged from 13 to 62 % in 2004, from 11 to 50 % in 2005, and from 17 to 67 % in 2006. The lesion area of the donor Owarihatamochi was less than 1 % in all the tests. The mean lesion area of the CSSL (genotype A) was significantly smaller than that of the susceptible Aichiasahi (abbreviated as AA). Those of genotypes B and I were comparable to that of the CSSL. In contrast, genotype F had the largest mean lesion area among the sub-CSSLs, with the same level as AA. The rest (genotypes C, D, E, G, and H) were intermediate. This ranking was consistent across the 3 years. Both the distal (genotypes C, D and E) and proximal regions (G and H) of the donor segment on their own gave a significant decrease in mean lesion area compared with the AA. Therefore, we hypothesized that two regions, spanning marker intervals RM3534–RM349 and RM317–RM3217, are associated with blast resistance. Furthermore, two genotypes carrying different lengths of the distal region (D and E) had small but significant differences in two of the 3 years. Therefore, we tentatively assigned two putative QTLs, qBR4-2a and qBR4-2b, in the marker interval RM3534–RM349. A QTL in the proximal region (RM317–RM3217), designated qBR4-2c, had a small effect that was significant only in 2004 (genotypes G and H).
Delimitation of qBR4-2a and qBR4-2b
In order to validate and delimit qBR4-2a and qBR4-2b, we screened recombinants in a mapping population in which both loci segregated, and which lacked the resistance allele at qBR4-2c. We evaluated 30 sub-CSSLs representing 12 genotypes selected from 3,103 individuals in the field for 2 years. The mean lesion area of the sub-CSSLs ranged from 19 to 64 % in 2008 at the AARC and from 19 to 51 % in 2009 at the NIAS. The mean lesion area of the original sub-CSSL (genotype J) was significantly smaller than that of AA in both years, and two other genotypes (K and Q) were comparable to it (Fig. 3). In contrast, five genotypes (N, O, P, T, and U) had the largest mean lesion area among the sub-CSSLs. The rest (genotypes L, M, R, and S) were intermediate. The ranking was consistent across the 2 years. We concluded that this chromosomal region includes two loci: qBR4-2a (between markers ID20-t12 and ID20-t07) and qBR4-2b (between ID20-70 and ID01-37). qBR4-2a had a slightly larger effect than qBR4-2b.

Physical map and graphical genotypes of sub-CSSLs around qBR4-2a and qBR4-2b. Black bars indicate regions derived from the resistant Owarihatamochi; white bars indicate those derived from the susceptible Aichiasahi. The locations of qBR4-2a and qBR4-2b, indicated at the bottom, are based on the phenotypic data tabulated on the right
Validation of qBR4-2c by QTL pyramiding
In order to validate the effect of qBR4-2c, we crossed a sub-CSSL carrying this QTL (genotype H) with one carrying qBR4-2a and qBR4-2b (D) and one carrying qBR4-2a only (E), and selected progeny homozygous for the resistance alleles at two or three loci. 2 years’ evaluation suggested that the addition of qBR4-2c significantly reduced the lesion area in the genetic background of genotypes D and E, and further confirmed the partial resistance conferred by genotype H (Table 1).
Candidate genes for qBR4-2a and qBR4-2b
We delimited qBR4-2a and qBR4-2b to within regions of 113 and 32 kb, respectively, in the Nipponbare sequence (IRGSP 2005), where the Rice TOGO Browser (http://agri-trait.dna.affrc.go.jp/index.html; Nagamura et al. 2011) identified 24 and 2 putative CDSs (Supp. Table S2). The two putative CDSs in the qBR4-2b region show similarity to previously reported disease resistance proteins containing a nucleotide-binding site (NBS) and leucine-rich repeats (LRRs) (McHale et al. 2006), either or both of which are candidates for qBR4-2b. To sequence the qBR4-2a locus, we constructed a BAC library of Owarihatamochi and fully sequenced two clones, Owa28H01 and Owa28A18, containing qBR4-2a. Sequencing and RiceGAAS analysis revealed that the 113-kb sequence between markers ID20-t12 and ID20-t07 contains 27 putative CDSs (Supp. Table S3). Sequence comparison of the qBR4-2a region between susceptible Nipponbare and resistant Owarihatamochi revealed overall similarity, but several regions showed lower similarity owing to chromosomal reorganization (insertions or deletions and duplication) in the Owarihatamochi genome in a region containing putative CDSs with similarity to three genes encoding NBS-LRR disease resistance proteins (Fig. 4): CDSs O16, O22, and O23. O22 and O23 lack an NBS domain, leaving only O16 as the most probable candidate for qBR4-2a. A phylogenetic tree of deduced amino acid sequences of the NBS domain of O16 and of previously cloned disease resistance genes in rice and other crops obtained from public databases grouped O16 with a bacterial blight resistance gene of rice, Xa1 (Yoshimura et al. 1998), and rice blast resistance genes Pib, Pi37, and Pish (Lin et al. 2007; Takahashi et al. 2010; Wang et al. 1999) (Fig. 5).

