
Abstract
Streptococcus pyogenes (group A Streptococcus (GAS)) causes ∼700 million human infections each year, resulting in over 500,000 deaths. The development of a commercial GAS vaccine is hampered by the occurrence of many unique GAS serotypes, antigenic variation within the same serotype, differences in serotype geographical distribution, and the production of antibodies cross-reactive with human tissue that may lead to autoimmune disease. Several independent studies have documented a number of GAS cell wall-associated or secreted metabolic enzymes that contain neither N-terminal leader sequences nor C-terminal cell wall anchors. Here, we applied a proteomic analysis of serotype M1T1 GAS cell wall extracts for the purpose of vaccine development. This approach catalogued several anchorless proteins and identified two protective vaccine candidates, arginine deiminase and trigger factor. These surface-exposed enzymes are expressed across multiple GAS serotypes exhibiting ≥99% amino acid sequence identity. Vaccine safety concerns are alleviated by the observation that these vaccine candidates lack human homologs, while sera from human populations suffering repeated GAS infections and high levels of autoimmune complications do not recognize these enzymes. Our study demonstrates anchorless cell surface antigens as promising vaccine candidates for the prevention of GAS disease.
Similar content being viewed by others


Introduction
The Gram-positive bacterium group A Streptococcus ((GAS), S. pyogenes) is a major human pathogen causing the ubiquitous infections of pharyngitis and impetigo and, less commonly, life-threatening invasive diseases including streptococcal toxic shock-like syndrome and necrotizing fasciitis [1]. Repeated infections may trigger a suite of non-suppurative immune sequelae including acute rheumatic fever, rheumatic heart disease, and acute post-streptococcal glomerulonephritis [1]. Globally, GAS causes ∼700 million human infections each year, resulting in over 500,000 deaths [2].
A safe and efficacious commercial vaccine to prevent GAS infection has yet to be developed, and this critical public health goal is fraught with pitfalls posed by the pathogen’s unique phenotypic features. In contrast to Streptococcus pneumoniae, where commercial vaccines have been developed using conjugated capsular polysaccharides (CPS) of the most common serotypes, the CPS of all GAS strains is a homopolymer of hyaluronic acid, a common sugar in human connective tissues, and therefore immunologically inert. The antigenically diverse surface-anchored GAS M proteins are capable of eliciting protective immunity; however, comparison of the encoding (emm) genes indicates sequence diversity in the N-terminal hypervariable region, and adjacent ‘A’ repeat domains generate over 100 distinct GAS emm types [3]. Regions of the M protein dimeric coiled-coil structure are implicated in eliciting cross-reactive antibodies against human cardiac and basal ganglia tissue that are key to the immuno-pathogenesis of rheumatic fever, thereby raising important concerns regarding vaccine safety. Nonetheless, immune sera raised against 30 defined amino-terminal M protein fragments in the 30-valent experimental M protein vaccine did not cross-react with human brain, kidney, or heart tissue in indirect immunofluorescence assays. These authors also demonstrated considerable cross-protection of the 30-valent vaccine against non-vaccine M serotypes [4].
Recently, a series of independent studies have documented a class of GAS cell wall-associated or secreted metabolic enzymes that contain neither N-terminal leader sequences nor C-terminal cell wall anchors [5–9]. Perhaps because these molecules do not fit common categories of bacterial vaccine antigen, e.g., carbohydrate capsule, surface-anchored protein, or secreted toxin, their potential as immunogens has not been pursued. Here, we adopt a post-proteomic approach to reveal the vaccine efficacy of selected highly conserved and non-human cross-reactive anchorless cell wall-associated enzymes against lethal GAS infection.
Materials and methods
GAS strains used in this study
The S. pyogenes strains utilized in this study have been described previously: 5448 (emm1) [8], 5448AP (emm1) [10], HSC5 (emm14) [11], NS88.2 (emm98.1) and NS192 (emm100) [12, 13], pM1 (emm1) [14], 2036 (emm6) [15], DSM2071 (emm23) [16], NV-2728 (emm12) (Novartis Vaccines, Sienna, Italy), pL1 (emm54) (M. Batzloff, unpublished), and 20174 (emm3) (M. Kotb, unpublished).
Protein identification by two-dimensional electrophoresis
To identify the major cell wall-associated proteins of invasive M1T1 clonal 5448 S. pyogenes isolate [8] grown in the presence of the cysteine protease inhibitor E64, a two-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) proteomic analysis of a mutanolysin cell wall extract was performed as previously described [17].
Construction of expression plasmids and recombinant protein purification
The genes sagP (Genbank accession number AF468045), ropA (AAM80241), and fba (AAK36400) encode arginine deiminase (ADI), trigger factor (TF), and fructose bisphosphate aldolase (FBA), respectively. Primers were designed (Table 1) to amplify the genes from GAS strain 5448 for recombinant gene expression and the resultant recombinant proteins purified as described previously for ropA [18]. The production of recombinant M1 protein is described elsewhere [19].
Preparation of pepsin M protein extracts
Extracts were prepared as previously described [20].
ELISA
ELISA was performed as previously described [15], and the titer designated the highest serum dilution with an absorbance (450 nm) cut-off greater than 0.2.
