
Abstract
Congenital long QT syndrome (LQTS) caused by compound mutations is usually associated with more severe clinical phenotypes. We identified a LQTS family harboring three compound mutations in different genes (KCNQ1-R174C, hERG-E1039X and SCN5A-E428K). KCNQ1-R174C, hERG-E1039X and SCN5A-E428K mutations and/or relevant wild-type (WT) cDNAs were respectively expressed in mammalian cells. IKs-like, IKr-like, INa-like currents and the functional interaction between KCNQ1-R174C and hERG-E1039X channels were studied using patch-clamp and immunocytochemistry techniques. (1) Expression of KCNQ1-R174C alone showed no IKs. Co-expression of KCNQ1-WT + KCNQ1-R174C caused a loss-of-function in IKs and blunted the activation of IKs in response to isoproterenol. (2) Expression of hERG-E1039X alone and co-expression of hERG-WT + hERG-E1039X negatively shifted inactivation curves and decelerated the recovery time from inactivation. (3) Expression of SCN5A-E428K increased peak INa, but had no effect on late INa. (4) IKs and IKr interact, and hERG-E1039X caused a loss-of-function in IKs. (5) Immunocytochemical studies indicated that KCNQ1-R174C is trafficking defective and hERG-E1039X is defective in biosynthesis/degradation, but the abnormities were rescued by co-expression with WT. Thus, KCNQ1-R174C and hERG-E1039X disrupted IKs and IKr functions, respectively. The synergistic lesion, caused by KCNQ1-R174C and hERG-E1039X in IKs, is very likely why patients showed more severe phenotypes in the compound mutation case.
Similar content being viewed by others

Introduction
Congenital long QT syndrome (LQTS) is a life-threatening condition characterized by an abnormally prolonged QT interval on the electrocardiogram (ECG) and torsades de pointes (TdP)-triggered cardiac events such as syncope, cardiac arrest and sudden cardiac death1,2. Molecular genetic studies have revealed that congenital LQTS is linked to mutations in genes encoding for cardiac ion channels or their regulatory proteins. To date at least 15 genes have been identified to be responsible for different subtypes of the syndrome3,4, with the first three LQTS (LQT1-3, caused by mutations in KCNQ1, KCNH2, and SCN5A genes, respectively) being the most common and accounting for approximately 75% of genotype-positive LQTS population3.
The KCNQ1 (Kv7.1) and KCNH2 (hERG or Kv11.1) genes respectively encode α-subunits of slow (IKs) and rapid (IKr) components of channels mediating delayed rectifier potassium (K+) currents. IKs and IKr are two major outward repolarizing K+ currents during the plateau and repolarization phases of the cardiac action potential (AP), and play a critical role in controlling the ventricular AP duration (APD)5,6. The SCN5A gene encodes the α-subunit of the predominant cardiac sodium channel (NaV1.5) that conducts the depolarizing sodium inward current and is mainly responsible for the initial depolarization in cardiomyocytes. Mutations in KCNQ1, hERG and SCN5A can cause LQTS through either a loss-of-function of potassium channels (IKs and IKr) or a gain-of-function of sodium channel leading to an increase in the late INa, lengthening the cardiac APD and manifesting as a prolonged QT interval7.
About 4–11% of LQTS patients host multiple mutations and typically present at a younger age with a more severe cardiac phenotype compared with individuals carrying a single mutation8,9,10. Patients with compound mutations were found to be associated with longer QTc, more frequent cardiac events, and earlier onset of cardiac events. However, the underlying mechanisms remain unclear.
We identified a LQTS family harboring three compound mutations in different genes: one missense mutation in KCNQ1 (R174C), one nonsense mutation in hERG (E1039X) and another missense mutation in SCN5A (E428K). To the best of our knowledge, this is the first report of LQTS associated with three different rare variants. We characterized the functional consequences of the IKs, IKr and INa channels reconstituted with these three mutations in mammalian cells and provide important insight into molecular mechanisms underlying the LQTS associated with compound mutations. Specifically, we found that recombinant channel ‘IKs’ and ‘IKr’ interact and mutations in the two α subunits might produce a synergistic lesion in cardiac channel function. These findings may explain why patients with compound mutations show a more severe phenotype than those carrying a single mutation and suggest that the management of such patients should be tailored to their increased risk for arrhythmias8,9,10,11.
Results
Case description
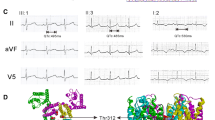
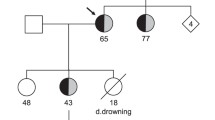
The index patient was a 9-year-old boy (indicated by arrow in family pedigree of Fig. 1a), who experienced repetitive syncope while playing at school. He was identified as carrying three heterozygous mutations in three different genes: p.R174C (c.520 C > T) in KCNQ1, p.E1039X (c.3115 G > T) in hERG, and p.E428K (c.1282 G > A) in SCN5A. Figure 1b shows locations of three mutations in the relevant ion channel protein. The proband was admitted to a nearby hospital and diagnosed with LQTS. The Schwartz score was 4.5 points (T-wave alternans, notched T wave in three leads, low HR for age and syncope with stress). His basal ECG showed a negative T wave in lead III, aVF and V1-V3, and treadmill stress test uncovered a greater QT prolongation and the appearance of biphasic T wave on exercise (Fig. 1c).