Sequence comparison of qBR4-2a between susceptible Nipponbare and resistant Owarihatamochi. Putative coding sequences (CDSs) are indicated by boxes; black boxes represent those that encode proteins with similarity to proteins containing a nucleotide-binding site (NBS) and leucine-rich repeats (LRRs). Shading indicates regions with very high sequence identity (>98% DNA identity overall) between genotypes. Position zero on the scale corresponds to 32,280,568 bp on the International Rice Genome Sequencing Project (IRGSP) build 5 pseudomolecules of the rice genome (Supp. Table S2)

Phylogenetic analysis of the putative qBR4-2a with 30 other plant R genes. Deduced amino acid sequences of the putative nucleotide-binding site (NBS) site of qBR4-2a (O16) and of R genes obtained from GenBank were aligned, and a neighbor-joining phylogenetic tree was generated using CLUSTALW (http://clustalw.ddbj.nig.ac.jp/top-e.html) and Treeview software (http://taxonomy.zoology.gla.ac.uk/rod/treeview.html). Numbers on branches indicate the percentage of 1,000 bootstrap replicates which support the adjacent node. The unit branch length is 0.5 nucleotide substitutions per site (bar)
Discussion
Recent progress in genomics has enhanced understanding of the genetic basis of agronomic traits, including those controlled by multiple loci in rice (Yamamoto et al. 2009; Yano and Sasaki 1997; Yonemaru et al. 2010). Yet, despite the great potential of marker-assisted selection in breeding programs, the use of beneficial QTLs from exotic germplasms is still a challenge.
One reason concerns the lower reliability of detection and the lower resolution of mapping of QTLs with minor effects in analysis using primary mapping populations, such as backcrossed inbred lines and recombinant inbred lines (Fukuoka et al. 2010). Advanced backcross progeny can be used to cope with this problem (Fukuoka et al. 2010), as we showed here by validating the effect of qBR4-2 through the use of a CSSL. Importantly, our results suggest that qBR4-2 is a gene complex comprising three loci, qBR4-2a, qBR4-2b, and qBR4-2c, which cumulatively enhance disease resistance. The effect of qBR4-2c was stably detected in the presence of the other two loci, although it was almost undetectable in the genetic background of the susceptible cultivar Aichiasahi. Such evidence partly explains the complicated genetic control of QTL-mediated resistance to blast (Rao et al. 2005; Wu et al. 2005). Understanding the isolate/race specificity of each individual QTL for blast resistance is crucial for deciding which resistance genes are to be used in rice breeding programs. Our methods show how such specificity can be identified.
Another reason concerns linkage drag, which is frequently observed in cross-breeding using exotic germplasms (Brown 2002; Ruge-Wehling et al. 2006), and which explains the difficulties in introducing QTL alleles for blast resistance into elite cultivars (Higashi 1995; Morimoto 1980; Saka 2006). Precise map information for a gene or QTL associated with traits of agricultural value is indispensable to marker-assisted elimination of undesirable traits tightly linked with the gene or QTL (Fukuoka et al. 2009). We determined the precise map location of two of the three QTLs detected here. Interestingly, these QTLs lie in the same region as a cluster of QTLs for traits that are strongly associated with productivity, including morphology and photosynthesis (Courtois et al. 2003; Ikeda et al. 2007; Saito et al. 2004; Sardesai et al. 2002; Takai et al. 2010). This observation highlights the importance of this region as a target for selection in breeding. Such findings will allow us to combine QTLs for blast resistance with QTLs for other agronomic traits while using marker-assisted selection to remove linkage drag.
