
Abstract
An association panel consisting of 185 accessions representative of the barley germplasm cultivated in the Mediterranean basin was used to localise quantitative trait loci (QTL) controlling grain yield and yield related traits. The germplasm set was genotyped with 1,536 SNP markers and tested for associations with phenotypic data gathered over 2 years for a total of 24 year × location combinations under a broad range of environmental conditions. Analysis of multi-environmental trial (MET) data by fitting a mixed model with kinship estimates detected from two to seven QTL for the major components of yield including 1000 kernel weight, grains per spike and spikes per m2, as well as heading date, harvest index and plant height. Several of the associations involved SNPs tightly linked to known major genes determining spike morphology in barley (vrs1 and int-c). Similarly, the largest QTL for heading date co-locates with SNPs linked with eam6, a major locus for heading date in barley for autumn sown conditions. Co-localization of several QTL related to yield components traits suggest that major developmental loci may be linked to most of the associations. This study highlights the potential of association genetics to identify genetic variants controlling complex traits.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Yield increase represents the Holy Grail of most breeding programmes. Breeding for complex traits, such as yield for drought conditions, a common feature of several Mediterranean environments, is extremely difficult because of the relative magnitude of Genotype × Environment (G.E) interactions compared to Genotype main effect. High G.E interactions mean that genotypes grown in multi-environmental trials tend to react substantially differently to varying environmental conditions. Additional layers of complexity arise from a spectrum of other abiotic stresses that may occur together with drought at different plant developmental stages and modify the drought stress impact on growth and metabolism, e.g. maximum and minimum temperatures, radiation, soil characteristics, and precipitation. Another complication is the unknown, and possibly large, number of quantitative trait loci (QTL), which control yield and its component traits, QTL × QTL interactions and the independent sensitivities of each QTL to the environmental conditions.
As the numbers of molecular markers for crop plants such as barley has increased and their cost has decreased the number of QTL studies have increased exponentially (Thomas 2003; Rae et al. 2007). Despite this increase, marker assisted selection (MAS) is not a major activity in current barley breeding programmes. This may reflect the limited scope of bi-parental mapping, the traditional QTL analysis that uses two parental genotypes, limiting allelic diversity to two alleles in a reduced genetic background compared to that being exploited in a breeding programme. Furthermore, increased mapping resolution for gene cloning can only be obtained in biparental populations by using large progeny numbers; for example, cloning vrs1 and Rpg1 genes in barley involved 9,831 and 8,518 gametes, respectively (Brueggeman et al. 2002; Komatsuda et al. 2007), and most QTL studies use small population sizes that result in poor map resolution (Kearsey and Farquhar 1998). Therefore, the markers developed from these studies span a large genetic interval of the genes controlling the trait, and are not entirely diagnostic. This constitutes an additional obstacle to the implementation of molecular markers in breeding programmes by marker assisted selection, particularly where there is close linkage to QTL for other traits.
Progress in high throughput molecular marker platforms providing good genome coverage (from hundreds to thousands) together with decreasing genotyping costs have awakened the interests of plant geneticists in using naturally occurring variation for identifying genomic regions involved in complex traits (Close et al. 2009). Association genetics offers a potentially powerful approach to identify genetic variants which control complex traits and promises to overcome several of the issues hindering the adoption of QTL-MAS in breeding programmes (Mackay and Powell 2007). In that sense, and because association mapping of QTL can be done in naturally occurring germplasm collections as well as representative groupings, association panels benefit from decades of recombination events accumulated in an heterogeneous genetic background. Hence, association studies promise high QTL mapping resolution over a wide range of genetic diversity, i.e. the results should be applicable to all the genetic backgrounds surveyed. Thus, if the association panel is representative of the current breeding gene pool, markers associated with the trait can be used in MAS. This would allow early selection for high yielding lines avoiding the potential elimination of lines with good yield potential in early generations.
Hordeum vulgare is an ideal candidate for association mapping. It is highly autogamous, has a long history of recombination events and conserved linkage disequilibrium (Caldwell et al. 2006). This means that fewer markers are required to survey the whole genome compared to outbreeding species such as maize (Remington et al. 2001). Nevertheless, although association genetics has become more popular for mapping purposes in crop plants, few papers have been published reporting whole genome association analyses. In one of the few examples, Kraakman et al. (2004), reported several marker-trait associations for yield and yield stability in a 146 modern 2 row spring barley collection of elite lines representing material that had been evaluated in official Danish variety trials from 1993 to 2000. Although, marker technologies have improved there are still very few publications examining complex traits at the whole genome level, with most emphasis on simple trait associations (Waugh et al.2009).