Molecular discovery and clinical characterization of three-mutation carriers. (a) The pedigree of three-mutation carriers. Square and circle symbols represent male and female subjects, respectively. Left solid symbols (◧ and ◐) indicate syncope carriers, right solid symbols (◐ and ◧) indicate QTc prolongation, arrow indicates proband, N mark indicates a family member whose information is not available, and QTc = corrected QT interval. (b) Representative result of deoxyribonucleic acid sequence analysis and predicted topology of IKs (I), IKr (II), and INa (III) channels. Red arrows and solid circle symbols indicate the location of three mutations in sequence and topology, respectively (c) Twelve-lead electrocardiograms of proband at rest and after exercise.
The proband had a family history of syncope and QT prolongation (Fig. 1a). Clinical and genetic analysis of his familial members revealed that his 2-year-old sister also carried all three mutations and another younger sister had both KCNQ1-R174C and hERG-E1039X mutations. Both sisters showed QT prolongation, but were asymptomatic. All members with one (KCNQ1-R174C) or two compound mutations (KCNQ1-R174C + SCN5A-E428K) in his mother’s family were asymptomatic and showed no QT prolongation. In LQT1 patients, QT prolongation can be sometimes detected along with the increase of heart rate12, however, they did not agreed for the exercise stress test. In his father’s family (hERG-E1039X carriers), the grandmother (17-year-old at onset time) suffered syncope by a telephone ringing while she was sleeping, which is typical for LQT2. One of the proband’s uncles experienced syncope, and 3/6 mutation carriers showed QT prolongation. Compared with QTc intervals in single hERG-E1039X carriers (440.2 ± 12.2 ms, n = 6), those in hERG-E1039X carriers with additional mutations (500.3 ± 16.7 ms, n = 3) were significantly longer (P < 0.05).
Electrophysiological study
The KCNQ1-R174C mutation produced a mild inhibitory effect on ‘I Ks’
Figure 2a–c show representative whole-cell current traces recorded from CHO cells expressing KCNE1 with KCNQ1-WT, KCNQ1-WT + KCNQ1-R174C and KCNQ1-R174C, respectively. Both the steady state and tail IKs amplitude in WT + R174C conditions were mildly decreased compared to WT alone, whilst R174C KCNQ1 alone produced no currents. Figure 2d shows the current-voltage (I - V) relations for IKs tails elicited after the voltage-step to −50 mV from various test potentials. Figure 2e summarizes IKs tail densities measured at +30 mV. Compared with WT, WT +R174C KCNQ1 significantly decreased IKs densities for voltages between −30 mV and +30 mV (Fig. 2d,e and Table 1). Voltage-dependent activation was quantified by fitting a Boltzmann equation to the I-V relations, and resultant data show that WT + R174C KCNQ1 significantly increased the Vh value (Table 1).

KCNQ1-R174C mutation produced a mild inhibitory effect on ‘IKs’. Representative current traces recorded from CHO cells expressing KCNE1 with KCNQ1-WT (a), KCNQ1-WT + KCNQ1-R174C (b) and KCNQ1-R174C (c). (d) Current-voltage (I-V) relations for tail IKs elicited after the voltage-step to −50 mV from various test potentials. Protocol is shown as inset to ‘A’. (e) Mean peak IKs tail densities recorded on repolarization to −50 mV following 2-s depolarization to +30 mV for the different transfection conditions. (f) Deactivation time constants (τ) obtained by fitting the tail IKs decay to a single exponential function for voltages between −60 mV and −30 mV. (g) Superimposition of IKs traces recorded from HEK 293 cells expressing Yotiao + KCNE1 with KCNQ1-WT or KCNQ1-WT + KCNQ1-R174C before and after bath application of 100 nmol/L isoproterenol. *P < 0.05 vs WT.
Deactivation rates for IKs were measured by depolarizing cells to +30 mV for 2 s, followed by repolarizing steps from −60 mV to −30 mV in 10-mV increments. Figure 2f shows the time constant for deactivation plotted as a function of repolarization potential. Compared with WT, WT + R174C significantly accelerated the deactivation rates between −60 mV and −50 mV (Table 1). Overall, the R174C mutation exerted a mild inhibitory effect on KCNQ1-WT channel13.
As the proband experienced syncopal episodes while playing with his classmates and his QTc interval was prolonged by exercise (Fig. 1c), we further tested whether KCNQ1-R174C might impair the response of IKs to adrenergic stimulation in HEK293 cells co-expressing WT + R174C KCNQ1 with KCNE1 and Yotiao12. As typically shown in Fig. 2g, 100 nM isoproterenol increased IKs by 93.5 ± 15.8% (n = 18) in cells expressing WT alone, but only mildly increased IKs by 50 .4 ± 7.6%, (n = 13, P < 0.05 vs WT) in cells expressing WT + R174C KCNQ1. The result suggests that WT + R174C KCNQ1 partially blunted the activation of IKs in response to isoproterenol, which is consistent with a previous report on the response of this mutant channel to forskolin in Xenopus laevis oocytes13.
HERG-E1039X mutation caused an incomplete loss-of-function in ‘I Kr’
Figure 3a–c show representative whole-cell current traces recorded from CHO cells expressing hERG-WT, hERG-WT + hERG-E1039X and hERG-E1039X, respectively. I-V relations in Fig. 3d indicates that, although the steady state and tail IKr amplitudes in E1039X hERG alone were notably decreased, the IKr amplitudes in WT + E1039X hERG were not significantly changed (Table 2).

HERG-E1039X mutation caused an incomplete loss-of-function in ‘IKr’. Representative current traces recorded from CHO cells expressing hERG-WT (a), hERG-WT + hERG-E1039X (b) and hERG-E1039X (c), respectively. Protocol shown as inset to ‘A’. (d) I-V relations for tail IKr elicited after the voltage-step to −50 mV from various test potentials. *P < 0.05 vs WT, **P < 0.01 vs WT. (e) Normalized activation and steady-state inactivation were determined by means of the voltage protocols shown in the inset. (f) Deactivation time constants (τ) obtained by fitting the tail IKr decay (the inset in the upper panel) to a bi-exponential function for voltages between −70 mV and −40 mV.
