
Abstract
The plasma proteins of the complement system are essential in the innate immune response against bacteria. Complement labels bacteria with opsonins to support phagocytosis and generates chemoattractants to attract phagocytes to the site of infection. In turn, bacterial human pathogens have evolved different strategies to specifically impair the complement response. Here, we review the large arsenal of complement inhibitors produced by the gram-positive pathogens Staphylococcus aureus and Group A Streptococcus. We discuss how these bacterial molecules provide us with new tools to treat both infectious and inflammatory disease conditions in humans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
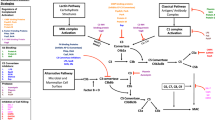
The innate immune system plays a critical role in our host defense against invading pathogens. This system consists of three different parts: (a) the complement system, (b) phagocytes, and (c) antimicrobial peptides. The three major functions of the complement cascade in innate immunity are (1) to label pathogens or immunogenic particles with C3b and iC3b to facilitate phagocytic uptake via complement receptors, (2) to attract phagocytes by producing chemoattractant C5a, and (3) to directly lyse gram-negative bacteria through membrane attack complex (MAC; C5b-9) formation (Fig. 1) [1]. Complement is initiated by two specific recognition pathways, the classical and lectin pathway, which are amplified by the alternative pathway. All three pathways converge at the formation of the C3 convertases. These bimolecular surface-bound enzyme complexes catalyze the key reaction in complement activation: cleavage of complement protein C3 into C3a, a chemoattractant with bactericidal activity [2–4], and C3b [5]. Convertase formation is pivotal in complement activation since C3b and its inactive derivative iC3b facilitate phagocytosis. Furthermore, the deposited C3b can form new convertases, thereby amplifying the opsonization process. Subsequently, the high concentrations of locally deposited C3b induce a shift in substrate specificity of the convertase to complement protein C5. The cleavage products of C5 are C5a, a potent chemoattractant, and C5b that initiates the lytic pathway. Due to the resulting C5a gradient, neutrophils migrate toward the site of infection and phagocytose the invaders. Neutrophils kill ingested particles intracellularly by the production of reactive oxygen species and the release of granule constituents such as proteolytic enzymes and antimicrobial peptides. These peptides are also released into the extracellular space to kill microorganisms extracellularly [6]. Together, these complement-mediated events are responsible for the efficient elimination of invading bacteria.

Schematic overview of the complement system. The complement cascade is activated by recognition of microbe-bound antibodies or microbial sugars by the C1 complex (CP) or the MBL- and ficolin-MASP-2 complex (LP), respectively. Both C1s and MASP-2 cleave complement proteins C4 and C2 to generate the CP/LP C3 convertase: C4b2a. The Alternative Pathway (AP) C3 convertase C3bBb is generated after binding of fB to surface-bound C3b and subsequent cleavage by fD. The AP C3 convertase is stabilized by the positive regulator human properdin [47, 48]. Both C3 convertases (C4b2a and C3bBb) cleave C3 into C3a and C3b. C3b covalently binds to bacterial surfaces, thereby amplifying complement activation through formation of new C3 convertases and contributes to phagocytosis, antigen presentation, and formation of C5 convertases (C4b2a3b and C3bBb3b). C5 convertases cleave C5 into the anaphylatoxin C5a, which attracts neutrophils to the site of infection, and C5b, which forms a complex with complement proteins C6–C9 to generate the membrane attack complex
It has become increasingly evident that pathogens have developed various ways to evade the constant attack of the complement system. Pathogens produce proteins that modulate all stages of the complement cascade. These proteins inhibit complement recognition and amplification, cleave complement proteins, or interfere with complement receptor interactions on phagocytes. A number of reviews have been published on these specific topics [7–9]. In this paper, we give a broad overview of the complement evasion strategies of the two most important gram-positive human pathogens Staphylococcus aureus and group A Streptococcus (GAS), and we discuss the potential of these bacterial complement modulators as therapeutic agents.