The main objective of this work was to investigate whether association genetic approaches could be used to examine complex traits such as yield and its components. The data set for this analysis comprised 185 barley lines grown in distinct Mediterranean environments and including landraces, old and modern cultivars that we genotyped with 1,536 SNP loci, and phenotyped in 24 Mediterranean environments for yield, days to heading, harvest index, height and thousand kernel weight. Patterns of linkage disequilibrium, genetic diversity and single trial associations for yield, have been described in previous studies using the same germplasm set genotyped with 1,130 polymorphic Diversity Array Technology (DArT®, Wenzl et al. 2004) markers (Comadran et al. 2008, 2009). Here, we have utilised a mixed model approach (Van Eeuwijk et al. 2010) as a solution to the problems posed by both the diverse nature of the germplasm and the presence of extensive G.E interactions in an over-sites analysis to identify the most robust and stable QTL across environments. Analysis of an extensive multi-environmental trial (MET) data should allow for large G.E interactions, find main effects which are stable in most of the environments, and increase statistical power by increasing available number of observations per sample. Used appropriately, mixed model association scans of MET data have the potential to overcome reproducibility problems and identify genetic variants that are stable across the environments of interest (Piepho 2000; Van Eeuwijk et al. 2010). Lastly, the possibilities for exploiting significant associations in terms of prioritizing QTL and exploiting SNP genotyping platforms and barley genome synteny conservation to fully sequenced organisms, such as rice or Brachypodium dystachion, to delimitate the genomic intervals containing causal variants behind the regulation of a positive marker-trait association are discussed.
Materials and methods
Plant material and phenotypic evaluation
Phenotypic data was collected on a diverse collection of 192 spring and winter barley lines that encompassed a range of cultivated forms representing the present and past of the cultivated barley germplasm around the Mediterranean basin (supplementary data). Plant material utilised in this study have been described in more in a previous manuscript (Comadran et al. 2009).
The germplasm collection was evaluated in 24 trials grown for harvest years 2004 and 2005 (Comadran et al. 2008). Each trial was grown in a cyclical design using a block size of 60 including 4 checks, each repeated three times in each block. Five blocks were sown for each trial thus providing 1.25 replicates sown together in a rectangular grid of 15 rows and 20 columns. During the growing season, each plot was assessed for the date at which the ears had a quarter emerged from the auricles (GS53)—days to heading—and, once growth had ceased, distance from the ground to the tip of the ear—height (cm). Prior to harvest, a grab sample was taken from each plot, dried, weighed and threshed and the grain weighed to derive harvest index. Each plot was harvested by a small plot combine and the plot weight together with the plot area used to estimate yield in t/ha. Finally, thousand grain weight (g) was estimated from a cleaned sample of seed from each plot. Best Linear Unbiased Predictors (BLUPs) for each one of the traits in each environment were estimated using REstricted Maximum Likelihood (REML) directive in GENSTAT 11 (Payne et al. 2008) using fixed effects for checks, and random effects for columns, rows and test entry. Repeatabilities for each single trait and trial were calculated as:
where the genetic variance was estimated from the test entries and the residual variance from the replicated entries and checks.
Genotypic analysis and population structure
DNA was isolated from a single plant of each accession included in the trial series and was sent to the Southern California Genotyping Consortium at the University of California, Los Angeles for genotyping with a 1536-plex Oligo Pool Assay (OPA) (barley OPA1-BOPA1) (Close et al. 2009). Scores were collected for 185 of the 192 genotypes, as 7 samples were omitted because they were either duplicates, DNA quality was poor, or they were mislabeled. From the genotypic data collected for the remaining 185 accessions, ambiguous calls were coded as ‘missing data’ in all subsequent analysis. A subset of 1,307 SNPs with minimum allele frequency (MAF) >10% and ‘missing data’ <10% was collected for all 185 barley accessions providing a data matrix of 1,307 × 185 allele calls which was used for further analyses. To explore population structure, principal coordinate analysis, PCO, based on simple matching coefficients of SNP genotypic data of all markers was performed with GENSTAT 11 (Payne et al. 2008).