Figure 3e and f show normalized voltage dependence of activation/inactivation curves and time constants for deactivation under three different conditions, respectively. Numerical data pertaining to the biophysical properties therein are summarized in Table 2. Compared with those of WT hERG (Vh: −52.50 ± 3.04 mV; τ of recovery from inactivation at −40 mV: 2.90 ± 0.20 ms; n = 18), however, the Vh for the steady state inactivation of both E1039X (−64.63 ± 4.07 mV, n = 14) and WT + E1039X (−64.62 ± 3.75 mV, n = 14) showed a marked (P < 0.05 vs WT) negative shift, and the time course (τ) of recovery from inactivation at −40 mV was significantly (P < 0.05 vs WT) slower for both E1039X (3.85 ± 0.39 ms, n = 14) and WT + E1039X (3.70 ± 0.35 ms, n = 14). The pronounced hyperpolarizing shift of inactivation and slowed recovery from inactivation are likely to decrease the IKr channel availability during excitation and to cause an incomplete loss-of-function in IKr. The parameters for activation and time constants for deactivation were not significantly different between WT, E1039X and WT + E1039X hERG (Table 2).
SCN5A-E428K increased the peak ‘I Na’ currents but produced no late ‘I Na’
Figure 4a shows representative whole-cell current traces recorded from CHO cells expressing hβ1 with SCN5A-WT or SCN5A-E428K. Figure 4b shows I-V relations for peak INa elicited by the protocol shown in the inset, and Fig. 4c summarizes peak INa densities. Compared with WT, E428K SCN5A significantly increased peak INa densities between −70 mV and −10 mV. The peak INa density of E428K was 943.9 ± 93.8 pA/pF (n = 21) at −55 mV, which is significantly (P < 0.05) larger than that of WT (625.4 ± 124.4 pA/pF, n = 19,) at −50 mV (Fig. 4c). Figure 4d and f show conductance-voltage and steady state inactivation curves, representative late INa traces recorded in the presence of 30 μM tetrodotoxin (TTX), and the properties of INa recovery from inactivation for WT and E428K SCN5A. The mutation caused no significant changes to these parameters.

SCN5A-E428K mutation decreased the peak currents in ‘INa’. (a) Representative current traces recorded from CHO cells expressing hβ1 with SCN5A-WT or SCN5A-E428K. Inset shows voltage protocol. (b) I-V relations for peak INa densities of WT and E428K channels. (c) Mean peak INa densities of WT (at −50 mV) and E428K (at −55 mV) channels. (d) Voltage dependence of relative INa conductance activation and steady-state inactivation were determined by means of the voltage protocols shown in the inset. (e) Representative late INa currents recorded in the absence or presence of 30 μM TTX. (f) The time course of recovery from inactivation of INa was elicited with a double pulse protocol. *P < 0.05 vs WT.
Co-expression of KCNQ1/KCNE1 with hERG
Based on the above electrophysiological findings on the three mutations, two loss-of-function mutations of potassium channels appeared to cause the clinical phenotype in this relatively large LQTS family. The proband’s two sisters carrying the same combination of heterozygous compound KCNQ1 and hERG mutations showed long QT features very similar to those of the proband, suggesting that the pathogenesis of triple mutation carriers was mainly associated with the KCNQ1-R174C and hERG-E1039X. We therefore examined the interaction between KCNQ1-R174C and hERG-E1039X by co-expressing the two mutations into CHO cells.
Figure 5a shows representative current traces recorded from a cell co-expressing KCNQ1-WT/KCNE1 and hERG-WT. In the presence of 1 μM E4031 (Kv11.1/IKr inhibitor) and 2 μM HMR1556 (Kv7.1/IKs inhibitor), the current was totally blocked, which confirms that the current was exclusively composed of KCNQ1 + KCNE1 and hERG channel currents. Figure 5b1–b4 and c1–c4 show the representative whole-cell current traces recorded from CHO cells expressing KCNE1 + KCNQ1-WT/KCNQ1-R174C with hERG-WT/ hERG-E1039X in the presence of 1 μM E4031 or 2 μM HMR1556, respectively.

Interaction between ‘IKs’ and ‘IKr’ when KCNQ1/KCNE1 and hERG were co-expressed together. (a) Representative current traces recorded from CHO cells expressing with hERG-WT + KCNQ1-WT/KCNE1 in the presence of 2 μM HMR1556 and 1 μM E4031. (b) and (c) show representative current traces recorded from CHO cells expressing KCNQ1-WT/KCNE1 + hERG-WT, KCNQ1-R174C/KCNE1 + hERG-WT, KCNQ1-WT/KCNE1 + hERG-E1039X, and KCNQ1-R174C/KCNE1 + hERG-E1039X, in the presence of 1 μM E4031 (b 1 –b 4 ) or 2 μM HMR1556 (c 1 –c 4 ). Protocols shown as insets to ‘B1’ and ‘C1’, respectively. (d) I-V relations for tail currents elicited after the voltage-step to −50 mV from various test potentials in the presence of E4031 (except for KCNQ1-WT). **P < 0.01 vs KCNQ1-WT. (e) Deactivation time constants (τ) obtained by the same method as Fig. 2f in the presence of E4031. #P < 0.05 vs KCNQ1-WT + hERG-WT. (f) I-V relations for tail currents elicited after the voltage-step to −50 mV from various test potentials in the presence of 2 μM HMR1556. *P < 0.05 vs hERG-WT + KCNQ1-WT/KCNE1; #P < 0.05 vs hERG-WT + KCNQ1-WT/KCNE1, ##P < 0.01 vs hERG-WT + KCNQ1-WT/KCNE1.