Modulation of complement recognition and activation
The classical and lectin pathways of complement ensure immediate recognition of invading bacteria as nonself. The C1-complex (classical pathway) recognizes bacterium-bound IgG and IgM while mannose-binding lectins and ficolins (lectin pathway) bind bacterial sugar moieties. S. aureus expresses two surface-anchored proteins that impair IgG function (Fig. 2a): staphylococcal protein A (SpA) and staphylococcal immunoglobulin-binding protein (Sbi). SpA is a surface protein with four or five immunoglobulin-binding repeat domains. Each domain can bind the Fc-part of IgG, thereby blocking the interaction with Fc receptors on neutrophils in vitro [10, 11]. Sbi consists of four small domains, of which two (Sbi-I and Sbi-II) can bind IgG [12]. Next to blocking Fc-receptor-mediated phagocytosis, Sbi has been suggested to block binding of C1q and subsequent activation of the classical pathway.

Bacterial evasion of complement recognition (a) and opsonization (b). Illustrated are proteins of S. aureus (red) and GAS (yellow) that modulate complement recognition and opsonization. a To prevent recognition by the classical pathway, S. aureus expresses the surface proteins ProtA and Sbi. Both proteins bind the Fc tail of IgG preventing its interaction with FcR on neutrophils. Another mechanism to prevent recognition is by inactivating IgG, either directly by proteases IdeS, Mac-2, and SpeB, or indirectly by SAK-activated plasmin. b There are three ways by which S. aureus and GAS modulate opsonization: (1) C3 cleavage; directly by SpeB or indirectly by SAK-mediated activated plasmin ClfA binds human fI, thereby enhancing C3 cleavage. (2) Convertase modulation; S. aureus inactivates the C3 convertases by secreting SCIN, SCIN-B, SCIN-C, Efb, and Ecb. (3) Modulating host regulators; Sbi and the streptococcal M-protein bind the negative convertase regulators (human FH, FHL1, C4BP, and CD46) while SpeB cleaves the positive convertase regulator, properdin
Another strategy to prevent recognition is to eliminate opsonic molecules from the bacterial surface by proteolytic degradation. Staphylokinase (SAK) is a secreted protein that binds and activates surface-bound plasminogen into plasmin, which may enhance bacterial invasion through host tissues. Interestingly, it has been shown that SAK is anti-opsonic as well. SAK-mediated plasmin deposition on the bacterial surface can cleave IgG and C3b and thereby inhibit phagocytosis in vitro [13]. GAS expresses several proteases that directly cleave IgG: the Endoglycosidase in Streptococcus pyogenes (EndoS) specifically hydrolyzes the asparagine-linked glycan in the CH2 domain of IgG; the IgG-degrading enzyme of S. pyogenes (IdeS or Mac-1), Mac-2, and streptococcal pyrogenic exotoxin B (SpeB) all cleave IgG in the hinge region [14, 15].
Modulation of complement amplification
Formation of the C3 convertases is elemental for amplification of complement activation and downstream immune responses. There are three ways by which S. aureus and GAS modulate this central step in the complement cascade (Fig. 2b):
-
1.
Cleavage of C3
The abundant GAS protease SpeB is, next to cleaving IgG, involved in breakdown of C3. Comparison of wild-type GAS and a SpeB knockout showed that SpeB blocks neutrophil recruitment to the site of infection and subsequent phagocytosis and bacterial clearance in vivo [16, 17]. The S. aureus surface protein clumping factor A (ClfA) can bind the human C3b protease factor I (fI), thereby enhancing cleavage of surface-bound C3b into iC3b in vitro [18].
-
2.
