
Abstract
Within the Dutch genomics initiative the “Centre for Biosystems Genomics” (CBSG) a major research effort is directed at the identification and unraveling of processes and mechanisms affecting fruit quality in tomato. The basis of this fruit quality program was a diverse set of 94 cultivated tomato cultivars, representing a wide spectrum of phenotypes for quality related traits. This paper describes a diversity study performed on these cultivars, using information of 882 AFLP markers, of which 304 markers had a known map position. The AFLP markers were scored as much as possible in a co-dominant fashion. We investigated genome distribution and coverage for the mapped markers and conclude that it proved difficult to arrive at a dense and uniformly distributed coverage of the genome with markers. Mapped markers and unmapped markers were used to investigate population structure. A clear substructure was observed which seemed to coincide with a grouping based on fruit size. Finally, we studied amount and decay of linkage disequilibrium (LD) along the chromosomes. LD was observed over considerable (genetic) distances. We discuss the feasibility of marker-trait association studies and conclude that the amount of genetic variation in our set of cultivars is limited, but that there exists scope for association studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Five years ago, the Dutch plant genomics initiative the “Centre for Biosystems Genomics” (CBSG) was set up to stimulate plant genomics research in general and to investigate more specifically for potato and tomato the phenotypic and genetic variation in consumer quality, environmental quality, technological aspects and societal aspects (e.g., public acceptance of genomics methodologies). To provide a benchmark for research advances in those agricultural crops, Arabidopsis was included as a model crop for which state of the art resources were available.
A considerable part of the CBSG resources was concentrated on the improvement of tomato fruit quality. As part of that research, diversity and fruit quality was assessed for a panel of 94 greenhouse cultivated tomatoes, containing both older as well as current tomato cultivars. Breeding companies that are partners within CSBG were asked to provide a sample of their germplasm taking care to include contrasts on relevant quality aspects. Tomato quality was investigated in terms of metabolic profiling and in terms of sensory and consumer appreciation studies on ripe tomato fruits. This paper focuses on genetic diversity, as sampled by molecular markers. The results of the quality assessments and marker—trait association studies for quality will be presented elsewhere.
Sufficient genetic variation, or diversity, in target traits is a condition for progress in plant breeding. Genetic diversity can be investigated in many ways, for example, through morphological and biochemical traits, pedigree analysis or by molecular markers (Franco et al. 2001; Zhao et al. 2005a). Among these methods, morphological traits are intuitive and practical, but as they are subject to environmental influences and selection pressure during domestication and breeding the interpretation of the results of diversity studies based on such traits can be difficult. Pedigree records are often incomplete or have limited availability. The advent of DNA markers stimulated diversity studies in plants, as DNA markers allow extensive sampling at the DNA level. Within tomato (Solanum lycopersicum), many studies have been performed on genetic diversity and variety identification, using molecular markers such as RFLP (Miller and Tanksley 1990), RAPD (Williams and St. Clair 1993; Villand et al. 1998; Noli et al. 1999; Carelli et al. 2006), AFLP (Park et al. 2004) and SSR (Bredemeijer et al. 2002).
For the diversity study in this paper, we used AFLP markers (Vos et al. 1995). AFLP genotyping has the advantage of sampling a large number of informative fragments with random genomic origin (Mueller and LaReesa Wolfenbarger 1999). Although other choices of marker types would have been possible, AFLP markers have some desirable properties in the light of application in statistical diversity analyses (see Meudt and Clarke 2007; Bonin et al. 2007 for a more elaborate discussion on this subject). We investigated the distribution of AFLP markers across the genome, and assessed genotypic frequencies for the marker loci. Bayesian and nearest neighbor clustering methods were used for identifying possible population structure. Finally, as a precursor to later association studies, we studied the magnitude of linkage disequilibrium (LD) and the decay of LD as a function of genetic (map) distance.
Materials and methods
Germplasm
Five internationally operating vegetable breeding companies (Enza, De Ruiter Seeds, Rijk Zwaan, Seminis Vegetable Seeds and Syngenta Seeds), all participating in the research consortium CBSG (see Van Berloo et al. 2007 for more details), were asked to provide tomato cultivars from their collections of current and historic cultivated tomato germplasm. These companies together collected a set of ~150 cultivars. Based on previously gathered sensory and morphological data, a set of 94 cultivars was selected from this collection. This set can be considered representative, with regard to phenotypic diversity, for the available germplasm used in commercial European greenhouse tomato cultivation. The phenotypic diversity of the sampled germplasm was compared to other studies in tomato for two traits: Brix (soluble solid content, expressed as a percentage) and average fruit weight. Thus, we first compared the observed ranges in our germplasm for the two traits to the observations of Chaïb et al. (2006), who investigated tomato fruit quality aspects in a set of 144 RILs derived from an inter-specific cross and in approximately 100 derived backcross lines. In addition, we looked at the observations of Helyes et al. (2003), who described phenotypic variation in an experiment involving 18 tomato cultivars.
