
Abstract
The tryptophan-catabolizing enzyme indoleamine-2,3-dioxygenase (IDO) initiates the first and rate-limiting step of the kynurenine pathway. It is induced by proinflammatory cytokines such as interferon-β and interferon-γ and has established effects in the control of intracellular parasites. The recent detection of its decisive function in immune tolerance at the maternal–fetal interface stimulated various studies unraveling its regulatory effect on T cells in many pathologies. In the brain, IDO can be induced in microglia by interferon-γ-producing T helper (Th) 1 cells, thereby initiating a negative feedback loop which downmodulates neuroinflammation in experimental autoimmune encephalomyelitis (EAE), the animal model of multiple sclerosis (MS). This protective effect could to be counteracted by the production of neurotoxic metabolites of the kynurenine pathway such as quinolinic acid, which are produced upon IDO induction. Some metabolites of the kynurenine pathway can pass the blood–brain barrier and thus could act as neurotoxins, e.g., during systemic infection. In this paper, we give a brief overview on established immune regulatory functions of IDO, review recent data on IDO expression in the brain, and propose that autoimmune neuroinflammation and the increasingly appreciated neuronal damage in MS are linked by Th1-mediated IDO induction through subsequent synthesis of toxic metabolites of tryptophan.
Similar content being viewed by others

Introduction
Mammals have two different oxygenases for the degradation of the essential amino acid tryptophan (Trp). The tryptophan dioxygenase (TDO), which is primarily expressed within the liver, catabolizes the main part of dietary Trp for the maintenance of serum levels. The second enzyme is the indoleamine 2,3-dioxygenase (IDO), which represents the first and rate-limiting enzyme of the kynurenine pathway in extrahepatic tissues. This enzyme was first described by Higuchi et al. [1]. IDO is a heme-containing enzyme that catalyzes the oxidative cleavage of the Trp pyrrol ring, thereby producing N-formyl-kynurenine, which is then further degraded along the kynurenine pathway (Fig. 1).

The kynurenine pathway. Enzymatic degradation of tryptophan to kynurenic acid or quinolinic acid via the kynurenine pathway. Neurotoxic metabolites are labeled in italics
IDO expression is inducible by the proinflammatory cytokine interferon-γ (IFN-γ) and to a lower extent by interferon-β (IFN-β) in several cell types including macrophages, dendritic cells (DC), and fibroblasts [2–5]. In the brain, murine and human microglia have been shown to express IDO upon treatment with IFN-γ [6, 7]. In peripheral tissues, IDO expression is a common mechanism to suppress the proliferation of infectious parasites such as Chlamydia trachomatis [8] and Toxoplasma gondii [9, 10] through Trp depletion. Moreover, metabolites of the kynurenine pathway also exhibit immune modulatory functions, e.g., during tumor maintenance [11] and allograft acceptance/rejection [12, 13].
During experimental autoimmune encephalomyelitis (EAE), IDO induction has been shown to downmodulate neuroinflammation [14–16]. However, the induction of IDO in the central nervous system (CNS) is delicate because several metabolites of the kynurenine pathway have well-established neurotoxic effects [17]. Given that multiple sclerosis (MS) is characterized by the massive influx of activated T helper (Th) 1 cells and loss of neurons, an increasingly appreciated hallmark of this disease [18, 19], we propose that neuroinflammation and neurodegeneration are linked by IFN-γ-mediated IDO induction and the accompanying production of toxic Trp metabolites.
IDO in infection
Over the last decades, many studies with human cells showed that IFN-γ-induced IDO expression represents an important mechanism of antimicrobial resistance to parasites [9, 20, 21] and bacteria [8, 22, 23]. In all these cases, the functional expression of IDO and the subsequent degradation of Trp were identified as the effector mechanisms of microbial suppression. Induction of the kynurenine pathway was recently shown to be involved in downmodulation of a fungal infection of the gastrointestinal tract with Candida albicans [24]. In vivo, inhibition of IDO exacerbated the infection and its associated inflammatory pathology. In extension of previous studies, Montagnoli et al. [25] demonstrated a reduced number of CD4+ CD25+regulatory T cells (Tregs) in C. albicans-infected animals after IDO inhibition. This cell type is capable of downmodulating inflammatory and antifungal Th1 immunity in C. albicans-infected mice. In fact, the strain of C. albicans used in these experiments was Trp prototrophic. Therefore, the antifungal effect of IDO expression is likely to be mediated through the modification of the host’s T cell.
Recent data demonstrate that IDO also plays a role in viral infections. The replication of cytomegalovirus and herpes simplex type I and II has been shown to be restricted by IFN-γ-induced IDO expression [26–28]. Opposite effects have been observed in the course of CNS infection with the human immunodeficiency virus (HIV). IDO activity is also increased in response to this virus [29], but IDO inhibition by 1-methyl Trp does not increase but rather decrease the viral burden. Remarkably, IDO inhibition amplified the number of HIV-specific cytotoxic T cells in HIV-infected severe combined immunodeficiency mice [30]. The HIV virus-infected cells seem to protect themselves from killer cell-mediated lysis by immunomodulation, thereby providing a niche hiding the virus from the immune system. Thus, during infection, IDO induction can exert divergent effects: On the one hand, it limits growth of infectious agents but also the strength of the immune response. The latter may be important to limit loss of infected cells in organs of poor regenerative capacity at the prize of viral persistence [31, 32].