Direct inactivation of C3 convertases
Convertases are the major complement target among S. aureus immune evasion strategies. S. aureus secretes five different molecules that directly inhibit these central enzyme complexes. Staphylococcal complement inhibitor (SCIN) and its homologues SCIN-B and SCIN-C are highly effective C3 convertase inhibitors that block conversion of C3 and subsequent phagocytosis and C5a formation in vitro at low concentrations [19]. The alternative pathway C3 convertase consists of a cofactor (C3b) which is loosely bound to the protease subunit (Bb). Recent structural studies revealed that the small 10-kD SCIN protein fixates the convertase conformation and as such hampers a critical rearrangement of the protease subunit Bb in relation to substrate C3 [20, 21]. The action of SCIN on the classical pathway convertase remains to be resolved but seems to be caused by a stabilizing mechanism as well [19]. Extracellular fibrinogen-binding protein (Efb) and extracellular complement-binding protein (Ecb) can modulate the alternative pathway convertase by binding to the C3b molecule directly [22]. The crystal structures of both molecules in complex with the C3d domain of C3 have revealed their exact binding sites [23, 24]. Interestingly, since the C3d fragment of C3 is involved in stimulation of adaptive immune responses, it was recently suggested that Efb functions as an adaptive immunity modulator as well [25].
-
3.
Binding or cleavage of human convertase regulators
To protect host tissues from excessive complement activity, humans express complement regulators that downregulate convertase activity. A large number of pathogens express molecules that attract these regulators to their surface. The staphylococcal IgG-binding molecule Sbi has a diverse role in complement modulation. Next to its two IgG-binding domains, Sbi-III and IV, it can also bind to C3 [26]. Furthermore, Sbi binds the human complement regulators factor H (FH) and factor H-related proteins and can form a stable tripartite complex with C3 and FH [27]. Altogether, these actions result in inhibition of the alternative pathway in vitro. The streptococcal M protein is a multifunctional surface molecule that binds four different human convertase regulators: FH, factor H like-1 (FHL1), membrane cofactor protein (CD46), and C4-binding protein (C4BP). M protein consists of two polypeptide chains configured in an alpha-helical coiled coil complex that is anchored to the cell membrane via its C terminus [28–30]. From the N to the C terminus, the protein comprises four repeat regions: hypervariable (a), variable (b), and conserved (c and d). Depending on the M type, the hypervariable region can bind FH, FHL-1 [31], and C4BP [32]. By binding these complement regulators, M protein limits deposition of C4b and C3b increasing GAS resistance to phagocytosis [33]. In vivo studies with strains expressing mutated M protein indicate that protein is an important GAS virulent factor [34]. Finally, SpeB also digests the positive convertase regulator properdin, making the streptococci more resistant to neutrophil killing in vitro [35].
Modulation of the complement effector functions: inhibiting C5 activation and neutrophil migration
In the final step of the complement cascade, the C5 convertases cleave C5 to generate the important chemoattractant C5a that directs neutrophils to the site of infection. S. aureus and GAS use various approaches to evade the C5a-mediated immune responses (Fig. 3). Staphylococcal superantigen-like 7 (SSL7) is a secreted protein that specifically binds to C5 [36] and thereby prevents C5 activation into C5b and C5a in vitro (J. Bestebroer, personal communication). Efb and Ecb are very potent inhibitors of C5a responses in vitro and in vivo [22]. These molecules elicit their immune modulating capacity by binding the C3b part of the C5 convertase complex. GAS uses the cell-associated peptidase ScpA to directly target C5a. ScpA cleaves C5a and blocks chemotaxis of phagocytes toward the site of infection [37]. In a mouse pneumonia model, it was shown that the ScpA knockout mutant was less virulent than wild-type GAS [38]. Modulation of C5a responses by S. aureus proceeds through inhibition of the C5a receptor (C5aR). The chemotaxis inhibitory protein of S. aureus (CHIPS) is a 14-kD excreted protein that effectively blocks neutrophil recruitment toward C5a in vitro by binding the C5aR with high (nanomolar) affinity [39]. The recent nuclear magnetic resonance (NMR) structure of CHIPS in complex with the N terminus of the C5aR showed two sulfated tyrosine (residues 11 and 14) of the C5aR to be essential for the interaction in between the C5aR and the inhibitor CHIPS [40].