Figure 5d shows I-V relations for tail currents elicited using the protocol in inset of Fig. 5b1 in the presence of 1 μM E4031. Open circles indicate those measured for KCNQ1-WT alone. Compared with KCNQ1-WT, both KCNQ1-WT + hERG-WT (solid squares) and KCNQ1-WT + hERG-E1039X (solid triangles) significantly increased the tail current amplitudes (Fig. 5d and Table 1). In addition, KCNQ1-WT + hERG-WT caused a significant negative shift of both Vh and K values of activation curve, and deactivation rates of KCNQ1-WT + hERG-WT were significantly slower than those of KCNQ1-WT between −60 mV and −50 mV (Table 1). These data suggest that hERG exerted a gain-of-function effect on IKs when co-expressed with KCNQ1.
On the other hand, Fig. 5e shows that KCNQ1-WT + hERG-E1039X (solid triangles) significantly accelerated deactivation times between −60 mV and −50 mV (Table 1) compared with KCNQ1-WT + hERG-WT (solid squares), which suggests that hERG-E1039X led to altered IKs kinetics when co-expressed with KCNQ1, although tail current densities were not significantly affected (Table 1).
Figure 5f shows I-V relations for tail currents elicited using the protocol in inset of Fig. 5c1 in the presence of 2 μM HMR1556. Compared with those of hERG-WT, tail currents and the Vh values for the steady state inactivation of both hERG-WT + KCNQ1-WT and hERG-WT + KCNQ1-R174C were significantly lower, but the Vh values for voltage-dependent activation and the recovery time from inactivation of both hERG-WT + KCNQ1-WT and hERG-WT + KCNQ1-R174C were significantly higher at −40 mV (Table 2). Taken together, KCNQ1 (including KCNQ1-R174C mutant channels) attenuated IKr when co-expressed with hERG. In the meantime, Fig. 5f and Table 2 show that the tail current of hERG-E1039X + KCNQ1-WT (solid squares) was lower than that of hERG-WT + KCNQ1-WT (open squares) and the tail current of hERG-E1039X + KCNQ1-R174C (solid diamonds) was lower than that of hERG-WT + KCNQ1-R174C (open diamonds), which supports the above data that hERG-E1039X caused a loss-of-function in IKr even in the condition of co-expression with KCNQ1. However, we failed to detect any significant changes in parameters between hERG-WT + KCNQ1-WT and hERG-WT + KCNQ1-R174C or between hERG-E1039X + KCNQ1-WT and hERG-E1039X + KCNQ1-R174C, which implicates that KCNQ1-R174C did not affect the function of IKr when co-expressed with hERG.
Expression of channel tetramers on cell membrane was disrupted by either KCNQ1-R174C or hERG-E1039X but rescued by co-expression of WT
Figure 6A shows confocal images of CHO cells expressing KCNQ1/KCNE1 (upper panels) and hERG (lower panels). Both of KCNQ1-WT and hERG-WT proteins were amply transported to the cell membrane. In cells expressing KCNQ1-R174C alone, mutant proteins were mostly distributed in the cytosol but scarcely on the cell membrane, suggesting the trafficking defect of channel protein. While the hERG-E1039X mutant proteins were less presented both in cytosol and on cell membrane, implicating the inhibited protein synthesis and/or its potentiated degradation by hERG-E1039X. However, in cells expressing KCNQ1-WT + KCNQ1-R174C or hERG-WT + hERG-E1039X, channel proteins were expressed both on the cell membrane and in the cytosol, suggesting that the cell membrane expression of channel proteins was increased by the co-expression of WT subunits.

Cellular localization of KCNQ1 and hERG proteins in CHO cells. Confocal images of upper panels in (A) are shown of KCNQ1 (labelled by green) in representative CHO cells co-expressing KCNE1 with KCNQ1-WT, KCNQ1-WT + KCNQ1-R174C, and KCNQ1-R174C, respectively. The lower panels in (a) are confocal images of hERG (labelled by red) in representative CHO cells co-expressing hERG-WT, hERG-WT + hERG-E1039X and hERG-E1039X, respectively. (B) Confocal images of KCNQ1 and hERG proteins in CHO cells co-expressing KCNQ1-WT/KCNE1 + hERG-WT, KCNQ1-WT/KCNE1 + hERG-E1039X, KCNQ1-R174C/KCNE1 + hERG-WT, and KCNQ1-R174C/KCNE1 + hERG-E1039X, respectively. Upper panels in (b) are images of KCNQ1 (labelled by green), middle panels are images of hERG (labelled by red), and lower panels are merged images of KCNQ1 + hERG (labelled by yellow). Cells were immunostained with an anti-KCNQ1 or an anti-hERG antibodies. Nuclei were immunostained with DAPI.
Figure 6B shows confocal images of CHO cells co-expressing (from left to right) KCNQ1-WT/KCNE1 + hERG-WT, KCNQ1-WT/KCNE1 + hERG-E1039X, KCNQ1-R174C/KCNE1 + hERG-WT, and KCNQ1-R174C/KCNE1 + hERG-E1039X. The upper (or middle) panels show that in cells co-expressing KCNQ1-WT/KCNE1 + hERG-WT and KCNQ1-WT/KCNE1 + hERG-E1039X (or middle panels: KCNQ1-R174C/KCNE1 + hERG-WT), KCNQ1 (or middle panels: hERG) proteins were expressed both on the cell membrane and in the cytosol. But, in cells (upper panels) co-expressing KCNQ1-R174C/KCNE1 + hERG-WT (or -E1039X), KCNQ1-R174C proteins were mostly distributed in the cytosol. On the other hand, in cells (middle panels) expressing hERG-E1039X + KCNQ1-WT (or -R174C)/KCNE1, hERG-E1039X proteins were less present both in cytosol and on cell membrane. These data are consistent with those in Fig. 6A, and suggest that KCNQ1 and hERG did not affect the expression pattern of proteins one-another, which is also confirmed by the merged figures in lower panels.