Possible mechanisms of IDO-mediated tolerance induction
In contrast to the liver, Trp degradation is restricted to pathologic conditions in all other organs. The functional expression of IDO initiates the kynurenine pathway during which the degradation progresses along several enzymatic reactions (Fig. 1). Intermediates such as kynurenic acid (KA), quinolinic acid (QUIN) and 3-hydroxyanthranilic acid (3-HAA) have strong effects on many cell types including lymphocytes and antigen-presenting cells (APC). Munn et al. [33] were the first to demonstrate that blocking the IDO during pregnancy in mice causes tolerance breakdown and fetus rejection. Since then, several studies have confirmed a major role of IDO in the maintenance of immune tolerance [16, 34]. Two mechanisms have been proposed to explain the downmodulatory effect of IDO activation on T cells:
-
1)
Munn et al. [33] hypothesized that in analogy of its function during infection, IDO activity creates a Trp-depleted microenvironment limiting the proliferation of T cells.
-
2)
Fallarino et al. [4] showed that Trp degradation products enhance the susceptibility of T cell to apoptosis.
In fact, IDO activity depletes Trp from the culture medium of human macrophages and activated T cells cultured in such medium arrest in a late G1 phase [35]. This Trp starvation-mediated arrest has been shown to be caused in part by the stress-activated GCN2 kinase [36]. However, T cells not only stop proliferation when they are kept under Trp-depleted conditions, they also become highly sensitive to CD95L (FasL)-induced apoptosis [37]. CD95L-mediated deletion of activated T cells is a common mechanism of self-limitation of inflammation [38] and important to minimize inflammation-mediated damage in sensitive tissues such as the brain and the eye [39].
While IDO is inducible in many cell types, its regulation in DC turns out to be decisive for shifting the balance between tolerance and immunity. DC are professional APC. The type of DC presenting an antigen determines the T cell-polarizing signals and thus the T cell differentiation into Th1, Th2, or Treg. In the case of tolerance induction, the cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) plays a crucial role. CTLA-4 blocks the CD28-B7 costimulatory signaling, which is essential for functional T cell activation [40]. CTLA-4 immonoglobulin (CTLA-4-Ig) induces IFN-γ expression in DC and thereby an auto- or paracrine induction of IDO expression in the local microenvironment, providing the conditions for long-term survival of allogeneic islet transplants [41]. In cell cultures, DC expressing B220 or CD8α upregulate IDO expression when they are cocultured with CTLA-4-expressing T cells [42].
IDO-expressing DC inhibit T cell proliferation in vitro even when Trp is still available in the medium [43]. This effect is caused by kynurenine, 3-hydroxyanthranilin, and 3-HAA, which are all Trp metabolites produced downstream of the kynurenine pathway [44]. Moreover, 3-HAA and QUIN induce CD95L-independent apoptosis via caspase 8 in activated Th1 but not Th2 cells [4]. This effect can be increased by lowering Trp concentrations in cell cultures [43]. The restriction of these effects to Th1 cells may represent an IDO-dependent mechanism of immune deviation during inflammation.
Interestingly, in CD123+DC, IDO expression is induced by interleukine 10 (IL-10), an anti-inflammatory cytokine expressed by Treg cells [45]. While in most investigated in vitro models, IFN-γ is the main inducer of IDO in macrophages, DC, fibroblasts, and microglia [2–5, 46], its expression can also be triggered by lipopolysaccharide (LPS) through an IFN-γ-independent mechanism [47]. The induction of IDO expression without IFN-γ signaling is not dependent on signal transducer and activator of transcription 1α and interferon regulatory factor-1 but requires p38 mitogen-activated protein kinase and nuclear factor-κB [48]. Thus, IDO induction is not restricted to Th1 cells secreting INF-γ but alternate triggers involving IL-10 and LPS. This is in line with the observation that Th2-mediated experimental asthma is also abrogated by functional expression of IDO [49].
An undesirable case of tolerance induction is the manifestation of tumors. The first recognition of tumor antigens by T cells occurs in the tumor draining lymph nodes. Within such lymph nodes, there is a population of plasmocytoid DC (PDC) expressing B220, CD11c, and CD19. These PDC induce T cell anergy and immunosuppression in vivo by the constitutive expression of IDO [50]. CD19+PDC are also found in neighboring lymph nodes and spleen, but in contrast to the PDC from tumor draining sentinal lymph nodes, they do not express IDO constitutively. The tumor itself is therefore likely to trigger IDO expression in draining lymph nodes by an as yet unknown mechanism. One possible way of induction could be the binding of CTLA-4 expressed by Tregs, which have been shown to induce IDO expression in DC in vitro [44, 51]. The adoptive transfer of IDO expressing PDC in vivo induces not only a systemic unresponsiveness to antigens [52] but also antigen-specific anergy of T cells within lymph nodes [50].
Tolerogenic mechanisms are not only active in sentinal lymph nodes but were also found within the tumor itself, and IDO expression may represent one of such mechanisms [34]. In fact, the tumor cell line P815 becomes resistant against immunological deletion when the cells were transfected for constitutive IDO expression. In ovarian and colorectal cancer, IDO expression within the tumors correlates to malignancy [53]. In support of this concept, colorectal tumors exhibiting a high IDO activity have a significantly reduced number of CD3+ infiltrating T cells and show an increased frequency of metastases [54]. Thus, determining IDO expression in tumors may be used for clinical prognostic in the future. Moreover, IDO inhibitors could increase the success of antitumor treatment [11].