Cleavage of C5 also results in formation of C5b, the molecule that initiates the formation of the membrane attack complex (C5b-9). This complex can damage target cells such as gram-negative bacteria. MAC formation is often monitored in vitro by a so-called hemolytic assay in which erythrocytes (often sheep or rabbit) are used as a direct target for complement activation and hemolysis. The S. aureus proteins SCIN, Efb, Ecb, and SSL7 block MAC-mediated erythrocyte hemolysis since they inhibit the complement cascade upstream of MAC formation. The highly variable streptococcal inhibitor of complement (SIC) from GAS was found to specifically block formation of the MAC by binding to the C5b-7 complex and preventing erythrocyte hemolysis in vitro [41]. The relevance of MAC inhibition by gram-positive bacteria remains unclear since they are naturally protected from lysis via their thick cell wall. The SIC molecule also interacts with several human antimicrobial peptides (LL-37, defensins) [42], and this might be more relevant for protecting GAS from host immunity.

Bacterial evasion of C5-mediated responses. Illustrated are proteins of S. aureus (red) and GAS (yellow) that modulate C5-mediated responses. Efb and Ecb block the formation of C5 convertases and thereby inhibit C5a and MAC generation. SSL7 binds to C5 and prevents the cleavage of C5 into C5a and C5b. Neutrophil influx and phagocytosis are inhibited by CHIPS, which binds the C5aR, and ScpA that cleaves C5a. SIC inhibits the formation of the MAC, the final step in the complement cascade
Complement evasion molecules as tools and targets in therapy
Bacterial complement modulators have important applications for the treatment of inflammatory diseases where complement is directed against our own cells [43]. Per definition, molecules that counteract acute innate immune mechanisms are anti-inflammatory compounds. As opposed to several bacterial toxins, the high specificity and the mode of action that depends entirely on protein–protein interactions makes these nontoxic proteins suitable anti-inflammatory drugs. It is tempting to speculate that these molecules could be used as injectables in acute inflammatory disorders. However, since these molecules are derived from common human pathogens, pre-existing antibodies severely complicate this approach. Furthermore, a number of the described molecules appear to be human-specific which complicates functional studies in vivo [19, 44, 45] However, the immunological molecules that are attacked by these factors might prove to be the essential targets to tackle in anti-inflammatory therapy. We feel that the bacterial products could be used as an indicator of the “Achilles heel” of the human immune system—almost at the atomic level. Especially, since the 3-D structures of a number of molecules in context with their target are now elucidated in several crystallographic and NMR studies [19, 20, 25, 40, 46], this opens new possibilities for smart design of small-molecule therapeutics. Thus, we can copy these evolutionary-designed molecules to counteract the detrimental effects of complement activation in inflammatory disease states.
Next to their potential as anti-inflammatory compounds, we believe that targeting of complement evasion molecules should be included in future strategies to fight bacterial infections. The evasion molecules described above not only suppress natural immunity during an infection but also hamper the current attempts to create effective vaccines. The efficacy of vaccines is held back by the existing bacterial immune evasion strategies since they prevent neutrophils or complement to reach the site of infection. Even with sufficient opsonic or bactericidal antibodies on the bacterium, nothing will happen if the innate immune effectors are absent. Since immune evasion molecules are essentially vaccine evasion molecules, we propose that these molecules should be specifically targeted in the vaccine. Alternatively, we could think of ways to neutralize these molecules and use that in novel strategies to fight infectious diseases. In fact, we propose to inhibit the inhibitor and thereby decrease the advantage of the bacterium over the host.
So, taken together, the smart attack of the bacteria on our complement system might hold clues for future drugs in both inflammatory as well as infectious diseases. In this way, we could turn this powerful escape mechanism into our own advantage.