Discussion
Although LQTS caused by two compound mutations is relatively common, the arrhythmia associated with three different compound mutations is a rare case, which accounts for 0.2% of our LQTS cohort. The present study on the three-compound mutation case indicated that KCNQ1-R174C produced a mild inhibitory effect on IKs and hERG-E1039X caused an incomplete loss-of-function in IKr. In addition, the present study showed that IKs and IKr interact: it is striking that hERG-E1039X caused a loss-of-function in IKs when co-expressed with KCNQ1, which is likely to exacerbate the dysfunction of IKs caused by KCNQ1-R174C. This result might reveal why compound mutations are associated with increased arrhythmic risk.
Of the three pathogenic mutations in the present study, KCNQ1-R174C was previously reported to be associated with both heterozygous LQT114 and homozygous autosomal-recessive LQT115, in which the homozygous KCNQ1-R174C carrier displayed extreme QT prolongation and suffered multiple breakthrough cardiac events before succumbing to his malignant LQTS phenotype. Our clinical data show that patients with heterozygous KCNQ1-R174C or KCNQ1-R174C/SCN5A-E428K compound mutation were asymptomatic and their QTc intervals were not prolonged (Fig. 1a), suggesting that the phenotype caused by heterozygous KCNQ1-R174C is not severe and individuals carrying the same mutation exhibit diverse cardiac phenotypes clinically13,14,15. These results are well explained with our electrophysiological data: KCNQ1-WT + KCNQ1-R174C produced a mild inhibitory effect on IKs channel and KCNQ1-R174 alone produced no IKs current (Fig. 2b–d and Table 1). Immunocytochemical study show that, same as the cell surface expression pattern of another KCNQ1 mutation G269S12, the trafficking-deficiency in the homologous KCNQ1-R174C channel was greatly rescued by co-expression with the WT subunit (upper panels in Fig. 6A), resulting in the increased expression of channel proteins on the cell membrane. This result further explains clinical phenotypes of KCNQ1-R174C mutation carriers. Consistent with a previous finding that KCNQ1-R174C blunted the increase in IKs with forskolin in Xenopus laevis oocytes13, we found that KCNQ1-R174C blunted the increase in IKs with isoproterenol, which further confirms our previous speculation: a patient with KCNQ1 mutation showing an excessive prolongation of QT intervals on exercise is likely due to an adrenergic up-regulation of ICa,L without concomitant up-regulation of IKs12.
SCN5A-E428K was reported to be linked to atrial fibrillation (AF)16,17. The present clinical data show that two mutation carriers (harboring KCNQ1-R174C simultaneously) in proband’s mother family were asymptomatic and did not exhibit QTc interval prolongation (Fig. 1a). Electrophysiological study revealed that SCN5A-E428K increased peak INa but did not affect the late INa, which indicates that this mutation might be associated with such genetic disorders as AF rather than LQTS because the increase of the late INa is a characteristic indicator for LQT318,19. In addition to above data, we predicted the pathogenicity of substitutions in SCN5A-E428K and KCNQ1-R174C mutations through the PolyPhen-2 system20. The results show that SCN5A-E428K is relatively benign, whereas the KCNQ1-R174C is strongly considered to be damaging.
HERG-E1039X is a novel nonsense mutation in distal C-terminus. Clinical data show that two of hERG-E1039X mutation carriers experienced syncope and half of the mutation carriers showed QT prolongation in proband’s father family (Fig. 1a). Functional analysis indicates that hERG-E1039X mutation shifted the inactivation curve of IKr in the hyperpolarizing direction and decelerated the time of recovery from inactivation (Table 2), while other gating kinetics and the current density were not significantly affected. Previous studies postulated that nonsense mutations in hERG cause abnormal transcription/translation of IKr21. The present immunocytochemical data show that, in cells expressing hERG-WT + hERG-E1039X, channel proteins were amply expressed both on the cell membrane and in the cytosol (lower panels in Fig. 6A), suggesting that channel protein expression in heterozygous channels was very similar to that in WT channels although E1039X mutant alone disrupted the biosynthesis and/or degradation of hERG channel protein. The distinguishing features of IKr kinetics are the rapid voltage-dependent inactivation and recovery from inactivation, subsequently coupling with a slow deactivation21,22. Most LQT2-causing mutations associated with abnormal channel gating/kinetics are involved in the accelerated deactivation21,23,24. Only a few studies reported that the loss-of-function in IKr caused by hERG channel pore missense mutations (V644L24, G584S25, V603L and A614V26) was associated with channel inactivation. The present study provides evidence that E1039X, a nonsense mutation located in hERG’s distal C-terminus, caused LQT2 through inactivation mechanism, which gives us two notions: (1) in addition to affecting gene transcription/translation, a nonsense mutation in hERG can lead to LQT2 through disrupting inactivation gating of IKr; (2) the distal C-terminus is also involved in the inactivation in IKr. In the present study, we cannot rule out the possibility that a nonsense-mediated mRNA decay (NMD) is involved in the phenotype of patients carrying hERG-E1039X. The position of E1039X is close to the other two hERG nonsense mutations (W1001 × and R1014X) which were reported to degrade mutant mRNA by NMD and to be associated with LQT221.