Seeds of the tomato germplasm discussed in this paper are available on request. Please contact the CBSG consortium secretariat (see http://www.cbsg.nl for contact info).
Morphological and fruit observations
The chosen cultivars were grown in replicated trials in the greenhouse, and fruits were harvested and subjected to a number of observations and analyses, which are discussed in van Berloo et al. (2007), Ursem et al. (2008) and other papers in preparation. Twelve morphological and anatomical plant characteristics were scored: total plant length at the end of the cultivation period; total weight of vegetative tissues (leaves, stem) at the end of the cultivation period; accumulated weight of all harvested fruits during cultivation; average number of flowers per inflorescence; average stem inter-node length; fresh weight and dry weight of the vegetative system (dry weight measured after storage in an oven at 80°C for 48 h); total leaf area (which was measured at the end of the cultivation period using a dedicated apparatus with semi-automatic pass-through similar to the device described by Murata and Hayashi 1967); shape of the fruits (expressed as a length/width ratio); total number of harvested fruits during cultivation; and average fruit weight and fruit firmness (which was measured as the amount of deformation of the fruit when submitted to a force of 3 N). These observations can be used to assess the phenotypic diversity among the set of cultivars, and compare this type of diversity with the other types of diversity like that based on molecular markers, the latter being the main theme of this paper. In addition to the above traits, Brix (soluble solid content of the tomato fruit) was measured using a refractometer (GMK-701R; Nie-Co Products, Aalsmeer, NL).
AFLP genotyping
Fifty AFLP primer combinations were run on DNA samples of the 94 tomato cultivars. This large number of primer combinations was used because it is known that the amount of marker polymorphism in cultivated tomato is generally low (Miller and Tanksley 1990; Archak et al. 2002). However, when polymorphism rate is low the AFLP technology is often still capable of uncovering genetic differences (Spooner et al. 2005; Meudt and Clarke 2007). Furthermore, once interesting AFLP markers have been identified, they can often be converted to single-locus markers in an efficient way (Brugmans et al. 2003). AFLP genotyping was performed at Keygene NV using their standard in-house developed protocols (Vos et al. 1995). EcoRI-Mse and PstI-Mse primer combinations were used. Selective nucleotide adaptors were selected, based on previous experience in tomato, in such a way that the expected number of polymorphic loci was optimal. Genotyping resulted in the scoring of 882 polymorphic AFLP fragments. For 304 AFLP fragments the map position could be established from an integrated inter-specific linkage map.
AFLP scoring is standardly done by registration of presence and absence of bands, i.e., in a dominant fashion. We developed a co-dominant scoring methodology which is based on the application of mixture models to the quantitative gel intensity values for the fragments. This method extends the mixture model methods described by Piepho and Koch (2000) and Jansen et al. (2001) for biparental populations to germplasm panels of arbitrary composition. The essence of our mixture model is that we estimated the fractions of homozygotes and heterozygotes from the distribution of band intensity data itself, where the earlier methods for biparental populations fixed those fractions in relation to the type of population. For example, for an F2 population, the fractions will be 0.25 − 0.50 − 0.25, respectively, for homozygote band present, heterozygote band present, and homozygote band absent.
For quantification and investigation of genetic diversity, we used both the co-dominant genotypic classification on the basis of AFLP band intensities explained above, and log transformed numerical band intensity values. The reason for using both types of marker derived information is mainly one of convenience in later calculations whose results will be presented elsewhere. For the present paper, the choice of log band intensity or discrete (band) allele number did not influence results or interpretations related to diversity.
Linkage map
Since the marker data were scored on a set of cultivars, and not in a segregating population, we could not create a genetic linkage map from the data at hand. Therefore, we used an existing high density linkage map created from integrating three inter-specific linkage maps. Identity of newly scored AFLP fragments and previously mapped fragments followed from equality of their mobility numbers (Rouppe van der voort et al. 1997; Vuylsteke et al. 1999). The integrated map we used has not been published, but can be considered an extension of the previously published integrated map by Haanstra et al. (1999).
Genome coverage, marker distribution and test for Hardy–Weinberg equilibrium
Only a minority of the scored AFLP markers could be assigned map positions: 304 out of 882. Through our estimate of the decay of LD we could estimate the fraction of the genome sampled by the available set of mapped markers. The allocation of markers to genome positions was analyzed by looking at the distribution of distances between neighboring markers. We assessed the general distribution of markers over the chromosomes in the form of box plots of inter marker distances as well as the uniformity of markers along the chromosome through Chi-square tests that compared the observed numbers of markers in 10 cM chromosome intervals with expected numbers assuming a uniform distribution. Estimated allele frequencies were used to calculate expected genotype frequencies, which were compared to observed genotype frequencies in tests for Hardy–Weinberg (HW) equilibrium (Falconer and Mackay 1996, Chap. 1).
