
Abstract
Osthole, also known as osthol, is a coumarin derivative found in several medicinal plants such as Cnidium monnieri and Angelica pubescens. It can be obtained via extraction and separation from plants or total synthesis. Plenty of experiments have suggested that osthole exhibited multiple biological activities covering antitumor, anti-inflammatory, neuroprotective, osteogenic, cardiovascular protective, antimicrobial, and antiparasitic activities. In addition, there has been some research done on the optimization and modification of osthole. This article summarizes the comprehensive information regarding the sources and modification progress of osthole. It also introduces the up-to-date biological activities of osthole, which could be of great value for its use in future research.

Similar content being viewed by others

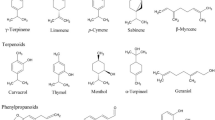

Introduction
Coumarin, with a 2H-benzopyran-2-one core structure, is considered the simplest form within a huge class of naturally occurring phenolic substances. It was derived from a French term “Coumarou” and first isolated as a natural product from tonka beans by Vogel in 1820 [1]. Coumarins are widely distributed in higher plants (mostly from Umbelliferae and Rutaceae families), as well as bacteria and fungi. Natural coumarins are mainly divided into six types according to the chemical structure of the most common cores. These include simple coumarins, furanocoumarins/dihydrofuranocoumarins (linear and angular), pyranocoumarins/dihydropyranocoumarins (linear and angular), biscoumarins/dicoumarins, benzocoumarins, and coumestans [2].
Both natural and synthetic coumarin-based compounds have attracted much attention due to their various function. The function of coumarins in plants as secondary metabolites has been estimated to protect against herbivores and microorganisms [3, 4]. Owing to the aromatic odor, coumarin has been used in perfumes since 1882 [5]. By the way, they have been used as additives in food and cosmetics because of their fungicide and antioxidant activities [6]. Moreover, plenty of coumarins have been considered as drug candidates with diverse pharmacological activities, benefiting from their attractive features, such as low molecular weight, simple structure, high solubility in most of the organic solvents, low side effects, fewer drug resistance, high bioavailability, broad-spectrum, and better curative effects [7]. Various pharmacological effects of coumarins have been reported. They exhibit anti-inflammatory, antibacterial, antiviral, inhibition of cyclooxygenases, antioxidant, antithrombotic, inhibition of lipoxygenase, inhibition of xanthine oxidase, anti-Alzheimer’s disease (AD), antidiabetic, anticonvulsant, as well as antitumor effects [8]. Apart from their biological activities, coumarins have also been used as photocleavable protecting groups or fluorescent probes due to their luminescent properties [9]. Osthole (7-methoxy-8-(3-methyl-2-butenyl) coumarin), also known as osthol, is a coumarin derivative first obtained from the Cnidium plant (Fig. 1). It is mainly found in the 14 genera of Umbelliferae (Angelica, Archangelica, Cachrys, Cnidium, Selinum, Ferula, Heracleum, Libanotis, Petroselinum, Pastinaca, Peucedanum, Pimpinella, Prangos, and Seseli) and 17 genera of Rutaceae (Citrus, Clausena, Feronia, Flindersia, Haplophyllum, Limonia, Melicope, Micromelum, Murraya, Myrtopsis, Pentaceras, Phebalium, Pilocarpus, Poncirus, Skimmia, Thamnosma, and Ticorea). It can also be found in Compositae and Leguminosae [10, 11]. Osthole is so named due to its highest content found in Fructus Cnidii, which is the dried and mature fruit of Cnidium monnieri (L.) Cusson and commonly applied in the clinical practice of Traditional Chinese Medicine (TCM) to treat male sexual dysfunction and other symptoms. A number of studies have shown that osthole exhibited various pharmacological and biological effects, including antitumor, anti-inflammation, neuroprotection, immunomodulator, and hepatitis suppressor [12, 13].

Structure of osthole
Although osthole has attracted a great deal of attention in recent years, only few reviews have been conducted on this important active ingredient of TCM [12, 13], focusing on its bioactivities. This review aimed to provide updated and comprehensive information about osthole, covering its sources, biological activities, and analogs in recent years. It will provide some helpful reminders for future research on this valuable natural product.
Sources of osthole
Plant sources
Osthole belongs to simple coumarins, which originate from the phenylpropanoid pathway [14]. The biosynthesis of coumarins starts with the formation of L-phenylalanine [15, 16], which is transformed into trans-cinnamic acid catalyzed by phenylalanine ammonia-lyase (PLA). Cinnamtae-4-hydroxylase (C4H) catalyzes the conversion of trans-cinnamic acid to 4-coumaric acid, which is further converted into 4-coumaroyl-CoA catalyzed by 4-coumarate-CoA ligase (4CL). The subsequent reaction of 4-coumaroyl-CoA to 2,4-dihydroxycinnamoyl CoA is performed in the presence of 4-coumaoyl-CoA 2’-hydroxylase. Then the 2,4-dihydroxycinnamoyl CoA undergoes spontaneous lactonization to form the critical intermediate, umbelliferone. Among the above enzymes involved in coumarin biosynthesis, PLA, C4H, and 4CL have been verified in Cnidium monnieri (L.) Cuss by Wu’s research group [15]. They also identified the genes encoding these enzymes using quantitative real-time PCR. As for C2’H, Kong et al. identified it from Peucedanum praeruptorum Dunn and characterized its function in the biosynthesis of coumarin skeleton [16]. Although the 6-C- and 7-O-prenylation of umbelliferone has been described in vitro, the enzymatic step of umbelliferone prenylation at 8-position still needs further study. Karamat et al. characterized a membrane-bound prenyltransferase (PcPT) in parsley. It was found that PcPT had strict substrate specificity toward umbelliferone and dimethylallyl diphosphate (DMAPP), affording predominant 6-C-prenylated product (demethylsuberosin) and rare 8-C-prenylated derivative (osthenol) [17]. O-methylation is one of the most important reactions in coumarin biosynthesis. However, the methylation mechanism has been little known and few enzymes participating in the O-methylation of specific hydroxylated coumarins have been described. The first enzyme specific to bergaptol O-methylation (BMT) was identified in 2004 [18]. Another enzyme similar to caffeic acid O-methyltransferase (COMT-S) involved in catalyzing the hydroxylated coumarins in Peucedanum praeruptorum Dunn was identified in 2019 [19]. Despite the unclear methylation mechanism, it is to be expected that a similar O-methyltransferase (OMT) catalyzes the methylation of osthenol to produce osthole (Fig. 2).

Proposed pathway of osthole biosynthesis. PLA phenylalanine ammonia-lyase, C4H cinnamate-4-hydroxylase, 4CL 4-coumarate-CoA ligase, C2’H 4-Coumaroyl CoA 2′-hydroxylase, PT prenyltransferase, DMAPP dimethylallyl diphosphate, OMT O-methyltransferase
There has been a lot of research done on the extraction and separation of osthole from Fructus Cnidii, and most research has involved the process of organic solvent extraction. However, other various extraction methods have been developed, including microwave-assisted extraction, supercritical CO2 extraction, ultrasonic-assisted extraction, alkali-solution and acid-isolation, aqueous two-phase extraction, percolation extraction, and enzymolysis extraction (Table 1).
Synthesis
There are two synthetic strategies for osthole: One is to initially synthesize a coumarin skeleton and subsequently introduce 8-isopentenyl. The other strategy involves the simultaneous ring closing of coumarin with the introduction of isopentenyl in the same reaction.
Liu et al. reported the synthetic method used to synthesize osthole through the olefin metathesis procedure of 8-allyl-7-methoxycoumarin [20]. Compound 2 was obtained by heating the solution of 2,4-dihydroxybenzaldehyde (1) and ethyl 2-chloroacetate together with (2-ethoxy-2-oxoethyl) triphenylphosphonium in ethanol. Treatment of 2 with 3-chloropropene in dry acetone in the presence of potassium carbonate and potassium iodide afforded compound 3, which was then heated with ethylene glycol in a sealed tube to primarily afford the C-8 Claisen rearrangement product 4. Methylation of compound 4 with dimethyl sulfate in acetone resulted in compound 5, which reacted with 2-methyl-2-butylene catalyzed by a Grubbs 2nd catalyst in dichloromethane according to the alkenes metathesis procedure to produce osthole (Scheme 1).

Synthesis of osthole. Reagents and conditions: a ClCH2COOC2H5, Ph3P+-CH2COOC2H5, CH3CH2OH, 80 °C, 2 h, 72%; b 3-Chloropropene, K2CO3, KI, acetone, 60 °C, 22 h, 90.6%; c ethylene glycol, reflux, 6 h; d (CH3O)2SO2, K2CO3, KI, acetone, rt, 5 h, 94.6%; e 2-methyl-2-butylene, Grubbs 2nd catalyst, DCM, 45 °C, 2 h, 77.6%
In another work, Yin et al. developed a general and efficient palladium-catalyzed protocol for the synthesis of an array of C-allyl coumarins by using allyl tributyltins as multi-coupling nucleophiles. The functionalized 7-hydroxy-8-iodocoumarin (6) was prepared by treating 7-hydroxycoumarin (2) with I2/KI/20% NH3.H2O (Scheme 2). It was then transformed into 7-alkoxy-8-iodocoumarin (7) in the condition of MeI/K2CO3/acetone. The 8-iodocoumarin derivative underwent a Stille cross-coupling reaction with tributyl (3-methylbut-2-en-1-yl) stannane in the presence of a catalytic amount of Pd(PPh3)4 in DMF at 80 °C, resulting in productive yields of osthole [21]. Similarly, an inexpensive and eco-friendly method to synthesize osthole with a Grignard reagent assisted by non-precious metals was developed by Liu et al. This method employed CuI and LiCl as promoters and covered 7-methoxyl-8-iodocoumarin (7) to result in osthole with a yield of 80% (Scheme 2) [22].

Synthesis of osthole. Reagents and conditions: a I2/KI, 20% NH3.H2O, rt; b MeI, K2CO3, acetone; c for reference [21]: tributyl(3-methylbut-2-en-1-yl)stannane (1.5 equiv), Pd(PPh3)4 (5 mol%), PPh3(5 mol%), LiCl (4.0 equiv), DMF, 80 °C; for reference [22]: (1) i-PrMgCl, THF, −20 °C; (2) 1-bromo-3-methylbut-2-ene, CuI, LiCl, −20 °C to rt, 80%
Gulías et al. reported a practical procedure to synthesize the scaffold of coumarin through a Rhodium(III)-catalyzed C–H activation/annulation reaction [23]. The compound 2-hydroxy-4-methoxybenzaldehyde (8) was placed in a reaction with N-iodosuccinimide to introduce iodine to the 8-position of coumarin in the presence of aluminum chloride. The aldehyde group of compound 9 was converted into vinyl through a Wittig reaction rendering compound 10, which was then transformed into the cyclization product 7 catalyzed by Rhodium (III). Osthole was then synthesized from compound 7 via a Stille cross-coupling reaction similar to the method Yin reported (Scheme 3).

Synthesis of osthole. Reagents and conditions: a (1) AlCl3, DCM, −20 °C; (2) NIS, −20 °C to rt, overnight, 88%; b PPh3MeBr, NaH, THF, rt, 12 h, 46%; c [Cp*RhCl2]2 (2.5 mol%), Cu(OAc)2.H2O (1.2 equiv), MeCN, 85 °C, 16 h, 37%; d tributyl(3-methylbut-2-en-1-yl)stannane, Pd(dppf)Cl2, DMF, 125 °C, 5 h, 32%
A microwave-promoted tandem Claisen rearrangement/olefination/cyclization approaches to osthole was performed by Schmidt and co-workers [24]. Compound 11 was synthesized from compound 8 via a palladium-catalyzed allylation with tert-butyl (2-methylbut-3-en-2-yl) carbonate, and then made to react with ethyl 2-(triphenylphosphoranylidene) acetate to impart osthole via a Claisen rearrangement/olefination/cyclization in one pot (Scheme 4). Similarly, Konrádová et al. reported a microwave-assisted synthesis of coumarins. Osthole was prepared with an overall yield of 78 % by employing compound 8 in a two-pot protocol (Scheme 4) [25].