References
Walport MJ (2001) Complement. First of two parts. N Engl J Med 344:1058–1066
Hartmann K, Henz BM, Krüger-Krasagakes S, Köhl J, Burger R, Guhl S, Haase I, Lippert U, Zuberbier T (1997) C3a and C5a stimulate chemotaxis of human mast cells. Blood 89:2863–2870
Daffern PJ, Pfeifer PH, Ember JA, Hugli TE (1995) C3a is a chemotaxin for human eosinophils but not for neutrophils. I. C3a stimulation of neutrophils is secondary to eosinophil activation. J Exp Med 181:2119–2127
Nordahl EA, Rydengård V, Nyberg P, Nitsche DP, Mörgelin M, Malmsten M, Bjårck L, Schmidtchen A (2004) Activation of the complement system generates antibacterial peptides. Proc Natl Acad Sci USA 101:16879–16884
Gros P, Milder F, Janssen B (2008) Complement driven by conformational changes. Nat Rev Immunol 8:48–58
Borregaard N, Sørensen OE, Theilgaard-Mönch K (2007) Neutrophil granules: a library of innate immunity proteins. Trends Immunol 28:340–345
Blom AM, Hallström T, Riesbeck K (2009) Complement evasion strategies of pathogens—acquisition of inhibitors and beyond. Mol Immunol 46:2808–2817
Lambris JD, Ricklin D, Geisbrecht BV (2008) Complement evasion by human pathogens. Nat Rev Microbiol 6:132–142
Geisbrecht BV (2008) Staphylococcal complement inhibitors: biological functions, recognition of complement components, and potential therapeutic implications. Adv Exp Med Biol 632:221–236
Forsgren A, Sjoquist J (1966) "Protein A" from S. aureus. I. Pseudo-immune reaction with human gamma-globulin. J Immunol 97:822–827
Goward CR, Scawen MD, Murphy JP, Atkinson T (1993) Molecular evolution of bacterial cell-surface proteins. Trends Biochem Sci 18:136–140
Atkins KL, Burman JD, Chamberlain ES, Cooper JE, Poutrel B, Bagby S, Jenkins ATA, Feil EJ, Van den Elsen JMH (2008) S. aureus IgG-binding proteins SpA and Sbi: host specificity and mechanisms of immune complex formation. Mol Immunol 45:1600–1611
Rooijakkers SHM, van Wamel WJB, Ruyken M, van Kessel KPM, van Strijp JAG (2005) Anti-opsonic properties of staphylokinase. Microbes Infect 7:476–484
Collin M, Olsen A (2001) Effect of SpeB and EndoS from Streptococcus pyogenes on human immunoglobulins. Infect Immun 69:7187–7189
Collin M, Olsen A (2001) EndoS, a novel secreted protein from Streptococcus pyogenes with endoglycosidase activity on human IgG. EMBO J 20:3046–3055
Lukomski S, Burns EH, Wyde PR, Podbielski A, Rurangirwa J, Moore-Poveda DK, Musser JM (1998) Genetic inactivation of an extracellular cysteine protease (SpeB) expressed by Streptococcus pyogenes decreases resistance to phagocytosis and dissemination to organs. Infect Immun 66:771–776
Terao Y, Mori Y, Yamaguchi M, Shimizu Y, Ooe K, Hamada S, Kawabata S (2008) Group A streptococcal cysteine protease degrades C3 (C3b) and contributes to evasion of innate immunity. J Biol Chem 283:6253–6260
Hair PS, Ward MD, Semmes OJ, Foster TJ, Cunnion KM (2008) Staphylococcus aureus clumping factor A binds to complement regulator factor I and increases factor I cleavage of C3b. J Infect Dis 198:125–133
Rooijakkers SHM, Ruyken M, Roos A, Daha MR, Presanis JS, Sim RB, van Willem JB, Wamel KPM, van Kessel J, van Strijp AG et al (2005) Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat Immunol 6:920–927