Immunization and intraperitoneal challenge of mice with GAS
Three immunization and intraperitoneal challenge experiments were performed. For the first experiment, 4- to 6-week-old (Animal Resource Centre, Australia) female BALB/c mice (n = 20 for test antigens; n = 30 for controls) were subcutaneously immunized with 10 μg recombinant protein adjuvanted with complete Freund’s adjuvant (CFA) in the primary immunization. Two mice immunized with recombinant ADI died of incidental causes prior to challenge. Mice were challenged via the intraperitoneal route on day 56 with approximately 8 × 106 CFU of GAS M1 serotype strain pM1 [14]. Results of independent replicates (n = 10) were combined for each test antigen. Mice were monitored twice a day for 10 days after challenge and euthanized when they exhibited defined humane endpoints approved by University of Wollongong and Queensland Institute of Medical Research Animal Ethics Committees. For the second experiment, 5-week-old (Charles River, Italy) female CDI mice (n = 48 for ADI, ADI and TF combination, and Alum negative control; n = 42 for TF; n = 24 for M protein positive control) were intraperitoneally immunized with 10 μg recombinant protein combined with Alum on days 0, 21, and 35. Groups of mice were challenged via the intraperitoneal route on day 55 with 1.35 × 106 CFU of GAS serotype M1 strain pM1. Results of three independent replicates were combined and presented for each test antigen. For the third experiment, 5-week-old (Charles River, Italy) female CDI mice (n = 48 for ADI and TF combination, and Alum negative control; n = 24 for M protein positive control) were intraperitoneally immunized with 10 μg recombinant protein combined with Alum on days 0, 21, and 35. Groups of mice were challenged via the intraperitoneal route on day 55 with 1.2 × 102 CFU of GAS serotype M12 strain M12-2728. Results of three independent replicates were combined and presented for each test antigen. In experiments 2 and 3, mice were monitored on a daily basis for at least 1 week after treatment and euthanized when they exhibited defined humane endpoints that had been pre-established for the study in agreement with Novartis Animal Welfare Policies. Experimental protocols described in this report complied with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (National Health and Medical Research Council, Australia).
Intraperitoneal immunization and subcutaneous challenge of mice with a hypervirulent covS mutant GAS strain
Four- to six-week-old female C57BL/J6 mice (Animal Resource Centre, Australia) (n = 40 for ADI and TF; n = 21 for ADI and TF combination; n = 80 for M1 protein positive control; n = 79 for PBS negative control) were immunized on days 0, 21, and 28 with 10 μg recombinant protein adjuvanted with CFA in the primary immunization. One PBS negative control mouse died of incidental causes prior to challenge. Mice were challenged via the subcutaneous route on day 56 with approximately 2 × 108 CFU of hypervirulent covS mutant GAS strain 5448AP of serotype 1 [10]. Results of independent replicates (n = 10) were combined for each test antigen. Mice were monitored twice a day for 10 days after challenge and euthanized when they exhibited defined humane endpoints approved by the University of Wollongong Animal Ethics Committee.
Opsonophagocytosis killing assay
GAS strain 5448 was grown to stationary phase and the opsonophagocytosis killing assay performed with the specific polyclonal mouse serum raised against M1 protein, ADI, or TF (adjuvanted with CFA), as previously described [19]. Two biological replicates were undertaken using quadruplicate samples.
Detection of cell wall-associated proteins in cell wall extracts
SDS-PAGE and Western blotting was performed as described previously [21]. The primary serum was polyclonal mouse anti-serum directed against each recombinant protein (adjuvanted with CFA), while the secondary anti-serum was goat anti-mouse IgG–horseradish peroxidise (HRP) conjugate (Chemicon). Antibodies were diluted 1:2,000.
Detection of proteins on GAS cell surface using flow cytometry
Flow cytometry was utilized to determine whether candidate antigens were localized on the surface of GAS isolates 5448 (M1T1) [8], pM1 (M1) [14], and pL1 (M54) using pooled polyclonal mouse anti-sera (n = 10) directed against each recombinant protein (adjuvanted with CFA) and M12-2728 (M12) using pooled polyclonal mouse anti-sera (n = 10) directed against each recombinant protein (adjuvanted with Alum). Incubations were performed for either 1 h on ice or overnight at 4°C. A 1-mL volume of logarithmic-phase bacteria (OD600 = 0.6) was washed twice in filtered phosphate buffered saline (PBS) and non-specific binding sites and Ig-binding proteins blocked with 3% (w/v) bovine serum albumin (BSA)/PBS with Tween 20 (PBST) followed by normal human IgG diluted 1:150 in 0.3% (w/v) BSA/PBST, respectively. Cells were washed and incubated with primary antibody diluted 1:50 in 0.3% (w/v) BSA/PBST. Anti-M Protein or anti-pep M extract serum and serum from mice immunized with PBS were utilized as the positive and negative controls, respectively. Bacteria were washed and incubated with the secondary antibody, goat anti-mouse fluorescein isothiocyanate (FITC)-conjugated IgG (Zymed, USA) diluted 1:150 in 0.3% (w/v) BSA/PBST. Finally, cells were washed and fixed with 4% (w/v) paraformaldehyde/PBS and either analyzed in a FACSort flow cytometer (Becton Dickson, USA) with CellQuest software (version 3.1f) or a BD Accuri C6 flow cytometer (BD Accuri Cytometers, USA) with BD CFlow software. Cell-surface-associated FITC-fluorescence (excitation, 488 nm; emission, 520 nm) was analyzed using a uniform population of gated cells. Histograms displaying FL-1 (FITC) fluorescence were prepared using FlowJo software (version 7.2.5). Specific antibody binding was calculated by subtracting the average geometric mean of the no primary antibody control from the test sample. Each antigen was tested in triplicate.