Synthesis of osthole. Reagents and conditions: a tert-butyl (2-methylbut-3-en-2-yl) carbonate, [Pd(PPh3)4] (1.0 mol%), THF, 0 °C, 87%; b 2-methylbut-3-yn-2-ol, TFAA, Cu(acac), DBU, MeCN, −5 to 0 °C, 5 h; c H2 (1 atm), Pd/CaCO3, EtOAc, rt; d for reference [24]: ethyl 2-(triphenylphosphoranylidene) acetate, N,N-diethylaniline (0.15 M), MW, 250 °C, 1 h, 68%; for reference [25]: methyl 2-(triphenylphosphoranylidene)acetate, MW (300 W), toluene, 185 °C, 1 h. (TFAA = trifluoroacetic anhydride, acac = acetylacetonate, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene)
Biological activities of osthole
The biological activities of osthole discussed here were mainly reported from relative research activities performed since 2015.
Cytotoxic and antitumor activity
Recent research regarding the cytotoxic and antitumor activity of osthole has involved various kinds of tumor cells/models as showed in Table 2. These include breast cancer cells [26,27,28,29], ovarian carcinoma (OC) cells [30,31,32], endometrial carcinoma (EC) cells [33], head and neck squamous cell carcinoma (HNSCC) cells [34], hepatocellular carcinoma (HCC) cells [35,36,37], human gastric cancer cells [38, 39], cervical cancer cells [40,41,42], rhabdomyosarcoma (RMS) cells [43], intrahepatic cholangiocarcinoma (ICC) cells [44], esophageal squamous cell carcinoma (ESCC) cells [45], renal cell carcinoma (RCC) cells [46, 47], nasopharyngeal cancer (NPC) cells [48], bladder cancer cells [49], lung cancer cells [50,51,52], leukemia cells [53, 54], osteosarcoma (OS) cells [55], human melanoma cells [56], and glioma cells [57].
The antitumor effects of osthole and its relevant molecular mechanism are still being researched. The antitumor effects mainly cover inhibiting the proliferation, promoting apoptosis, inducing cell cycle arrest, inhibiting migration and invasion, overcoming chemoresistance, inducing the production of reactive oxygen species, promoting antitumor immune responses, triggering DNA damage, and inducing autophagy and pyroptosis. The application of osthole has been suggested to suppress the phosphatidylinositol-3 kinase (PI3K)/protein kinase B (AKT) signaling pathway in HNSCC cells [34], human gastric cancer cells [38], ICC cells [44], ESCC cells [45], K562 cells [54], MG-63cells, and SAOS-2 cells [40]; regulate peroxisome proliferator-activated receptor γ (PPARγ) and recombinant farnesoid X receptor (FXR)-mediated target genes involved in cell growth and metabolism in MCF-7 cells [28]; suppress signal transducer and activator of transcription 3 (STAT3) in TNBC cells [29]; regulate Bcl-2, Bax and Caspase 3/9 in A2780 cells, OV2008 cells [31], HeLa cells [41]; decrease cytoplasmic poly (A) denylation element binding protein (CPEB2) expression via upregulating miR-424 in Ishikawa and KLE cells [33]; and downregulate c-FLIP expression in Caki cells [44], leading to its proliferation-inhibition and apoptosis-induction effects. In addition, Park et al. demonstrated that osthole inhibited the progression of human breast cancer BT-474 and MCF-7 cells via the induction of cell cycle arrest through modulation of cell cycle regulatory genes, induction of loss of mitochondrial membrane potential (MMP), and endoplasmic reticulum (ER) stress [27]. It was also reported that osthole could significantly suppress OC cellular growth via mitochondria-mediated apoptosis, trigger cell autophagy, and induce pyroptosis [30]. Notably, Lin et al. found that osthole inhibited HCC cell proliferation, induced cell cycle G2/M phase arrest via the downregulation of Cdc2 and cyclin B1 levels, and triggered DNA damage via an increase in excision repair cross-completion 1 gene (ERCC1) expression [35]. Furthermore, osthole was able to reverse the chemoresistance of cisplatin-resistant cervical cancer to cisplatin (CDDP) [42] and overcome P-glycoprotein (P-gp)-mediated multi-drug resistance (MDR) in human myelogenous leukemia K562/ADM cells [53] through inhibiting the PI3K/AKT signaling pathway. Osthole also exhibited synergistic and additive interactions with CDDP in human melanoma cell FM55P and FM55M2 [56]. Moreover, osthole has been shown to inhibit cancer cell invasion and transition through the suppression of matrix metalloproteinase-2 (MMP-2), matrix metalloproteinase-9 (MMP-9), and inhibiting epithelial-mesenchymal transition (EMT) [31, 35, 47, 50]. Furthermore, Liu et al. revealed that, through the inhibitory effect on epidermal growth factor receptor tyrosine kinase (EGFR-TPK), aminopeptidase N (APN) and MMP-2, osthole could decrease cyclooxygenase 2 (COX-2), vascular endothelial growth factor (VEGF), and nuclear factor kappa-B (NF-κB) expressions, eventually inhibiting the growth and invasion of bladder cancer cells [49]. Osthole combined with CDDP was capable of causing the apoptosis of human lung cancer cell NCI-H460 by inducing production of reactive oxygen species (ROS) [52]. Osthole has been shown to trigger glioma cell necroptosis via ROS overproduction in glioma cells U87 and C6 [57].
The antitumor activities of osthole have been investigated in vivo. Results indicated that it was effective at restraining the progression of AKT/c-Met-driven HCC while alleviating hepatic steatosis in AKT/c-Met mice by reducing the expression of phosphor-extracellular signal-regulated kinase 1/2 (ERK1/2), proliferating cell nuclear antigen (PCNA) and Ki67 [36], and repressing the phospho-AKT (Thr308) /ribosomal protein S6 (RPS6) / fatty acid synthase (FASN) signaling. Zhang et al. announced that osthole enhanced the T-cell mediated antitumor immune responses in tumor-bearing mice with HCC [37].
Anti-inflammatory activity
The anti-inflammatory activities of osthole have been investigated in various cells. These include SH-SY5Y cells subjected to scratch injury [58], peripheral blood mononuclear cells (PBMC) from children with autistic spectrum disorders (ASD) [59], lipopolysaccharide (LPS)-induced mouse peritoneal macrophages [60], LPS-induced mouse monocyte-macrophage RAW 264.7 cells [61], IL-1β-stimulated SW982 cells [62], oleic acid (OA)/LPS induced non-alcohol steatohepatitis (NASH) cells [63], LPS-stimulated BV2 mouse microglia [64], OA-induced FL83B cells [65], histamine-stimulated PBMC [66, 67], LPS-stimulated 3T3-L1 adipocytes [68], and LPS-treated Caco-2 cells [69]. Meanwhile, the anti-inflammatory effect of osthole was also confirmed in animal models, such as LPS-induced acute lung injury mice [70], 2,4-dinitrochlorobenzene-induced atopic dermatitis mice [71], cortical stab wound brain injury mice [58], septic mice [60], dextran sulfate sodium (DSS)-induced ulcerative colitis (UC) mice [61], collagen-induced arthritis rats [62], formalin-induced hyperalgesia mice [72], carrageenan-induced paw edema mice [72], monocrotaline (MCT)-induced pulmonary arterial hypertension (PAH) rats [73], adenine-induced chronic kidney failure (CKF) rats [74], renal ischemia-reperfusion (I/R) injury rats [75], 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis mice [76], lumbar disk herniation (LDH) rats [77], retina I/R injury rats [78], and stab wound injury mice [79].
Janus kinase-signal transduction and activator of transcription (JAK-STAT), mitogen-activated protein kinase (MAPK), and nuclear factor KB (NF-κB) are three important cellular signaling pathways that play pivotal roles in inflammatory responses. Osthole showed an anti-inflammatory activity in LPS-induced acute lung injury mice [70] and renal I/R injury rats [75] by suppressing the JAK2/STAT3 signaling pathway. Osthole exerted anti-inflammatory effects by blocking the activation of MAPK/p38 pathways [61, 65]. In addition, it was confirmed in vitro and in vivo that osthole exerted anti-inflammatory effects via the blocking of NF-κB pathways [58, 60, 61, 63,64,65, 68, 73, 74]. Furthermore, osthole protected against inflammation in a rat model of CKF associated with the activation of the PI3K/AKT/nuclear factor (erythroid-derived 2)-like 2 (Nrf2) signaling [74]. Withal, osthole treatment significantly inhibited atopic dermatitis via directly inhibiting thymic stromal lymphopoietin (TSLP) expression level in keratinocytes [71]. Kordulewska et al. revealed that COX-2 might be involved in the pathogenesis of ASD and/or allergies, and osthole could decrease the effects of COX-2 in inflammation and ASD development [59]. Zhao et al. demonstrated that osthole could inhibit OA/LPS-induced lipid accumulation and inflammatory response through activating PPARα signaling pathway in cultured hepatocytes [63]. It was also demonstrated that osthole might potentially inhibit the activity of histamine H1 receptors and reverse the increase of COX-2 mRNA expression induced by histamine [66, 67].
Neuroprotective activity
The neuroprotective effect of osthole has been investigated in various Alzheimer’s disease (AD) models. Osthole may improve learning and memory functions, inhibit neuronal apoptosis, and elevate the expression of glial cell line derived neurotrophic factor (GDNF), synaptophysin (SYP), and postsynaptic density protein 95 (PSD95). In addition, osthole could remarkably enhance the proliferation of neural stem cells (NSCs) and increase the amount of mature neurons in amyloid precursor protein/presenilin 1 (APP/PS1) transgenic mice [80]. Chu et al. promulgated that osthole exhibited a neuroprotection effect on glutamate-induced apoptotic cells and an AD mouse model via modulating oxidative stress; especially Nrf2 signaling [81]. Lin et al. revealed that miR-101a-3p was the osthole-mediated miRNA in AD, and APP was the target gene. Osthole has protective effects on APP/PS1 mice and APP-SH-SY5Y cells, which might be due to the fact that oshtole is capable of raising the miR-101a-3p expression, inhibiting expression of APP mRNA/protein, and preventing the formation of Aβ protein [82]. Yao et al. found that osthole could decrease the tau protein phosphorylation level via activation of the PI3K/AKT/GSK-3β signaling pathway [83]. Osthole also exerted a neuroprotective effect on neuron synapses in an AD cell model via the upregulation of miR-9 [84]. It protected the neuron synapses against infection with the APP gene and related to inhibition of the recombinant calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2)/adenosine 5’-monophosphate-activated protein kinase (AMPK) signaling pathway [85].
Osthole exhibited a protective effect against ischemia or mechanical injury in the brain. Osthole may prevent cerebral I/P injury via activating the Notch 1 signaling pathway [86], suppressing cell apoptosis [87], reducing oxidative stress injury and reserving the disruption of blood-brain barrier (BBB) [88]. Yan et al. revealed that osthole improved learning and memory functions in mechanical brain injury (MBI) mice and increased the number of neurons in the regions of brain injury, hippocampus DG, and CA3 regions, which were partially mediated by the activation of the Notch signaling pathway [89]. Xia et al. revealed that osthole reduced secondary brain damage by suppressing inflammation and apoptosis in stab wound injury mice models [90]. Kong et al. published findings showing that osthole had protective effects on the nerves in mice with craniocerebral injuries. This data might be related to improving the neurological functions, promoting wound healing, inhibiting the production of inflammatory factors, and increasing the expression of neurotrophic factors in mice [91].
Osthole may delay senescence in NSCs and enhance NSCs proliferation and neuronal differentiation. Yao et al. indicated that osthole delayed tert-butyl hydroperoxide (t-BHP)-induced premature senescence in NSCs, probably through the p16-pRb signaling pathway [92, 93]. Yan et al. discovered that osthole could protect bone marrow-derived NSCs against oxidative stress injury through PI3K/AKT pathway [94]. Kong et al. documented that osthole stimulated adult NSCs, promoted neurogenesis in the hippocampus, and ameliorated cognitive impairment in APP/PS1 transgenic mice [95]. Yao et al. submitted that osthole enhanced NSCs proliferation and neuronal differentiation while suppressing apoptosis via the activation of Wnt/β-catenin signaling [96]. Moreover, osthole combined with NSC transplantation is superior than either method alone for the treatment of traumatic brain injuries caused by neuroendoscopy [97].
Osthole was shown to attenuate Parkinson’s disease symptoms in mice induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) through the suppression of the Notch signaling pathway [98]. Mei et al. demonstrated that osthole had a protective effect against 6-hydroxydopamine (6-OHDA) induced ER stress and oxidative stress injuries in PC12 cells, which involved the AktiGSK-311 pathway [99]. Mao et al. reported that osthole protected hippocampal HT22 cells from glutamate excitotoxicity through a mechanism involving the activation of the PI3K/AKT signaling pathway [100].
Osteogenic activity
Plenty of studies have shown that osthole might promote bone formation and differentiation of osteoblasts, suppress formation and activity of osteoclasts, increase bone density, and be used as a potential agent for osteoporosis treatments. The study of Yu et al. primarily demonstrated that osthole could rescue the ethanol-induced osteonecrosis of the femoral head (ONFH) via the promotion of bone formation. This data may be related to the effects of modulating Wnt/β-catenin pathway, driving vascularization and retarding adipogenesis [101]. Ma et al. documented that osthole inhibited osteoclast formation and bone resorption by regulating NF-κB signaling and nuclear factor of activated T-cells cytoplasmic 1 (NFATc1) activation stimulated by receptor activator of nuclear factor κB ligand (RANKL) [102]. Similar results were reported by Li et al.; that is, osthole inhibited osteoclast formation and partially reversed bone loss induced by 5/6 nephrectomy in mice through the upregulation of osteoprotegerin (OPG)/RANKL [103]. It was also found that osthole could stimulate the expression of OPG via the activation of β-catenin signaling pathway and enhance the bone mass of senile osteoporosis [104]. Zhang et al. discovered that osthole-mediated osteogenesis was related to the activation of the cAMP/CREB signaling pathway and downstream transcription factor osterix expression [105]. Zheng proved that the anti-proliferation and pro-differentiation effects of osthole on osteoblasts were relevant with the activation of the ERS and Wnt/β-catenin signaling pathway, respectively [106]. In addition, Zheng et al. reported that osthole-treated bone marrow mesenchymal stem cells (BMMSCs) were more effective than BMMSCs in osteoporosis treatments. One hypothesis suggestes that the upregulation of protein and mRNA expression level of autophagy-associated genes, Beclin1, and LC3 may be the cause [107]. Yang et al. found osthole could restore the immunosuppressive ability of osteoporotic BMMSCs and enhance the immunosuppressive effects by promoting the Fas/FasL system [108]. Osthole can also promote bone strength and enhance fracture healing through the activation of BMP2 signaling in chondrocytes [109].
Cardiovascular protective activity
Osthole has been shown to possess protective effect on the heart and circulatory system. Liu et al. found osthole significantly inhibited myocardial damage in a myocardial I/R injury model, and this may be achieved by enhancing the autophagy partially through the inhibition of a miR-30a expression [110]. Yang et al. demonstrated that osthole pretreatment prevented myocardial I/R injury from mitochondrial mediating apoptosis by suppressing the activation of the Wnt/β-catenin/p53 signaling pathway [111]. Osthole was also proposed to suppress the right ventricular remodeling process induced by monocrotaline (MCT) in rats. This process may show a relationship to the reduction of myocardial apoptosis and inflammation [112] and the upregulation of the expression of PPARα and PPARγ [113]. Duan et al. revealed that osthole markedly reduced the infarct size and the levels of casein kinase (CK), the MB isoenzyme of creatine kinase (CKMB), lactate dehydrogenase (LDH), and cardiac troponin T (cTnT) in rats with acute myocardial infarction (AMI). These cardioprotective effects might be associated with the inhibition of inflammatory reactions, the reduction in MMP-2 activity, and the activation of MAPK cascades [114].
Antimicrobial and antiparasitic activity
Osthole exerted inhibitory activity against various viruses, bacteriae and parasites. Chen et al. reported that osthole provided stronger antiviral activity on tobacco mosaic virus (TMV) infections as it appears in Nicotiana glutinosa than that of eugenol and ningnanmycin, with inhibitory, protective, and curative effects of 72.57, 70.26, and 61.97%, respectively. It can directly affect the viral particles and correspondingly reduce the level of coat protein [115]. It was suggested that osthole can be used as an inhibitor of MCR-1 enzyme and restore the antibacterial activity of polymyxin B against MCR-1 positive enterobacteriaceae (mainly including Escherichia coli and Klebsiella pneumoniae) [116]. In addition, osthole showed remarkable anti-Leishmania activity in vitro and in vivo [117]. Yuan et al. reported osthole exhibited a high activity against Echinococcus granulosus in vitro and Echinococcus multilocularis in vivo [118].
Development of osthole analogs
Modification of the coumarin lactone ring
Inspired by the O-acyl-hydroxylamine groups within new pesticidal agents (Aldicarb, Methomyl, and Oxamyl), Guo et al. prepared a series of osthole-based esters containing O-acyl-hydroxylamine moiety and evaluated their biological activities including insecticidal, hemolytic activities, and cytotoxicity [119]. Intermediate 14 was synthesized via a two-step method because a direct synthesis was unsuccessful. Osthole was initially reacted with a Lawesson reagent in THF to obtain thio-osthole 13. This derivative was then converted into 14 with hydroxylamine hydrochloride treatment. Finally, intermediate 14 was condensed with a corresponding acid to afford the target derivatives 15 (Scheme 5). The SARs evaluation showed that the replacement of carbonyl gourp with thiocarbonyl group did not enchance the growth inhibition (GI) effect against Mythimna separata (M. separata). The derivatives with electron-withdrawing groups (EWGs), on the benzene ring at 1-position, exhibited higher GI activity than that of electron-donating groups (EDGs). In addition, compounds 15a, 15b, 15c, and 15d exhibited more efficient GI effect against M. separata than the botanical insecticide, toosendanin (TSN). Notably, compound 15c with a 2,4-di-Cl-phenyl group at 1-position had the highest corrected mortality rate (CMR) value of 85.7%. Additionally, compound 15e displayed a pronounced larvicidal effect with a lethal concentration 50 (LC50) value of 0.64 μM, lower than that of TSN (Tables 3 and 4).