Rooijakkers SHM, Wu J, Ruyken M, van Domselaar R, Planken KL, Tzekou A, Ricklin D, Lambris JD, Janssen BJC, van Strijp JAG, Gros P (2009) Structural and functional implications of the alternative complement pathway C3 convertase stabilized by a staphylococcal inhibitor. Nat Immunol 10:721–727
Ricklin D, Tzekou A, Garcia BL, Hammel M, McWhorter WJ, Sfyroera G, Wu Y, Holers M, Herbert AP, Barlow PN, Geisbrecht BV, Lambris JD (2009) A molecular insight into complement evasion by the staphylococcal complement inhibitor protein family. J Immunol 183:2565–2574
Jongerius I, Köhl J, Pandey MK, Ruyken M, Van Kessel KPM, Van Strijp JAG, Rooijakkers SHM (2007) Staphylococcal complement evasion by various convertase-blocking molecules. J Exp Med 204:2461–2471
Hammel M, Sfyroera G, Ricklin D, Magotti P, Lambris JD, Geisbrecht BV (2007) A structural basis for complement inhibition by Staphylococcus aureus. Nat Immunol 8:430–437
Hammel M, Sfyroera G, Pyrpassopoulos S, Ricklin D, Ramyar KX, Pop M, Jin Z, Lambris JD, Geisbrecht BV (2007) Characterization of Ehp, a secreted complement inhibitory protein from Staphylococcus aureus. J Biol Chem 282:30051–30061
Ricklin D, Ricklin-Lichtsteiner SK, Markiewski MM, Geisbrecht BV, Lambris JD (2008) Cutting edge: members of the Staphylococcus aureus extracellular fibrinogen-binding protein family inhibit the interaction of C3d with complement receptor 2. J Immunol 181:7463–7467
Burman JD, Leung E, Atkins KL, O'Seaghdha MN, Lango L, Bernardo P, Bagby S, Svergun DI, Foster TJ, Isenman DE, van den Elsen JM (2008) Interaction of human complement with Sbi, a staphylococcal immunoglobulin-binding protein: indications of a novel mechanism of complement evasion by Staphylococcus aureus. J Biol Chem 283:17579–17593
Haupt K, Reuter M, van den Elsen J, Burman J, Halbich S, Richter J, Skerka C, Zipfel PF (2008) The Staphylococcus aureus protein Sbi acts as a complement inhibitor and forms a tripartite complex with host complement Factor H and C3b. PLoS Pathog 4:e1000250
Robinson JH, Kehoe MA (1992) Group A streptococcal M proteins: virulence factors and protective antigens. Immunol Today 13:362–367
Fischetti VA (1989) Streptococcal M protein: molecular design and biological behavior. Clin Microbiol Rev 2:285–314
McNamara C, Zinkernagel AS, Macheboeuf P, Cunningham MW, Nizet V, Ghosh P (2008) Coiled-coil irregularities and instabilities in group A Streptococcus M1 are required for virulence. Science 319:1405–1408
Kotarsky H, Hellwage J, Johnsson E, Skerka C, Svensson HG, Lindahl G, Sjobring U, Zipfel PF (1998) Identification of a domain in human factor H and factor H-like protein-1 required for the interaction with streptococcal M proteins. J Immunol 160:3349–3354
Johnsson E, Thern A, Dahlback B, Heden LO, Wikstrom M, Lindahl G (1996) A highly variable region in members of the streptococcal M protein family binds the human complement regulator C4BP. J Immunol 157:3021–3029
Carlsson F, Berggard K, Stalhammar-Carlemalm M, Lindahl G (2003) Evasion of phagocytosis through cooperation between two ligand-binding regions in Streptococcus pyogenes M protein. J Exp Med 198:1057–1068
Ashbaugh CD, Warren HB, Carey VJ, Wessels MR (1998) Molecular analysis of the role of the group A streptococcal cysteine protease, hyaluronic acid capsule, and M protein in a murine model of human invasive soft-tissue infection. J Clin Invest 102:550–560