Testing the reactivity of human endemic serum with recombinant proteins
The reactivity of recombinant protein antigens with pooled serum was tested by ELISA as described previously [22]. Human serum was obtained from two populations (1) school-aged Aboriginal children (n = 30) living in remote communities in the Northern Territory, Australia; (2) Indian children and young adults (9-22 years old) living in Chandrigarh, India, pooled into the following categories: individuals without disease symptoms (n = 10); patients with pharyngitis (n = 8); patients with acute rheumatic fever (n = 9); patients with rheumatic heart disease (n = 10). Test serum was diluted twofold starting with a 1:100 dilution, and the secondary antibody, goat anti-human IgG-HRP (Bio-Rad), was diluted 1:2,000. Recombinant M1 protein was incubated with a 1:200 dilution of Aboriginal serum or Indian rheumatic heart disease serum on plates as a control to normalize data. Two experimental biological replicates were performed.
Production of polyclonal rabbit serum
Rabbit polyclonal serum was raised against ADI, TF, or M1 protein according to previously stated methods [22].
Cross-reactivity of candidate antigen polyclonal sera with human heart extract
Human heart tissue (5 g) was homogenized in 5 mL of PBS containing complete protease inhibitor cocktail (Roche) using a PolyTron PT2100. Debris was removed by centrifugation at 12,000×g for 30 min at 4°C. The reactivity between human heart proteins and test serum from rabbits immunized with individual recombinant antigens was tested by ELISA. Ninety-six-well plates were coated with 20 μg/mL of the human heart extract in 0.1 M carbonate coating buffer (pH 9.6) and blocked with 1% (w/v) gelatin in PBS. The test serum was diluted twofold starting with a 1:10 dilution in 1% (w/v) gelatin. Naive rabbit serum was utilized as a negative control. The secondary antibody, goat anti-rabbit IgG-HRP (Dianova), was diluted 1:3,000 in 1% (w/v) gelatine. Two biological replicates were undertaken using duplicate samples.
Transmission immunogold electron microscopy
GAS cells were grown to stationary phase and then immuno-gold-labeled and visualized as previously described [21].
Detection of proteins on the cell surface using immunofluorescence microscopy
Confocal immunofluorescence microscopy was performed as previously described [23] using a 100X/1.0 FLUOTAR PL oil immersion objective lens with a Leica TCS SP confocal microscope mounted on a Leica DM IRBE inverted microscope (Leica Microsystems, Germany) configured for the detection of FITC.
Statistical analyses
Differences in survival of mice immunized with recombinant protein antigens compared with sham-immunized mice were determined by the log-rank test. Differences were considered statistically significant at P < 0.05. Statistical analysis of human serum ELISA and human heart extract cross-reactivity ELISA were performed using a one-way analysis of variance. To determine the shift in cell surface fluorescence, the geometric mean of the flow cytometry histograms resulting from the surface labelling of the test serum were compared with the geometric mean of the sham-immunized serum histogram using an unpaired t test with a confidence interval of 95%. All statistical tests were performed using GraphPad™ Prism software version 4.2.
Results
Proteomic analysis of GAS cell wall extracts
Proteomic analysis of GAS cell wall extract was undertaken using isolate 5448, a representative of the globally disseminated M1T1 clonal serotype [8]. The cell wall extract was separated by two-dimensional gel electrophoresis and the landmark proteins identified by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. In total, 31 distinct cell-wall-associated proteins were identified (Fig. 1a; Supplementary Table 1). Among these were metabolic enzymes that share significant amino acid sequence identity with human homologs (e.g., GAS α-enolase shares 49% identity with human α-enolase), which raises concern for autoimmune consequences [24]. For further characterization, we chose three candidates that are not represented in the human proteome and do not share significant amino acid sequence identity with other human proteins as determined by BLAST-P interrogation (Supplementary Table 1): ADI [25, 26] and TF [27, 28], which are potential GAS virulence determinants, and FBA, which affords vaccine protection against S. pneumoniae [29]. All three enzymes lack N-terminal signal sequences and C-terminal cell wall anchors [7]. The genes encoding these three candidate antigens are highly conserved, sharing ≥99% amino acid sequence identity. Each enzyme is detected in cell wall fractions across a range of disparate GAS M serotypes (Fig. 1b).