Synthesis of osthole analogs 15. Reagents and conditions: a Lawesson reagent, THF, reflux, 24 h, 76%; b NH2OH.HCl, pyridine, reflux, 5 h, 70%; c RCOOH, DCC, DMAP, DCM, rt, 1–3 h, 32–98%
Using a click chemistry approach, Farooq et al. synthesized a series of diverse triazoles linked through the hydroxyl group of lactone ring opened osthole and evaluated their cytotoxicity against seven different human cancer cell lines. Namely, these were Colo-205, HCT-116, T47D, NCIH322, A549, PC-3, and A-431 cell lines [120]. Osthole was subjected to lactone ring opening in DMSO using NaOH as a base yielding a cis (Z) product which simultaneously underwent alkylation of the hydroxyl group in the presence of propargyl bromide to form compound 16. Reacting with various aromatic azides under Sharpless click chemistry conditions (CuSO4.5H2O, and sodium ascorbate in t-BuOH/H2O (1/1)), compound 16 was regioselectivly transformed into 1,4-disubstituted-1,2,3-triazoles (17) through 1,3-dipolar cycloaddition reaction typically called a Huisgen cycloaddition with satisfactory yields (Scheme 6). Most of the synthesized analogs displayed broad-spectrum cytotoxic effects. Not only a particular substituent but also its position played a significant role on the bioactivity profile. It was demonstrated that the broad spectrum cytotoxic efficiency of compounds bearing EWGs, whereas the selective activity of analogs bearing EDGs. Among all the tested triazolyl analogs, compound 17a showed the best activity with half-maximal inhibitory concentration (IC50) values of 1.3, 4.9, 3.6, 41.0, 35.2, 26.4, and 7.2 mM against Colo-205, HCT-116, T47D, NCI-H322, A549, PC-3, A-431 cell lines, respectively (Table 5). It induced potent apoptotic effects in Colo-205 cells by inducing a decrease in mitochondrial membrane potential.

Synthesis of osthole analogs 17. Reagents and conditions: a NaOH/DMSO, propargyl bromide, rt, 2 h; b RN3, t-butanol/H2O, CuSO4.5H2O, sodium ascorbate, rt, 0.5–1 h
Yang et al. synthesized NBM-T-L-BMX-OS01 (BMX, 20) and identified it as a potent inhibitor of histone deacetylase 8 (HDAC8) [121]. Compound 20 was synthesized from osthole utilizing 3 steps of a semi-synthesis (Scheme 7), including ring opening of coumarin lactone in alkaline solution, nucleophilic substitution reaction of hydroxyl group with 4-methoxyl benzyl chloride, and formation of hydroxamic acid. Compound 20 showed an obvious inhibition of HDAC8 with a concentration of 50% of the maximal effect (EC50) value of 0.831 μM and exhibited obviously positive activity in enhancing learning and memory in rats (Table 6).

Synthesis of osthole analog 20. Reagents and conditions: a (1) NaOH, EtOH, Δ, N2, 6 h; (2) Dis-H2O, 1 N HCl, pH 4-5; b 4-methoxy benzyl chloride, K2CO3, acetone, Δ, N2, overnight; c (1) NH2OH.HCl, 10% KOH/MeOH, reflux, N2, overnight; (2) Dis-H2O, 1 N HCl, pH 5–6
Yang et al. studied the anti-angiogenic activities of compound 20 and found that it could inhibit VEGF-induced proliferation, migration, and endothelial tube formation in human umbilical endothelial cells (HUVECs), attenuate VEGF-induced micro-vessel sprouting from aortic rings ex vivo, reduce HCT116 colorectal cancer cells-induced angiogenesis in vivo, inhibit the phosphorylation of VEGFR2, focal adhesion kinase (FAK), AKT, and ERK in HUVECs exposed to VEGF, and inhibit HCT116 cell proliferation [122]. Chen et al. investigated the anti-cancer activities of compound 20. It was found that compound 20 could synergize the efficacy of CDDP in inhibiting proliferation and colony formation, apoptosis induction, and cell cycle arrest. The function of compound 20 might be due to the possibility that it induces mitochondrial ROS generation and attenuates the phosphorylation of ERK and AKT [123].
Modification of the 7-substituent of coumarin
A series of 7-substituted osthole derivatives were synthesized and evaluated for their calcium rivalry activities by Wu [124]. Osthole was allowed to undergo demethylation in the presence of Cys/NaH to afford compound 21. Intermediate 21 was reacted with 1,2-dibromoethane and 1-bromo-3-chloropropane catalyzed by K2CO3, giving the corresponding substitute products 22 and 23, respectively. These were then reacted with aryl piperazine to produce target compounds (Scheme 8). Derivatives of osthole linked with aryl piperazine through three methylene groups were superior to two methylenes. EDGs were beneficial for activity, especially on 2-position of the aryl. Among them, compounds 24a and 24b showed a similar activity as verapamil (EC50 = 6.85 M, 1.03 μM, respectively) (Table 7).

Synthesis of osthole analogs 24–27. Reagents and conditions: a Cys, NaH, DMF, reflux, 3 h, 82.38%; b 1,2-dibromoethane, K2CO3, DMF, rt, 48 h, 42.3%; c 1-bromo-3-chloropropane, K2CO3, DMF, 60 °C, 48 h, 82.3%; d 1-arylpiperazine, K2CO3, KI, DMF, rt, 48 h; e 1-arylpiperazine, K2CO3, KI, DMF, 70 °C, 48 h
Ding et al. synthesized two osthole derivatives by introducing 4-piperidinopiperidine and 2-(1-piperazinyl) pyrimidine moieties to the 7-O position [125]. The 7-hydroxyl intermediate (21) was prepared from osthole using BBr3 and then was reacted with 28 and 29 to afford the corresponding compounds 30 and 31 (Scheme 9). The results of the antitumor activity evaluation by the MTT method indicated that an introduction of large substitutes into the 7-O position could improve inhibitory activity on HCT116 cells (Table 8).

Synthesis of osthole analogs 30 and 31. Reagents and conditions: a BBr3, DCM, −40 °C to rt, 3 h, 92%; b 2-bromoacetyl bromide, K2CO3, DCM, rt, 3 h; c K2CO3, DMF, 90 °C, 4 h, 65% (30), 59% (31)
Zhang et al. synthesized two series of 7-substitued osthole derivatives and evaluated their neuroprotective activity [126]. The demethylation of osthole was catalyzed by Cys/NaH to afford the intermediate 21, which was then reacted with bis(trichloromethyl) carbonate (BTC) and amines in the presence of N,N-diisopropylethylamine (DIPEA), resulting in compound 32. If reacted with 2-chloroacetamide in the presence of potassium carbonate, intermediate 21 was transformed into compounds 33 (Scheme 10). Most synthesized compounds exhibited cytoprotective properties against oxidative stress induced by H2O2 in SH-SY5Y cells. The SARs study indicated that direct introduction of a carbamate into osthole did not show enhanced cell viabilities. Introduction of a piperazine group, tetrahydropyrrole group, and aromatic amine group might be beneficial for the enhancement of osthole’s neuroprotective properties. Compounds 32a and 33a showed better antioxidative and anti-inflammatory activities compared with osthole (Table 9). The docking analysis of compound 33a with Aβ42 (1IYT) revealed the compound bound to C-terminus region of the peptide in U-shape binding gesture. It could interact with residues Met35, Leu34, Ile31, Gly25, Val24 Ala21, and Phe20. An interaction with Gly25, Val24, Ala21, and Phe20 might interrupt the production of salt bridge, which is formed during the aggregation of Aβ42. The isopentenyl group and 4-(pyrrolidin-1-yl)phenyl structure can make compound 33a suitable for the surface of the α-helix of Aβ42 (Fig. 3).

Synthesis of osthole analogs 32 and 33. Reagents and conditions: a Cys, NaH, DMF; b R1NH2, BTC, DIPEA; c R2COCH2Cl, K2CO3, MeCN, reflux

Predicted binding modes of compound 33a with Aβ42 (1IYT)
Analogs modified with an 8-substituent of coumarin
Osthole derivatives containing imine structures and their activities against Plutella xylostella(L.), Tetranychus telarius, 3 kinds of storage pests, 15 kinds of pathogenic fungi, and 6 kinds of pathogenic bacteria were reported in Wei’s dissertation [127]. Osthole was allowed to undergo oxidation in the presence of selenium dioxide to produce intermediate 34, which was then reacted with different amines, affording target compounds (Scheme 11). Compounds 35b and 35c, containing fluorine atoms, exhibited relatively higher activity (EC50 = 25.48 μg/mL, 20.76 μg/mL) against Thantephorus cucumris than osthole (EC50 = 43.99 μg/mL).

Synthesis of osthole analogs 35a–35e. Reagents and conditions: a SeO2, DMSO, EtOH, 90 °C, 3 h; b RNH2/hydroxylamine hydrochloride, EtOH, reflux, 2–5 h
Inspired by the structure of Combretastatin A-4, a stilbene compound isolated from the trunk of Combretum caffrum, Yang et al. designed and synthesized 7 stilbene compounds from osthole and investigated their effects on HUVEC cell proliferation in vitro [128]. Osthole was treated with 3-chloroperbenzoic acid (m-CPBA), NaIO4, and a Jones reagent successively to produce intermediate 38. Intermediate 38 was then allowed to undergo a Friedel-Crafts reaction, reduction, and dehydration, affording target compounds 41a–41d and 41f. Compounds 41d and 41f were hydrolyzed using acid to form compounds 41e and 41g, respectively (Scheme 12). The biological activity assay revealed that these compounds showed lower cytotoxicity against HUVEC. The best compound 41g showed some cytotoxicity against HUVEC with an IC50 value of 12.135 μg/mL, higher than that of CA-4 (IC50 value of CA-4 was 0.0154 μg/mL). This may due to its poor solubility or the cis structure of stilbene.