Association genetics methodology
GENSTAT 11 software (Payne et al. 2008) was first used to estimate a relative kinship matrix (K) on the basis of simple matching coefficients from 1,307 of the 1,536 BOPA1 SNPs with MAF > 10%. For the combined analysis across environments, the adjusted means per environment were first computed as described above, and these were included in a mixed model analysis over all 24 trials. Genetic polymorphisms (SNPs) and environmental main effects were considered as fixed terms of the model. We used the EXPERIMENTS option of the REML directive in GENSTAT 11 and residual terms for each trial were allowed to be independent. Genotypes were included in the random term of the model, so that a correlation structure generated by their relatedness could be imposed on the genotypes’ variance–covariance structure (Yu et al. 2006; Malosetti et al. 2007). The genetic variance–covariance structure, G = 2 Kσ 2g , where σ 2g is the genetic variance and K is an N × N matrix containing relative kinship coefficients, was included in the random term of the model to reduce type I error in the association (Comadran et al. 2009). Variance components were estimated by using the REML directive in GENSTAT 11 with the significance of the association of each SNP marker being determined by a Wald test, the standard test for testing fixed effects in mixed models (Payne et al. 2008). Shrinkage of total genetic variance after adding each SNP into the model was used to estimate the percentage of total σ 2g accounted by the QTL. QTL effects and their standard errors were estimated from the table of effects from the REML analysis. In addition to the ‘K model’ described above, we also tested an alternative mixed model (‘K + Q model’), where the resulting STRUCTURE output matrix of probabilities of belonging to each of k groups when k = 6 (Q) (Comadran et al. 2009) was directly added as co-factor in the random term of the ‘K model’ as described in the literature (Yu et al. 2006). Both ‘K model’ and ‘K + Q model’ yielded similar Akaike ans Schwarz information coefficients suggesting that adding the extra Q co-variate does not improve the model. Based on these results, we chose to adopt the simpler of the two models (Supplementary text 1). Moreover, as a contrast to just QTL main effects, a mixed model was fitted to test G.E interactions for yield. Environmental main effects and SNP.E interaction terms were considered as fixed terms of the models. Random and residual terms were the same as for the main effects mixed model. The purpose of G.E analysis for yield was not to explain QTL.E effects in terms of candidate genes, but to estimate their frequency and magnitude for the discussion.
The choice of a threshold –log10 (p value) above which to consider marker-trait associations to be significant was determined by using the GRAMDOM directive in GENSTAT 11 to generate random trait data following normal distribution with mean 0 and variance 1 for each trial to test for significance thresholds under the null model. Ten independent simulations, applying the mixed models described above, were performed using the random trait data to identify the appropriate –log10 (p value).
Results
Phenotypic variation
Phenotypic data were gathered from 28 trials (Comadran et al. 2008), although, for each phenotype, we only considered trials with complete data for all the genotype entries. Therefore, we considered 24 trials for yield, 19 for heading date, 24 for height, 15 for grains per spike, 16 for spike number, 23 for thousand kernel weight and 23 for harvest index. Repeatability values and trait means are summarized in Table 1. In general, repeatability values were high, with heading date and thousand kernel weight having the highest values and exceeded 0.9 at some trial sites. In contrast, harvest index and spike number proved to be less heritable with average repeatability values of 0.4 and 0.42, respectively. The other component of yield, grains per spike, was highly repeatable with the result that yield itself had a high repeatability value (Table 1).
Genotypic diversity and population structure
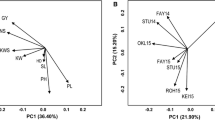
Principal coordinate analysis divided the germplasm into five population clusters corresponding to 59 South-West Mediterranean accessions, 18 Turkish accessions, 15 Syrian and Jordan landraces, 42 Northern European 6 row winter accessions, and 51 Northern European 2 row spring accessions (Fig. 1). These groupings were similar to those observed in a previous study using 1,130 DArT® molecular markers (Comadran et al. 2009). The number of polymorphic markers (MAF > 0.1) per group ranged from 106 and 458 for the Turkish and the Syrian and Jordan accessions to 788, 806 and 813 for the Northern European 6 row winter, Northern European 2 row spring and South-West Mediterranean accessions (Fig. 2). Low levels of polymorphism in the Turkish and the Syrian and Jordan landraces may be related to the low number of lines from both geographical regions utilized in the study and the high similarity of the Turkish lines we sampled due to the narrow genepool managed within the Turkish breeding program with Tokak featuring strong in their pedigrees (Comadran et al. 2009) (Fig. 3).

Hordeum vulgare diversity. Principal coordinates analysis. Lines are coloured according to population structure clusters described in Comadran et al. 2009

Venn diagram showing polymorphic marker overlap. A three-way Venn diagram for the major population clusters present in our sample: North-Europe 2 row springs, North-Europe 6 row winters and South-West Mediterranean accessions comprising 51, 42 and 59 accessions, respectively. Accessions from Turkey and Jordan were not included because their low line numbers (18 and 15) may not be representative of the genetic diversity present in their cultivation regions

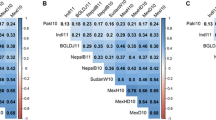
Whole genome scan profiles (examples). Significances of the calculated P values are shown as −log10 (P value). Genetic map position, and percentage of the variance accounted for are shown for the largest QTL
Mixed model association scans of MET data and QTL main effects
The maximum –log10 (p value) obtained in the simulations with random variation following normal distribution with mean 0 and variance 1 had a –log10 (p value) was 2.9, which we therefore utilised as a threshold for declaring significant QTL. Main genetic effect QTL for yield, heading date, harvest index, height, grains per spike, spikes per square meter and thousand kernel weight, were identified by analysing all the available trial data together and are summarized in Table 2.