Sequence comparison of qBR4-2a between Nipponbare and Owarihatamochi implies a complicated evolutionary history of this region, as suggested in other resistance gene complexes (Dixon et al. 1996; Wang et al. 1998; Xiao et al. 2001). The involvement of the qBR4-2 region in blast resistance has been reported in several rice cultivars and wild relatives (Goto 1988; Hirabayashi et al. 2010; Miyamoto et al. 2001; Terashima et al. 2008; Wang et al. 1994; Xu et al. 2008). To determine whether the resistance is based on allelic differences at one or more of the qBR4-2 loci is an important issue to clarify for the use of natural variation in blast resistance. The accumulation of rice genome sequence and haplotype information and the use of DNA genotyping technology will be helpful in clarifying this point (Ebana et al. 2010; Huang et al. 2010; Nagasaki et al. 2010; Yamamoto et al. 2010).
Our results suggest that allelic variation in one or more NBS-LRR genes is responsible for differences in blast resistance in rice. NBS-LRR genes are an important component in the evolution of plant resistance, mostly in race specificity (Bai et al. 2002; Bennetzen and Hulbert 1992; Hayashi and Yoshida 2009; Liu et al. 2007; Michelmore and Meyers 1998; Qu et al. 2006; Richter and Ronald 2000; Wei et al. 2002). Recent evidence suggests that tightly linked blast resistance genes epistatically control race-specific resistance at the Pikm, Pi5, and Pia loci (Ashikawa et al. 2008; Lee et al. 2009; Okuyama et al. 2011), but difficulties in validating quantitative differences have limited the number of reports on the additive effect of resistance genes. Our results confirm the effect of three loci in multiple trials and suggest that multiple QTLs contribute to the differences in the magnitude of effect.
NBS-LRR genes were not found in the qBR4-2c region in the Nipponbare genome. Sequence comparison between Nipponbare and Owarihatamochi and further fine genetic analysis of qBR4-2c will aid our understanding of QTL-mediated resistance to blast and the natural variation in the defense responses of plants. The characterization of lines combining QTLs, including the qBR4-2 complex, will help us to better understand rice–blast interaction.
References
Abe S, Suga R, Ono S (1976) Inheritance of the blast-resistance of upland rice varieties. Part 3. Test for genotype of the true resistance. (in Japanese). Bull Ibaraki Agric Exp Stn 17:77–82
Akagi H, Yokozeki Y, Inagaki A, Fujimura T (1996) Microsatellite DNA markers for rice chromosomes. Theor Appl Genet 93:1071–1077
Ali ML, Sanchez PL, Yu S-B, Lorieux M, Eizenga GC (2010) Chromosome segment substitution lines: a powerful tool for the introgression of valuable genes from Oryza wild species into cultivated rice (O. sativa). Rice 3:218–234
Ashikawa I, Hayashi N, Yamane H, Kanamori H, Wu J, Matsumoto T, Ono K, Yano M (2008) Two adjacent nucleotide-binding site-leucine-rich repeat class genes are required to confer pikm-specific rice blast resistance. Genetics 180:2267–2276
Bai J, Pennill LA, Ning J, Lee SW, Ramalingam J, Webb CA, Zhao B, Sun Q, Nelson JC, Leach JE, Hulbert SH (2002) Diversity in nucleotide binding site-leucine-rich repeat genes in cereals. Genome Res 12:1871–1884
Bennetzen JL, Hulbert SH (1992) Organization, instability and evolution of plant disease resistance genes. Plant Mol Biol 20:575–578
Bonman JM, Khush GS, Nelson RJ (1992) Breeding rice for resistance to pests. Annu Rev Phytopathol 30:507–528
Brown JK (2002) Yield penalties of disease resistance in crops. Curr Opin Plant Biol 5:339–344
Courtois B, Shen L, Petalcorin W, Carandang S, Mauleon R, Li Z (2003) Locating QTLs controlling constitutive root traits in the rice population IAC 165 × Co39. Euphytica 134:335–345