Synthesis of osthole analogs 41a–41g. Reagents and conditions: a m-CPBA, DCM, rt, 24 h; b NaIO4, acetone/H2O, 80 °C, 6 h; c Jones reagent, acetone, rt, 1 h; d (1) SOCl2, reflux, 2 h; (2) R1R2C6H4, AlCl3, nitrobenzene, 10–15 °C, 1 h; (3) 10% HCl solution; e solution of NaBH4 in EtOH, pyridine, DCM, 60 °C, 6 h; f P2O5, toluene/CHCl3, 15 °C; g 12 mol/L HCl, MeOH/CHCl3; h HOAc, H2O
Yu et al. invented and disclosed a kind of osthole hydrazone/acylhydrazone/sulfonylhydrazone derivative, as well as the preparation method and application in the treatment of Alzheimer’s disease [129]. After oxidation catalyzed by selenium dioxide, osthole was converted to intermediate 34, which was then reacted with various substituted hydrazines/hydrazides/sulfonyl hydrazines to give corresponding osthole derivatives. Most derivatives showed preferable inhibitory activities against acetylcholinesterase and had a potential application value in the development of acetylcholinesterase depressants. Among hydrazone derivatives, EDGs on the substituent of aryl demonstrated to be beneficial. The inhibition rate of compound 42b on acetylcholinesterase was 66.1% at the concentration of 0.5 mg/mL. The substituent of 3-pyridyl in acylhydrazone derivative showed the second strongest inhibition against acetylcholinesterase. (Scheme 13, Table 10).

Synthesis of osthole analogs 42–44. Reagents and conditions: a SeO2, dioxane, EtOH, 60–80 °C, 1.5 h; b R1NHNH2, AcOH, EtOH, reflux, 0.5–1 h; c R2CONHNH2, AcOH, EtOH, reflux, 0.5–1 h; d R3SO2NHNH2, EtOH, rt, 1–2 h
Ren et al. also developed a series of osthole derivatives containing hydrazone/acylhydrazone/sulfonylhydrazone moieties at the C-8 position and evaluated their insecticidal activities [130]. This synthetic strategy was similar to which Yu reported with the exception of a slight change in the reaction conditions. Evaluation of their insecticidal activity against pre-third-instar larvae of M. separata at 1 mg/mL revealed that introduction of certain substituted benzoyl hydrazones on the isopentenyl fragment at the C-8 position of osthole was essential for the enhancement of insecticidal activity, especially when the benzoyl group bearing a EWG. The 4-substituted compounds had better insecticidal activity than the 3-substituted ones. Compounds 43f, 43h, 43k, and 43l showed potent insecticidal activity compared with osthole. Notably, compound 42b exhibited the most potential insecticidal activity with the final mortality rate (FMR) measured at 55.2%; whereas the FMRs of osthole and TSN were 34.5 and 44.8%, respectively. The insecticidal activity was not improved when benzoyl hydrazones was replaced with different phenylacetyl hydrazones or 1-naphthylacetyl hydrazone. Furthermore, hydrazones and phenylsulfonylhydrazones products did not exhibit increased activity. (Table 11).
Additionally, Ren et al. reported a series of osthole derivatives containing oxime ester fragments at the C-8 position and their pesticidal activities [131]. Aldehyde intermediate 34 was synthesized as above, which was reacted with hydroxylamine hydrochloride in the presence of sodium hydroxide to give the oxime intermediate 45. After condensing with various carboxylic acids, intermediate 45 was converted into target compounds 46 (Scheme 14). The acaricidal activities of compounds against the female adults of Tetranychus cinnabarinus (T. cinnabarinus) at 0.5 mg/mL showed that introduction of some EWGs on the benzene ring led to potent compounds, whereas introduction of EDGs on the benzene ring resulted in less active compounds. Compound 46a showed three-fold more potent acaricidal activity of the precursor osthole against T. cinnabarinus. Compounds 46a and 46b showed the most pronounced growth inhibitory activity against M. separata (Table 12). Unsimilarly, introduction of EWGs or EDGs on the pyridyl ring cannot afford potent compounds.

Synthesis of osthole analogs 46. Reagents and conditions: a NH2OH.HCl, NaOH, EtOH/H2O, rt, 24 h, 74%; b RCOOH, DCC or EDC/DMAP, DCM, rt, 8 h, 15–95%
Yang et al. disclosed a series of carboxylate, sulfonate, and amide derivatives of osthole and their antibacterial activities [132,133,134]. Osthole was oxidized into intermediate 34, which was then further oxidized by NaClO2 to afford the carboxylic acid 47, or reduced by sodium borohydride to afford alcohol intermediate 49. Intermediate 47 was condensed with various amines giving target amide derivatives 48. Reacted with carboxylic acids or sulfonyl chlorides, alcohol 49 was transformed into carboxylate and sulfonate derivatives, respectively (Scheme 15). The antibacterial activity assay revealed several osthole derivatives exhibited more potent activity against Staphylococcus aureus (S. aureus), Escherichia coli (E.coli), and Methicillin-resistant Staphylococcus aureus (MRSA) than the precursor and sulfadiazine (Table 13). In general, the amide derivatives of osthole exhibited less antibacterial activity than that of carboxylate and sulfonate derivatives. Among all these three kinds of compounds, EWGs were essential for enhancement of activity. Sulfonate derivatives bearing EWGs on 2- or 4-position of the benzene ring showed higher potency. As for carboxylate derivatives, a EWG-substituted benzyl group was better than a EWG-substituted phenyl group for increasing antibacterial activity. Phenylacetates retained a good activity, whereas aliphatic carboxylates did not display any antibacterial activity.

Synthesis of osthole analogs 48, 50, and 51. Reagents and conditions: a NaClO2, NaH2PO4, t-BuOH, 2-methyl-2-butene, 40 °C; b R1NH2, DCC, DCM, rt; c NaBH3, EtOH, rt; d R2COOH, DCC, DMAP, DCM, rt; e R3SO2Cl, TEA, DCM, rt
Lai et al. disclosed the synthesis of 2′-substituted derivatives of osthole [135]. Reacted with hydrogen bromide in carbon tetrachloride or sodium bromide in ethanol, osthole was converted into the 2′-brominated derivative 52a. Similarly, osthole was transformed into the 2′-iodinated derivative 52b when reacted with hydrogen iodide in carbon tetrachloride or sodium iodide in ethanol (Scheme 16).

Synthesis of osthole analogs 52a and 52b. Reagents and conditions: a HBr in CCl4 or NaBr/EtOH for 52a, HI in CCl4 or NaI/EtOH for 52b
Qian et al. disclosed a series of 8-arylosthole derivatives and their antibacterial activities [136]. Intermediate 7 was synthesized in a similar method as above. Through a Suzuki cross coupling, the 8-iodo intermediate 7 was allowed to react with various arylboronic acids catalyzed by triphenylphosphine palladium chloride to afford target compounds 53 (Scheme 17). The disclosed compound has certain inhibitory effects on various plant pathogens. Compounds 53a and 53c had obvious inhibitory effects on strawberry anthracnose and potato blight, compared with osthole. Compounds 53a, 53b, and 53c showed activity against kiwifruit brown spot and botrytis cinerea (Table 14).

Synthesis of osthole analogs 53. Reagents and conditions: a I2, KI, K2CO3, H2O, 30 °C; b MeI, K2CO3, acetone, 55 °C; c ArB(OH)2, Pd(PPh3)Cl2, Cs2CO3, toluene, 110 °C
Analogs modified with multiple sites of coumarin
Hitotsuyanagi et al. reported a series of osthole derivatives with modifications on the 3,4-olefinic bond, the substituent at 7-position, and the isopentenyl group at 8-position. He then studied their cytotoxic activity [137]. Catalytic hydrogenation of osthole over palladium on carbon gave dihydro-(54) and tetrahydro-(55) derivatives in a ratio of 1:3. They underwent des-O-methylation catalyzed by boron tribromide giving phenols 56 and 57, respectively. Intermediate 56 was converted into compound 58 via a Mitsunobu reaction with the corresponding alcohols or an O-acylation with the corresponding acyl chlorides. The dehydroxylation of 54 occurred through the reduction of the intermediary triflate using 1,1-bis(diphenylphosphino)ferrocene, Pd(OAc)2 and formic acid to afford compound 59. The epoxidation of osthole using m-CPBA yielded epoxide 36, which was subjected to acid hydrolysis affording diol compound 60. The acid-catalyzed hydration and the bromination of osthole yielded tertiary alcohol 61 and dibromide 62, respectively (Scheme 18). The SARs study revealed that the 3,4-olefinic bond and the C5 unit attached at the 8-position were essential for its cytotoxic activity. The unsaturation of the prenyl group is not essential to the activity and the dibromide compound can retain activity. However, introduction of a hydrophilic moiety into the prenyl group resulted in a loss in activity. The substitution of the methyl group by a longer alkyl chain (C2-C7) at the 7-position showed higher cytotoxicity than osthole. The ethoxy (58a) and the n-propoxy (58b) analogs exhibited potent cytotoxicity against V-79 cells with IC50 values of 5.8 and 5.6 μM, respectively; lower than that of osthole as 57 μM.

Synthesis of osthole analogs 57–62. Reagents and conditions: a H2, Pd/C, DCM/EtOH, 21%; b Pd/C, mesitylene, reflux, 72%; c BBr3, DCM, −78 °C, 69%; d ROH, Ph3P, diethyl azodicarboxylate, toluene. rt, e RCOCl, Et3N, DCM, rt; f (1) (CF3SO2)2O, Et3N, DCM, 0 °C; (2)1,1-bis(diphenylphosphino)ferrocene, Pd(OAc)2, HCOOH, DMF, 60 °C, 50%; g m-CPBA, DCM, 0 °C, 88%; h H2SO4, THF/H2O, rt, 73%; i BF3.OEt2, H2O/DCM, rt, 52%; j Br2, DCM, 0 °C, 34%
Gopalakrishnan et al. reported that the use of AlCl3/EtSH reagent for demethylation of osthole at room temperature resulted in direct cyclization by a trans-etherification reaction yielding pyranocoumarin (64). Reaction of osthole with AlCl3-DMS complex yielded the demethylated product osthenol (21) (Scheme 19) [138].

Synthesis of osthole analog 64. Reagents and conditions: a AlCl3, ethanethiol, DCM, 0 °C to rt, 24 h, 76%; b dimethyl sulfide, AlCl3, DCM, 0 °C to rt, 24 h, 62%
You et al. developed a series of osthole derivatives bearing aryl substituents at the 3-position of coumarin and evaluated their growth inhibitory activity against human breast cancer cell lines (MCF-7 and MDA-MB-231) [139]. The hydrogenation of isopentenyl of osthole was catalyzed by platinum oxide to afford the reduced osthole derivative 54, which was subjected to bromination in the presence of NBS under microwave irradiations to yield selectively 3-bromo intermediate 65. Compound 65 was employed to couple with various arylboronic acids to yield compounds 66, which were converted into demethylated derivatives (67) when treated with BBr3 (Scheme 20). The SARs study showed the methoxyl at 7-position was critical for maintaining the antitumor activity, and the modification at 3-position and the hydrogenation on the double bond of prenyl group could obviously improve its antitumor activity. Some derivatives exhibited good cytotoxicity; among them trifluoromethoxy phenyl derivative 66d was found to be the most potent compound with IC50 values of 0.24, 0.31 μM against MCF-7 and MDA-MB-231, respectively, which was improved more than 100-fold compared with its parent compound osthole (Table 15).

Synthesis of osthole analogs 66 and 67. Reagents and conditions: a PtO2, H2, 24 h, 80%; b NBS, NaOAc, CH3CN, MW, 2 h, 60%; c Pd(PPh3)4, phenylboronic acid, K3PO4, dioxane, 60–90%; d BBr3, DCM, −78 °C, 40–70%
Huang et al. synthesized 9 novel, osthole-based N-hydroxycinnamides and investigated their effectiveness as the surface recognition cap in hydroxamate-based compounds as inhibitors of HDACs [140]. Through reduction and demethylation, osthole was converted into intermediate 57, which was reacted with 2-chloroethyl, 3-chloropropyl, and 4-chlorobutyl bromide to afford intermediates 68. Coupling of the methyl coumarates with the previously prepared chlorides 68 provided intermediate 69. Hydrolysis of 69 in the presence of lithium hydroxide gave the corresponding cinnamic acids 70, which were reacted with ethyl chloroformate followed by hydroxylamine to give N-hydroxycinnamides 71 (Scheme 21). The enzymatic assay revealed that either meta- or para- substitution of the N-hydroxycinnamide had a positive contribution to enzyme inhibition, whereas ortho-substituted compounds had much lower activity. It was indicated that a long linker chain with more than three carbons weakened the binding affinity. The long linker may prevent the osthole group from effectively interacting with the potential binding pockets at the rim of the active site. Compounds 71d, 71e, and 71g exhibited potent inhibitory activities (IC50 = 24.5, 20.0, 19.6 nM) against nuclear HDACs in HeLa cells comparable to that of suberoylanilide hydroxamic acid (SAHA; IC50 = 18.7 nM), a potent inhibitor clinically used for the treatment of cutaneous T-cell lymphoma (CTCL). Compounds 71d, 71e, and 71g showed slightly lower growth inhibitory activity against HepG2 cells compared with SAHA (Table 16). The docking analysis of compound 71d with the enzyme was performed using Autodock4 and its accompanying graphical user interface AutoDockTools. Because of the significant conformational plasticity around the active site in HDAC8-inhibitor complex, the crystal structure of HDLP, which has been employed extensively to investigate the binding modes of various HDAC inhibitors, was chosen as a preferred target. The results demonstrate the hydroxamate moiety of 71d binds to the catalytic zinc ion, and that the coumarin ring interacts with the same surface pocket with which the phenyl ring of SAHA binds (Fig. 4). Furthermore, the dimethylpropyl arm of osthole is oriented toward an adjacent enlarged hydrophobic pocket pocket. This indicates modified osthole derivatives with aliphatic handle may be potential selective inhibitors of class IIa HDACs.