Yield QTL main effects and QTL.E interaction
Three main effect QTL for grain yield were detected in our data set. One is located in the centromeric region of chromosome 2H, and the other two on the long arm of chromosome 7H (Table 2). The two on the long arm of 7H are less than 15 cM apart and only one SNP is significantly associated with yield in each case. SNPs 11_11348 and 11_11445 are A/G and A/T polymorphisms, respectively, with the A allele of the former and the T allele of the latter being associated with increased yield. Just 24 of the 185 lines have both increasing alleles and 8 of these are all of the selections from a Spanish landrace. Just three modern cultivars (Massine, Oussama and Siberia) carry both increasing alleles for yield compared to 9 and 12 for the old cultivars and landraces, respectively. As a contrast QTL.E analysis for yield identified 47 SNP markers that exceeded the threshold of 2.9, which defined 27 genomic regions at least 5 cM apart from the next positive SNP marker: 4, 2, 5, 4, 7, 2, 1 and 2 QTL for chromosomes 1H, 2H, 3H, 4H, 5H, 6H, 7H and unmapped SNPs, respectively. BOPA SNP markers closely linked to barley vernalization genes Sgh2 and sgh1 responsible for the winter/spring growth habit co-segregate with SNP.E QTL.
Heading date
As noted previously, the population clearly segregates for winter and spring growth habit (Comadran et al. 2008), controlled by the combined effects of Sgh2 and sgh1 on chromosomes 5H and 4H, respectively, which might be expected to have a major effect upon heading date. All our trials were autumn-sown and, effects of the two genes for growth habit on heading date are, however, not normally observed under these conditions (Cuesta-Marcos et al. 2008) and neither did we detect any associations with SNPs located in the vicinity of the two vernalisation genes. We did detect five main effect QTL for heading date with single QTL located on chromosomes 1H, 3H and 5H and two QTL on chromosome 2H, separated by over 60 cM (Table 2). By exploring synteny conservation with rice, we found that two QTL map in the region of genes known to affect flowering. SNP 11_10357 located on chromosome 1H at100.69 cM is approximately 30 gene models away from a rice homologue of the cloned barley gene HvFT3 (Faure et al. 2007), which has a major affect on heading date in short days such as those experienced in our trials. The biggest effect was located in the centromeric region of chromosome 2H, where a major gene affecting heading date, eam6, is located (Cuesta-Marcos et al. 2009). This gene has yet to be cloned but the effect we detected of 3.4 days is half of what has been reported for an eam6 mutant under field cultivation in Sweden (Gustafsson et al. 1960), and in two winter × spring crosses: Nure × Tremois (Tondelli, A. Personal communication), and Beka × Mogador (Cuesta-Marcos et al. 2008). SNP 11_20438, which is associated with eam6, is a C/G polymorphism with the C allele being associated with earlier heading. The G allele of SNP 11_20438 is found predominantly in NW European spring and late-heading winter types such as Maris Otter and thus our results are consistent with previous studies. The reduced effect that we detected could be due to one or more of the following factors: (a) the different genetic backgrounds that exist amongst our association panel, (b) some of the effect being absorbed by the kinship matrix, (c) the stress levels in the environments that we used reducing the range in heading date, (d) the tendency to over-estimate effects in bi-parental QTL mapping. SNP 11_10357 is an A/G polymorphism with the A allele being associated with later heading and carried mostly by spring cultivars, although it is also found in Nure and several other winter cultivars. The other three detected QTL are located on 2H at 128.26 cM, 3H at 58.01 cM, The QTL on the long arm of chromosome 5H is 74 and 80 gene models away from the rice genes homologous to the barley phytochrome C and Sgh2 genes, respectively (Szucs et al. 2006; Szucs et al. 2007), although the associated SNP is not the closest SNP to these genes, and it may well be that variation in co-segregating loci also affects flowering irrespective of habit. The associated SNP, 11_11090, is a C/G polymorphism with the C allele having the second strongest effect upon heading date (Table 2). It is, however, a relatively minor allele being found in just 23 of the lines, many of which are winter types, although it is also found in Tremois and not in Nure and may reflect some of the differences in heading date detected in the Nure × Tremois population at this region.
Harvest index
We detected three QTL for Harvest Index with the most significant being located in the same region on chromosome 2H as QTL for heading date and yield. The second QTL was located on 1HL and accounts for slightly more of the phenotypic variation than the one on 2H but has a smaller effect (Table 2). The SNP on 1H is an A/C polymorphism with the A allele decreasing harvest index. The QTL on 2H is associated with SNP 11_10191, the same SNP as that associated with yield but different from that associated with heading date, although it is co-located with SNP 11_20438 on the consensus map (Close et al. 2009). SNP 11_10191 is an A/C polymorphism with the A allele being associated with increased harvest index and also increased yield. The A allele is generally associated with the G allele of SNP 11_20438 so that the later heading lines tend to have the higher harvest index and yield, as one would expect under non-stressed conditions. There are, however, 13 recombinants and all of these belong in the Syrian/Jordanian grouping. A third QTL was associated with SNP 11_10918, located on chromosome 3H at 123.68 cM. SNP 11_10918 is a C/G polymorphism with the C allele being associated with increased harvest index and it is noticeable that the G allele is frequently found in the Syrian Jordanian landraces and some of the North West European cultivars included in the study.