The functional interaction between IKr and IKs is still in dispute27,28,29. Ren et al. showed that transiently expressed WT or mutant KCNQ1 downregulated Kv11.1 in both CHO and HEK 293 cells stably expressing hERG and the interactions of two channels occurred via the C terminus30, whose findings are consistent with the present study: both KCNQ1-WT and KCNQ1-R174C mutant decreased hERG channel currents when co-expressed with hERG in CHO cells. When co-expressed with KCNQ1, C-terminal mutation hERG-E1039X disrupted KCNQ1/KCNE1 currents, whist hERG-WT produced a different effect in KCNQ1/KCNE1 currents. These data also support the above study that hERG C-terminus is involved in the interaction between IKr and IKs30.
It is well known that compound mutation carriers exhibit a more severe phenotype than those with a single mutation8,9,10,11. Westenskow at al found that a KCNQ1 mutant and a KCNE1 mutant could lead to cumulative lesion in IKs8. Biliczki et al. reported that IKs blocker chromanol 293B alone did not markedly lengthen dog ventricular APD, however, when repolarization had already been prolonged by IKr blocker dofetilide, inhibition of IKs with same concentration of chromanol 293B substantially delayed repolarization31. Their data suggest a synergistic prolongation of repolarization produced by IKr and IKs blockade. In the present experiment, hERG-E1039X caused a loss-of-function in KCNQ1/KCNE1 channels. This result indicates that a mutation in hERG not only can disrupt IKr but can worsen IKs function and superimpose to cause a synergistic lesion to the defective IKs encoded with a mutant KCNQ1, leading to further prolongation of APD and the QT interval. Our in silico study also confirms that the synergistic effects of KCNQ1-R174C and hERG-E1039X in IKs could prolong APD markedly (see Supplementary Material). Therefore, although phenotype of heterozygous KCNQ1-R174C carriers are mild, patients harboring additive pathogenic mutation hERG-E1039X showed more severe QT interval prolongations because of superimposed IKs lesion caused by hERG-E1039X mutation and the proband even experienced a syncope evoked by exercise32. Based on the above functional consequence, we suggest that compound pathogenic mutation carriers should be tailored to their increased risks for arrhythmias because these patients are more readily to be predispose to fatal arrhythmias. For example, a hERG-KCNQ1 compound mutation carrier should avoid QT-prolonging medications and swimming.
Conclusion
We identified a LQTS family harboring three pathogenic mutations in different genes and characterized the functional consequences of related three mutant channels. The synergistic lesion caused by different pathogenic mutation is very likely why patients with compound mutations showed a relatively more severe phenotype.
Methods
Clinical investigation and genetic testing
The study population consisted of 1,015 consecutive LQTS probands whose diagnosis was referred to the criteria of Schwartz et al.1. The protocol for genetic analysis was approved by the Institutional Ethics Committee of Shiga University of Medical Science and performed under its guidelines. Written informed consent was obtained from every subject before analysis, in accordance with the last version of the Declaration of Helsinki and with recommendations by the local ethics committee. Genomic deoxyribonucleic acid (DNA) used for genetic evaluation was isolated from venous blood lymphocytes. Genetic screening for mutations in LQTS-related genes including KCNQ1, hERG, SCN5A, KCNE1, KCNE2, KCNJ2 and CACNA1C was conducted by denaturing high-performance liquid chromatography (WAVE system, Transgenomic Inc., Omaha, Nebraska). For abnormal screening patterns, sequencing was performed with an automated sequencer (ABI PRISM 3100×, Applied Biosystems, Foster City, California).
Heterologous expression of cDNA in CHO cells
Full-length complementary deoxyribonucleic acid (cDNA) encoding human wild-type (WT) KCNQ1 (GenBank AF000571, kind gift from Dr. J. Barhanin, Institut de Pharmacologie Moleculaire et Cellulaire, CNRS, Valbonne, France) was subcloned into a pIRES2-EGFP expression vector. Full-length cDNA encoding human KCNE1 (GenBank M26685) subcloned into the pcDNA3.1 expression vector was obtained by polymerase chain reaction from human heart cDNA library (Clontech Laboratories, Mountain View, CA, USA). Full-length cDNA encoding human WT-hERG (GenBank AF363636, kind gift from Dr. M. Sanguinetti, University of Utah, Salt Lake City, UT, USA) was subcloned into pRc/CMV expression vector. Full-length cDNA encoding human WT-SCN5A (GenBank AB158469) was subcloned into pcDNA3.1 expression vector. Full-length cDNA encoding human sodium channel β1 subunit (hβ1) was subcloned into a bicistronic plasmid (pEGFP-IRES). KCNQ1-R174C, hERG-E1039X and SCN5A-E428K mutants were constructed using a Quick Change II XL site-directed mutagenesis kit according to the manufacturer’s instructions (Stratagene, La Jolla, California), and they were also subcloned into the pIRES2-EGFP, pRc/CMV and pcDNA3.1 expression vectors, respectively. All mutants were fully sequenced to ensure the fidelity. To constitute IKs, IKr or INa channels, KCNQ1-WT and/or KCNQ1-R174C + KCNE1 cDNAs (0.5 µg WT and/or 0.5 µg R174C + 0.5 µg KCNE1), hERG-WT and/or hERG-E1039X + green fluorescent protein (GFP) cDNAs (1 µg WT and/or 1 µg E1039X + 0.5 µg GFP), or SCN5A-WT and/or SCN5A-E428K + hβ1 cDNAs (1.0 µg WT and/or 1.0 µg E428K + 1.0 µg hβ1) were transiently transfected into CHO cells using Lipofectamine (Invitrogen Life Technologies, Inc. Carlsbad, CA, USA) according to the manufacturer’s instructions. In one set of experiments, we also co-transfected KCNQ1, KCNE1 and Yotiao cDNAs (0.5 µg WT and/or 0.5 µg R174C + 0.5 µg KCNE1 + 2 µg Yotiao) into human embryonic kidney 293 (HEK293).