Tsao N, Tsai W, Lin Y, Chuang W, Wang C, Kuo C (2006) Streptococcal pyrogenic exotoxin B cleaves properdin and inhibits complement-mediated opsonophagocytosis. Biochem Biophys Res Commun 339:779–784
Langley R, Wines B, Willoughby N, Basu I, Proft T, Fraser JD (2005) The staphylococcal superantigen-like protein 7 binds IgA and complement C5 and inhibits IgA-Fc alpha RI binding and serum killing of bacteria. J Immunol 174:2926–2933
Wexler DE, Chenoweth DE, Cleary PP (1985) Mechanism of action of the group A streptococcal C5a inactivator. Proc Natl Acad Sci USA 82:8144–8148
Husmann LK, Yung DL, Hollingshead SK, Scott JR (1997) Role of putative virulence factors of Streptococcus pyogenes in mouse models of long-term throat colonization and pneumonia. Infect Immun 65:1422–1430
Postma B, Kleibeuker W, Poppelier MJJG, Boonstra M, Van Kessel KPM, Van Strijp JAG, De Haas CJC (2005) Residues 10–18 within the C5a receptor N terminus compose a binding domain for chemotaxis inhibitory protein of Staphylococcus aureus. J Biol Chem 280:2020–2027
Ippel JH, De Haas CJC, Bunschoten A, Van Strijp JAG, Kruijtzer JAW, Liskamp RMJ, Kemmink J (2009) Structure of the tyrosine-sulfated C5a receptor N terminus in complex with chemotaxis inhibitory protein of Staphylococcus aureus. J Biol Chem 284:12363–12372
Fernie-King BA, Seilly DJ, Davies A, Lachmann PJ (2001) Streptococcal inhibitor of complement (SIC) inhibits the membrane attack complex by preventing uptake of C567 onto cell membranes. Immunology 103:390–398
Frick IM, Akesson P, Rasmussen M, Schmidtchen A, Björck L (2003) SIC, a secreted protein of Streptococcus pyogenes that inactivates antibacterial peptides. J Biol Chem 278:16561–16566
Sjoberg AP, Trouw LA, Blom AM (2009) Complement activation and inhibition: a delicate balance. Trends Immunol 30:83–90
de Haas CJC, Veldkamp KE, Peschel A, Weerkamp F, Van Wamel WJB, Heezius EC, Poppelier MJ, Van Kessel KP, Van Strijp JAG (2004) Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J Exp Med 199:687–695
Gladysheva IP, Turner RB, Sazonova IY, Liu L, Reed GL (2003) Coevolutionary patterns in plasminogen activation. Proc Natl Acad Sci USA 100:9168–9172
Haas PJ, de Haas CJC, Kleibeuker W, Poppelier MJ, Van Kessel KPM, Kruijtzer JA, Liskamp RM, Van Strijp JAG (2004) N-terminal residues of the chemotaxis inhibitory protein of Staphylococcus aureus are essential for blocking formylated peptide receptor but not C5a receptor. J Immunol 173:5704–5711
Kimura Y, Miwa T, Zhou L, Song WC (2008) Activator-specific requirement of properdin in the initiation and amplification of the alternative pathway complement. Blood 111:732–740
Hourcade DE (2006) The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem 281:2128–2132
Acknowledgments
A.L., J.v.S., and S.H.M.R. are supported by grants of the Netherlands Organization for Scientific Research (NWO-TOP and NWO-Veni). S.H.M.R. is also funded by the European Molecular Biology Organization.
Conflict of interest statement
The authors declare that they have no conflict of interests.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Laarman, A., Milder, F., van Strijp, J. et al. Complement inhibition by gram-positive pathogens: molecular mechanisms and therapeutic implications. J Mol Med 88, 115–120 (2010). https://doi.org/10.1007/s00109-009-0572-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-009-0572-y