Population structure analyses
Possible structure in the population was investigated in various ways. First, to the co-dominant genotypes we applied the Bayesian clustering method implemented in the Structure 2.1 package (Pritchard et al. 2000). This method is based on the creation of groups that are internally characterized by HW equilibrium and absence of LD, while between groups LD exists. A subset of the markers was used for this analysis. This subset covered the genome with evenly spaced markers at approximately 10 cM inter marker distance, thereby reducing the bias following from sampling closely linked markers. Structure settings were left at their default values and 10.000 cycles were applied for burn-in, followed by 100.000 cycles for the actual analysis, assuming admixture of populations. Secondly, we evaluated population structure by inspection of a neighbor joining dendrogram built from genotypic distances, which were calculated as Euclidean distances using the log band intensities. Genotypic distances were calculated by the Genstat statistical package (Genstat 2005). The MEGA 3 package (Kumar et al. 2004) was used for Neighbor Joining cluster analysis (Saitou and Nei 1987). A complementary analysis studying between group separation was done for within and between group distances, taking the a priori sets of cherry, beef, and round tomatoes as groups.
Relationships between genotypes were further studied with network methodology following Ursem et al. (2008). The log band intensities of the markers selected for use in the Structure analysis were now used to calculate Euclidean distances between genotypes. These Euclidean distances were transformed to similarities which served to join genotypes in a network representation created with the Pajek package (Bagatelj and Mrvar 2003), whenever the observed similarity exceeded the 99 percentile of the null distribution for the similarities. This null distribution was obtained from permutation of the log band intensities with respect to the genotypes. The network was inspected for connectivity. Clustering coefficients were calculated to quantify the density of the network around individual genotypes (Watts and Strogatz 1998). A clustering coefficient expresses the number of observed links between neighboring genotypes in relation to the total number of possible links between neighbors. Neighboring genotypes are pairs of genotypes that are both connected to a third genotype. Neighboring genotypes need not themselves be connected again, i.e., they might be linked to a third genotype without being directly linked themselves.
For comparison with the marker information, a second neighbor joining analysis was performed on distances obtained from morphological plant characteristics. Euclidean distances were derived from the set of 12 numerically scored morphological observations introduced in a previous paragraph.
LD analyses
LD between markers was assessed by calculation of squared correlation coefficients (R 2; Zhao et al. 2005b) between marker intensity patterns, using the Ggt 2.0 software package (Van Berloo 2008, 1999). LD statistics were calculated per chromosome for all marker pairs and subsequently, aggregated over all 12 chromosomes, the decay of LD with map distance was evaluated. As a threshold for significance, a 5% false discovery rate was chosen, following the two-step procedure described by Benjamini et al. (2005).
The strength of LD between marker pairs across the full genome was plotted in a heatmap, using the Ggt 2.0 package. Separate heatmaps were created for the beef and round tomatoes on the one hand and the cherry tomatoes on the other hand. In addition to the heatmaps, correlations between markers on different chromosomes were calculated.
Results
Germplasm phenotypic diversity
The ranges of observations for Brix and fruit weight in our experiments and in three populations from literature are summarized in Table 1. The set of germplasm discussed in this paper is indicated as CBSG—94 cultivars. Observations from the three different populations in literature are indicated by Helyes—18 cultivars (see Helyes et al. 2003); Chaïb—144 RILs and Chaïb—100 BC3S1 (see Chaïb et al. 2006). In their paper Chaïb et al. present data on three roughly equally sized BC3S1 populations. As the values reported were comparable we have chosen to present average values of these three populations. From Table 1 we observe that the range of observations on our set of cultivars exceeds the ranges reported in the other papers. It appears our set of cultivated germplasm is at least as diverse as the unadapted set of RILs derived from an inter-specific cross, and more diverse than the set of Helyes et al. for which the authors claim that the observations in that set were “extremely diverse”.
Linkage map
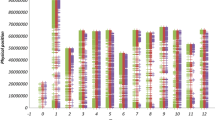
Figure 1 shows the mapped markers of our study on the genetic linkage map. The dark colored regions indicate areas sufficiently covered by markers, according to our estimate of the extent of LD, which was 15 cM (see below). The lack of markers at the start and end regions of some chromosomes, and the strong clustering of markers in the presumably centromeric region (and segments that are integrated from wild relatives into cultivated tomato) are clearly visible on this map.