Identification, serotype distribution, and surface localization of novel GAS antigens. a Representative two-dimensional gel electrophoresis profile of GAS strain 5448 mutanolysin cell wall extract. Protein spots identified by peptide mass mapping following trypsin digest and MALDI-TOF MS are denoted by numbered arrows. Arrows numbered 18 correspond to ADI, arrow number 13 corresponds to TF, arrow number 10 corresponds to FBA. Molecular mass markers are indicated in kilo-Daltons (kDa). b Western blot detection of cell surface-associated proteins (ADI, TF, and FBA) in cell wall extracts of eight representative GAS isolates (emm type provided here in brackets); from left to right, 5448 (emm1), NS88.2 (emm98.1), NS192 (emm100), DSM2071 (emm23), HSC5 (emm14), 20174 (emm3), pM1 (emm1), 2036 (emm6). Multiple blots were used to compile image. The size of each protein is indicated in kilo-Daltons (kDa). c Quantification of antigens on the surface of GAS M1 isolate 5448 using flow cytometry. Histogram for test antigens and M1 protein control indicated with solid line, histogram for sham-immunized serum denoted by shading. ADI, TF, and M1 protein were observed to have a significant difference in shift (P < 0.05) in comparison to the sham-immunized serum, which is indicated with an asterisk. Samples were analyzed in triplicate. d Detection of anchorless surface proteins on the surface of GAS strain 5448 using immuno-electron microscopy. Specific polyclonal rabbit anti-serum (upper panel) shows accumulation of gold particles (indicated by arrows) at the cell surface in comparison to the corresponding pre-immune serum (lower panel). Scale bars represent 200 nm. (e) Detection of M1 protein, ADI, TF, and FBA via immunofluorescence on the surface of GAS strain pM1 using anti-sera from BALB/c mice (n = 10) immunized with the cell surface-associated antigens and stained with goat anti mouse IgG-FITC. Fluorescence image (upper panel) and transmission image (lower panel). Scale bars indicate 5 μm. For each experiment (a–e), two independent replicates were prepared, and representative data are presented
Surface localization of candidate antigens
To investigate the presentation of the three vaccine candidates on the GAS cell surface, immunofluorescence microscopy, flow cytometry, and immuno-electron microscopy were undertaken utilizing antigen-specific polyclonal anti-serum. Anti-serum raised against the well-characterized surface-bound M protein was used as the positive control, while serum from mice immunized with PBS, or animal matched pre-immune serum was used as the negative controls for this series of experiments. Polyclonal anti-serum specific for M1 protein, ADI, TF, and FBA clearly bound to the GAS cell surface, whereas pre-immune serum (Fig. 1d), or sham immunized serum did not react (Fig. 1e). When tested on GAS isolates 5448 (M1), pM1 (M1), and pL1 (M54) using flow cytometry, in comparison to sham-immunized serum, polyclonal sera raised against full-length M protein or pepsin M extract, ADI or TF were observed to produce a significant shift in fluorescence (P < 0.05; Figs. 1c and 3a). Following surface binding to the M12-2728 (M12) GAS isolate, anti-TF and anti-M protein sera produced a significant shift in fluorescence compared with PBS sham sera in flow cytometry (P < 0.05; Fig. 3a). Anti-ADI serum also resulted in a shift, which was not statistically significant (P > 0.05; Fig. 3a).
Anchorless proteins elicit protective immunity
Conjecture exists in the scientific literature regarding the biological relevance and certitude of surface expression of anchorless proteins in GAS [5, 30]. To validate our findings and to test protective vaccine efficacy of the selected anchorless surface proteins, full-length M1 protein, ADI, TF, and FBA were adjuvanted with CFA and used to subcutaneously immunize BALB/c mice and the survival recorded following lethal intraperitoneal M1 GAS challenge. Immunization of mice with anchorless proteins adjuvanted with CFA resulted in high titer serum IgG antibodies (Fig. 2a), demonstrating that robust antibody responses are engendered following vaccination. Following lethal intraperitoneal M1 GAS challenge, BALB/c mice immunized with full-length M1 protein, ADI, and TF were observed to have significantly increased survival (P < 0.05), compared with sham-immunized mice (Fig. 2c). As experimental immunization with FBA (P > 0.05) failed to protect against lethal intraperitoneal M1 GAS challenge (Fig. 2c), the protective efficacy of FBA was not further evaluated. Next, we explored the capacity of ADI and TF to elicit protective immunity using Alum, an adjuvant approved for human use. Immunization of CD1 mice with anchorless proteins adjuvanted with Alum were again observed to have significantly increased survival (P < 0.05), compared with sham-immunized mice (Fig. 2d).