Synthesis of osthole analogs 71. Reagents and conditions: a H2, EtOAc, rt, 16 h, 92%; b BBr3, DCM, N2, 0 °C to rt, 2.5 h, 92%; c 1-bromo-2-chloroethane/1-bromo-3-chloropropane/1-bromo-4-chlorobutane, K2CO3, acetone, N2, 56 °C, 16 h, 81–85%; d methyl 4/3/2-hydroxycinnamate, K2CO3, DMF, N2, rt, 16 h, 51–60%; e LiOH, MeOH, N2, 63 °C, 16 h, 93–97%; f (1) ClCO2Et, Et3N, THF, rt, 1 h; (2) NH2OH.HCl, KOH, MeOH, rt, 3 h, 52–64%

Molecular modeling results of compounds 71d (blue) and SAHA (magenta) docked to HDLP (PDB code: 1C3R; surface representation)
Moreover, Huang et al. designed and evaluated a series of aliphatic-chain hydroxamates capped with osthole derivatives as HDACs inhibitors [141]. Oxidation of 55 and demethylation of 54 and 55 were carried out according to the reported methods [137]. Treatment of compound 56 with appropriate bromo-substituted methyl aliphatic acid esters with five to eight carbon chain length by using NaH as well as a catalytic amount of 18-crown-6 and tetra-n-butylammonium iodide (TBAI) yielded the corresponding intermediates 72. Under a similar condition reaction, compound 57, upon replacing NaH with K2CO3, gave corresponding intermediates 73. Hydrolysis of 72 and 73 in the presence of LiOH produced 74 and 75, respectively. Compounds 76 and 77 were synthesized from 74 and 75 via activation with ethyl chloroformate as reactive mixed anhydrides in situ and subsequent treatment with hydroxylamine. Compounds 75 were converted into bishydroxamates (78) when treated with 8 equiv of hydroxylamine (Scheme 22). In spite of three different recognition cap structures, monohydroxamates and bishydroxamates showed similar activity profile concerning chain length of the linker. The enzymatic assay revealed that a six-carbon linker resulted in optimal potency, whereas a four-carbon linker produced the poorest performers. Compounds 76c, 76d, 78c, and 78d exhibited comparable inhibitory activities against nuclear HDACs in HeLa cells to that of SAHA. Compound 76c showed higher cytotoxicity against PC-3, A549 and HepG3B cells compared with SAHA (Table 17). The molecular docking analysis of HDAC8 with compounds 76c and 78c revealed that they were most potent against nuclear extract HDACs, using Autodock and its graphical user interface AutoDockTools (ADT). They can interact with the same surface region of HDAC8 as SAHA (PDB code 1T69). In addition, the higher inhibition of compounds 76c and 78c than that of SAHA may due that the osthole-based caps possessing dimethyl propyl groups are capable of making additional hydrophobic contacts with the HDAC8 (Fig. 5).

Synthesis of osthole analogs 76–78. Reagents and conditions: a Pd-C, mesitylene, Δ, 16 h, 81%; b BBr3, DCM, N2, 0 °C to rt, 2.5 h, 89–92%; c for 72, bromo-substituted methyl aliphatic acid esters, NaH, 18-crown-6, TBAI, DMF, rt, 74–85%; for 73, bromo-substituted methyl aliphatic acid esters, K2CO3, 18-crown-6, TBAI, acetone, Δ, 2 h, 81–89%; d LiOH, MeOH, Δ, 2 h, 91–95%; e (1) ClCO2Et, Et3N, THF, rt, 1 h; (2) 2 equiv NH2OH, KOH, MeOH, rt, 3 h, 38–65%; f (1) ClCO2Et, Et3N, THF, rt, 1 h; (2) 8 equiv NH2OH, KOH, MeOH, rt, 3 h, 59–65%

Molecular modeling results of compounds 76c (magenta) and 78c (green) docked to HDAC8. SAHA is shown in gray sticks
In addition to osthole, Liu et al. also synthesized a series of osthole analogs and evaluated their anthelmintic activity [20]. Compounds 79 and 80 were synthesized from coumarin 2 using the method for synthesizing compound 3 as described above. Compound 3 was heated with ethylene glycol in a sealed tube at 60 °C to mainly afford the C-8 Claisen rearrangement product compound 4 together with its C-6 isomer compound 81 in a ratio of 4:1. Methylation of compound 4 with dimethyl sulfate in acetone resulted in compound 5 and alkylation of compound 4 with alkyl halide together with potassium carbonate and potassium iodide in acetone produced compounds 82a–82c (Scheme 23). The biological assay revealed that the activity of coumarin derivatives have a great relationship with the position of substituted group and the type of substituted group. The 7,8-disubstituted compounds were more effective than 7-position substituent compounds in removing the parasites. The substituent at 8-position was an important functional site in the structure of coumarin compound and its modification could contribute better anthelmintic activity against Dactylogyrus intermedius (D. intermedius). The substituent at 4-position may not play an important role in increasing anthelmintic activity of coumarin analogs. Of all the compounds, compound 82a showed better anthelmintic activity than other compounds against D. intermedius infestation with an EC50 value of 1.81 mg/L (Table 18).

Synthesis of osthole analogs 82 and 84. Reagents and conditions: a ClCH2COOC2H5, Ph3P+-CH2COOC2H5, CH3CH2OH, 80 °C, 2 h, 72%; b 3-chloropropene/3-chloro-3-methyl-1-butylene/3-chloro-2-methyl-1-propene, K2CO3, KI, acetone, 60 °C, 22–24 h, 83–90%; c ethylene glycol, reflux, 6 h; d 3-chloropropene/3-chloro-2-methyl-1-propene/3-chloro-3-methyl-1-butylene/(CH3O)2SO2, K2CO3, KI, acetone, rt - 60 °C, 5–22 h, 60–98%; e ethyl acetoacetate, H2SO4, ice bath, 16 h, 90%
Lee et al. measured a series of N-hydroxycinnamide derivatives (Fig. 6) and examined their hypoglycemia activities in vitro and in vivo [142]. It was found that compounds 84a and 84f had great potential for increasing glucose uptake in L6 skeletal muscle cells, increased glucose transporter 4 (GLUT4) translocation to plasma membranes and counteracted hyperglycemia in mice with streptozotocin-induced diabetes. This was associated with the activation of AMPK and p38 MAPK by compounds 84a and 84f and subsequently led to an amelioration of hyperglycemia.

Structure of compounds 83 and 84
Liu et al. developed an efficient method using Grignard reagents to synthesize osthole derivatives and evaluated their larvicidal activities on mosquitoes [22]. Synthesis of compounds 86 were similar to that of osthole described above. A bio-activity evaluation showed that several derivatives of osthole exhibited good to excellent larvicidal activities (from 35 to 100%) at 10 mg/L. It was noted that compound 80j showed excellent larvicidal activities even at 2 mg/L, which were far better than the precursor osthole as a control (Scheme 24, Table 19).

Synthesis of osthole analogs 86a–86k. Reagents and conditions: a (1) i-PrMgCl, THF, −20 °C; (2) 1-bromo-3-methylbut-2-ene, CuI, LiCl, −20 °C to rt, 43–83%
Above all, the modification progress of osthole was mainly about the lactone ring, 7-methoxyl, 8-isopentenyl, 3,4-double bond and 3-hydrogen, individually or in combination (Fig. 7). The lactone ring was opened and further introduced triazoles or hydroxamic acids, or changed into a lactam ring based on bioisosterism. The 7-methoxyl group of osthole was replaced with ethers or esters after the methyl group was removed. Plenty of research has focused on the 8-isopentenyl group, which was oxidized and subsequently converted into hydrazones, acylhydrazones, sulfonylhydrazones, oxime esters, amides, carboxylate esters, and sulfonate esters. The isopentenyl group was also reduced and halogenated or replaced with aromatic groups. Some osthole derivatives with 7,8-substituents within a ring were also investigated. The 3-hydrogen of osthole was replaced with bromine or aromatic groups.