The kynurenine-pathway and neurotoxic metabolites in the brain
Two Trp degradation products, QUIN and 3-HAA, exhibit neurotoxic properties. QUIN is an endogen N-methyl-d-aspartate (NMDA) receptor agonist [55]. At micromolar concentrations, the excitotoxic effect of QUIN can be mimicked in primary cortical neuronal cell cultures [56]. The same effect is found in vivo where intracerebral injection of QUIN induces excitotoxic lesions [17]. The second neurotoxic Trp metabolite is 3-HAA, which is unstable under physiological conditions. Upon spontaneous auto-oxidation, 3-HAA produces reactive radical species, which in turn induce oxidative stress and apoptosis in neurons [57–59].
In a dead end side branch of the kynurenine pathway, KA is synthesized by kynurenine aminotransferases (KATs). KA is known as a noncompetitive NMDA receptor antagonist [60]. Therefore, KA might counteract the neurotoxic effect of QUIN. Indeed, blocking of the kynurenine pathway at the kynurenine hydroxylase stage reduced the neuronal damage after cerebral ischemia in vivo [61] and postischemic neuronal death in slice cultures [62].
As anticipated, such treatment forced Trp degradation to the KA branch [59, 63]. To analyze the biological role of KA, Yu et al. [64] created a knockout mouse deficient for KAT2 expression. KAT2 is the aminotransferase substantially contributing to the KA formation in the CNS [65]. These mice exhibit a decreased KA formation within the CNS for the first 3 weeks of life, which afterward returns to control levels as seen in wild-type mice. No significant differences in the production of QUIN or 3-HAA were observed at any age [64]. The delayed compensation was assumed to be caused by the alternative KAT1 enzyme or by other enzymes that exhibit KAT activity. To analyze a neuroprotective effect of endogenous KA in vivo, Sapko et al. [66] induced excitotoxic lesions by the injection of QUIN in 14-day-old KAT2−/− mice. In comparison to wild-type mice, the lesion volumes were significantly increased in the knockout. If the same experiment was performed in 2-month-old mice, the lesion volumes were similar in knockout and wild-type animals. This suggests that the CNS-specific synthesis of KA by KATs represents a neuroprotective mechanism, which at least in part counteracts the neurotoxic effects of QUIN.
The neurotoxic effects caused by chronical induction of the kynurenine pathway have been analyzed in several diseases. Mackay et al. [67] reported an increased Trp katabolism to kynurenine but not to QUIN in the serum from patients with brain injuries even several years after injury. They proposed that this might be a result of increased activity of IDO and/or TDO. QUIN accumulates within the cerebrospinal fluid (CSF) of humans after traumatic brain injuries [68] and is increased in the CNS but not in the blood of gerbils after cerebral ischemia [69]. It is therefore tempting to speculate that trauma-induced IDO activation induces secondary neuronal damage via accumulation of neurotoxic metabolites. First evidence for this hypothesis derives from studies of spinal cord injury. Inhibition of 3-HAA oxygenase attenuated QUIN accumulation after spinal cord injury and reduced the severity of injury-related functional deficits [70, 71]. A similar mechanism may also be active in HIV encephalopathy. HIV-infected macrophages within the CNS express IDO. The way of its induction by the virus is currently not clear, but it is plausible that IFN-γ or even virus particles themselves trigger it [72]. However, HIV-1 is known to persist within the CNS [73], and as discussed above, its persistence seems to involve IDO-mediated immune deviation [30]. Thus, the chronic production of neurotoxic substances such as QUIN may cause part of the damage leading to HIV dementia [74].
Increasing data show the involvement of the kynurenine pathway in several neurodegenerative diseases such as Parkinson’s, Huntington’s, and Alzheimer’s disease, epilepsy, and amyotrophic lateral sclerosis and in mental disorders such as schizophrenia and depression [75]. It is remarkable that even an acute injury of the brain induces long-lasting alterations in Trp degradation with a shift toward detrimental metabolites [67]. The respective enzymes thus are promising therapeutic targets for the future. However, it is noteworthy that many human cells respond to stimulation with IFN-γ, Tumor necrosis factor-α (TNF-α), and LPS by much higher IDO activities than their murine counterparts. On the other hand, only the latter synthesize high amounts of reactive nitrogen species via inducible nitric oxide synthase induction in response to stimulation [76] rendering it difficult to transfer results from animal models to the human situation.
While the detrimental effects of QUIN and 3-HAA to neurons are well described, the cell types producing these metabolites under pathologic conditions are ill defined. In primary cell cultures, microglia, astrocytes, and neurons have been shown to express IDO upon IFN-γ stimulation. Mass spectrometry of QUIN in these cultures revealed its degradation by astrocytes and neurons, suggesting that they do not contribute to neurotoxicity but to neuroprotection. On the other hand, stimulated microglia synthesize high amounts of QUIN [46]. This in vitro observation is in line with the immune histological identification of IDO-positive microglia/macrophages in EAE and viral encephalitis [15, 72]. Unfortunately, no marker exists to differentiate between intrinsic microglia and recruited macrophages. In activated macrophages, the kynurenine pathway is much more effective than in activated microglia [6, 46], and therefore, macrophages may provide significantly more harm for neurons.