Solutions and chemicals
The pipette solution for potassium channel current recording contained (in mM): 70 potassium aspartate, 40 KCl, 10 KH2PO4, 1 MgSO4, 3 Na2-ATP, 0.1 Li2-GTP, 5 EGTA and 5 HEPES; pH was adjusted to 7.2 with KOH. The pipette solution for sodium channel current recording contained (in mM): 10 NaF, 110 CsF, 20 CsCl, 10 EGTA and 10 HEPES; pH was adjusted to 7.35 with CsOH. The extracellular solution contained (in mM) 140 NaCl, 5.4 KCl, 1.8 CaCl2, 0.5 MgCl2, 0.33 NaH2 PO4, 5.5 glucose, and 5.0 HEPES; pH was adjusted to 7.4 with NaOH. IKr blocker E-4031 (Wako, JAPAN) was dissolved in distilled water to yield 5 mM stock solution. The IKs blocker HMR1556 (Aventis Pharma Deutchland GmbH) was dissolved in dimethyl sulfoxide (DMSO, Sigma) to yield a 10 mM stock solution. Isoproterenol (Sigma) was dissolved in distilled water (containing 1 mM ascorbic acid) to yield a 10 mM stock solution and was kept in the dark at 4 °C.
Electrophysiological recordings and data analysis
Forty eight hours after transfection, cells attached to a glass coverslip were transferred to a 0.5-ml bath chamber perfused with extracellular solution and maintained at 25 °C (for IKs and IKr) or at 22–23 °C (for INa). Patch-clamp experiments were conducted on GFP positive cells. Whole-cell membrane currents were recorded with an EPC-8 patch-clamp amplifier (HEKA, Lambrecht, Germany) and a resistance of 2.5 to 3.5 MΩ (IKs and IKr) or 1.5 to 2.0 MΩ (INa) electrodes.
Currents were evoked by depolarizing voltage-clamp steps given from a holding potential of −80 mV (IKs and IKr) or −120 mV (INa) to various test potentials. Amplitudes of both IKs and IKr were determined by measuring the amplitude of tail current. All currents were normalized to the cell membrane capacitance to obtain current densities (pA/pF). The protocols used for the assessment of the voltage dependence of activation/inactivation and recovery from inactivation are provided as insets in the relevant figures. Voltage-dependence of activation/inactivation were evaluated by fitting the I-V relation of currents to a Boltzmann as previously described12. The deactivation kinetics of IKs after depolarization and the recovery from inactivation data of IKr were determined by a single exponential equation: Y(t) = A0 + A exp(−t/τ). The deactivation kinetics of IKr after depolarization, the recovery from inactivation of INa and decay phase of INa data were fitted with a bi-exponential function of the form: Y(t) = A0 + Af exp(−t/τf) + As exp(−t/τs), where Af and As are the fractions of fast and slow relevant components, respectively. The persistent inward (late) INa, a hallmark of biophysical abnormality in LQT3, was determined as the tetrodotoxin (TTX, 30 µM)-sensitive current measured after 800 ms of depolarization at −20 mV.
Immunocytochemistry
Forty-eight hours after transfection, CHO cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS, pH 7.4) for 20 min and permeabilized with 0.2% Triton X-100 in PBS (PBST) for 10–20 min at room temperature. Cells were blocked with 10% Blocking One (Nacalai, Japan) in PBST for 30 min and then incubated overnight at 4 °C with rabbit polyclonal anti-Kv7.1 antibody (1:5000, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and goat polyclonal anti-Kv11.1 antibody (1:1000, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Following incubation, cells were labeled with an AlexaFluor® 488-conjugated donkey anti-rabbit IgG (Molecular Probes, Eugene, Oregon) at 1:400 dilution for KCNQ1 or with an AlexaFluor® 568-conjugated donkey anti-goat IgG (Molecular Probes, Eugene, Oregon) at 1:400 dilution for hERG. Nuclei were stained with 4′-6-diamino-2-phenylindole (DAPI). Immunofluorescence stained cells were captured using a confocal laser scanning system Clsi (Nikon) on an Eclipse TE2000-E inverted microscope (Nikon). In the present study, if a cell showed KCNQ1 expression, above 90% of such cells simultaneously showed hERG expression in co-expression experiments.
Statistical analysis
All data are expressed as mean ± SE, with the number of experiments in parentheses. Statistical comparisons were analyzed using unpaired Student t-test or 1-way ANOVA with Newman-Keuls post hoc test. A P value of < 0.05 was considered statistically significant.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Schwartz, P. J., Crotti, L. & Insolia, R. Long-QT syndrome: from genetics to management. Circ. Arrhythm. Electrophysiol. 5, 868–877 (2012).
Roden, D. M. Clinical practice: long-QT syndrome. N. Engl. J. Med. 358, 169–176 (2008).
Bohnen, M. S. et al. Molecular pathophysiology of congenital long QT syndrome. Physiol Rev. 97, 89–134 (2017).
Schwartz, P. J., Ackeman, M. J., George, A. L. & Wilde, A. A. Impact of genetics on the clinical management of channelopathies. J. Am. Coll. Cardiol. 62, 169–180 (2013).
Sanguinetti, M. C. & Long, Q. T. syndrome: ionic basis and arrhythmia mechanism in long QT syndrome type 1. J. Cardiovasc. Electrophysiol. 11, 710–712 (2000).