ADI, TF, and FBA antigens produce high-titer antibodies and protect against lethal intraperitoneal GAS challenge. a Serum-specific IgG antibody titers in BALB/c mice (n = 10) following subcutaneous immunization of antigen on days 0, 21, and 28. Mice serum was tested for specific reactivity on days 0, 20, 27, and 41. The absorbance cut-off for the determination of titer was 0.2. Red bars represent the arithmetic mean; two independent experimental repeats were performed. Serum obtained from sham-immunized mice did not produce measurable titers against ADI, TF, FBA, or M1 protein. b Percent survival of the serotype M1 GAS strain 5448 following opsonophagocytosis killing assay. GAS were pre-incubated with specific polyclonal mouse serum raised against M1 protein, ADI, or TF (serum from PBS sham-immunized mice was included as a negative control) and subsequently incubated with normal heparinized human blood. Quadruplicate data from two repeat experiments are presented. Percent survival of GAS was calculated in comparison to the number of GAS (colony-forming units per milliliter) surviving in the PBS sham-immunized serum control. Asterisk indicates a significant difference (P < 0.01) between the test antiserum and the PBS sham-immunized control serum. c Kaplan–Meier survival curves for intraperitoneal challenge of immunized BALB/c mice with a lethal dose of pM1 GAS, 8 × 106 colony-forming units (CFU) (n = 18 for ADI; n = 20 for TF and FBA). M1 protein (n = 30) was selected as a positive control, while PBS was the negative control (n = 30). Antigens were co-administered with CFA in the primary immunization. Two independent replicates were performed for ADI, TF, and FBA; three independent replicates were performed for the controls, M1 and PBS; each repeat contained n = 10 mice for each antigen or control. d Kaplan–Meier survival curves for intraperitoneal challenge of CD1 mice with 1.35 × 106 CFU of pM1 GAS (n = 48 for ADI, n = 42 for TF). M1 protein (n = 24) was used as a positive control, while PBS was the negative control (n = 46). Antigens were co-administered with Alum. Three independent replicates were performed; all data are presented
Invasive GAS infections are known to be caused by both wild-type and hypervirulent covRS forms with differing transcriptional profiles [31]. Thus, an alternate GAS challenge model was explored in which CFA-adjuvanted full-length M1 protein, ADI, TF, or a combination of ADI and TF were used to immunize C57BL/J6 mice prior to subcutaneous challenge with the hypervirulent covS mutant M1 strain, 5448AP [10]. Immunization with ADI alone resulted in a trend toward increased survival (P > 0.05), while immunization with M1 protein or the 1:1 combination of ADI and TF engendered protective immunity against subcutaneous challenge with 5448AP (P < 0.05) (Fig. 3b). The ability of ADI and TF to confer protective immunity against heterologous GAS challenge was investigated. Intraperitoneal immunization of CD1 mice with M12 protein or a 1:1 cocktail of ADI and TF adjuvanted with Alum resulted in significant protection against intraperitoneal challenge with the M12 GAS strain M12-2728 (P < 0.05) (Fig. 3c). The observed protection of mice may be due to a concert of factors including antibody-mediated opsonophagocytic killing of GAS (Fig. 2b), antibody-mediated neutralization of the enzymatic activity of ADI or TF, or non-antibody-mediated mechanisms.

a Quantification of antigens on the surface of GAS isolates pM1, pL1, and M12-2728 using flow cytometry. Histogram for test antigens and positive control indicated with solid line; histogram for sham-immunized serum denoted by shading. Sera raised against M protein, pepsin M protein extracts, ADI, and TF produced a shift in cell surface FITC fluorescence compared with PBS sham serum. A significant shift in fluorescence (P < 0.05) is indicated with an asterisk. Samples were analyzed in triplicate. b Kaplan–Meier survival curves for subcutaneous challenge of C57BL/J6 mice (co-immunized with Freund’s complete adjuvant) with a lethal dose of 5448AP GAS, 2 × 108 CFU (n = 40 for ADI, n = 40 for TF, n = 21 for ADI + TF cocktail). M1 protein (n = 80) was selected as a positive control, while PBS was the negative control (n = 79). At least two independent repeats were performed; all data are presented. c Kaplan–Meier survival curves for intraperitoneal challenge of CD1 mice (co-immunized with Alum) with 1.2 × 102 CFU of M12-2728 GAS (n = 48 for ADI + TF cocktail). M12 protein (n = 24) was selected as a positive control, while Alum was the negative control (n = 48). Three independent replicates were performed; all data are presented
Reactivity of candidate antigens with human sera and cross-reactivity with human heart tissue
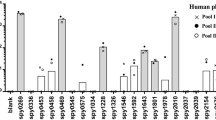
The Aboriginal population of the Northern Territory of Australia suffers some of the highest reported rates of GAS disease and autoimmune sequelae [32]. We examined whether members of this population have pre-existing immunity against ADI and TF, hypothesizing that an immune response against these protective antigens may be lacking in a human population that suffers repeated GAS infections [33]. The Aboriginal immune response was determined using a pool of serum (n = 30) obtained from Aboriginal children living in remote communities of the Northern Territory suffering endemic GAS infection. In stark comparison to M1 protein (P < 0.001), there were minimal serum antibody responses directed against ADI and TF (Fig. 4a). The human immune response mounted against the anchorless proteins was also investigated using four pools of serum isolated from individuals living in Chandigarh, India, classified into the following clinical disease categories: individuals living in endemic area showing no symptoms (n = 10), pharyngitis (n = 8), acute rheumatic fever (n = 9), and rheumatic heart disease (n = 10). The serum antibody response detected against ADI and TF for each of these four pools of sera was significantly less than the response engendered by M1 protein (P < 0.01) (Fig. 4b–e). Finally, polyclonal M1 protein antiserum strongly cross-reacted with human heart extract, in contrast to anti-ADI and anti-TF polyclonal sera (Fig. 4f).