Dixon MS, Jones DA, Keddie JS, Thomas CM, Harrison K, Jones JD (1996) The tomato Cf-2 disease resistance locus comprises two functional genes encoding leucine-rich repeat proteins. Cell 84:451–459
Ebana K, Yonemaru J, Fukuoka S, Iwata H, Kanamori H, Namiki N, Nagasaki H, Yano M (2010) Genetic structure revealed by a whole-genome single-nucleotide polymorphism survey of diverse accessions of cultivated Asian rice (Oryza sativa L.). Breed Sci 60:390–397
Ebitani T, Takeuchi Y, Nonoue Y, Yamamoto T, Takeuchi K, Yano M (2005) Construction and evaluation of chromosome segment substitution lines carrying overlapping chromosome segments of indica rice cultivar ‘Kasalath’ in a genetic background of japonica elite cultivar ‘Koshihikari’. Breed Sci 55:65–73
Eshed Y, Zamir D (1995) An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics 141:1147–1162
Ezuka A (1972) Field resistance of rice varieties to rice blast disease. Rev Plant Prot Res 5:1–21
Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM et al (1995) Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269:496–512
Fukuoka S, Okuno K (2001) QTL analysis and mapping of pi21, a recessive gene for field resistance to rice blast in Japanese upland rice. Theor Appl Genet 103:185–190
Fukuoka S, Saka N, Koga H, Ono K, Shimizu T, Ebana K, Hayashi N, Takahashi A, Hirochika H, Okuno K, Yano M (2009) Loss of function of a proline-containing protein confers durable disease resistance in rice. Science 325:998–1001
Fukuoka S, Nonoue Y, Yano M (2010) Germplasm enhancement by developing advanced plant materials from diverse rice accessions. Breed Sci 60:509–517
Goto I (1988) Genetic studies on resistance of rice plant to blast fungus (VII). Blast resistance genes of Kuroka. Ann Phytopath Soc 54:460–465
Greenberg JT, Yao N (2004) The role and regulation of programmed cell death in plant–pathogen interactions. Cell Microbiol 6:201–211
Hayashi K, Yoshida H (2009) Refunctionalization of the ancient rice blast disease resistance gene Pit by the recruitment of a retrotransposon as a promoter. Plant J 57:413–425
Higashi T (1995) Genetic studies on field resistance of rice to blast disease. (In Japanese). Bull Tohoku Natl Agric Exp Stn 35:438–448
Higashi T, Kushibuchi K (1978) Genetics analysis of field resistance to leaf blast (Pyricularia oryzae) in rice. Jpn J Breed 28:277–286
Hirabayashi H, Sato H, Nonoue Y, Kuno-Takemoto Y, Takeuchi Y, Kato H, Nemoto H, Ogawa T, Yano M, Imbe T, Ando I (2010) Development of introgression lines derived from Oryza rufipogon and O. glumaepatula in the genetic background of japonica cultivated rice (O. sativa L.) and evaluation of resistance to rice blast. Breed Sci 60:604–612
Howell PM, Lydiate DJ, Marshall DF (1996) Towards developing intervarietal substitution lines in Brassica napus using marker-assisted selection. Genome 39:348–358
Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, Li C, Zhu C, Lu T, Zhang Z, Li M, Fan D, Guo Y, Wang A, Wang L, Deng L, Li W, Lu Y, Weng Q, Liu K, Huang T, Zhou T, Jing Y, Lin Z, Buckler ES, Qian Q, Zhang QF, Li J, Han B (2010) Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet 42:961–967
Huang H, Huang L, Feng G, Wang S, Wang Y, Liu J, Jiang N, Yan W, Xu L, Sun P, Li Z, Pan S, Liu X, Xiao Y, Liu E, Dai L, Wang GL (2011) Molecular mapping of the new blast resistance genes Pi47 and Pi48 in the durably resistant local rice cultivar Xiangzi 3150. Phytopathology 101:620–626
Ikeda H, Kamoshita A, Manabe T (2007) Genetic analysis of rooting ability of transplanted rice (Oryza sativa L.) under different water conditions. J Exp Bot 58:309–318