Under physiological conditions, most Trp metabolites of the brain are primarily produced outside of the CNS. The first substrate for the kynurenine pathway, Trp, is transported into the CNS by large neutral amino acid transporters [77]. However, in the absence of local inflammatory signals, the vast majority of Trp is not degradative to neurotoxic substances [78]. l-Kynurenine is also imported into the CNS by large neutral amino acid transporters and subsequently taken up by astrocytes and maybe microglia [79]. 3-Hydroxykynurenine is incorporated in the same way as l-kynurenine. Both substrates are then degradative depending on the distribution of downstream enzymes and the glial subtype.
Activated microglia secrete high amounts of neurotoxic 3-HAA and QUIN, while astrocytes synthesize but do not release significant quantities of QUIN [46]. In cell culture of human astrocytes, IFN-γ induces not only the degradation of neurotoxic 3-HAA and QUIN but also enhances the production of neuroprotective KA. [46]. Thus, astrocytes might counteract the production of neurotoxins by microglia.
In the CNS of gerbils, 85% of extracellular QUIN is imported from the blood into the brain under normal conditions, while almost all (96%) QUIN is produced within the CNS after intracerebral LPS stimulation. After systemic immune activation, almost all QUIN within the CNS is imported from the blood raising the intriguing question of whether peripheral QUIN production, e.g., during infection, may provide harm to the brain [78].
In summary, kynurenines produced in the periphery can enter the CNS through the blood–brain barrier, where they can be taken up and degraded by glial cells in an IDO-independent way. It is currently unclear whether this capacity of glial cells can fully protect neurons under all conditions of peripheral pathology. In case of local damage, astrocytes seem to eliminate neurotoxins produced by microglia. If this delicate balance is deranged by infiltrating macrophages remains to be evaluated.
The kynurenine pathway in EAE and MS
Like many autoimmune diseases, MS is characterized by waxing and waning inflammation in the target organ. What initiates the onset of individual attacks is poorly understood, but there is some insight from animal models into what drives their termination and what cause tissue damage. In EAE, the animal model for MS, autoimmunity to myelin epitopes is induced by immunization with myelin epitopes or transfer of myelin-specific Th1 cells. Subsequently, leukocytes accumulate in perivascular cuffs around brain blood vessels. Many of these cells are Th1 lymphocytes, but there is also a high percentage (depending on the model, approx. 50%) of macrophages [80]. As a result of this inflammation, myelin is phagocytosed by macrophages and activated microglia. In addition, there is a significant degeneration of axons and neurons, which for long has been neglected [18, 19].
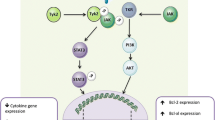
During the acute phase of EAE, tissue levels of the neurotoxin QUIN are increased in the lumbar and sacral parts of the spinal cord. Interestingly, the clinical disease severity and the QUIN concentration in the cervicolumbar spinal cord correlate well [81]. Immunohistochemical studies have shown that enzymes of the kynurenine pathway such as the IDO and kynurenine 3-mono-xygenase are mainly expressed by infiltrating macrophages/activated microglia in the perivascular/juxtavascular area during the acute phase of EAE [15, 56]. IDO expression and activity is increased in the acute and remission phase of EAE. The main inducer of IDO, the proinflammatory cytokine IFN-γ, is the key cytokine of encephalitogenic Th1 cells. TNF-α is also secreted by these Th1 cells and acts synergistically with IFN-γ on the induction of IDO in macrophages and microglia [2, 6].
In analogy to the experiments by Munn et al. [33] in the placenta, the net effect of IDO induction during neuroinflammation has been tested by in vivo inhibition experiments. Daily application of the IDO inhibitor 1-methyl-Trp clearly exacerbated disease development and reduced the clinical recovery [14, 15]. The effects of IDO inhibition were similar when the treatment started in the preclinical phase [14] or at the onset of the acute phase of disease [15]. This observation suggests that the inhibition of IDO in the periphery is not crucial for disease development. Consequently, the local induction of the kynurenine pathway and the accompanying synthesis of Trp metabolites appear to downmodulate autoimmune CNS inflammation.
Recently, the anti-inflammatory effect of the kynurenine pathway reaction products was shown by intraperitoneally injection of 3-HAA and oral administration of its synthesized derivate N-3,4-dimethoxycinnamoyl anthranilic acid (DAA). Both substances significantly reduced the relapse phase in immunized SJL mice [16]. Splenocytes from animals that had been treated with 3-HAA or 2,4-DAA showed decreased proliferation of T cells and expression of IFN-γ and TNF-α. Moreover, treatment of immunized mice with 3-HAA or 3,4-DAA also shifted inflammatory lymph node cells to a potentially regulatory cell type with a decreased IFN-γ secretion and an increased production of IL-10 [16].
