
Abstract
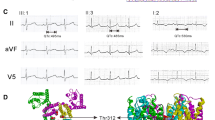
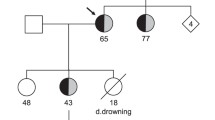
We have found a novel nonsense mutation in the C-terminus of HERG in a four-generation Chinese family with long QT syndrome and investigated the molecular mechanism of this mutation in vitro. Six family members, including the proband, were clinically affected. Syncope and ventricular tachycardia of torsades de pointes were triggered by startling or emotional stress, and β-adrenergic blockade treatment was ineffective. Haplotype analysis showed that only LQT2 markers cosegregated with the disease, and sequence analysis revealed a substitution of T with C at nucleotide position 2770 of the HERG gene (U04270), which creates a stop codon at amino acid position 863 (R863X) of the HERG protein, leading to a deletion of 296 amino acids. Whole cell patch clamp studies showed that the R863X HERG could not induce time-dependent current. Coexpression of R863X with wild-type HERG showed reduced current densities and accelerated voltage-dependent inactivation of HERG channels. Subcellular localization of R863X-EGFP revealed that the mutant did not traffic to the cell surface. These data suggest that R863X failed to form functional HERG channels, contributing to a prolongation of the QT interval and long QT syndrome with a dominant phenotype. These findings provide new insights into the structure-function relationships of the HERG C-terminus.





Similar content being viewed by others

References
Priori SG, Barhanin J, Hauer RN, Haverkamp W, Jongsma HJ, Kleber AG, McKenna WJ, Roden DM, Rudy Y, Schwartz K, Schwartz PJ, Towbin JA, Wilde AM (1999) Genetic and molecular basis of cardiac arrhythmias: impact on clinical management parts I and II. Circulation 99:518–528
Keating MT, Sanguinetti MC (2001) Molecular and cellular mechanisms of cardiac arrhythmias. Cell 104:569–580
Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogne K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V (2003) Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421:634–639
Sanguinetti MC, Jiang C, Curran ME, Keating MT (1995) A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81:299–307
Trudeau MC, Warmke JW, Ganetzky B, Robertson GA (1995) HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 269:92–95
Kupershmidt S, Snyders DJ, Raes A, Roden DM (1998) A K+ channel splice variant common in human heart lacks a C-terminal domain required for expression of rapidly activating delayed rectifier current. J Biol Chem 273:27231–27235
London B, Trudeau MC, Newton KP, Beyer AK, Copeland NG, Gilbert DJ, Jenkins NA, Satler NA, Robertson GA (1997) Two isoforms of the mouse ether-a-go-go-related gene coassemble to form channels with properties similar to the rapidly activating component of the cardiac delayed rectifier K+ current. Circ Res 81:870–878
Lees-Miller JP, Kondo C, Wang L, Duff HJ (1997) Electrophysiological characterization of an alternatively processed ERG K+ channel in mouse and human hearts. Circ Res 81:719–726
Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SAN (1999) MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97:175–187
Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT (2000) Spectrum of mutations in long-QT syndrome genes KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102:1178–1185
Paulussen A, Raes A, Matthijs G, Snyders DJ, Cohen N, Aerssens J (2002) A novel mutation (T65P) in the PAS domain of the human potassium channel HERG results in the long QT syndrome by trafficking deficiency. J Biol Chem 277:48610–48616
Furutani M, Trudeau MC, Hagiwara N, Seki A, Gong Q, Zhou Z, Imamura S, Nagashima H, Kasanuki H, Takao A, Momma K, January CT, Robertson GA, Matsuoka R (1999) Novel mechanism associated with an inherited cardiac arrhythmia: defective protein trafficking by the mutant HERG (G601S) potassium channel. Circulation 99:2290–2294
Zhou Z, Gong Q, Epstein ML, January CT (1998) HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. J Biol Chem 273:21061–21066