IRGSP (2005) The map-based sequence of the rice genome. Nature 436:793–800
Jia Y, Liu G (2011) Mapping quantitative trait loci for resistance to rice blast. Phytopathology 101:176–181
Kato T, Endo I, Yano M, Sasaki T, Inoue M, Kudo S (2002) Mapping of quantitative trait loci for field resistance to rice blast in upland rice, ‘Sensho’ (in Japanese). Breed Res 3:119–124
Kiyosawa S (1982) Genetic and epidemiological modeling of breakdown of plant disease resistance. Annu Rev Phytopathol 20:93–117
Koide Y, Telebanco-Yanoria MJ, Dela Pena F, Fukuta Y, Kobayashi N (2011) Characterization of application for mapping a resistance gene, Pi19(t). J Phytopathol 159:85–93
Kubo T, Aida Y, Nakamura K, Tsunematsu H, Doi K, Yoshimura A (2002) Reciprocal chromosome segment substitution series derived from Japonica and Indica cross of rice (Oryza sativa L.). Breed Sci 52:319–325
Kurata N, Nagamura Y, Yamamoto K, Harushima Y, Sue N, Wu J, Antonio BA, Shomura A, Shimizu T, Lin SY et al (1994) A 300 kilobase interval genetic map of rice including 883 expressed sequences. Nat Genet 8:365–372
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newberg LA (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Lee SK, Song MY, Seo YS, Kim HK, Ko S, Cao PJ, Suh JP, Yi G, Roh JH, Lee S, An G, Hahn TR, Wang GL, Ronald P, Jeon JS (2009) Rice Pi5-mediated resistance to Magnaporthe oryzae requires the presence of two CC-NB-LRR genes. Genetics 181:1627–1638
Lin F, Chen S, Que Z, Wang L, Liu X, Pan Q (2007) The blast resistance gene Pi37 encodes a nucleotide binding site leucine-rich repeat protein and is a member of a resistance gene cluster on rice chromosome 1. Genetics 177:1871–1880
Liu J, Liu X, Dai L, Wang G (2007) Recent progress in elucidating the structure, function and evolution of disease resistance genes in plants. J Genet Genomics 34:765–776
Liu Y, Zhu XY, Zhang S, Bernardo M, Edwards J, Galbraith DW, Leach J, Zhang G, Liu B, Leung H (2011) Dissecting quantitative resistance against blast disease using heterogeneous inbred family lines in rice. Theor Appl Genet 122:341–353
McCouch SR, Teytelman L, Xu Y, Lobos KB, Clare K, Walton M, Fu B, Maghirang R, Li Z, Xing Y, Zhang Q, Kono I, Yano M, Fjellstrom R, DeClerck G, Schneider D, Cartinhour S, Ware D, Stein L (2002) Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res 9:199–207
McHale L, Tan X, Koehl P, Michelmore RW (2006) Plant NBS-LRR proteins: adaptable guards. Genome Biol 7:212
Michelmore RW, Meyers BC (1998) Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res 8:1113–1130
Miyamoto M, Yano M, Hirasawa H (2001) Mapping of quantitative trait loci conferring blast field resistance in the Japanese upland rice variety. Kahei Breed Sci 54:257–261
Morimoto T (1980) Breeding by Chinese upland rice Sensho. (in Japanese). In: Yamasaki Y, Kozaka T (eds) Rice blast disease and breeding for resistance to blast. Hakuyusha, Tokyo, pp 25–34
Nagamura Y, Antonio BA, Sato Y, Miyao A, Namiki N, Yonemaru J, Minami H, Kamatsuki K, Shimura K, Shimizu Y, Hirochika H (2011) Rice TOGO Browser: a platform to retrieve integrated information for rice functional and applied genomics. Plant Cell Physiol 52:230–237
Nagasaki H, Ebana K, Shibaya T, Yonemaru J, Yano M (2010) Core single-nucleotide polymorphisms—a tool for genetic analysis of the Japanese rice population. Breed Sci 60:648–655
Nguyen TT, Koizumi S, La TN, Zenbayashi KS, Ashizawa T, Yasuda N, Imazaki I, Miyasaka A (2006) Pi35(t), a new gene conferring partial resistance to leaf blast in the rice cultivar Hokkai 188. Theor Appl Genet 113:697–704