From all these studies in EAE, one can interpret IDO induction by IFN-γ-secreting Th1 cells as a protective negative feedback loop eventually terminating neuroinflammation [15]. However, this interpretation by no means excludes significant bystander damage through toxic Trp metabolites as the downside of such self-limitation (Fig. 2). Conversely, the apparent tight control over IDO expression in the brain supports this view. Currently, little is known as to the induction of IDO and the subsequent synthesis of bioactive kynurenines within the CNS during MS. Kepplinger et al. [82] described an increase in KA in the CSF of MS patients during relapse, and Barans et al. [83] described low activities of the KA synthesizing enzymes KAT I and KAT II in postmortem MS brains. The latter results were supported by the analysis of CSF probes from MS patients where a decrease in KA was found in the relapse phases [84]. However, changes in the synthesis of KA do not necessarily lead to an alteration in the synthesis of QUIN or 3-HAA. Consequently, modulation of the kynurenine pathway in human MS remains to be further investigated.

The dual role of IDO: immune regulation and bystander damage. Immune regulation (left side): Infiltrating Th1 cells (T) secrete high amounts of interferon-γ inducing IDO expression in microglia (MG). Through the subsequent Trp depletion and production of toxic metabolites, T cell growth is inhibited, and apoptosis is supported. This negative feedback loop may underlie the self-limitation of inflammation not only in MS. Bystander damage (right side): IDO induction causes enhanced production of neurotoxins such as QUIN and 3-HAA. Excessive production during neuroinflammation is likely to contribute to neurodegeneration
The overall beneficial effects of IFN-β treatment in MS are poorly understood but often explained by a shift from Th1 to Th2-mediated immune responses. Interestingly, IFN-β induces the kynurenine pathway and the synthesis of QUIN in human macrophages, although to a lesser extent than IFN-γ [5]. This is in line with the observation that treatment with IFN-β increased the relative IDO activity in blood serum samples (Kyn/Trp) from MS patients [85]. However, increased IDO activity was not found in specimens deriving from patients who received long-term treatment with IFN-β. This is of note, as QUIN and 3-HAA induce secondary degeneration of neurons and potentially of oligodendrocytes [86]. This must be taken into account when testing therapeutical strategies targeting the kynurenine pathway in MS. Moreover, the contribution of Trp metabolites to the increasingly appreciated axonal and neuronal damage in MS [19] must be further explored.
References
Higuchi K, Kuno S, Hayaishi O (1963) Enzymic formation of D-kynurenine. Fed Proc 22:243
Carlin JM, Borden EC, Sondel PM, Byrne GI (1989) Interferon-induced indoleamine 2,3- dioxygenase activity in human mononuclear phagocytes. J Leukoc Biol 45:29–34
Currier AR, Ziegler MH, Riley MM, Babcock TA, Telbis VP, Carlin JM (2000) Tumor necrosis factor-alpha an lipopolysaccharide enhances interferon-induced antichlamydial indoleamine dioxygenase activity independently. J Interferon Cytokine Res 20:369–376
Fallarino F, Grohmann U, Vacca C, Bianchi C, Orabona C, Spreca A, Fioretti MC, Lucetti P (2002) T cell apoptosis by tryptophan catabolism. Cell Death Differ 9:1069–1077
Guillemin GJ, Kerr SJ, Pemberton LA, Smith DG, Smythe GA, Armati PJ, Brew BJ (2001) IFN-beta1b induces kynurenine pathway metabolism in human macrophages: potential implications for multiple sclerosis treatment. J Interferon Cytokine Res 21:1097–1010
Alberati-Giani D, Ricciardi-Castagnoli P, Kohler C, Cesura AM (1996) Regulation of the kynurenine metabolic pathway by interferon-gamma in murine cloned macrophages and microglial cells. J Neurochem 66:996–1004
Heyes MP, Achim CL, Wiley CA, Major EO, Saito K, Markey SP (1996) Human microglia convert L-tryptophan into the neurotoxin quinolinic acid. Biochem J 320:595–597
Nettelnbreker E, Zeidler H, Bartels H, Dreses-Werringloer U, Daubener W, Holtmann H, Kohler L (1998) Studies of persistent infection by Chlamydia trachomatis serovar K in TPA-differentiated U937 cells and the role of IFN-gamma. J Med Microbiol 47:141–149
Pfefferkorn ER (1984) Interferon gamma blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cell to degrade tryptophan. Proc Natl Acad Sci 81:908–912
Daubener W, MacKenzie CR (1999) IFN-gamma activated indoleamine 2,3- dioxygenase activty in human cells is an antiparasitic and an antibacterial effector mechanism. Adv Exp Med Biol 467:517–524
Muller AJ, Prendergast GC (2005) Marrying immunotherapy with chemotherapy: why say IDO? Cancer Res 65:8065–8068
Bauer TM, Jiga LP, Chuang JJ, Randozzo M, Opelz G, Terness P (2005) Studying the immunosuppressive role of indoleamine 2,3-dioxygenase: tryptophan metabolites suppress rat allogeneic T-cell responses in vitro and in vivo. Transpl Int 18:95–100