Reactivity of antigens with human serum and cross-reactivity with human heart extract. The human IgG response against M1 protein, ADI, and TF was determined using a a pool of serum (n = 30) from Aboriginal children living in remote communities of the Northern Territory (Australia) suffering endemic GAS infection, b a pool of serum (n = 10) from individuals with no disease symptoms in Chandigarh (India), c a pool of serum (n = 8) from patients with pharyngitis in Chandigarh (India), d a pool of serum (n = 9) from patients with acute rheumatic fever in Chandigarh (India), and e a pool of serum (n = 10) from patients with rheumatic heart disease in Chandigarh (India). f Reactivity of rabbit polyclonal antisera raised against M1 protein, ADI, or TF with human heart extract. Asterisk: response against M1 protein is significantly higher than test antigens (P < 0.01). Error bars represent the standard error of the mean. For each component of figure (a–f), duplicate data from two independent replicate experiments are presented
Discussion
GAS is a major human pathogen causing ∼700 million human infections each year, resulting in over 500,000 deaths [2]. Although several surface proteins and virulence factors have been previously assessed as vaccine candidates [34], there is no commercial vaccine available to prevent GAS infection. Following a proteomic analysis of an M1T1 cell wall extract, we selected three anchorless proteins: ADI, FBA, and TF for further characterization as putative GAS vaccine candidates.
Anchorless proteins are highly conserved and are localized on the surface of GAS
Several previously characterized GAS vaccine candidates: M protein, SfbI, SOF, and FbaA [34] are not conserved between GAS serotypes or expressed by all serotypes of GAS. The genes encoding each of the anchorless vaccine antigens of this study were ubiquitous among sequenced GAS genomes with ≥99% conservation among serotypes. In addition, the anchorless vaccine antigens were detected in mutanolysin-derived cell wall extracts of eight representative clinical GAS isolates (Fig. 1b). While the anchorless proteins lack signal sequence(s) for export and the traditional LPXTG motif known to anchor Gram-positive surface proteins to the cell wall, they were detected on the GAS cell surface using immunofluorescent microscopy (Fig. 1e), flow cytometry (Figs. 1c and 3a), and immuno-EM (Fig. 1d). At this stage, their mechanism of export is unknown. Anchorless GAS cell surface-associated proteins of cytoplasmic origin is by no means a new phenomenon. The glycolytic surface-exposed plasminogen-binding virulence factors of GAS, α-enolase [35], and GAPDH [36] are established examples of two traditionally cytoplasmic proteins lacking a LPXTG motif located on the surface of GAS possessing a dual biochemical and virulence-associated functionality. In addition to ADI, TF, and FBA, both α-enolase and GAPDH were among the 31 proteins identified in the proteomic analysis of GAS cell wall extracts (Fig. 1a).
Anchorless proteins protect against lethal GAS challenge
When adjuvanted with CFA or the human-approved adjuvant Alum, ADI and TF elicited a protective immune response against intraperitoneal challenge with a homologous M1 isolate (Fig. 2c, d). Anti-ADI and anti-M1 protein sera generated following immunization of mice resulted in the killing of M1 GAS in whole human blood (Fig. 2b). Opsonophagocytic killing of GAS is hypothesized to play a role, at least in part, in the efficacy of protective GAS antigens by facilitating bacterial clearance in the infected host. When administered in a two-antigen cocktail, the combination ADI and TF was observed to act synergistically, conferring significant protection against lethal subcutaneous M1 GAS challenge (P < 0.05; Fig. 3b) and against intraperitoneal challenge with a heterologous M12 isolate (P < 0.05; Fig. 3c). It is possible that vaccine preparations containing a number of protective GAS antigens (or fragments thereof) may result in complete protection. The addition of ubiquitiously expressed antigens such as ADI or TF to multi-antigen GAS vaccine preparations may help overcome issues of serotype-dependent protection in cases where the expression of the individual antigens is limited to a subset of serotypes.
Anchorless proteins do not react with human sera or heart tissue
During early immunization studies of human subjects with GAS vaccine preparations, the administration of partially purified M3 protein was reported to result in three cases of acute rheumatic fever [37]. It has been subsequently established that the host immune response to M protein can produce antibodies that react with host tissue such as cardiac myosin [38]. As a consequence, there has been a heightened concern that host antibodies generated against GAS vaccine antigens may cross-react with human proteins and tissues potentially leading to auto-immune disease. To address the issue of cross-reactivity, we tested the capacity of human serum collected from individuals living in an area of endemic GAS disease to react with the anchorless antigens. The lack of an immune response against ADI and TF in both these populations, which have high rates of autoimmune sequelae [32], suggest that these proteins are not involved in triggering autoimmune disease. Moreover, the lack of an immune response against these potentially protective antigens, despite repeated infection, suggests that GAS may have evolved a mechanism to shield these antigens from the normal host immune response. When incubated directly with human heart extract, polyclonal anti-ADI and anti-TF serum antibodies did not significantly react (P > 0.05; Fig. 4f), in contrast to anti-M1 protein serum. These results suggest that, unlike full-length M protein, the B cell epitopes contained within ADI and TF do not cross-react with human cardiac proteins.
In conclusion, two anchorless cell surface proteins of GAS, ADI, and TF, were identified in this study and found to confer protection against lethal GAS challenge in a variety of mouse models. These protective vaccine candidates are highly conserved, are expressed across multiple GAS serotypes, and lack significant amino acid sequence identity with any known human protein. Furthermore, sera from human populations suffering repeated GAS infections and high levels of autoimmune complications did not recognize these enzymes, and polyclonal antiserum raised against ADI and TF did not cross-react with human heart extract. This novel class of antigen warrants further investigation as vaccine candidates for the prevention of GAS disease, a major cause of morbidity and mortality worldwide.