Okuyama Y, Kanzaki H, Abe A, Yoshida K, Tamiru M, Saitoh H, Fujibe T, Matsumura H, Shenton M, Galam DC, Undan J, Ito A, Sone T, Terauchi R (2011) A multifaceted genomics approach allows the isolation of the rice Pia-blast resistance gene consisting of two adjacent NBS-LRR protein genes. Plant J 66:467–479
Osoegawa K, Woon PY, Zhao B, Frengen E, Tateno M, Catanese JJ, de Jong PJ (1998) An improved approach for construction of bacterial artificial chromosome libraries. Genomics 52:1–8
Ou S-H (1985) Rice diseases, 2nd edn. Commonwealth Agricultural Bureaux, Slough
Parlevliet JE (1979) Components of resistance that reduce the rate of epidemic development. Annu Rev Phytopathol 17:203–222
Qu S, Liu G, Zhou B, Bellizzi M, Zeng L, Dai L, Han B, Wang GL (2006) The broad-spectrum blast resistance gene Pi9 encodes a nucleotide-binding site-leucine-rich repeat protein and is a member of a multigene family in rice. Genetics 172:1901–1914
Rao ZM, Wu JL, Zhuang JY, Chai RY, Fan YY, Leung H, Zheng KL (2005) Genetic dissections of partial resistances to leaf and neck blast in rice (Oryza sativa L.). Yi Chuan Xue Bao 32:555–565
Richter TE, Ronald PC (2000) The evolution of disease resistance genes. Plant Mol Biol 42:195–204
Ruge-Wehling B, Linz A, Habekuss A, Wehling P (2006) Mapping of Rym16Hb, the second soil-borne virus-resistance gene introgressed from Hordeum bulbosum. Theor Appl Genet 113:867–873
Saito K, Hayano-Saito Y, Maruyama-Funatsuki W, Sato Y, Kato A (2004) Physical mapping and putative candidate gene identification of a quantitative trait locus Ctb1 for cold tolerance at the booting stage of rice. Theor Appl Genet 109:515–522
Saka N (2006) A rice (Oryza sativa L.) breeding for field resistance to blast disease (Pyricularia oryzae) in Mountainous Region Agricultural Research Institute, Aichi Agricultural Research Center of Japan. Plant Prod Sci 9:3–9
Sakata K, Nagamura Y, Numa H, Antonio BA, Nagasaki H, Idonuma A, Watanabe W, Shimizu Y, Horiuchi I, Matsumoto T, Sasaki T, Higo K (2002) RiceGAAS: an automated annotation system and database for rice genome sequence. Nucleic Acids Res 30:98–102
Sardesai N, Kumar A, Rajyashri R, Nair S, Mohan M (2002) Identification and mapping of an AFLP marker linked to Gm7, a gall midge resistance gene and its conversion to a SCAR marker for its utility in marker aided selection in rice. Theor Appl Genet 105:691–698
Shi X, Wang J, Bao Y, Li P, Xie L, Huang J, Zhang H (2010) Identification of the quantitative trait loci in Japonica rice landrace Heikezijing responsible for broad-spectrum resistance to rice blast. Phytopathology 100:822–829
Suh JP, Roh JH, Cho YC, Han SS, Kim YG, Jena KK (2009) The Pi40 gene for durable resistance to rice blast and molecular analysis of Pi40-advanced backcross breeding lines. Phytopathology 99:243–250
Takahashi A, Hayashi N, Miyao A, Hirochika H (2010) Unique features of the rice blast resistance Pish locus revealed by large scale retrotransposon-tagging. BMC Plant Biol 10:175
Takai T, Kondo M, Yano M, Yamamoto T (2010) A quantitative trait locus for chlorophyll content and its association with leaf photosynthesis in rice. Rice 3:172–180
Terashima T, Fukuoka S, Saka N, Kudo S (2008) Mapping of a blast field resistance gene Pi39(t) of elite rice strain Chubu 111. Plant Breed 127:485–489
Wang GL, Mackill DJ, Bonman JM, McCouch SR, Champoux MC, Nelson RJ (1994) RFLP mapping of genes conferring complete and partial resistance to blast in a durably resistant rice cultivar. Genetics 136:1421–1434
Wang GL, Ruan DL, Song WY, Sideris S, Chen L, Pi LY, Zhang S, Zhang Z, Fauquet C, Gaut BS, Whalen MC, Ronald PC (1998) Xa21D encodes a receptor-like molecule with a leucine-rich repeat domain that determines race-specific recognition and is subject to adaptive evolution. Plant Cell 10:765–779