Height
Despite being moderately repeatable (Table 1), just two significant associations were found for height on the long arm of chromosome 2H and on top of the short arm of chromosome 5H. The effects were relatively minor, accounting for just 2% of the overall height mean and thus would not represent the action of major dwarfing genes. Exploring syntenic relationships with rice, we found that the associated SNP 11_10376 on chromosome 2H co-locates with Flt-2L, a locus in barley controlling heading date and plant height (Chen et al. 2009). Interestingly, our analysis identified a QTL for heading date in the region as well (SNP 11_20366 located in chromosome 2H-128.26 cM). The interval delimited by both SNP markers encompasses 73 rice gene models. SNP 11_10376 is an A/G polymorphism with the A allele increasing height and found in many of the known taller lines. The SNP on chromosome 5H that is associated with height is 11_20894, which is a C/G polymorphism with the G allele increasing height and being found in many of the Syrian Jordanian landraces.
Grains per spike
QTL for grains per spike should reflect major genes determining 2- and 6-rowed barley phenotypes, with some minor genes affecting numbers within each major gene group. Historically, the 2/6 rows phenotype is under the control of two major genes (Komatsuda and Mano 2002), both of which have already been cloned: vrs1 on the long arm of chromosome 2H (Pourkheirandish et al. 2007) and int-c on the short arm of chromosome 4H (Waugh et al. 2009). The largest QTL that we detected explained over 40% of the phenotypic variance and the associated SNP is just 3 rice gene models away from the the candidate gene for int-c (L. Ramsay, Personal communication). The second most significant association was detected on chromosome 2H within 3.9 cM of the SNP closest to HvHox1, the candidate gene for vrs1 (Saisho et al. 2009). SNP 11_10287, an A/G polymorphism with the A allele increasing grains per spike, was the SNP located in the region of Vrs1 on chromosome 2H whereas SNP 11_20422, a C/G polymorphism with the C allele increasing grains per spike was located in the region of int-c on chromosome 4H. With the exception of Ager, the C allele of SNP 11_20422 and the G allele of SNP 11_2087 are found exclusively in 2 row types so that there is generally a dispersion of alleles increasing grains per spike amongst the two major gene phenotypes after correcting for kinship relationships.
We detected five additional QTL for grains per spike, one of which was located on chromosome 1H, two on chromosome 2H and the other two on chromosome 4H. SNP 11_20121 is an A/G polymorphism that was associated with the height effect on chromosome 1H with the A allele being associated with an increase in grains per spike. Notably, the G allele was preponderantly found in winter lines with a 6 row phenotype. SNP 11_10436, an A/G polymorphism, was located within 1 cM of the QTL for heading date on chromosome 2H that was associated with eam6. Here, the G allele, largely found in spring 2 row types, is associated with decreased grains per spike but is generally also coupled with the later flowering of the G allele of SNP 11_20438. SNP 11_20610, a C/G polymorphism located on chromosome 4H at 54.2 cM, is associated with grains per spike with the G allele being found mainly in 6 rows and decreasing the character. Similarly, the A allele of SNP 11_20732, which is associated with the grain per spike QTL at 92.4 cM on chromosome 4H, is also found mainly in 6 rows and decreases the character.
Spikes per square metre
Despite having a relatively low repeatability, we identified two QTL for spikes per square metre, one of which was highly significant and accounted for nearly 30% of the phenotypic variation in the character (Table 2). The highly significant association is with SNP 11_20422 on chromosome 4H, which is the same SNP as that we detected in association with grains per spike and in the region of int-c. This is therefore most likely to be a pleiotropic effect of int-c with a compensation effect of increased grains per spike in the 2 row types resulting in lower spikes per square metre. The other QTL is located distally on the short arm of chromosome 5H and accounts for over 12% of the phenotypic variation in the character. SNP 11_20553, which is associated with the character, is an A/G polymorphism with the G allele having a decreasing effect and largely being found amongst the spring 2 row lines.