Graphical representation of markers scored along the 12 tomato chromosomes. Filled areas of the chromosome bars indicate the region covered with the set of co-dominantly scored AFLP markers. These areas were defined by taking an interval of 15 cM, which was the estimate range of LD in this dataset, on both sides of each marker. Start and end of each chromosome was obtained from previous mapping results (Keygene NV, unpublished data). Length of longest chromosome (7) is 128.5 cM
Genome coverage
Table 2 shows details on the number of markers per chromosome, a Chi-square test for uniformity of marker distribution, the relative fraction of the genome covered by the scored markers and the fraction of markers in HW equilibrium. The Chi-square test on evenly distribution of markers over the chromosomes clearly indicated that the distribution of markers over the chromosomes was not uniform (P < 0.001) and that for chromosomes 4, 6, 9 and 11 the distribution of markers within the chromosome was not uniform either. Some deviations from HW equilibrium were observed for markers on all chromosomes, but for some chromosomes (4, 5 and 8) HW equilibrium was almost completely absent. The non-uniform dispersion of markers along the chromosomes becomes apparent when we look at the distribution of markers over the chromosomes in Fig. 1. To investigate the distribution of inter-marker distances per chromosome, we constructed box-plots that visualize the distribution of distances between consecutive markers (Fig. 2). Figure 2 and Table 2 show considerable variability for coverage and distribution of markers. Several regions are well or very well covered while other chromosomes are only sparsely covered. Chromosomes 4, 6 and 9 have an average distance between consecutive markers of less than 1 cM, and 75% of all marker distances between neighboring markers are less than 3 cM. On the other hand for chromosomes 2, 3 and 7 more than half of the distances between consecutive markers are larger than 3 cM.

Box plot showing the distribution of marker distances between consecutive markers, arranged per chromosome. For chromosomes 4, 5, 6, 9 and 10 marker distances are generally small, indicating sufficient marker coverage, for the other chromosomes sparsely or insufficiently covered regions are observed
Population structure analyses
The analysis with the Structure 2.1 software package indicated the presence of population structure, because the model likelihood increased considerably when more than one population was assumed. However, estimating the number of populations was difficult as the likelihood for three to six populations was very similar and only increased when seven or more populations were assumed (data not shown). A closer look at the membership probabilities for the genotypes showed us that, when four or more subpopulations were assumed, all genotypes became “distributed” over at least two populations. We therefore chose the simplest solution that still assigned a large part of the genotypes to a single population, which was at three (sub-) populations. A second Structure run using all 882 co-dominant markers was performed (data not shown) and confirmed the earlier results. Figure 3 gives subpopulation memberships in the form of a bar plot for the three cluster solution. To initiate the analysis, the tomato types (beef, cherry, and round) were used as prior population identifiers. These types are indicated by the numbers below the bars. The plot clearly demonstrates that one of the identified populations coincides almost completely with the cherry tomato type (cluster 1 and group C in the plot). Although most cultivars belonging to group B (beef tomatoes) have a high probability to belong to the population defined by cluster 2, and many cultivars belonging to group R (round tomatoes) have a high probability of belonging to the population defined by cluster 3, tomato type and subpopulation do not completely coincide for beef and round tomato types. Further evidence for this can be found in the membership proportions, as given in the structure output for each of the three a-priori defined types, presented in Table 3. Each of the three a priori tomato types coincides to a certain degree with one of the a posteriori clusters. This is most obvious for the cherry group, for which the a-posteriori membership probabilities were on average 0.87 for cluster 1. The beef and round tomato cultivars also coincide with proper clusters, but with more exceptions.

Bar plot showing subpopulation membership probabilities on vertical axis of the 94 cultivars assuming three subpopulations on the horizontal axis. Prior population membership based on tomato fruit type is indicated on the x-axis (beef type B, cherry type C, round type R)
A neighbor joining dendrogram (Fig. 4) derived from Euclidean distances on (log transformed) band intensities also indicated the presence of groups in the set of genotypes. Especially the cherry tomatoes stand out as a separate group. Beef and round tomatoes are not clearly separated and showed smaller distances among their cultivars than the cherry cultivars. To check the stability of the dendrogram we used bootstrapped cluster analysis in MEGA 3, using for convenience a dominant scoring of AFLP markers. We found that the main separation of subclusters was very stable (bootstrap values of 99% for the main subdividing nodes). We did not observe indications for clustering of genotypes related to the breeding origin of the cultivars, as most genotypes that originated from a particular company ended up scattered in a seemingly random fashion throughout the dendrogram (data not shown). Figure 5 gives the genetic distances within and between the groups (cherry vs beef + round) defined by the tomato types. The cherry group showed larger within-group genetic distances than the beef + round group, but the between group distances were clearly larger than the within group distances.