Reported modifications of osthole
Summary and outlook
Osthole (7-methoxy-8-(3-methyl-2-butenyl) coumarin), also known as osthol, which was first isolated from the Cnidium plant, has demonstrated its antitumor, anti-inflammatory, neuroprotective, osteogenic, cardiovascular protective, antimicrobial, and antiparasitic activities. Although most published studies have focused on the biological activities of osthole, some derivatives and analogs of osthole have also been obtained in recent years through structural modifications. The structural modifications of osthole typically focus on the lactone ring, 7-methoxyl, 8-isopentenyl, 3,4-double bond of coumarin, or simultaneous modification of multiple sites of osthole. Most of these compounds exhibit cytotoxicity, HDACs inhibitory activity, AChE inhibitory activity, antibacterial activity, and insecticidal effect. Several compounds show antagonistic effects on smooth muscle contractions of agonist. In the forthcoming years, with the development of the synthesis and extraction of osthole, as well as investigations of the pharmacological mechanism, the structural modification of osthole will be easier, more rational and more valuable. SARs will be supplemented and clear. From the above review, it’s clear to conclude that osthole and its analogs are promising candidates for the treatment of cancer, neurodegenerative diseases, osteoporosis, and bacterial/fungal/parasitic infections. They are worthy of further research. We hope this review will serve to stimulate research into this fascinating and very useful area, and that it will lead to the design and discovery of osthole-furnished drugs.
References
Al-Warhi T, Sabt A, Elkaeed EB, Eldehna WM. Recent advancements of coumarin-based anticancer agents: an up-to-date review. Bioorg Chem. 2020;103:104163. https://doi.org/10.1016/j.bioorg.
Zhu JJ, Jiang JG. Pharmacological and nutritional effects of natural coumarins and their structure–activity relationships. Mol Nutr Food Res. 2018;e1701073. https://doi.org/10.1002/mnfr.201701073.
Onder A. Anticancer activity of natural coumarins for biological targets. Stud Nat Products Chem. 2020;64:85–109.
Murray RDH, Mendez J, Brown SA. The natural coumarins. Occurrence, chemistry, and biochemistry. Chichester: John Wiley & Sons Ltd; 1982.
Matos MJ, Santana L, Uriarte E, Abreu OA, Molina E, Yordi EG. Coumarins-an important class of phytochemicals. INTECH Open. Science 2015;5:113–40.
Mark R, Lyu X, Lee JJL, Parra-Saldívar R, Chen WN. Sustainable production of natural phenolics for functional food applications. J Funct Foods. 2019;57:233–54.
Stefanachi A, Leonetti F, Pisani L, Catto M, Carotti A. Coumarin: a natural, privileged and versatile scaffold for bioactive compounds. Molecules. 2018;23:250. https://doi.org/10.3390/molecules23020250.
Annunziata F, Pinna C, Dallavalle S, Tamborini L, Pinto A. An overview of coumarin as a versatile and readily accessible scaffold with broad-ranging biological activities. Int J Mol Sci. 2020;21:4618. https://doi.org/10.3390/ijms21134618.
Tasior M, Kim D, Singha S, Krzeszewski M, Ahn KH, Gryko DT. π-Expanded coumarins: synthesis, optical properties and applications. R Soc Chem. 2014;3:1421–46. https://doi.org/10.1039/c4cc03078h.
Zhang QY, Chen HC, Qin LP. Distribution and pharmacological activity of osthole in plant kingdom. World Phytomedicines. 2002;17:16–18.
Yan J, Xuan WD, Bian J. Research progress of osthole. Chin Pharm. 2012;21:110–2.
You LS, Song F, An R, Wang XH. Osthole: a promising lead compound for drug discovery from a Traditional Chinese Medicine (TCM). Nat Prod Commun. 2009;4:297–302.
Zhang ZR, Leung WN, Cheung HY, Chan CW. Osthole: a review on its bioactivities, pharmacological properties, and potential as alternative medicine. Evid Based Complement Altern Med. 2015;2015:919616. https://doi.org/10.1155/2015/919616.
Yang SM, Shim GY, Kim BG, Ahn JH. Biological synthesis of coumarins in Escherichia coli. Micro Cell Fact. 2015;14:65. https://doi.org/10.1186/s12934-015-0248-y.
Shi YY, Zhang SX, Peng DY, Shan CM, Zhao LQ, Wang B, et al. De novo transcriptome analysis of Cnidium monnieri (L.) Cuss and detection of genes related to coumarin biosynthesis. PeerJ. 2020;8:e10157. https://doi.org/10.7717/peerj.10157.
Yao RL, Zhao YC, Liu TT, Huang CL, Xu S, Sui ZW, et al. Identification and functional characterization of a p-coumaroyl CoA 2’-hydroxylase involved in the biosynthesis of coumarin skeleton from Peucedanum praeruptorum Dunn. Plant Mol Biol. 2017;95:199–213. https://doi.org/10.1007/s11103-017-0650-4.
Karamat F, Olry A, Munakata R, Koeduka T, Sugiyama A, Paris C, et al. A coumarin-specific prenyltransferase catalyzes the crucial biosynthetic reaction for furanocoumarin formation in parsley. Plant J. 2014;77:627–38. https://doi.org/10.1111/tpj.12409.
Hehmann M, Lukacin R, Ekiert H, Matern U. Furanocoumarin biosynthesis in Ammi majus L. Cloning of bergaptol O-methyltransferase. Eur J Biochem. 2004;271:932–40. https://doi.org/10.1111/j.1432-1033.2004.03995.x.
Zhao YC, Wang NN, Sui ZW, Huang CL, Zeng ZX, Kong LY. The molecular and structural basis of O-methylation reaction in coumarin biosynthesis in Peucedanum praeruptorum Dunn. Int J Mol Sci. 2019;20:1533. https://doi.org/10.3390/ijms20071533.
Liu GL, Hao B, Liu SP, Wang GX. Synthesis and anthelmintic activity of osthol analogs against Dactylogyrus intermedius in goldfish. Eur J Med Chem. 2012;54:582–90. https://doi.org/10.1016/j.ejmech.2012.06.009.
Yin Q, Yan H, Zhang YQ, Wang YG, Zhang GJ, He YX, et al. Palladium-catalyzed synthesis of 8-allyl or 8-prenylcoumarins by using organotin reagents as multicoupling nucleophiles. Appl Organometal Chem. 2013;27:85–88.
Liu M, Liu Y, Hua XW, Wu CC, Zhou S, Wang BL, et al. Synthesis of osthole derivatives with Grignard reagents and their larvicidal activities on mosquitoes. Chin J Chem. 2015;33:1353–8.
Gulías M, Marcos-Atanes D, Mascareñas JL, Font M. Practical large-scale preparation of benzoxepines and coumarins through Rhodium(III)-catalyzed C–H activation/annulation reactions. Org Process Res Dev. 2019;23:1669–73. https://doi.org/10.1021/acs.oprd.9b00191.
Schmidt B, Riemer M. Synthesis of allyl- and prenylcoumarins via microwave-promoted tandem Claisen rearrangement/Wittig olefination. Synthesis 2016;48:141–9.
Konrádová D, Kozubíková H, Doležal K, Pospíšil J. Microwave-assisted synthesis of phenylpropanoids and coumarins: total synthesis of osthol. Eur J Org Chem. 2017;2017:5204–13. https://doi.org/10.1002/ejoc.201701021.
Mei J, Wang TJ, Zhao SJ, Zhang Y, Mei J. Osthole inhibits breast cancer progression through upregulating tumor suppressor GNG7. J Oncol. 2021;2021:6610511. https://doi.org/10.1155/2021/6610511.
Park W, Park S, Song G, Lim W. Inhibitory effects of osthole on human breast cancer cell progression via induction of cell cycle arrest, mitochondrial dysfunction, and ER stress. Nutrients. 2019;11:2777. https://doi.org/10.3390/nu11112777.
Zhang Y, Song HZ, Wen H, Zhang XH, Chen XT, Qi ZG. Osthole inhibits the proliferation of breast cancer MCF-7 cells via activating peroxisome proliferatoractivated receptor γ. γZhongliu Yanjiu Yu Linchuang. 2015;27:375–80.
Dai XX, Yin CT, Zhang Y, Guo GL, Zhao CG, Wang OC, et al. Osthole inhibits triple negative breast cancer cells by suppressing STAT3. J Exp Clin Canc Res. 2018;37:322. https://doi.org/10.1186/s13046-018-0992-z.
Liang J, Zhou JL, Xu YQ, Huang XF, Wang XF, Huang WH, et al. Osthole inhibits ovarian carcinoma cells through LC3-mediated autophagy and GSDME-dependent pyroptosis except for apoptosis. Eur J Pharm. 2020;874:172990. https://doi.org/10.1016/j.ejphar.2020.172990.
Jiang GQ, Liu J, Ren BY, Tang YW, Owusu L, Li M, et al. Anti-tumor effects of osthole on ovarian cancer cells in vitro. J Ethnopharmacol. 2016;193:368–76. https://doi.org/10.1016/j.jep.2016.08.045.
Bae H, Lee JY, Song J, Song G, Lim W. Osthole interacts with an ER-mitochondria axis and facilitates tumor suppression in ovarian cancer. J Cell Physiol. 2021;236:1025–42. https://doi.org/10.1002/jcp.29913.
Lu KN, Lin JJ, Jiang J. Osthole inhibited cell proliferation and induced cell apoptosis through decreasing CPEB2 expression via upregulating miR-424 in endometrial carcinoma. J Recept Signal Transduct Res. 2020;40:89–96. https://doi.org/10.1080/10799893.2019.1710846.
Yang J, Zhu XJ, Jin MZ, Cao ZW, Ren YY, Gu ZW. Osthole induces cell cycle arrest and apoptosis in head and neck squamous cell carcinoma by suppressing the PI3K/AKT signaling pathway. Chem Biol Interact. 2020;316:108934. https://doi.org/10.1016/j.cbi.2019.108934.
Lin ZK, Liu J, Jiang GQ, Tan G, Gong P, Luo HF, et al. Osthole inhibits the tumorigenesis of hepatocellular carcinoma cells. Oncol Rep. 2017;37:1611–8. https://doi.org/10.3892/or.2017.5403.
Mo YS, Wu Y, Li X, Rao H, Tian XX, Wu DN, et al. Osthole delays hepatocarcinogenesis in mice by suppressing AKT/FASN axis and ERK phosphorylation. Eur J Pharm. 2020;867:172788. https://doi.org/10.1016/j.ejphar.2019.172788.
Zhang LR, Jiang GR, Yao F, Liang GQ, Wang F, Xu H, et al. Osthole promotes anti-tumor immune responses in tumor-bearing mice with hepatocellular carcinoma. Immunopharm Immunotoxicol. 2015;37:301–7. https://doi.org/10.3109/08923973.2015.1035391.
Xu XJ, Liu XY, Zhang Y. Osthole inhibits gastric cancer cell proliferation through regulation of PI3K/AKT. PLoS ONE. 2018;13:e0193449/1–10. https://doi.org/10.1371/journal.pone.0193449. eCollection 2018
Yang Y, Ren F, Tian ZY, Song W, Cheng BF, Feng ZW. Osthole synergizes with HER2 inhibitor, trastuzumab in HER2-overexpressed N87 gastric cancer by inducing apoptosis and inhibition of AKT-MAPK pathway. Front Pharmacol. 2018;9:1392. https://doi.org/10.3389/fphar.2018.01392. eCollection 2018
Che YL, Li J, Li ZJ, Li J, Wang S, Yan Y, et al. Osthole enhances antitumor activity and irradiation sensitivity of cervical cancer cells by suppressing ATM/NF-κB signaling. Oncol Rep. 2018;40:737–47. https://doi.org/10.3892/or.2018.6514.
Zhao XJ. Effect mechanism of HeLa cells apoptosis induced by osthole. Zhongguo Yiyao Daobao. 2016;13:32–35.
Su J, Zhang F, Li X, Liu Z. Osthole promotes the suppressive effects of cisplatin on NRF2 expression to prevent drug-resistant cervical cancer progression. Biochem Biophs Res Commun. 2019;514:510–7. https://doi.org/10.1016/j.bbrc.2019.04.021.
Jarzab A, Luszczki J, Guz M, Skalicka-Wozniak K, Halasa M, Smok-Kalwat J, et al. Combination of osthole and cisplatin against rhabdomyosarcoma TE671 cells yielded additive pharmacologic interaction by means of isobolographic analysis. Anticancer Res. 2018;38:205–10. https://doi.org/10.21873/anticanres.12209.
Zhu XY, Song XL, Xie K, Zhang X, He W, Liu FB. Osthole induces apoptosis and suppresses proliferation via the PI3K/AKT pathway in intrahepatic cholangiocarcinoma. Int J Mol Med. 2017;40:1143–51. https://doi.org/10.3892/ijmm.2017.3113.
Zhu XB, Li ZZ, Li TT, Long F, Lv YS, Liu L, et al. Osthole inhibits the PI3K/AKT signaling pathway via activation of PTEN and induces cell cycle arrest and apoptosis in esophageal squamous cell carcinoma. Biomed Pharmacother. 2018;102:502–9. https://doi.org/10.1016/j.biopha.2018.03.106.
Min KJ, Han MA, Kim S, Park JW, Kwon TK. Osthole enhances TRAIL-mediated apoptosis through downregulation of c-FLIP expression in renal carcinoma Caki cells. Oncol Rep. 2017;37:2348–54. https://doi.org/10.3892/or.2017.5490.
Liu L, Mao J, Wang QF, Zhang ZW, Wu GZ, Tang QZ, et al. In vitro anticancer activities of osthole against renal cell carcinoma cells. Biomed Pharmacother. 2017;94:1020–7. https://doi.org/10.1016/j.biopha.2017.07.155.
Liu PY, Chang DC, Lo YS, His YT, Lin CC, Chuang YC, et al. Osthole induces human nasopharyngeal cancer cells apoptosis through Fas-Fas ligand and mitochondrial pathway. Environ Toxicol. 2018;33:446–53. https://doi.org/10.1002/tox.22530.
Liu J, Xu R, Zhao XK.Mechanisms for effect of osthole on inhibiting growth and invasion of bladder cancer cells. Zhong Nan Da Xue Xue Bao Yi Xue Ban.2016;41:345–52. https://doi.org/10.11817/j.issn.1672-7347.2016.04.002.
Feng HT, Lu JJ, Wang YT, Pei LX, Chen XP. Osthole inhibited TGFβ-induced epithelial-mesenchymal transition (EMT) by suppressing NF-κB mediated Snail activation in lung cancer A549 cells. Cell Adh Migr. 2017;11:464–75. https://doi.org/10.1080/19336918.2016.1259058.