Thousand kernel weight
Thousand kernel weight had the second highest repeatability value after heading date (Table 1). We detected three QTL but none of these accounted for more than 10% of the phenotypic variation, although the largest effect was over 5% of the trait mean. Two of the QTL are, however, located in the same region as QTL for grains per spike but associated with different SNPs. SNP 11_10213 is an A/C polymorphism on chromosome 2H, which is located in the region of vrs1 with the A allele associated with a decrease in thousand grain weight. Notably, the A allele of SNP 11_20287 is located in the same region and is associated with an increase in grains per spike in the same region but is generally associated with the C allele of SNP 11_10213, i.e. the net effect is to increase both grains per spike and individual grain weight after correcting for structural relationships. In contrast, the G allele of SNP 11_20680, which is located on chromosome 4H in the region of int-c, is largely found in the spring 2 row lines and is associated with smaller grains. It is generally also found with the C allele of SNP 11_20422, which as noted above, is also located in the region and is associated with increased grains per spike. Therefore, in this one, more normal compensatory mechanisms appear to be operating. The third QTL is associated with SNP 11_10796, which is located in the centromeric region of chromosome 2H approximately 5 cM from the QTL for heading date and grains per spike in the region of eam6. Here the A allele of an A/G polymorphism is associated with a decrease in the character and is largely found in the spring entries from around the Mediterranean region. Considering all three QTL in the region the associations are generally of earlier flowering types producing more grains per spike but of lower individual grain weights, indicating that compensatory mechanisms appear to be operating.
Discussion and conclusions
With the development of high throughput sequence based molecular markers, it is now possible to fast-track QTL to potential candidate genes using approaches based upon association mapping and synteny conservation with sequenced cereal genomes (Waugh et al. 2009). In this study, we have shown that the combination of MET data analysis, high throughput SNP genotyping and association mapping identified several important QTL for yield and yield components in barley. Associations for grains per spike identified SNPs tightly linked to known major genes determining spike morphology in barley (int-c), height (sdw1/denso), and flowering (HvFT3 and HvBM5A). In addition, the largest QTL for heading date co-locates with eam6, a major locus for heading date in barley for autumn sown conditions, which has been reported to have significant effects on yield (Cuesta-Marcos et al. 2009). Moreover, co-localization of several QTL related to yield components traits suggest that major developmental loci with pleiotropic effects may be linked to most of the associations which emphasises the value of this dataset for gene discovery. It is possible to exploit barley synteny conservation with rice and/or Brachypodium to identify a small set of candidate genes co-locating with QTL for a given trait. In the case of simple traits (regulated by few genes), re-sequencing the gene candidate in stocks of induced mutants is a simple way to confirm the candidate as the causal gene (Druka et al. 2010). Dealing with traits governed by several genes, validation of the gene candidates is more difficult due to the lack of obvious induced mutants and the usually unknown nature/function of the gene behind the association. However, mutant collections can still be divided into phenotypic classes related to distinct yield components such as heading date, plant height, spike morphology, or kernel shape. Genotypic data from NILs (near-isogenic lines) that carry induced/natural variants related to these phenotypic classes can be compared with the location of QTL to potentially identify candidate genes behind the control of quantitative variation (Druka et al. 2010).
Clearly, by using an association panel, we dramatically widen the window of genetic diversity over classical two parent approaches to QTL analysis, and have increased QTL mapping resolution. However, the use of populations with a heterogeneous genetic background has definite drawbacks; the usefulness of high genome coverage is decreased by the inability of bi-allelic SNP to efficiently capture genetic variation determined by more than 2 alleles. Moreover, presence of unaccounted alleles may introduce artificial recombination events thus decreasing the precision of the association mapping exercise. We found a clear example of this in our study: ear-type (2 or 6 row ear), a trait known to be largely controlled by two genes, vrs1 and int-c (Pourkheirandish et al. 2007; Waugh et al. 2009). In our data set, there are three clearly distinct phenotypes, 2 rows, 6 rows and deficiens types, with the last being classified as 2 rows but controlled by a third allele at vrs1. As each BOPA1 SNP marker represents one polymorphism in each of 1,536 independent genes, our analysis is most effective at detecting two class segregations and thus the presence of three alleles and phenotypes will result in a less significant association and poorer genetic mapping precision of the QTL. Thus, presence of more than two alleles in the vrs1 locus resulted in poor mapping precision of the QTL (vrs1 maps 3.9 cM away from the positive SNP) compared to that of int-c (3 gene models away in rice), although further removal of deficiens spike phenotypes from the analysis moved the QTL to only eight gene models away from HvHox1, the candidate gene for vrs1 (Saisho et al. 2009).
We found other major known genes that co-locate with QTL related to their putative function but their level of significance was too low to consider them positive. For example, a GA-20 oxidase that has been proposed as the gene candidate behind the green revolution semi-dwarfing gene sdw1, synonym denso (Jia et al. 2009), co-segregates with a QTL for harvest index (SNP 11_10918 on chromosome 3H 123.68 cM). The positive SNP for harvest index is bracketed by SNPs that flank sdw1 (WTB Thomas, unpublished data); however, the level of significance for an effect on plant height did not exceed the main effect threshold in this region of the genome. Including SNP 11_10918 in a QTL × environment analysis revealed the presence of a strong G.E height effect associated with the SNP. Under the severely stressed conditions of some of the environments that we utilized, the dwarfing effect of sdw1 is unlikely to be significantly expressed, which would explain the absence of main effect association with it.