Neighbor joining tree for the set of 94 tomato cultivars. Genetic distances are calculated as Euclidean distances based on log band intensity values of 882 AFLP markers. The different shapes indicate the three different tomato types (filled square beef tomato, filled circle round tomato, filled diamond cherry tomato). Main separation of branches was confirmed by bootstrap analysis (99%)

Box plots showing the genetic distances (see text for details) within and between the types of tomatoes. C the group containing cherry tomato genotypes, B + R the group containing all beef and round tomato types. Genetic distances within the groups were smaller than the genetic distances between the groups
The network constructed on the basis of band intensities in Fig. 6 emphasized once again the difference between the cherry tomatoes and the beef and round tomatoes. Figure 6 shows that the connectivity in the cherry cluster was extremely low with an average cluster coefficient of 0.24, while the connectivity in the beef and round cluster was very high with a clustering coefficient of 0.70. Both clusters were uniquely connected through the genotype pair R131 and C132. Interestingly, the single outlying genotype R068 was also connected to R131. These three genotypes happened to have in common an elongated Roma-like fruit shape. Other unique genotypes were R038 and B098. (Unique genotypes have cluster coefficient zero). Looking at the distributions of clustering coefficients for the genotypes stemming from a particular breeding company (Table 4), it was observed that the median clustering coefficient for company 4 was the lowest, and the coefficient for company 5 was the highest, with the companies 1, 2 and 3 having more or less the same intermediate value. This means that the amount of genotypic variation selected by company 4 was highest, that by company 5 lowest. Still, differences were very small.

Network representation of genetic diversity, representing Euclidean distances calculated using quantitative band intensity values for a selected subset of markers. Circular nodes represent genotypes, connecting lines represent similarities that exceed a predefined threshold (see text for details). Colors of the nodes and first character of node labels indicate tomato type: blue for beef, green for cherry and red for round. Numbers in the nodes indicate the breeding companies that provided the seeds of the tomato genotypes
The Euclidean distances, based on numerical trait values for 12 morphological and harvest related traits, were used to create another neighbor joining dendrogram (Fig. 7). This dendrogram shows a large cluster and a smaller cluster. The smaller cluster clearly stands out and is dominated by cherry cultivars, but no exclusive separation of cherry and other tomatoes can be observed.

Neighbor joining tree for the 94 tomato cultivars. Genotypic distances are calculated as Euclidean distances based on 12 morphological and harvest related traits (see text for details). The different shapes indicate the three different tomato types (filled square beef tomato, filled circle round tomato, filled diamond cherry tomato)
Our analyses of both types of genetic distances indicate the presence of at least two subgroups: a group containing all cherry type cultivars and a large group containing beef and round cultivars. This second group could still be subjected to further subdivision, but no obvious partitioning related to tomato type can be found, which confirms the results obtained using the Structure package.
Linkage disequilibrium analysis
As separate analyses of LD decay for the cherry and beef-round tomatoes yielded very similar results, only the decay for the cherry group is presented (Fig. 8). Extensive LD can be observed and reasonably strong LD can be found for markers up to 20 cM apart, indicating good possibilities to explore LD based marker-trait associations in this material. We used a slightly conservative estimate of the average extent of LD of 15 cM to determine the genome coverage presented in Fig. 1.

Linkage disequilibrium R 2, versus map distance in cM, for the cherry cultivars. The red line indicates a smoothing spline. The 0.05 false discovery rate threshold is R 2 = 0.37
Figure 9 shows separate LD heat maps for the beef-round (Fig. 9a) and cherry (Fig. 9b) tomato types. Both heat maps show that strong LD is limited to certain hotspots, and is mostly concentrated along the diagonal of these plots, as was expected. Strong LD between markers on different chromosomes is rare, although it does occur at some spots. In the heat map for the cherry group, considerable LD can be observed between markers on chromosomes 2 and 4, 4 and 8, 4 and 11, 1 and 9 and 4 and 9. The heat map for the beef-round tomato types shows inter-chromosome LD for markers on chromosomes 4 and 10, 4 and 11, 1 and 9 and 4 and 9.

Heatmap display of LD as R 2 for all marker-pairs. a Heatmap of LD between markers for the beef and round tomato types. b Heatmap of LD between markers for the cherry tomato type. In both figures on the x and y axis the 304 mapped markers are arranged in map order. LD between pairs of markers is plotted in the bottom diagonal part of the figure. Along the diagonal the chromosomes and marker positions are indicated. The color intensity reflects the amount of LD
Discussion
In this paper we have shown that AFLP markers are useful tools for genetic analysis, and that they allow us to get an estimate of the genetic diversity. The number of markers obtained using AFLP methodology is of course highly dependent on the genotyping and protocol conditions, but an average of 30–40 polymorphic markers per primer combination is often feasible (e.g. Haanstra et al. 1999; Isidore et al. 2003; Keygene, unpublished data). In our case on average only 18 polymorphic markers per primer combination could be scored co-dominantly, which is an indication that the amount of genotypic variability present in the tested set of cultivars is limited.