Nakajima T, Kurabayashi M, Ohyama Y, Kaneko Y, Furukawa T, Itoh T, Taniguchi Y, Tanaka T, Nakamura Y, Hiraoka M, Nagai R (2000) Characterization of S818L mutation in HERG C-terminus in LQT2. Modification of activation-deactivation gating properties. FEBS Lett 481:197–203
Huang FD, Chen J, Lin M, Keating MT, Sanguinetti MC (2001) Long-QT syndrome-associated missense mutations in the pore helix of the HERG potassium channel. Circulation 104:1071–1075
Nakajima T, Furukawa T, Hirano Y, Tanaka T, Sakurada H, Takahashi T, Nagai R, Itoh T, Katayama Y, Nakamura Y, Hiraoka M (1999) Voltage-shift of the current activation in HERG S4 mutation (R534C) in LQT2. Cardiovasc Res 44:283–293
Lees-Miller JP, Duan Y, Teng GQ, Thorstad K, Duff HJ (2000) Novel gain-of-function mechanism in K(+) channel-related long-QT syndrome: altered gating and selectivity in the HERG1 N629D mutant. Circ Res 86:507–513
Nakajima T, Furukawa T, Tanaka T, Katayama Y, Nagai R, Nakamura Y, Hiraoka M (1998) Novel mechanism of HERG current suppression in LQT2: shift in voltage dependence of HERG inactivation. Circ Res 83:415–422
Chen J, Zou A, Splawski I, Keating MT, Sanguinetti MC (1999) Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. J Biol Chem 274:10113–10118
Berthet M, Denjoy I, Donger C, Demay L, Hammoude H, Klug D, Schulze-Bahr E, Richard P, Funke H, Schwartz K, Coumel P, Hainque B, Guicheney P (1999) C-terminal HERG mutations: the role of hypokalemia and a KCNQ1-associated mutation in cardiac event occurrence. Circulation 99:1464–1470
Schwartz PJ, Moss AJ, Vincent GM, Crampton RS (1993) Diagnostic criteria for the long QT syndrome: an update. Circulation 88:782–784
Splawski I, Shen J, Timothy KW, Vincent GM, Lehmann MH, Keating MT (1998) Genomic structure of three long QT syndrome genes: KCNQ1, HERG and KCNE1. Genomics 51:86–97
Nakajima T, Kurabayashi M, Ohyama Y, Kaneko Y, Furukawa T, Itoh T, Taniguchi Y, Tanaka T, Nakamura Y, Hiraoka M, Nagai R (2000) Characterization of S818L mutation in HERG C-terminus in LQT2. Modification of activation-deactivation gating properties. FEBS Lett 481:197–203
Cui J, Kagan A, Qin D, Mathew J, Melman YF, McDonald TV (2001) Analysis of the cyclic nucleotide binding domain of the HERG potassium channel and interactions with KCNE2. J Biol Chem 276:17244–17251
Paulussen A, Yang P, Pangalos M, Verhasselt P, Marrannes R, Verfaille C, Vandenberk I, Crabbe R, Konings F, Luyten W, Armstrong M (2000) Analysis of the human KCNH2(HERG) gene: identification and characterization of a novel mutation Y667X associated with long QT syndrome and a non-pathological 9 bp insertion. Hum Mutat 15:483
Baroudi G, Pouliot V, Denjoy I, Guicheney, P (2001) Novel mechanism for Brugada syndrome: defective surface localization of an SCN5A mutant (R1432G). Circ Res 88:E78–83
Aydar E, Palmer C (2001) Functional characterization of the C-terminus of the human ether-a-go-go-related gene K+ channel (HERG). J Physiol 534:1–14
Akhavan A, Atanasiu R, Shrier A (2003) Identification of a C-terminal segment involved in maturation and stability of HERG potassium channel. J Biol Chem (in press)
Kagan A, Melman YF, Krumerman A, McDonald TV (2002) 14-3-3 amplifies and prolongs adrenergic stimulation of HERG K+ channel activity. EMBO J 21:1889–1898
Acknowledgements
We are grateful to the family members because this study would be impossible without their enthusiastic participation, and to Professors Michael C.Sanguinetti and Thomas V. McDonald for their gifts of plasmids, and Drs Jielin Pu and Dirk Isbrandt for their advice. This study was financially supported by the International Cooperation Department, the Ministry of Science and Technology, China (to R.H.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Teng, S., Ma, L., Dong, Y. et al. Clinical and electrophysiological characterization of a novel mutation R863X in HERG C-terminus associated with long QT syndrome. J Mol Med 82, 189–196 (2004). https://doi.org/10.1007/s00109-003-0504-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-003-0504-1