Viswanathan, P. C., Shaw, R. M. & Rudy, Y. Effects of I Kr and I Ks heterogeneity on Action potential duration and its rate dependence: a simulation study. Circulation. 99, 2466–2474 (1999).
Nerbonne, J. M. & Kass, R. S. Molecular physiology of cardiac repolarization. Physiol. Rev. 85, 1205–1253 (2005).
Westenskow, P., Splawski, I., Timothy, K. W., Keating, M. T. & Sanguinetti, M. C. Compound mutations: a common cause of severe long-QT syndrome. Circulation. 109, 1834–1841 (2004).
Schwartz, P. J., Priori, S. G. & Napolitano, C. How really rare are rare disease? The triguing case of independent compound mutations in the long QT syndrome. J. Cardiovasc. Electrophysiol. 14, 1120–1121 (2003).
Itoh, H. et al. Long QT syndrome with compound mutation is associated a more severe phenotype: A Japanese multicenter study. Heart Rhythm. 7, 1411–1118 (2010).
Priori, S. G. et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRS, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 10, 1932–1963 (2013).
Wu, J. et al. A molecular mechanism for adrenergic-induced long QT syndrome. J. Am. Coll. Cardiol. 63, 819–827 (2014).
Matavel, A., Medei, E. & Lopes, C. M. PKA and PKC partially rescue long QT type 1 phenotype by restoring channel-PIP2 interactions. Channels (Austin). 4, 3–11 (2010).
Chouabe, C. et al. Prooerties of KvLQT1 K+ channel mutations in Romano-Ward and Jervell and Lange-Nielsen inherited cardiac arrhythmias. EMBO. J 16, 5472–5479 (1997).
Giudicessi, J. R. & Ackerman, M. Prevalence and potential genetic determinants of sensorineural deafness in KCNQ1 homozygosity and compound heterozygosity. Circ. Cardiovasc. Genet. 6, 193–200 (2013).
Darbar, D. et al. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. 117, 1927–1935 (2008).
Ritchie, M. D. et al. Chromosome 4q25 variants are genetic modifiers of rare ion channel mutations associated with familial atrial fibrillation. J. Am. Coll. Cardiol. 60, 1173–1181 (2012).
Remme, C. A. Cardiac sodium channelopathy associated with SCN5A mutation: electrophysiological, molecular and genetic aspects. J. Physiol. 591, 4099–4116 (2013).
Bennett, P. B., Yazawa, K., Makita, N. & George, A. L. Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 376, 683–685 (1995).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. Chapter 7, Unit7.20 (2013).
Gong, Q., Zhang, L., Vincent, G. M., Horne, B. D. & Zhou, Z. Nonsense mutations in hERG cause a decrease min mutant mRNA transcripts by nonsense-medicated mRNA decay in human long QT syndrome. Circulation. 116, 17–24 (2007).
Vandenberg, J. I. et al. hERG K+ channels: structure, function, and clinical significance. Physiol. Rev. 92, 1393–1478 (2012).
Moss, A. J. et al. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation. 105, 794–799 (2002).
Anderson, C. L. et al. Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat. Commun. 5, 5535–5564 (2014).
Zhao, J. T. et al. Not all hERG pore domain mutations have a severe phenotype: G584S has an inactivation gating defect with mild phenotype compared to G572S, which has a dominant-negative trafficking defect and severe phenotype. J. Cardiovasc. Electrophysiol. 20, 923–930 (2009).
Nakajima, T. et al. Novel mechanism of HERG current suppression in LQT2: shift in voltage dependence of HERG inactivation. Circ. Res. 83, 415–422 (1998).
Ehrlich, J. R. et al. KvLQT1 modulates the distribution and biophysical properties of HERG. A novel alpha-subunit interaction between delayed rectifier currents. J. Biol. Chem. 279, 1233–1241 (2004).
Biliczki, P. et al. Trafficking-deficient long QT syndrome mutation KCNQ1-T587M confers severe clinical Phenotype by impairment of HERG membrane localization: evidence for clinically significant I Kr-I Ks alpha-subunit interaction. Heart Rhythm. 6, 1792–1801 (2009).
Grunnet, M. et al. Functional assessment of compound mutations in the KCNQ1 and HERG genes associated with long QT syndrome. Heart Rhythm. 2, 1238–1249 (2005).
Ren, X. Q. et al. Pore mutants of HERG and KvLQT1 downregulate the reciprocal currents in stable cell lines. Am. J. Physiol. Heart Circ. Physiol. 299, H1525–1534 (2010).
Biliczki, P., Virag, L., Iost, N., Papp, J. G. & Varro, A. Interaction of different potassium channels in cardiac repolarization in dog ventricular preparations: role of repolarization reserve. Br. J. Pharmacol. 137, 361–368 (2002).
Jost, N. et al. Restricting excessive cardiac action potential and QT prolongation: a vital role for I Ks in human ventricular muscle. Circulation. 112, 1392–1399 (2005).
Acknowledgements
This work was supported by National Natural Science Foundation of China (#81273501 and #81470378).
Author information
Authors and Affiliations
Contributions
J.W., Y.M., W.D., T.H., H.K. and Q.W. performed the experiments. J.W., H.I., F.T., and T.M. analysed the results. J.W., S.O., J.H., A.J., H.M. and M.H. conceived the experiments. J.W., Y.M. and M.H. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, J., Mizusawa, Y., Ohno, S. et al. A hERG mutation E1039X produced a synergistic lesion on IKs together with KCNQ1-R174C mutation in a LQTS family with three compound mutations. Sci Rep 8, 3129 (2018). https://doi.org/10.1038/s41598-018-21442-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-21442-6
- Springer Nature Limited