We used normalized gel intensity data to analyze individual marker behavior in more detail, and identified and scored a substantial number of markers co-dominantly. Especially in a situation like the one studied in this paper, where analyses are performed on plant material that is to a large extent (expected to be) heterozygous, the ability to discriminate between presence of a marker band in single or double quantity can be beneficial to increase power in diversity and association analyses.
A possible explanation for the uneven distribution of markers along the chromosomes may lie in the origin of the integrated genetic map that was at our disposal. The research that formed the basis for the construction of that map was directed at a number of specific tomato genomic regions, which contained genes of interest, like resistance loci (e.g., Van Berloo and Lindhout 2001). Therefore the region on chromosome 6, bearing resistance against tomato yellow leaf curling virus and the region on chromosome 9, harboring tomato mosaic virus resistance, have become more saturated with markers than other regions. From Table 2 we observe that especially chromosome 4 and to a lesser extent chromosomes 5 and 8 show strong deviation from HW equilibrium. Chromosome 4 also shows considerable LD with markers on other chromosomes. Recent breeding efforts focusing on (regions of) chromosome 4 could be responsible for these observations.
Even though we selected, among the available cultivated material for greenhouse cultivation, a set of varieties that was quite diverse at the phenotypic level, we discovered that the diversity at marker level was limited. The dendrogram based on phenotypic observations (Fig. 7) shows larger diversity than the dendrogram based on molecular marker data (Fig. 4). This observation is in line with other studies in cultivated tomato (e.g., Archak et al. 2002; Park et al. 2004; Garcia-Martinez et al. 2006) and could be a consequence of a strong selection pressure that was applied to the current cultivated germplasm. Also the fact that several regions of the genome were poorly covered by markers does not necessarily reflect a shortage of markers, but a lack of polymorphic markers, since many markers that are polymorphic when sampled in wider genetic backgrounds, like a population derived from an inter-specific cross, did not show polymorphism among our set of 94 cultivated varieties.
Based on fruit morphology we could clearly define three groups within our set of cultivars. A large fruited group (beef), a group with regular sized (round) table tomatoes and a group with much smaller fruited (cherry) tomatoes. The network analysis confirmed the classification in groups, but also revealed that this classification may be extended with a classification based on fruit shape, as cultivars with different fruit sizes but with a similar elongated shape remained closely connected in this diagram. At the genomic (AFLP) level the distinction between the cherry tomatoes and the round and beef tomatoes remained clear. Minor differences between the round and beef tomatoes were observed but these differences were far less striking than the dominating cherry versus beef + round division. Most likely this is caused by the shared breeding history of these tomato types, which is much longer for round and beef tomato cultivars (and parental breeding lines), while the cherry tomatoes have only recently become more popular and subjected to breeding efforts.
The magnitude of LD and the length of genome intervals with strong LD appear to be larger than that observed in studies dealing with other plant species like sugar beet (Kraft et al. 2000), Arabidopsis (Nordborg et al. 2002) and barley (Kraakman et al. 2004). A reason for the strong LD could be the self pollinated history of the tomato species, combined with a very limited variability, due to strong and continued selection by plant breeders and re-usage of donor lines from a limited gene pool. Although new donor segments have been introduced from more variable related species into the tomato germplasm, mainly to add resistance genes, the time for recombination and breaking down of LD within these donor segments has been rather short, and the process to achieve linkage equilibrium is still ongoing. Furthermore, new donor genome segments are continuously being introduced by breeders, thereby introducing new LD into the cultivated germplasm.
LD that was observed between markers that are located on different chromosomes could be caused by errors in the map positions of some of these markers, or by simultaneous selection of certain regions, that remain together because of advantageous epistatic effects, but further research would be needed to confirm this hypothesis.
The LD analyses presented in this paper indicate strong LD among markers, and LD extending over relatively large regions. These results may be biased by chromosome fragments that have been introgressed from wild germplasm in the past decades, i.e., fragments containing resistance genes. However, the high amount of LD that is found indicates that there is sufficient prospect for association mapping. Even regions that are only sparsely covered by markers may still show sufficient LD to be interesting for marker-trait association studies. The downside of the strong LD that is found over a considerable genetic distance will be that any results of association studies will only be roughly indicative for the genomic position of effects that are observed.