Li Y, Tredget EE, Ghaffari A, Lin X, Kilani RT, Ghahary A (2006) Local expression of indoleamine 2,3-dioxygenase protects engraftment of xenogeneic skin substitute. J Invest Dermatol 126:128–136
Sakurai K, Zou JP, Tschetter JR, Ward JM, Shearer GM (2002) Effect of indoleamine 2,3-dioxygenase on induction of experimental autoimmune encephalomyelitis. J Neuroimmunol 129:186–196
Kwidzinski E, Bunse J, Aktas O, Richter D, Mutlu L, Zipp F, Nitsch R, Bechmann I (2005) Indoleamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. FASEB J 19:1347–1349
Platten M, Ho PP, Youssef S, Fontoura P, Garren H, Hur EM, Gupta R, Lee LY, Kidd BA, Robinson WH, Sobel RA, Selly ML, Steinmann L (2005) Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science 310:850–855
Schwarcz R, Whetsell WO Jr, Mangano RM (1983) Quinolinic acid: an endogenous metabolite that produces axon-sparing lesions in rat brain. Science 219:316–318
Aktas O, Smordochenko A, Brocke S, Infante-Duarte C, Topphoff US, Vogt J, Prozorovski T, Meier S, Osmanova V, Pohl E, Bechmann I, Nitsch R, Zipp F (2005) Neuronal damage in autoimmune neuroinflammation mediated by the death ligand TRAIL. Neuron 46:421–432
Zipp F, Aktas O (2006) The brain as a target of inflammation: common pathways link inflammatory and neurodegenerative diseases. Trends Neurosci 29:518–527
Sanni LA, Thomas SR, Tattam BN, Moore DE, Chaudhri G, Stocker R, Hunt NH (1998) Dramatic changes in oxidative tryptophan metabolism along the kynurenine pathway in experimental cerebral and noncerebral malaria. Am J Pathol 152:611–619
Silva NM, Rodrigues CV, Santoro MM, Reis LF, Alvarez-Leite JI, Gazzinelli RT (2002) Expression of indoleamine 2,3-dioxygenase, tryptophan degradation and kynurenine formation during in vivo infection with Toxoplasma gondii: induction by endogenous gamma interferon and requirement of interferon regulatory factor 1. Infect Immun 70:859–868
Beatty WL, Belanger TA, Desai AA, Morrison RP, Byrne GI (1994) Tryptophan depletion as a mechanism of gamma interferon-mediated chlamydial persistence. Infect Immun 62:3705–3711
MacKenzie CR, Hadding U, Daubener W (1998) Interferon-gamma-induced activation of indoleamine 2,3-dioxygenase in cord blood monocyte-derived macrophages inhibits the growth of group B streptococci. J Infect Dis 178:875–878
Bozza S, Fallarino F, Pitzurra L, Zelante T, Montagnoli C, Bellochio S, Mosci P, Vacca C, Pucetti P, Romani L (2005) A crucial role for tryptophan catabolism at the host/Candida albicans interface. J Immunol 174:2910–2918
Montagnoli C, Bacci A, Bozza S, Gaziano R, Mosci P, Sharpe AH, Romani L (2002) B7/CD28-dependent CD4+CD25+ regulatory T cells are essential components of the memory-protective immunity to Candida albicans. J Immunol 169:6298–6308
Bodaghi B, Goureau O, Zipeto D, Laurent L, Virelizier JL, Michelson S (1999) Role of IFN-gamma-induced indoleamine 2,3 dioxygenase and inducible nitric oxide synthase in the replication of human cytomegalovirus in retinal pigment epithelial cells. J Immunol 162:957–964
Adams O, Besken K, Oberdörfer C, MacKenzie CR, Rüßing D, Däubener W (2004) Inhibition of human herpes simplex virus type 2 by interferon γ and tumor necrosis factor a is mediated by indoleamine 2,3-dioxygenase. Microbes and Infection 6:806–812
Adams O, Besken K, Oberdörfer C, MacKenzie CR, Takikawa O, Däubener W (2004) Role of indoleamine-2,3-dioxygenase in alpha/beta and gamma interferon-mediated antiviral effects against herpes simplex virus infections. J Virol 78:2632–2636
Sardar AM, Reynolds GP (1995) Frontal cortex indoleamine-2,3-dioxygenase activity is increased in HIV-1-associated dementia. Neurosci Lett 187:9–12
Potula R, Poluektova L, Knipe B, Chrastil J, Heilman D, Dou H, Takikawa O, Munn DH, Gendelman HE, Persidsky Y (2005) Inhibition of indoleamine 2,3-dioxygenase (IDO) enhances elimination of virus-infected macrophages in an animal model of HIV-1 encephalitis. Blood 106:2382–2390
Kwidzinski E, Mutlu LK, Kovac AD, Bunse J, Goldmann J, Mahlo J, Aktas O, Zipp F, Kamradt T, Nitsch R, Bechmann I (2003) Self-tolerance in the immune privileged CNS: lessons from the entorhinal cortex lesion model. J Neural Transm Suppl 65:29–49
Bechmann I (2005) Failed central nervous system regeneration: a downside of immune privilege? Neuromolecular Med 7:217–228
Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL (1998) Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 281:1122–1124
Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ (2003) Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med 9:1269–1274
Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL (1999) Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med 189:1363–1372
Munn DH, Sharma MD, Baban B, Harding HP, Thang Y, Ron D, Mellor AL (2005) GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 22:633–642