References
Tart AH, Walker MJ, Musser JM (2007) New understanding of the group A Streptococcus pathogenesis cycle. Trends Microbiol 15:318–325
Carapetis JR, Steer AC, Mulholland EK, Weber M (2005) The global burden of group A streptococcal diseases. Lancet Infect Dis 5:685–694
Johnson DR, Kaplan EL, VanGheem A, Facklam RR, Beall B (2006) Characterization of group A streptococci (Streptococcus pyogenes): correlation of M-protein and emm-gene type with T-protein agglutination pattern and serum opacity factor. J Med Microbiol 55:157–164
Dale JB, Penfound TA, Chiang EY, Walton WJ (2011) New 30-valent M protein-based vaccine evokes cross-opsonic antibodies against non-vaccine serotypes of group A streptococci. Vaccine 29:8175–8178
Severin A, Nickbarg E, Wooters J, Quazi SA, Matsuka YV, Murphy E, Moutsatsos IK, Zagursky RJ, Olmsted SB (2007) Proteomic analysis and identification of Streptococcus pyogenes surface-associated proteins. J Bacteriol 189:1514–1522
Lei B, Mackie S, Lukomski S, Musser JM (2000) Identification and immunogenicity of group A streptococcus culture supernatant proteins. Infect Immun 68:6807–6818
Cole JN, Ramirez RD, Currie BJ, Cordwell SJ, Djordjevic SP, Walker MJ (2005) Surface analysis and immune reactivity of major cell-wall associated proteins of group A Streptococcus. Infect Immun 73:3137–3146
Aziz RK, Pabst MJ, Jeng A, Kansal R, Low DE, Nizet V, Kotb M (2004) Invasive M1T1 group A Streptococcus undergoes a phase-shift in vivo to prevent proteolytic degradation of multiple virulence factors by SpeB. Mol Microbiol 51:123–134
Walker MJ, McArthur JD, McKay F, Ranson M (2005) Is plasminogen deployed as a Streptococcus pyogenes virulence factor? Trends Microbiol 13:308–313
Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, McArthur JD, Dinkla K, Aziz RK, Kansal RG et al (2007) DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med 13:981–985
Lyon WR, Caparon MG (2004) Role for serine protease HtrA (DegP) of Streptococcus pyogenes in the biogenesis of virulence factors SpeB and the hemolysin streptolysin S. Infect Immun 72:1618–1625
McKay FC, McArthur JD, Sanderson-Smith ML, Gardam S, Currie BJ, Sriprakash KS, Fagan PK, Towers RJ, Batzloff MR, Chhatwal GS et al (2004) Plasminogen binding by group A streptococcal isolates from a region of hyperendemicity for streptococcal skin infection and a high incidence of invasive infection. Infect Immun 72:364–370
Ramachandran V, McArthur JD, Behm CE, Gutzeit C, Dowton M, Fagan PK, Currie B, Sriprakash KS, Walker MJ (2004) Two distinct genotypes of prtF2, encoding a fibronectin binding protein, and the evolution of the gene family in Streptococcus pyogenes. J Bacteriol 186:7601–7609
Batzloff MR, Yan H, Davies MR, Hartas J, Lowell GH, White G, Burt DS, Leanderson T, Good MF (2005) Toward the development of an anti-disease, transmission-blocking intranasal vaccine for group A Streptococcus. J Infect Dis 192:1450–1455
Batzloff MR, Hayman WA, Davies MR, Zeng M, Pruksakorn S, Brandt ER, Good MF (2003) Protection against group A Streptococcus by immunisation with J8-diphtheria toxoid: contribution of J8- and diphtheria toxoid-specific antibodies to protection. J Infect Dis 187:1598–1608
Guzmán CA, Talay SR, Molinari G, Medina E, Chhatwal GS (1999) Protective immune response against Streptococcus pyogenes in mice after intranasal vaccination with fibronectin-binding protein SfbI. J Infect Dis 179:901–906
Ji Y, Schnitzler N, DeMaster E, Cleary PP (1998) Impact of M49, Mrp, Enn, and C5a Peptidase proteins on colonization of the mouse oral mucosa by Streptococcus pyogenes. Infect Immun 66:5399–5405
Cole JN, Aquilina JA, Hains PG, Henningham A, Sriprakash KS, Caparon MG, Nizet V, Kotb M, Cordwell SJ, Djordjevic SP et al (2007) Role of group A Streptococcus HtrA in the maturation of SpeB protease. Proteomics 7:4488–4498
Sanderson-Smith M, Batzloff MR, Sriprakash KS, Dowton M, Ranson M, Walker MJ (2006) Divergence in the plasminogen-binding group a streptococcal M protein family: functional conservation of binding site and potential role for immune selection of variants. J Biol Chem 281:3217–3226
Beachey EH, Stollerman G, Chiang EY, Seyer JM, Kang AH (1977) Purification and properties of M protein extracted from group A streptococci with pepsin: covalent structure of the amino terminal region of type 24 M antigen. J Exp Med 145:1469–1483
Cole JN, McArthur J, McKay FC, Sanderson-Smith M, Cork AJ, Ranson M, Rohde M, Itzek A, Sun H, Ginsburg D et al (2006) Trigger for group A streptococcal M1T1 invasive disease. FASEB J 20:E1139–E1145
Gillen CM, Towers RJ, McMillan DJ, Delvecchio A, Sriprakash KS, Currie B, Kreikemeyer B, Chhatwal GS, Walker MJ (2002) Immunological response mounted by Aboriginal Australians living in the Northern Territory of Australia against Streptococcus pyogenes serum opacity factor. Microbiology 148:169–178