We anticipated that detection of QTL at loci determining the major biotypes present in the sample, such as 2/6 row type and spring/winter growth habit (Fig. 1), would depend upon the degree of relatedness between the distinct biotypes present in our sample. If there is none, then all the differences will be absorbed by the kinship matrix, or any other form of population sub-structure correction, which can lead to false-negative associations. However, different geographic origins for row types and growth habits and presence of recombinants between these sample clusters (Comadran et al. 2009) should prevent statistical over-correction of these traits. We did identify SNP associations related to row type which have been discussed above. Referring to growth habit, it is important to note that in Autumn sown conditions the vernalization requirement of the accessions is satisfied and in consequence we should not be capturing genetic variants related to the vernalization loci on 4HL (sgh1) and 5HL (Sgh2). However, you certainly would expect to see an effect of both loci in trials where there was not enough cold to fulfil winter types’ vernalization requirements. Moreover, we know that Sgh2 and Fr-H1, a locus involved in cold resistance, are tightly linked and could even be a single gene with pleiotropic effects (Sutka et al. 1999). Thus, in very cold conditions there is the possibility to observe differences linked to Sgh2 just because the plant is better protected from cold damage. So, in an association, scan for main genetic effects on autumn sown barley over a wide range of environmental conditions we should not expect to observe effects associated to the vernalization genes as any lack of vernalisation will be ascribed to G.E interactions.
It has been well documented that G.E interactions reduce our ability to detect stable QTL. In this particular study, site mean yields ranged from 0.74 (Morocco dry site 2005) to 6.32 (Spain watered site 2004), and because of strong G.E interactions distinct years or sites resulted in different QTL in single trial analyses (Comadran et al. 2008). Yield G.E analysis, reporting 27 QTL regions at least 5 cM from each other, contrasts with the 3 QTL reported for yield main effects. The purpose behind the G.E analysis for yield was not to explain QTL.E effects in terms of candidate genes but to highlight the significance of G.E in Mediterranean environments and the importance of approaches that reduce the dimensionality/complexity from tens or hundreds of QTL to just a few key ones. In this study, a focus on SNP main effects in a MET data analysis of 24 years × site combinations provided valuable information that will help to identify several genomic regions with putatively stable effects across the Mediterranean mega-environment and to prioritize the most robust results.
The methods described in this study proved efficient for the detection of positive associations linked to most of the known barley structural major genes. The germplasm set which comprises 185 barley genotypes genotyped with 1,130 DArT® (Comadran et al., 2009) and 1307 SNP polymorphic molecular markers together with a large amount of phenotypic and environmental measures from 24 trials, constitutes a highly valuable tool for the barley community. This material will be particularly useful for targeted candidate gene approaches, where candidate gene re-sequencing can be use for allele mining on the germplasm set. Haplotype characterisation would overcome the limitations of bi-allelic SNP markers to cope with the possible multi-allelic (more than 2 alleles per locus) nature of the population.
References
Brueggeman R, Rostoks N, Kudrna D, Kilian A, Han F et al (2002) The barley stem rust-resistance gene Rpg1 is a novel disease-resistance gene with homology to receptor kinases. Proc Natl Acad Sci USA 99:9328–9333
Caldwell KS, Russell J, Langridge P, Powell W (2006) Extreme population-dependent linkage disequilibrium detected in an inbreeding plant species, Hordeum vulgare. Genetics 172:557–567
Chen A, Baumann U, Fincher GB, Collins NC (2009) Flt-2L, a locus in barley controlling flowering time, spike density, and plant height. Funct Integr Genom 9:243–254
Close TJ, Bhat PR, Lonardi S, Wu Y, Rostoks N et al (2009) Development and implementation of high-throughput SNP genotyping in barley. BMC Genom 10:582
Comadran J, Russell J, Eeuwijk FA, Ceccarelli S, Grando S et al (2008) Mapping adaptation of barley to droughted environments. Euphytica 161:35–45
Comadran J, Thomas WT, Van Eeuwijk FA, Ceccarelli S, Grando S et al (2009) Patterns of genetic diversity and linkage disequilibrium in a highly structured Hordeum vulgare association-mapping population for the Mediterranean basin. Theor Appl Genet 119:175–187
Cuesta-Marcos A, Igartua E, Ciudad F, Codesal P, Russell J et al (2008) Heading date QTL in a spring x winter barley cross evaluated in Mediterranean environments. Mol Breed 21:455–471