References
Archak S, Karihaloo JL, Jain A (2002) RAPD markers reveal narrowing genetic base of Indian tomato cultivars. Curr Sci 82:1139–1143
Bagatelj V, Mrvar A (2003) Pajek-analysis and visualization of large networks. In: Jünger M, Mutzel P (eds) Graph drawing software. Springer, Berlin, p 77
Benjamini Y, Krieger AM, Yekutieli D (2005) Adaptive linear step-up procedures that control the false discovery rate. URL: http://www.math.tau.ac.il/~ybenja/MyPapers/bkymarch9.pdf. Cited 6 March 2008
Bredemeijer GMM, Cooke RJ, Ganal MW, Peeters R, Isaac P, Noordijk Y, Rendell S, Jackson J, Röder M, Wendehake K, Dijcks M, Amelaine M, Wickaert V, Bertrand L, Vosman B (2002) Construction and testing of a microsatellite database containing more than 500 tomato varieties. Theor Appl Genet 105:1019–1026
Brugmans B, van der Hulst RGM, Visser RGF, Lindhout P, van Eck HJ (2003) A new and versatile method for the successful conversion of AFLP™ markers into simple single locus markers. Nucleic Acid Res 31:e55
Bonin A, Ehrich D, Manel S (2007) Statistical analysis of amplified fragment length polymorphism data: a toolbox for molecular ecologists and evolutionists. Mol Ecol 16:3737–3758
Carelli PM, Gerald LTS, Grazziotin GF, Echeverrigaray S (2006) Genetic diversity among Brazilian cultivars and landraces of tomato Lycopersicon esculentum Mill. revealed by RAPD markers. Genet Resour Crop Evol 53:395–400
Chaïb J, Lecomte L, Buret M, Causse M (2006) Stability over genetic background, generations and years of quantitative trait locus (QTLs) for organoleptic quality in tomato. Theor Appl Genet 112:934–944
Falconer DS, Mackay TFC (1996) Introduction to quantitative genetics, 4th edn. Longman, Harlow
Franco J, Crossa J, Ribaut JM, Betran J, Warburton ML, Khairallah M (2001) A method for combining molecular markers and phenotypic attributes for classifying plant genotypes. Theor Appl Genet 103:944–952
Garcia-Martinez S, Andreani L, Garcia-Gusano M, Geuna F, Ruiz JJ (2006) Evaluation of amplified length polymorphism and simple sequence repeats for tomato germplasm fingerprinting: utility for grouping together closely related cultivars. Genome 49:648–656
Genstat (2005) Genstat, 8th edn. VSN International Ltd, Hemel Hempstead
Haanstra JPW, Wye C, Verbakel H, Meijer-Dekens F, Van den Berg P, Odinot P, Van Heusden AW, Tanksley S, Lindhout P, Peleman P (1999) An integrated high-density RFLP-AFLP map of tomato based on two Lycopersicon esculentum × L. pennellii F2 populations. Theor Appl Genet 99:254–271
Helyes L, Dimény J, Pék Z (2003) Effect of the variety and growing methods as well as cultivation conditions on the composition of tomato [Lycopersicon lycopersicum (L.) Karsten] fruit. Acta Hortic 712:511–516
Isidore E, van Os H, Andrzejewski S, Bakker J, Barrena I, Bryan GJ, Caromel B, van Eck HJ, Ghareeb B, de Jong W, van Koert P, Lefebvre V, Milbourne D, Ritter E, Rouppe van der Voort J, Rousselle-Bourgeois F, Van Vliet J, Waugh R (2003) Toward a marker-dense meiotic map of the potato genome: lessons from linkage group I. Genetics 165:2107–2116
Jansen RC, Geerlings H, van Oeveren J, Van Schaik RC (2001) A comment on codominant scoring of AFLP markers. Genetics 158:925–926
Kraakman ATW, Niks RE, Van den Berg PMMM, Stam P, Van Eeuwijk FA (2004) Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics 168:435–446
Kraft T, Hansen M, Nilsson NO (2000) Linkage disequilibrium and fingerprinting in sugar beet. Theor Appl Genet 101:323–326
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5(2):150–163
Meudt HM, Clarke AC (2007) Almost forgotten or latest practice? AFLP applications, analyses and advances. Trends Plant Sci 12(3):106–117
Miller JC, Tanksley SD (1990) RFLP analysis of phylogenetic relationships and genetic variation in the genus Lycopersicon. Theor Appl Genet 80(4):437–448
Mueller UG, LaReesa Wolfenbarger L (1999) AFLP genotyping and fingerprinting. Trends Ecol Evol 14:389–394
Murata Y, Hayashi K (1967) On a new, automatic device for leaf-area measurement. Proc Crop Sci Soc Jpn 36:463–467
Noli E, Cont S, Maccaferri M, Sanguineti MC (1999) Molecular characterization of tomato cultivars. Seed Sci Technol 27:1–10
Nordborg M, Borevitz JO, Bergelson J, Berry CC, Chory J, Hagenblad J, Kreitman M, Maloof JN, Noyes T, Oefner PJ, Stahl EA D, Weigel D (2002) The extent of linkage disequilibrium in Arabidopsis thaliana. Nat Genet 30:190–193