Wang ZX, Yano M, Yamanouchi U, Iwamoto M, Monna L, Hayasaka H, Katayose Y, Sasaki T (1999) The Pib gene for rice blast resistance belongs to the nucleotide binding and leucine-rich repeat class of plant disease resistance genes. Plant J 19:55–64
Wei F, Wing RA, Wise RP (2002) Genome dynamics and evolution of the Mla (powdery mildew) resistance locus in barley. Plant Cell 14:1903–1917
Wu JL, Fan YY, Li DB, Zheng KL, Leung H, Zhuang JY (2005) Genetic control of rice blast resistance in the durably resistant cultivar Gumei 2 against multiple isolates. Theor Appl Genet 111:50–56
Xiao S, Ellwood S, Calis O, Patrick E, Li T, Coleman M, Turner JG (2001) Broad-spectrum mildew resistance in Arabidopsis thaliana mediated by RPW8. Science 291:118–120
Xu X, Chen H, Fujimura T, Kawasaki S (2008) Fine mapping of a strong QTL of field resistance against rice blast, Pikahei-1(t), from upland rice Kahei, utilizing a novel resistance evaluation system in the greenhouse. Theor Appl Genet 117:997–1008
Xu J, Zhao Q, Du P, Xu C, Wang B, Feng Q, Liu Q, Tang S, Gu M, Han B, Liang G (2010) Developing high throughput genotyped chromosome segment substitution lines based on population whole-genome re-sequencing in rice (Oryza sativa L.). BMC Genomics 11:656
Yamamoto T, Yonemaru J, Yano M (2009) Towards the understanding of complex traits in rice: substantially or superficially? DNA Res 16:141–154
Yamamoto T, Nagasaki H, Yonemaru J, Ebana K, Nakajima M, Shibaya T, Yano M (2010) Fine definition of the pedigree haplotypes of closely related rice cultivars by means of genome-wide discovery of single-nucleotide polymorphisms. BMC Genomics 11:267
Yano M, Sasaki T (1997) Genetic and molecular dissection of quantitative traits in rice. Plant Mol Biol 35:145–153
Yonemaru J, Yamamoto T, Fukuoka S, Uga Y, Hori K, Yano M (2010) Q-TARO: QTL annotation rice online database. Rice 3:194–203
Yoshimura S, Yamanouchi U, Katayose Y, Toki S, Wang ZX, Kono I, Kurata N, Yano M, Iwata N, Sasaki T (1998) Expression of Xa1, a bacterial blight-resistance gene in rice, is induced by bacterial inoculation. Proc Natl Acad Sci USA 95:1663–1668
Yoshimura A, Nagayama H, Sobrizal, Kurakazu T, Sanchez PL, Doi K, Yamagata Y, Yasui H (2010) Introgression lines of rice (Oryza sativa L.) carrying a donor genome from the wild species, O. glumaepatula Steud. and O. meridionalis Ng. Breed Sci 60:597–603
Zenbayashi K, Ashizawa T, Tani T, Koizumi S (2002) Mapping of the QTL (quantitative trait locus) conferring partial resistance to leaf blast in rice cultivar Chubu 32. Theor Appl Genet 104:547–552
Zenbayashi-Sawata K, Fukuoka S, Katagiri S, Fujisawa M, Matsumoto T, Ashizawa T, Koizumi S (2007) Genetic and physical mapping of the partial resistance gene, Pi34, to blast in rice. Phytopathology 97:598–602
Zhang H-B, Zhao X, Ding X, Paterson AH, Wing RA (1995) Preparation of megabase-size DNA from plant nuclei. Plant J 7:175–184
Acknowledgments
We thank the field managers of NIAS and the AARC for growing the rice and evaluating blast severity. This work was supported by grants from the Ministry of Agriculture, Forestry and Fisheries of Japan (QT4004, QTL2002, GB1004, RGB1101 and 2001).
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by T. Sasaki.
S. Fukuoka and R. Mizobuchi contributed equally to this manuscript.
An erratum to this article is available at http://dx.doi.org/10.1007/s00122-014-2295-x.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fukuoka, S., Mizobuchi, R., Saka, N. et al. A multiple gene complex on rice chromosome 4 is involved in durable resistance to rice blast. Theor Appl Genet 125, 551–559 (2012). https://doi.org/10.1007/s00122-012-1852-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-012-1852-4