Cuesta-Marcos A, Casas A, Hayes PM, Gracia MP, Lasa JM et al (2009) Yield QTL affected by heading date in Mediterranean grown barley. Plant Breed 128:46–53
Druka A, Franckowiak JD, Lundqvist U, Bonar N, Alexander J et al (2010) Exploiting induced variation to dissect quantitative traits in barley. Exp Plant Biol
Faure S, Higgins J, Turner AS, Laurie DA (2007) The FLOWERING LOCUS T-like gene family in barley (Hordeum vulgare). Genetics 176:599–609
Gustafsson A, Hagberg A, Lundqvist U (1960) The induction of early mutants in Bonus barley. Hereditas 675–699
Jia Q, Zhang J, Westcott S, Zhang XQ, Bellgard M et al (2009) GA-20 oxidase as a candidate for the semidwarf gene sdw1/denso in barley. Funct Integr Genom 9:255–262
Kearsey MJ, Farquhar AG (1998) QTL analysis in plants; where are we now? Heredity 80(Pt 2):137–142
Komatsuda T, Mano Y (2002) Molecular mapping of the intermedium spike-c (int-c) and non-brittle rachis 1 (btr1) loci in barley (Hordeum vulgare L.). Theor Appl Genet 105:85–90
Komatsuda T, Pourkheirandish M, He C, Azhaguvel P, Kanamori H et al (2007) Six-rowed barley originated from a mutation in a homeodomain-leucine zipper I-class homeobox gene. Proc Natl Acad Sci USA 104:1424–1429
Kraakman AT, Niks RE, Van PM, den Berg P, Stam FA, Eeuwijk Van (2004) Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics 168:435–446
Mackay I, Powell W (2007) Methods for linkage disequilibrium mapping in crops. Trends Plant Sci 12:57–63
Malosetti M, van der Linden CG, Vosman B, Van Eeuwijk FA (2007) A mixed-model approach to association mapping using pedigree information with an illustration of resistance to Phytophthora infestans in potato. Genetics 175:879–889
Payne RW, Murray DA, Harding SA, Baird DB, Soutar DM (2008) GenStat for windows, 11th edn. Introduction. VSN International, Hemel Hempstead
Piepho HP (2000) A mixed-model approach to mapping quantitative trait loci in barley on the basis of multiple environment data. Genetics 156:2043–2050
Pourkheirandish M, Wicker T, Stein N, Fujimura T, Komatsuda T (2007) Analysis of the barley chromosome 2 region containing the six-rowed spike gene vrs1 reveals a breakdown of rice-barley micro collinearity by a transposition. Theor Appl Genet 114:1357–1365
Rae SJ, Macaulay M, Ramsay L, Leigh F, Matthews D et al (2007) Molecular barley breeding. Euphytica 158:295–303
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR et al (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci USA 98:11479–11484
Saisho D, Purkheirandish M, Kanamori H, Matsumoto T, Komatsuda T (2009) Allelic variation of row type gene Vrs1 in barley and implication of the functional divergence. Breed Sci 59:621–628
Sutka J, Galiba G, Vaguifalvi A, Gill BS, Snape JW (1999) Physical mapping of the Vrn-A1 and Fr1 genes on chromosome 5A of wheat using deletion lines. Theor Appl Genet 199–202
Szucs P, Karsai I, von ZJ, Meszaros K, Cooper LL et al (2006) Positional relationships between photoperiod response QTL and photoreceptor and vernalization genes in barley. Theor Appl Genet 112:1277–1285
Szucs P, Skinner JS, Karsai I, Cuesta-Marcos A, Haggard KG et al (2007) Validation of the VRN-H2/VRN-H1 epistatic model in barley reveals that intron length variation in VRN-H1 may account for a continuum of vernalization sensitivity. Mol Genet Genom 277:249–261
Thomas WTB (2003) Prospects for molecular breeding of barley. Ann Appl Biol 142:1–12
Van Eeuwijk FA, Bink MC, Chenu K, Chapman SC (2010) Detection and use of QTL for complex traits in multiple environments. Curr Opin Plant Biol 13:193–205
Waugh R, Jannink JL, Muehlbauer GJ, Ramsay L (2009) The emergence of whole genome association scans in barley. Curr Opin Plant Biol 12:218–222
Wenzl P, Carling J, Kudrna D, Jaccoud D, Huttner E et al (2004) Diversity Arrays Technology (DArT) for whole-genome profiling of barley. Proc Natl Acad Sci USA 101:9915–9920
Yu J, Pressoir G, Briggs WH, BVroh I, Yamasaki M et al (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Acknowledgments
The above work was partially funded by the European Union-INCO-MED program (MABDE ICA3-CT2002-10026). SCRI received grant in aid from the Scottish Government Rural and Environment Research and Analysis Department. The Centre UdL-IRTA forms part of the Centre CONSOLIDER on Agrigenomics and acknowledges competitive grants AGL2008-05541/C02 and GEN2006-28560-E from the Spanish Ministry of Science and Innovation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by P. Hayes.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Comadran, J., Russell, J.R., Booth, A. et al. Mixed model association scans of multi-environmental trial data reveal major loci controlling yield and yield related traits in Hordeum vulgare in Mediterranean environments. Theor Appl Genet 122, 1363–1373 (2011). https://doi.org/10.1007/s00122-011-1537-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-011-1537-4