Lee GK, Park HJ, Macleod M, Chandler P, Munn DH, Mellor AL (2002) Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology 107:452–460
Dehin J, Walczak H, Baumler C, Debatin KM, Kramer PH (1995) Autocrine T cell suicide mediated by APO-1 (Fas/CD95). Nature 373:441
Bechmann I, Mor G, Nilsen J, Eliza M, Nitsch R, Naftolin F (1999) FasL (CD95L, Apo1L) is expressed in the normal rat and human brain: evidence for the existence of an immunological brain barrier. Glia 27:62–74
De Jong EC, Smits HH, Kapsenberg ML (2005) Dendritic cell-mediated T cell polarization. Semin Immun 26:289–307
Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, Candeloro P, Belladonna ML, Bianchi R, Fioretti MC, Pucetti P (2002) CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol 3:1097–1101
Mellor AL, Chandler P, Baban B, Hansen AM, Marshall B, Phikala J, Waldmann H, Cobbold S, Adams E, Munn DH (2004) Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int Immunol 16:1391–1401
Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB (2002) Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med 196:459–468
Terness P, Bauer TM, Rose L, Dufter C, Watzlik A, Simon H, Opelz G (2002) Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med 196:447–457
Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, Marshall B, Chandler P, Antonia SJ, Burgess R, Slingluff CL Jr, Mellor AL (2002) Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science 297:1867–1870
Guillemin GJ, Smythe G, Takikawa O, Brew BJ (2005) Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes and neurons. Glia 49:15–23
Fujigaki S, Saito K, Sekikawa K, Tone S, Takikawa O, Fujii H, Wada H, Noma A, Seishima M (2001) Lipopolysaccharide induction of indoleamine 2,3-dioxygenase is mediated dominantly by an IFN-gamma-independent mechanism. Eur J Immunol 31:2313–2318
Fujigaki H, Saito K, Fujigaki S, Takemura M, Sudo K, Ishiguro H, Seishima M (2006) The signal transducer and activator of transcription 1alpha and interferon regulatory factor 1 are not essential for the induction of indoleamine 2,3-dioxygenase by lipopolysaccharide: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB pathways, and synergistic effect of several proinflammatory cytokines. J Biochem 139:655–662
Hayashi T, Beck L, Rossetto C, Gong X, Takikawa O, Takabayashi K, Broide DH, Carson DA, Raz E (2004) Inhibition of experimental asthma by indoleamine 2,3-dioxygenase. J Clin Invest 114:270–279
Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, Messina JL, Chandler P, Koni PA, Mellor AL (2004) Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest 114:280–290
Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, Belladonna ML, Fioretti MC, Alegre ML, Lucetti P (2003) Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol 4:1206–1212
Grohmann U, Fallarino F, Silla S, Bianchi R, Belladonna ML, Vacca C, Micheletti A, Fioretti MC, Pucetti P (2001) CD40 ligation ablates the tolerogenic potential of lymphoid dendritic cells. J Immunol 166:277–283
Okamoto A, Nikaido T, Ochiai K, Takakura S, Saito M, Aoki Y, Ishii N, Yanaihara N, Yamada K, Takikawa O, Kawaguchi R, Isonishi S, Tanaka T, Urashima M (2005) Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin Cancer Res 11:6030–6039
Brandacher, G, Perathoner A, Ladurner R, Schneeberger S, Obrist P, Winkler C, Werner ER, Werner-Felmayer G, Weiss HG, Göbel G, Margreiter R, Königsrainer A, Fuchs D, Amberger A (2006) Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin Cancer Res 12:1144–1151
Stone TW, Perkins MN (1981) Quinolinic acid: a potent endogenous excitant at amino acid receptors in CNS. Eur J Pharmacol 72:411–412
Chiarugi A, Meli E, Moroni F (2001) Similarities and differences in the neuronal death processes activated by 3OH-kynurenine and quinolinic acid. J Neurochem 77:1310–1318
Eastman CL, Guilarte TR (1990) The role of hydrogen peroxide in the in vitro cytotoxicity of 3-hydroxykynurenine. Neurochem Res 15:1101–1107
Okuda S, Nishiyama N, Saito H, Katsuki H (1996) Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine. Proc Natl Acad Sci USA 93:12553–12558
Chiarugi A, Cozzi A, Ballerini C, Massacesi L, Moroni F (2001) Kynurenine 3-mono-oxygenase activity and neurotoxic kynurenine metabolites increase in the spinal cord of rats with experimental allergic encephalomyelitis. Neuroscience 102:687–695
Perkins MN, Stone TW (1982) An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res 247:184–187
Cozzi A, Carpendeo R, Moroni F (1999) Kynurenine hydroxylase inhibitors reduce ischemic brain damage: studies with (m-nitrobenzoyl)-alanine (mNBA) and 3,4-dimethoxy-[-N-4-(nitrophenyl)thiazol-2yl]-benzenesulfonamide (Ro 61–8048) in models of focal or global brain ischemia. J Cereb Blood Flow Metab 19:771–777
Carpenedo R, Meli E, Peruginelli F, Pellegrini-Giampietro DE, Morini F (2002) Kynurenine 3-mono-oxygenase inhibitors attenuate post-ischemic neuronal death in organotypic hippocampal slice cultures. J Neurochem 82:1465–1471