Brandt ER, Hayman WA, Currie B, Carapetis JR, Wood Y, Jackson DC, Cooper J, Melrose WD, Saul AJ, Good MF (1996) Opsonic human antibodies from an endemic population specific for a conserved epitope on the M protein of group A streptococci. Immunology 89:331–337
Fontán PA, Pancholi V, Noclari MM, Fischetti VA (2000) Antibodies to streptococcal surface enolase react with human a-enolase: implications in poststreptococcal sequelae. J Infect Dis 182:1712–1721
Degnan BA, Fontaine MC, Doebereiner AH, Lee JJ, Mastroeni P, Dougan G, Goodacre JA, Kehoe MA (2000) Characterisation of an isogenic mutant of Streptococcus pyogenes Manfredo lacking the ability to make streptococcal acid glycoprotein. Infect Immun 68:2441–2448
Degnan BA, Palmer JM, Robson T, Jones CED, Fischer M, Glanville M, Mellor GD, Diamond AG, Kehoe MA, Goodacre JA (1998) Inhibition of human peripheral blood mononuclear cell proliferation by Streptococcus pyogenes cell extract is associated with arginine deiminase activity. Infect Immun 66:3050–3058
Lyon WR, Caparon MG (2003) Trigger Factor-mediated prolyl isomerization influences maturation of the Streptococcus pyogenes cysteine protease. J Bacteriol 185:3661–3667
Yang X, Walters N, Robison A, Trunkle T, Pascual DW (2007) Nasal immunization with recombinant Brucella melitensis bp26 and trigger factor with cholera toxin reduces B. melitensis colonization. Vaccine 25:2261–2268
Ling E, Feldman G, Portnoi M, Dagan R, Overweg K, Mulholland F, Chalifa-Caspi V, Wells J, Mizrachi-Nebenzahl Y (2004) Glycolytic enzymes associated with the cell surface of Streptococcus pneumoniae are antigenic in humans and elicit protective immune responses in the mouse. Clin Exp Immunol 138:290–298
Rodreiguez-Ortega MJ, Norais N, Bensi G, Liberatori S, Capo S, Mora M, Scarselli M, Doro F, Ferrari G, Garaguso I et al (2006) Characterization and identification of vaccine candidate proteins through analysis of the group A Streptococcus surface proteome. Nat Biotechnol 24:191–197
Cole JN, Barnett TC, Nizet V, Walker MJ (2011) Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol 9:724–736
Carapetis JR, Wolff DR, Currie BJ (1996) Acute rheumatic fever and rheumatic heart disease in the top end of Australia’s Northern Territory. Med J Aust 164:146–149
Currie BJ, Carapetis JR (2000) Skin infections and infestations in Aboriginal communities in northern Australia. Australas J Dermatol 41:139–145
Cole JN, Henningham A, Gillen CM, Ramachandran V, Walker MJ (2008) Human pathogenic streptococcal proteomics and vaccine development. Proteomics Clin Appl 2:387–410
Pancholi V, Fischetti VA (1998) a-enolase, a novel strong plasmin(ogen) binding protein on the surface of pathogenic streptococci. J Biol Chem 273:14503–14515
Pancholi V, Fischetti VA (1992) A major surface protein on group A streptococci is glyceraldehyde-3-phosphate dehydrogenase with multiple binding activity. J Exp Med 176:415–426
Massell BF, Honikman LH, Amezcua J (1969) Rheumatic fever following streptococcal vaccination. Report of three cases. JAMA 207:1115–1119
Dale JB, Beachey EH (1985) Epitopes of streptococcal M proteins shared with cardiac myosin. J Exp Med 162:583–591
Acknowledgements and disclosure statement
The authors wish to thank M. Caparon for providing the HSC5 GAS strain, M. Kotb for providing the 20174 GAS strain, and B. Currie for providing Aboriginal serum. A.H. is a recipient of an Australian Postgraduate Award and a DAAD Research Grant for Doctoral Candidates, Young Academics and Scientists. A.J.C. is a recipient of a University of Wollongong Postgraduate Award. J.N.C. is the recipient of a National Health and Medical Research Council of Australia (NHMRC) Overseas Biomedical Fellowship. This work was supported by the NHMRC (M.J.W., B.K. and M.R.B.) and a DEST International Science Linkages Grant (M.J.W. and V.N.). B.K. and M.J.W. are NHMRC Research Fellows. The authors (A.H., E.C., J.N.C., C.M.G., V.R., K.S.S., I.Y.R.M., M.R.B., and M.J.W.) have an intellectual property or commercial interest in the antigens described in this study.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
Peptide mass fingerprinting identification of cell wall-associated proteins of S. pyogenes isolate 5448 and percent identity of cell wall-associated proteins with human homologs (PDF 60 kb)
Rights and permissions
About this article
Cite this article
Henningham, A., Chiarot, E., Gillen, C.M. et al. Conserved anchorless surface proteins as group A streptococcal vaccine candidates. J Mol Med 90, 1197–1207 (2012). https://doi.org/10.1007/s00109-012-0897-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-012-0897-9