Park YH, West MAL, St. Clair DA (2004) Evaluation of AFLPs for germplasm fingerprinting and assessment of genetic diversity in cultivars of tomato (Lycopersicon esculentum L.). Genome 47:510–518
Piepho HP, Koch G (2000) Codominant analysis of banding data from a dominant marker system by normal mixtures. Genetics 155:1459–1468
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rouppe van der voort JNAM, van Zandvoort P, van Eck HJ, Folkertsma RT, Hutten RCB, Draaistra J, Gommers FJ, Jacobsen E, Helder J, Bakker J (1997) Use of allele specificity of comigrating AFLP markers to align genetic maps from different potato genotypes. Mol Gen Genet 255:438–447
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Spooner DM, Peralta IE, Knapp S (2005) Comparison of AFLP with other markers for phylogenetic inference in wild tomatoes [Solanum L. section Lycopersicon (Mill.) Wettst]. Taxon 54:43–61
Ursem R, Tikunov Y, Bovy A, van Berloo R, van Eeuwijk FA (2008) A correlation network approach to metabolite data analysis for tomato fruits. Euphytica. doi:10.1007/s10681-008-9672-y
Van Berloo R (1999) GGT: software for the display of graphical genotypes. J Hered 90(2):328–329
Van Berloo R (2008) Computer note: GGT 2.0: versatile software for visualization and analysis of genetic data. J Hered 99(2):232–236. http://www.plantbreeding.wur.nl/UK/software_ggt.html. Accessed 6 March 2008
Van Berloo R, Lindhout P (2001) Mapping disease resistance genes in tomato. In: Zhu D, Hawtin G, Wang Y (eds) Int Symp Biotechnol Applic Hortic Crops, vol 12. China Agricultural Scientech Press, Beijing, pp 343–356
Van Berloo R, Van Heusden S, Bovy A, Meijer-Dekens F, Lindhout P, van Eeuwijk F (2007) Genetic research in a public–private research consortium: prospects for indirect use of Elite breeding germplasm in academic research. Euphytica. doi:10.1007/s10681-007-9519-y
Villand J, Skroch PW, Lai T, Hanson P, Kuo CG, Nienhuis J (1998) Genetic variation among tomato accessions from primary and secondary centers of diversity. Crop Sci 38:1339–1347
Vos P, Hogers R, Bleeker M, Reijans M, van der Lee TH, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23(21):4407–4414
Vuylsteke M, Mank R, Antonise R, Bastiaans E, Senior ML, Stuber CW, Melchinger AE, Lübberstedt T, Xia XC, Stam P, Zabeau M, Kuiper M (1999) Two high-density AFLP® linkage maps of Zea mays L.: analysis of distribution of AFLP markers. Theor Appl Genet 99:921–935
Watts DJ, Strogatz SH (1998) Collective dynamics in small world networks. Nature 393:440–442
Williams CE, St. Clair DA (1993) Phenetic relationships and levels of variability detected by restriction fragment length polymorphism and random amplified fragment length polymorphic DNA analysis of cultivated and wild accessions of Lycopersicon esculentum. Genome 36:619–630
Zhao JJ, Wang X, Deng B, Ping L, Wu J, Sun R, Xu Z, Vromans J, Koornneef M, Bonnema G (2005a) Genetic relationships within Brassica rapa as inferred from AFLP fingerprints. Theor Appl Genet 110:1301–1314
Zhao HD, Nettleton D, Soller M, Dekkers JCM (2005b) Evaluation of linkage disequilibrium measures between multi-allelic markers as predictors of linkage disequilibrium between markers and QTL. Genet Res Camb 86:77–87
Acknowledgments
The breeding companies De Ruiter Hybrid Seeds, Enza Zaden, Rijk Zwaan NV, Seminis Vegetable Seeds and Syngenta Seeds kindly provided the seeds of the tomato cultivars used in this experiment. Keygene NV was responsible for the AFLP genotyping and kindly assisted in the extraction of quantitative gel intensity scores from their in house databases. Bioseeds BV is acknowledged for providing information on map positions from their proprietary integrated high-density tomato genetic map. This research was funded by the Centre for Biosystems Genomics and the joint PhD program between Wageningen University and the Chinese Academy of Agricultural Sciences.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Yu.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van Berloo, R., Zhu, A., Ursem, R. et al. Diversity and linkage disequilibrium analysis within a selected set of cultivated tomatoes. Theor Appl Genet 117, 89–101 (2008). https://doi.org/10.1007/s00122-008-0755-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-008-0755-x