Moroni F, Russi P, Gallo-Mezo MA, Moneti G, Pellicari R (1991) Modulation of quinolinic and kynurenic acid content in the rat brain: effects of endotoxins and nicotinylalanine. J Neurochem 57:1630–1635
Yu P, Di Prospero NA, Sapko MT, Cai T, Chen A, Melendez-Ferro M, Du F, Whetsell WO Jr, Guidetti P, Schwarcz R, Tagle DA (2004) Biochemical and phenotypic abnormalities in kynurenine aminotransferase II-deficient mice. Mol Cell Biol 24:6919–6930
Guidetti P, Okuno E, Schwarcz R (1997) Characterization of rat brain kynurenine aminotransferases I and II. J Neurosci Res 50:457–465
Sapko MT, Guidetti P, Yu P, Tagle DA, Pellicari R, Schwarcz R (2006) Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: Implications for Huntington’s disease. Exp Neurol 197:31–40
Mackay GM, Forrest CM, Stoy N, Christofides J, Egerton M, Stone TW, Darlington LG (2006) Tryptophan metabolism and oxidative stress in patients with chronic brain injury. Eur J Neurol 13:30–42
Sinz EH, Kochanek PM, Heyes MP, Wisniewski SR, Bell MJ, Clarks RS, DeKosky ST, Blight AR, Marion DW (1998) Quinolinic acid is increased in CSF and associated with mortality after traumatic brain injury in humans. J Cereb Blood Flow Metab 18:610–615
Saito K, Nowak TS Jr, Markey SP, Heyes MP (1993) Mechanism of delayed increases in kynurenine pathway metabolism in damaged brain regions following transient cerebral ischemia. J Neurochem 60:180–192
Blight AR, Cohen TI, Saito K, Heyes MO (1995) Quinolinic acid and functional deficits following experimental spinal cord injuriy. Brain 118:735–752
Yates JR, Heyes MP, Blight AR (2006) 4-chloro-3-hydroxyanthranilate reduces local quinolinic acid synthesis, improves functional recovery and preserves white matter after spinal cord injury. J Neurotrauma 23:866–881
Depboylu C, Reinhardt TA, Takikawa O, Imai Y, Maeda H, Mitsuya H, Rausch D, Eiden LE, Weihe E (2004) Brain virus burden and indoleamine-2,3-dioxygenase expression during lentiviral infection of rhesus monkey are concomitantly lowered by 6-chloro-2′,3′-dideoxyguanosine. Eur J Neurosci 19:2997–3005
An SF, Groves M, Gray F, Scaravilli F (1999) Early entry and widespread cellular involvement of HIV-1 DNA in brains of HIV-1 positive asymptomatic individuals. J Neuropathol Exp Neurol 58:1156–1162
Lawrence DM, Major EO (2002) HIV-1 and the brain: connections between HIV-1-associated dementia, neuropathology and neuroimmunology. Microbes Infect 4:301–308
Nemeth H, Toldi J, Vecsei J (2006) Kynurenines, Parkinson’s disease and other neurodegenerative disorders: preclinical and clinical studies. J Neural Transm Suppl 70:285–304
Roshick C, Wood H, Caldwell HD, McClarty G (2006) Comparison of gamma interferon-mediated antichlamydial defense mechanisms in human and mouse cells. Infect Immun 74:225–238
Boado RJ, Li JY, Nagaya M, Zhang C, Pardridge WM (1999) Selective expression of the large neutral amino acid transporter at the blood-brain barrier. Proc Natl Acad Sci USA 96:12079–1284
Beagles KE, Morrison PF, Heyes MP (1998) Quinolinic acid in vivo synthesis rates, extracellular concentrations and intercompartmental distributions in normal and immune-activated brain as determined by multiple-isotope microdialysis. J Neurochem 70:281–291
Fukui S, Schwarcz R, Rapoport SI, Takada Y, Smith QR (1991) Blood–brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J Neurochem 56:2007–2017
Raine CS (1991) Multiple sclerosis: a pivotal role for the T cell in lesion development. Neuropathol Appl Neurobiol 17:265–274
Flanagan EM, Erickson JB, Viveros OH, Chang SY, Reinhard JF Jr (1995) Neurotoxin quinolinic acid is selectively elevated in spinal cords of rats with experimental allergic encephalomyelitis. J Neurochem 64:1192–1196
Kepplinger B, Baran H, Kainz A, Newcombe J, Nohl H (2001) Altered kynureninc acid levels in CSF and serum of patients with multiple sclerosis. ECTRIMS’2001. Mult Scler Abstr 118(Suppl 1)
Baran H, Kepplinger B, Newcombe J, Stolze K, Kainz A, Nohl H (2000) Lowered kynurenine aminotransferase activities in CNS of MS Patients. Society for Neuroscience 30th Annual Meeting, Abstract no. 1302
Rejdak K, Bartosik-Psujek H, Dobosz B, Grieb P, Giovannoni G, Turski WA, Stemasiak Z (2002) Decreased level of kynurenic acid in cerebrospinal fluid of relapsing-onset multiple sclerosis patients. Neurosci Lett 331:63–65
Amirkhani A, Rajda C, Arvidsson B, Bencsik K, Bodak K, Seres E, Markides KE, Vecsej L, Bergquist J (2005) Interferon-beta affects the tryptophan metabolism in multiple sclerosis patients. Eur J Neurol 12:625–631
Cammer W (2001) Oligodendrocyte killing by quinolinic acid in vitro. Brain Res 896:157–160
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kwidzinski, E., Bechmann, I. IDO expression in the brain: a double-edged sword. J Mol Med 85, 1351–1359 (2007). https://doi.org/10.1007/s00109-007-0229-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-007-0229-7