Abosharaf HA, Diab T, Mohamed TM, Atlam FM. Osthole extracted from a citrus fruit that affects apoptosis on A549 cell line by histone deacetylasese inhibition (HDACs). Biotechnol Rep. 2020;28:e00531. https://doi.org/10.1016/j.btre.2020.e00531.
Cheng LX, Zhang BH. Anticancer activity and mechanism of osthole combined with cisplatin in human lung cancer cells. Jianyan Yixue. 2015;30:631–4.
Wang H, Jia XH, Chen JR, Wang JY, You J. Osthole shows the potential to overcome P-glycoprotein-mediated multidrug resistance in human myelogenous leukemia K562/ADM cells by inhibiting the PI3K/AKT signaling pathway. Oncol Rep. 2016;35:3659–68. https://doi.org/10.3892/or.2016.4730.
Pan LP, Guo JM, Yuan W. Osthole induced proliferation inhibition and apoptosis of K562 cells by suppressing PI3K/AKT signal pathway. Linchuang Xueyexue Zazhi. 2016;29:232–4.
Wang L, Yang L, Lu Y, Chen YZ, Liu TH, Peng YL, et al. Osthole induces cell cycle arrest and inhibits migration and invasion via PTEN/AKT pathways in osteosarcoma. Cell Physiol Biochem. 2016;38:2173–82. https://doi.org/10.1159/000445573.
Wroblewska-Luczka P, Grabarska A, Florek-Luszczki M, Plewa Z, Luszczki JJ. Synergy, additivity, and antagonism between cisplatin and selected coumarins in human melanoma cells. Int J Mol Sci. 2021;22:537.
Huangfu MJ, Wei R, Wang J, Qin JL, Yu D, Guan X, et al. Osthole induces necroptosis via ROS overproduction in glioma cells. FEBS Open Bio. 2021;11:456–67. https://doi.org/10.1002/2211-5463.13069.
Kong L, Yao YJ, Xia Y, Liang XC, Ni YN, Yang JX. Osthole alleviates inflammation by down-regulating NF-κB signaling pathway in traumatic brain injury. Immunopharm Immunot. 2019;41:349–60. https://doi.org/10.1080/08923973.2019.1608560.
Kordulewska NK, Kostyra E, Chwala B, Moszynska M, Cieslinska A, Fiedorowicz E, et al. A novel concept of immunological and allergy interactions in autism spectrum disorders: Molecular, anti-inflammatory effect of osthole. Int Immunopharmacol. 2019;72:1–11. https://doi.org/10.1016/j.intimp.2019.01.058.
Jin YY, Qian JC, Ju X, Bao XD, Li L, Zheng SQ, et al. Osthole protects against acute lung injury by suppressing NF-κB-dependent inflammation. Mediators Inflamm. 2018;4934592/1–12. https://doi.org/10.1155/2018/4934592.
Fan HY, Gao ZF, Ji K, Li X, Wu JB, Liu Y, et al. The in vitro and in vivo anti-inflammatory effect of osthole, the major natural coumarin from Cnidium monnieri (L.) Cuss, via the blocking of the activation of the NF-κB and MAPK/p38 pathways. Phytomedicine. 2019;58:152864. https://doi.org/10.1016/j.phymed.2019.152864.
Xu RG, Liu Z, Hou JD, Huang T, Yang M. Osthole improves collagen-induced arthritis in a rat model through inhibiting inflammation and cellular Stress. Cell Mol Bio Lett. 2018;23:19/1–11. https://doi.org/10.1186/s11658-018-0086-0.
Zhao X, Xue J, Xie ML. Osthole inhibits oleic acid/lipopolysaccharide-induced lipid accumulation and inflammatory response through activating PPARα signaling pathway in cultured hepatocytes. Exp Gerontol. 2019;119:7–13. https://doi.org/10.1016/j.exger.2019.01.014.
Bao YX, Meng XL, Liu FN, Wang F, Yang JH, Wang HY, et al. Protective effects of osthole against inflammation induced by lipopolysaccharide in BV2 cells. Mol Med Rep. 2018;17:4561–6. https://doi.org/10.3892/mmr.2018.8447.
Huang WC, Liao PC, Huang CH, Hu S, Huang SC, Wu SJ. Osthole attenuates lipid accumulation, regulates the expression of inflammatory mediators, and increases antioxidants in FL83B cells. Biomed Pharmacother. 2017;91:78–87. https://doi.org/10.1016/j.biopha.2017.04.051.
Kordulewska NK, Kostyra E, Cieslinska A, Matysiewicz M, Fiedorowicz E, Sienkiewicz-Szlapka E. Changes in gene expression induced by histamine, fexofenadine and osthole: Expression of histamine H1 receptor, COX-2, NF-κB, CCR1, chemokine CCL5/RANTES and interleukin-1 in PBMC allergic and non-allergic patients. Immunobiology. 2017;222:571–81. https://doi.org/10.1016/j.imbio.2016.11.004.
Kordulewska NK, Kostyra E, Cieslinska A, Fiedorowicz E, Jarmolowska B. Cytokine production by PBMC and serum from allergic and non-allergic subjects following in vitro histamine stimulation to test fexofenadine and osthole anti-allergic properties. Eur J Pharm. 2016;791:763–72. https://doi.org/10.1016/j.ejphar.2016.
Wang XL, Shang X, Cui Y, Zhao X, Zhang Y, Xie ML. Osthole inhibits inflammatory cytokine release through PPARα/γ-mediated mechanisms in LPS-stimulated 3T3-L1 adipocytes. Immunopharm Immunotoxicol. 2015;37:185–92. https://doi.org/10.3109/08923973.2015.1009997.
Kordulewska NK, Topa H, Tanska M, Cieslinska A, Fiedorowicz E, Savelkoul HFJ, et al. Modulatory effects of osthole on lipopolysaccharides-induced inflammation in Caco-2 cell monolayer and cocultures with THP-1 and THP-1-derived macrophages. Nutrients. 2021;13:123. https://doi.org/10.3390/nu13010123.
Yang X, Zeng XJ, Fu J. Protective effect of osthole on LPS-induced acute lung injury in mice and its mechanism. Shiyong Yaowu Yu Linchuang. 2015;18:893–7.
Fu XP, Hong CH. Osthole attenuates mouse atopic dermatitis by inhibiting thymic stromal lymphopoietin production from keratinocytes. Exp Dermatol. 2019;28:561–7. https://doi.org/10.1111/exd.13910.
Singh G, Bhatti R, Mannan R, Singh D, Kesavan A, Singh P. Osthole ameliorates neurogenic and inflammatory hyperalgesia by modulation of iNOS, COX-2, and inflammatory cytokines in mice. Inflammopharmacology. 2019;27:949–60. https://doi.org/10.1007/s10787-018-0486-9.
Li YL, Wang YW, Li YQ, Qian ZQ, Zhu L, Yang DL. Osthole attenuates pulmonary arterial hypertension in monocrotaline-treated rats. Mol Med Rep. 2017;16:2823–9. https://doi.org/10.3892/mmr.2017.6876.
Huang T, Dong Z. Osthole protects against inflammation in a rat model of chronic kidney failure via suppression of nuclear factor-κB, transforming growth factor-β1 and activation of phosphoinositide 3-kinase/protein kinase B/nuclear factor (erythroid-derived 2)-like 2 signaling. Mol Med Rep. 2017;16:4915–21. https://doi.org/10.3892/mmr.2017.7125.
Luo LN, Xie DQ, Zhang XG, Jiang R. Osthole decreases renal ischemia-reperfusion injury by suppressing JAK2/STAT3 signaling activation. Exp Ther Med. 2016;12:2009–14. https://doi.org/10.3892/etm.2016.3603.
Sun W, Cai Y, Zhang XX, Chen H, Lin YD, Li H. Osthole pretreatment alleviates TNBS-induced colitis in mice via both cAMP/PKA-dependent and independent pathways. Acta Pharm Sin. 2017;38:1120–8. https://doi.org/10.1038/aps.2017.71.
Wu HX, Wang YM, Xu H, Wei M, He QL, Li MN, et al. Osthole, a coumarin analog from Cnidium monnieri (L.) Cusson, ameliorates nucleus pulposus-induced radicular inflammatory pain by inhibiting the activation of extracellular signal-regulated kinase in rats. Pharmacology. 2017;100:74–82. https://doi.org/10.1159/000475599.
Ding X, Liu H, Wang P. Protective effect of osthole on retina ischemia reperfusion injury in rats. Jiefangjun Yaoxue Xuebao. 2016;32:311–3. 317.
Kong L, Yao YJ, Jiao YN, Li SH, Tao ZY, Yan YH, et al. Anti-apoptosis and anti-inflammatory effect of osthole in mice following stab wound injury. Zhongguo Yaolixue Tongbao. 2015;31:999–1004.
Xiao HH, Wang YY, Wu YT, Li HY, Liang XC, Lin Y, et al. Osthole ameliorates cognitive impairments via augmenting neuronal population in APP/PS1 transgenic mice. Neurosci Res. 2021;164:33–45. https://doi.org/10.1016/j.neures.2020.04.001.
Chu QB, Zhu YF, Cao TJ, Zhang Y, Chang ZC, Liu Y, et al. Studies on the neuroprotection of osthole on glutamate-induced apoptotic cells and an Alzheimer’s disease mouse model via modulation oxidative stress. Appl Biochem Biotech. 2020;190:634–44. https://doi.org/10.1007/s12010-019-03101-2.
Lin Y, Liang XC, Yao YJ, Xiao HH, Shi Y, Yang JX. Osthole attenuates APP-induced Alzheimer’s disease through up-regulating miRNA-101a-3p. Life Sci. 2019;225:117–31. https://doi.org/10.1016/j.lfs.2019.04.004.
Yao YJ, Wang YM, Kong L, Chen YQ, Yang JX. Osthole decreases tau protein phosphorylation via PI3K/AKT/GSK-3βsignaling pathway in Alzheimer’s disease. Life Sci. 2019;217:16–24. https://doi.org/10.1016/j.lfs.2018.11.038.
Li SH, Yan YH, Jiao YN, Gao Z, Xia Y, Kong L, et al. Neuroprotective effect of osthole on neuron synapses in an Alzheimer’s disease cell model via upregulation of microRNA-9. J Mol Neurosci. 2016;60:71–81. https://doi.org/10.1007/s12031-016-0793-9.
Li SH, Jiao YN, Yao YJ, Kong L, Tao ZY, Yan YH, et al. Neuroprotective effect of osthole on neuron synapses infected with APP gene. Zhongguo Yaolixue Tongbao. 2015;31:1383–8.
Guan JH, Wei XT, Qu ST, Lv T, Fu Q, Yuan Y. Osthole prevents cerebral ischemia-reperfusion injury via the Notch signaling pathway. Biochem Cell Bio. 2017;95:459–67. https://doi.org/10.1139/bcb-2016-0233.
Li K, Ding D, Zhang M. Neuroprotection of osthole against cerebral ischemia/reperfusion injury through an anti-apoptotic pathway in rats. Bio Pharm Bull. 2016;39:336–42. https://doi.org/10.1248/bpb.b15-00699.
Chen ZW, Mao XX, Liu AM, Gao XY, Chen XH, Ye MZ, et al. Osthole, a natural coumarin improves cognitive impairments and BBB dysfunction after transient global brain ischemia in C57 BL/6J mice: involvement of Nrf2 pathway. Neurochem Res. 2015;40:186–94. https://doi.org/10.1007/s11064-014-1483-z.
Yan YH, Kong L, Xia Y, Liang WB, Wang LT, Song J, et al. Osthole promotes endogenous neural stem cell proliferation and improved neurological function through Notch signaling pathway in mice acute mechanical brain injury. Brain Behav Immun. 2018;67:118–29. https://doi.org/10.1016/j.bbi.2017.08.011.
Xia Y, Kong L, Yao YJ, Jiao YN, Song J, Tao ZY, et al. Osthole confers neuroprotection against cortical stab wound injury and attenuates secondary brain injury. J Neuroinflammation. 2015;12:155/1–11. https://doi.org/10.1186/s12974-015-0373-x.
Kong L, Yao YJ, Jiao YN, Li SH, Tao ZY, Yang JX. Protective effects of osthole on the nerves of model mice with craniocerebral injury. Zhongguo Yaofang. 2015;26:3046–9.
Yao YJ, Liang XC, Shi Y, Lin Y, Yang JX. Osthole delays tert-butyl hydroperoxide-induced premature senescence in neural stem cells. Cell Reprogram. 2018;20:268–74. https://doi.org/10.1089/cell.2018.0010.
Yao YJ, Kong L, Jiao YN, Li SH, Tao ZY, Yan YH, et al. Role of osthole in delaying the senescence of neural stem cells induced by tert-butylhydroperoxide. Zhongguo Yaolixue Yu Dulixue Zazhi. 2015;29:565–72.
Yan YH, Li SH, Li HY, Lin Y, Yang JX. Osthole protects bone marrow-derived neural stem cells from oxidative damage through PI3K/AKT pathway. Neurochem Res. 2017;42:398–405. https://doi.org/10.1007/s11064-016-2082-y.
Kong L, Hu Y, Yao YJ, Jiao YN, Li SH, Yang JX. The coumarin derivative osthole stimulates adult neural stem cells, promotes neurogenesis in the hippocampus, and ameliorates cognitive impairment in APP/PS1 transgenic mice. Bio Pharm Bull. 2015;38:1290–301. https://doi.org/10.1248/bpb.b15-00142.
Yao YJ, Gao Z, Liang WB, Kong L, Jiao YN, Li SH, et al. Osthole promotes neuronal differentiation and inhibits apoptosis via Wnt/β-catenin signaling in an Alzheimer’s disease model. Toxicol Appl Pharm. 2015;289:474–81. https://doi.org/10.1016/j.taap.2015.10.013.
Tao ZY, Gao P, Yan YH, Li HY, Song J, Yang JX. Osthole enhances the therapeutic efficiency of stem cell transplantation in neuroendoscopy caused traumatic brain injury. Bio Pharm Bull. 2017;40:1043–54. https://doi.org/10.1248/bpb.b17-00072.
Wang Y, Zhou Y, Wang X, Zhen F, Chen R, Geng DQ, et al. Osthole alleviates MPTP-induced Parkinson’s disease mice by suppressing Notch signaling pathway. Int J Neurosci. 2019;129:833–41. https://doi.org/10.1080/00207454.2019.1573171.
Mei H, Shen M, Xie SS, Yang SL, Chen HR, Jin Y, et al. Osthole prevents 6-hydroxydopamine (6 OHDA) induced endoplasmic reticulum stress and cytotoxicity through activating AktiGSK3 beta signaling pathway in PC12 cells. Lat Am J Pharm. 2017;36:1631–41.
Mao XY, Wang ZB, Zhou HH, Liu ZQ, Zhou Y. Osthole ameliorates glutamate-induced toxicity in HT22 cells via activating PI3K/AKT signaling pathway. Zhongnan Daxue Xuebao. Yixueban. 2015;40:955–9.
Yu HP, Zhu DY, Liu P, Yang QH, Gao JJ, Huang YG, et al. Osthole stimulates bone formation, drives vascularization and retards adipogenesis to alleviate alcoholinduced osteonecrosis of the femoral head. J Cell Mol Med. 2020;24:4439–51. https://doi.org/10.1111/jcmm.15103.
Ma Y, Wang LN, Zheng SY, Xu JK, Pan YL, Tu PC, et al. Osthole inhibits osteoclasts formation and bone resorption by regulating NF-κB signaling and NFATc1 activations stimulated by RANKL. J Cell Biochem. 2019;120:16052–61. https://doi.org/10.1002/jcb.28886.
Li XF, Xue CC, Wang LB, Tang DZ, Huang J, Zhao YJ, et al. Osteoprotective effects of osthole in a mouse model of 5/6 nephrectomy through inhibiting osteoclast formation. Mol Med Rep. 2016;14:3769–76. https://doi.org/10.3892/mmr.2016.5687.
Jin ZX, Liao XY, Da WW, Zhao YJ, Li XF, Tang DZ. Osthole enhances the bone mass of senile osteoporosis and stimulates the expression of osteoprotegerin by activating β-catenin signaling. Stem Cell Res Ther. 2021;12:154. https://doi.org/10.1186/s13287-021-02228-6.
Zhang ZR, Leung WN, Li G, Kong SK, Lu X, Wong YM, et al. Osthole enhances osteogenesis in osteoblasts by elevating transcription factor osterix via cAMP/CREB signaling in vitro and in vivo. Nutrients. 2017;9:588/1–15. https://doi.org/10.3390/nu9060588.
Zheng SY, Ma Y, Guo Y, Wang LN, Pan YL. Osthole promote differentiation and inhibit proliferation of osteoblast by activating wnt signaling and endoplasmic reticulum stress. Pharmacogn Mag. 2018;14:641–6.
Zheng XD, Yu Y, Shao BY, Gan N, Chen L, Yang DQ. Osthole improves therapy for osteoporosis through increasing autophagy of mesenchymal stem cells. Exp Anim. 2019;68:453–63. https://doi.org/10.1538/expanim.18-0178.
Yu Y, Chen M, Yang SY, Shao BY, Chen L, Dou L, et al. Osthole enhances the immunosuppressive effects of bone marrow-derived mesenchymal stem cells by promoting the Fas/FasL system. J Cell Mol Med. 2021;25:4835–45. https://doi.org/10.1111/jcmm.16459.
Wang PE, Ying J, Luo C, Jin X, Zhang SX, Xu TT, et al. Osthole promotes bone fracture healing through activation of BMP signaling in chondrocytes. Int J Bio Sci. 2017;13:996–1007. https://doi.org/10.7150/ijbs.19986. eCollection 2017
Liu SZ, He Y, Shi J, Liu LL, Ma H, He L et al. Downregulation of miRNA-30a enhanced autophagy in osthole-alleviated myocardium ischemia/reperfusion injury. J Cell Physiol. 2019; https://doi.org/10.1002/jcp.28556.
Yang X, Zeng XJ, Li Y, Fu J. Osthole attenuated myocardial ischemia/reperfusion by suppressing apoptosis via a mitochondrial pathway. Linchuang Xinxueguanbing Zazhi. 2016;32:517–21.
Li YL, Li YQ, Shi FG, Wang LN, Li LS, Yang DL. Osthole attenuates right ventricular remodeling via decreased myocardial apoptosis and inflammation in monocrotaline-induced rats. Eur J Pharm. 2018;818:525–33. https://doi.org/10.1016/j.ejphar.2017.11.006.
Li YL, Wang YW, Li YQ, Wang JY, Yang DL. Effect of osthole in attenuating right ventricle remodeling in monocrotaline-treated rats by up-regulating the expression of PPARα and PPARγ. Zhongguo Yaolixue Tongbao. 2015;31:1270–3.
Duan J, Yang Y, Liu H, Dou PC, Tan SY. Osthole ameliorates acute myocardial infarction in rats by decreasing the expression of inflammatoryrelated cytokines, diminishing MMP-2 expression and activating p-ERK. Int J Mol Med. 2016;37:207–16. https://doi.org/10.3892/ijmm.2015.2402.
Chen YH, Guo DS, Lu MH, Yue JY, Liu Y, Shang CM, et al. Inhibitory effect of osthole from Cnidium monnieri on tobacco mosaic virus (TMV) infection in Nicotiana glutinosa. Molecules. 2020;25:65. https://doi.org/10.3390/molecules25010065.
Deng XM, Zhou YL, Wang JF, inventors; Application of osthole in preparation of MCR-1 enzyme inhibitor. CN 109464439 A. 2019 January 10.
Kermani EK, Sajjadi SE, Hejazi SH, Arjmand R, Saberi S, Eskandarian AA. Anti-leishmania activity of osthole. Pharmacogn Res. 2016;8:S1–4. https://doi.org/10.4103/0974-8490.178650.
Yuan MM, Luo YP, Xin Q, Gao HJ, Zhang GC, Jing T. Efficacy of osthole for Echinococcus granulosus in vitro and Echinococcus multilocularis in vivo. Vet Parasitol. 2016;226:38–43. https://doi.org/10.1016/j.vetpar.2016.05.016.
Guo Y, Liu ZY, Hou EH, Ma NN, Gen YP, Che PC, et al. Application of natural products as insecticide candidates: semisynthesis and biological evaluation of some novel osthole-based esters. Bio Med Chem Lett 2020;30:127260. https://doi.org/10.1016/j.bmcl.2020.127260.
Farooq S, Shakeel-u-Rehman, Hussain A, Hamid A, Qurishi MA, Koul S. Click chemistry inspired synthesis and bioevaluation of novel triazolyl derivatives of osthol as potent cytotoxic agents. Eur J Med Chem. 2014;84:545–54. https://doi.org/10.1016/j.ejmech.2014.07.069.
Yang YC, Chen CN, Wu CI, Huang WJ, Kuo TY, Kuan MC, et al. NBM-T-L-BMX-OS01, semisynthesized from osthole, is a novel inhibitor of histone deacetylase and enhances learning and memory in rats. Evid Based Complement Altern Med. 2013;2013:514908. https://doi.org/10.1155/2013/514908.
Yang HY, Hsu YF, Chiu PT, Ho SJ, Wang CH, Chi CC, et al. Anti-cancer activity of an osthole derivative, NBM-T-BMX-OS01: targeting vascular endothelial growth factor receptor signaling and angiogenesis. PLoS ONE. 2013;8:e81592. https://doi.org/10.1371/journal.pone.0081592.
Chen TJ, Zhou YF, Ning JJ, Yang T, Ren H, Li Y, et al. NBM-T-BMX-OS01, an osthole derivative, sensitizes human lung cancer A549 cells to cisplatin through AMPK-dependent inhibition of ERK and AKT pathway. Cell Physiol Biochem 2015;36:893–906. https://doi.org/10.1159/000430264.
Wu JZ. Structural modification and calcium antagonistic activity of osthole [Dissertation]. China: Guizhou University; 2006.
Ding JL, Zhou WX, Zhang ZP, Xie SS. Synthesis and antitumor evaluation of osthole derivatives. Guangdong Huagong. 2020;47:21–22.
Zhang L, Wu YH, Yang GX, Gan HX, Sang DY, Zhou JY, et al. Design, synthesis and biological evaluation of novel osthole-based derivatives as potential neuroprotective agents. Bio Med Chem Lett. 2020;30:127633.
Wei WJ. Synthesis and bioactivity of osthol derivatives [Dissertation]. China: Huazhong Agricultural University; 2008.
Yang JB, Chen L, Su GQ, Ren Y. Synthesis of osthole stilbene derivatives and their inhibition on tubulin polymerization. J China Pharm Univ. 2011;42:107–11.
Yu Y, Chen YF, inventors; Osthole hydrazine/hydrazide/sulfonyl hydrazine derivatives with acetylcholinesterase inhibition activity and its preparation method. CN 110003160 A. 2019 May 14.
Ren ZL, Sun ZQ, Li TZ, Lv M, Zhang SY, Xu H. Regioselective hemisynthesis and insecticidal activity of C8-hydrazones/acylhydrazones/sulfonylhydrazones coumarin-type derivatives of osthole. Bio Med Chem Lett 2021;40:127962. https://doi.org/10.1016/j.bmcl.2021.127962.
Ren ZL, Lv M, Li TZ, Hao M, Li SC, Xu H. Construction of oxime ester derivatives of osthole from Cnidium monnieri, and evaluation of their agricultural activities and control efficiency. Pest Manag Sci. 2020;76:3560–7. https://doi.org/10.1002/ps.6056.
Yang JQ, An JL, Zhou XR, Zheng J, inventors; Osthole sulfonate compound, and its application in preparation of methicillin-resistant Staphylococcus aureus drug or drug composition. CN 111808062 A. 2020a October 23.
Yang JQ, Yang H, Zhang Y, Zheng J, inventors; Preparation of osthole amides as antibacterial agents. CN 111747917 A. 2020b July 28.
Yang JQ, An JL, Wu XJ, Zheng J, inventors; Preparation method of osthole ester compound and its application in preparation of drug for treating and/or preventing bacterial infection. CN 111732565 A. 2020c July 28.
Lai XP, Huang S, Jiang DX, Zhao AG, Qing HM, Xie YL, inventors; Cnidium lactone derivant and preparation method and application thereof. CN 1995029 A. 2006 December 14.
Qian S, Wang ZY, Yang LL, Lai M, Gao C, He QM et al, inventors; Osthole derivatives and preparation method and application thereof. CN 110396078 A. 2018 April 25.
Hitotsuyanagi Y, Kojima H, Ikuta H, Takeya K, Itokawa H. Identification and structure-activity relationship studies of osthol, a cytotoxic principle from Cnidium monnieri. Bio Med Chem Lett. 1996;6:1791–4.
Gopalakrishnan G, Kasinath V, Singh NDP, Thirumurugan R, Raj SSS, Shanmugam G. A new synthetic route to dihydrobenzopyran via tandem demethylation cyclisation. Molecules 2000;5:880–5.
You LS, An R, Wang XH, Li YM. Discovery of novel osthole derivatives as potential anti-breast cancer treatment. Bio Med Chem Lett. 2010;20:7426–8. https://doi.org/10.1016/j.bmcl.2010.10.027.
Huang WJ, Chen CC, Chao SW, Lee SS, Hsu FL, Lu YL, et al. Synthesis of N-hydroxycinnamides capped with a naturally occurring moiety as inhibitors of histone deacetylase. Chem Med Chem. 2010;5:598–607. https://doi.org/10.1002/cmdc.200900494.
Huang WJ, Chen CC, Chao SW, Yu CC, Yang CY, Guh JH, et al. Synthesis and evaluation of aliphatic-chain hydroxamates capped with osthole derivatives as histone deacetylase inhibitors. Eur J Med Chem. 2011;46:4042–9. https://doi.org/10.1016/j.ejmech.2011.06.002.
Lee WH, Wu HH, Huang WJ, Li YN, Lin RJ, Lin SY, et al. N-hydroxycinnamide derivatives of osthole ameliorate hyperglycemia through activation of AMPK and p38 MAPK. Molecules. 2015;20:4516–29. https://doi.org/10.3390/molecules20034516.
Zhang R, Fang Q. Study on the extraction and preparation of osthole. Stud J Tradit Chin Med. 2003;21:1336. https://doi.org/10.13193/j.archtcm.2003.08.118.zhangr.068.
Shang F, Wang YX, Chen FX, Qian CM. Optimization of extracting process of osthol with orthogonal design. China Pharmacist. 2006;9:621–22.
Deng H, Gu DD, Yang Y, Qin Y, Lan B, Wang AP, et al. Optimization of extraction process of osthole from Fructus Cnidii. West Chin J Pharm Sci. 2013;28:106–7. https://doi.org/10.13375/j.cnki.wcjps.2013.01.004.
Su XQ, Yang JH. Optimization of extraction process of osthole. Tianjin Pharm. 2009;21:3–4.
Wang XK, Li WD, Gao SY, Fan T. Cnidicin extraction technology. Appl Chem Ind. 2011;40:1237–8.
Lu Q, Wang YM, Wang XM. Study on extraction method of Cnidium Monnieri. J Inn Mong Agric Univ. 2010;31:298–330.
Li F, Zhu Y, Cheng W. Optimization of extraction technology of osthole by orthogonal test. Chin J Mod Drug Appl. 2011;5:82–83.
Yan ZF, Liu BS, Zhao SP. Study of CO2 supercritical technology on osthole extraction from Common Cnidium Fruit. World J Integr Tradit West Med. 2009;4:20–22.
Gong ZY, Zhang GY, Nie YL, Zhang XX, Gu HX. Influence of supercritical CO2 extraction technology on osthol content from fruits of Cnidium monnieri. Chin Tradit Herb Drugs 2006;17:2156–9.
Zhang YY, Gao YZ, Dian LH, Wu T. Study on extracting technique of osthole in Cnidium monnieri (L.) Cusson. Chin Med Mod Dist Educ Chin. 2008;6:726–7.
Chen H, Li XH, Lu XH, Yang XP, Liang JP. Optimization of Ultrasonic-assisted extraction of osthole from Radix Angelicae Pubescentis using response surface methodology. Nat Prod Res Dev. 2013;25:834–40.
Xie GL, Wang R, Yang J, Meng YM. Optimization of extraction process of osthol by quadratic general rotary combination design. Inf Tradit Chin Med. 2014;31:95–96.
Wang R, Yang J, Xie GL, Jiang JM, Wang YH. Extraction of osthole from Fructus Cnidii by ultrasonic method combined with surfactant. Lishizhen Med Mat. Med Res. 2013;24:603–4.
Xie L, Zhou J, Tian QQ, Wang QF. Aqueous two phase extraction and separation of osthole from Cnidium monnieri with analysis by high performance liquid chromatography. Pharm J Chin PLA. 2010;26:310–4.
Liu YJ, Hu YG, Niu XF. Study on extraction technology of osthole from Fructus Cnidii. Mod Tradit Chin Med. 2016;36:79–81.
Zhang L, Weng LD, Liu L, Zheng F, Liu Q. Optimization of percolation extraction conditions of Fructus Cnidii by orthogonal test. Chin J Exp Tradit Med Form. 2010;16:12–13.
Ning N, Han JJ, Hu YL, Zou ZY, Yu JS. Optimization of enzymatic extraction of osthole from Fructus Cnidii by Response Surface Methodology. Jiangsu Agric Sci. 2016;44:169–73. https://doi.org/10.15889/j.issn.1002-1302.2016.08.046.
Acknowledgements
We thank for the financial support from National Key R&D Program of China (2021YFE0202000), Traditional Chinese Medicine Bureau of Guangdong Province (Grants No. 202106070838012350) and Discipline Project of the 14th five-year plan of Guangzhou Medical University (06-410-2107218).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sun, M., Sun, M. & Zhang, J. Osthole: an overview of its sources, biological activities, and modification development. Med Chem Res 30, 1767–1794 (2021). https://doi.org/10.1007/s00044-021-02775-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-021-02775-w