
Abstract
Immune checkpoint blockade (ICB) therapies have achieved remarkable clinical responses in patients with many different types of cancer; however, most patients who receive ICB monotherapy fail to achieve long-term responses, and some tumors become immunotherapy-resistant and even hyperprogressive. Type I interferons (IFNs) have been demonstrated to inhibit tumor growth directly and indirectly by acting upon tumor and immune cells, respectively. Furthermore, accumulating evidence indicates that endo- and exogenously enhancing type I IFNs have a synergistic effect on anti-tumor immunity. Therefore, clinical trials studying new treatment strategies that combine type I IFN inducers with ICB are currently in progress. Here, we review the cellular sources of type I IFNs and their roles in the immune regulation of the tumor microenvironment. In addition, we highlight immunotherapies based on type I IFNs and combination therapy between type I IFN inducers and ICBs.
Similar content being viewed by others

Introduction
In the 1950s, in the course of their studies on the structures and properties of influenza A and other viruses, Alick Isaacs and Jean Lindenmann discovered a soluble factor that was produced by virus-infected cells and could inhibit viral infection; they named this compound “interferon” (IFN) for its capacity to “interfere” with viral replication [1, 2]. There are currently three known types of IFN: I, II, and III, which are classified by their sequence and cellular receptors [3, 4]. The type I IFN family comprises members encoded by multiple genes, including 14 highly homologous subtypes of IFN-α, IFN-β, and other lesser-known single gene products, such as IFN-ε, IFN-κ, IFN-ω, IFN-τ, IFN-δ, and IFN-ζ [3, 5,6,7]. The genes encoding human type I IFNs, including 13 IFN-α subtypes, as well as IFN-β, IFN-ε, IFN-κ, and IFN-ω, are found on the human chromosome 9p [3, 5, 7]. Of type I IFNs, IFN-α and IFN-β are the best understood (Fig. 1).

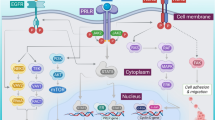
Type I IFNs signal transduction pathway. Type I IFNs bind to IFNAR1/IFNAR2 heterodimers to drive the activation of TYK2 and JAK1, which results in the accumulation of activated STAT1 and in the subsequent formation of STAT1–STAT2 heterodimers. Then, the dimerized STATs are combined with IRF9 to form ISGF3, which interacts with ISREs to induce the synthesis of various proteins from ISGs. Meanwhile, “non-classical” pathways, such as the MAPK, mTOR, and GCD pathways, are activated by type I IFNs and trigger the expression of additional ISGs
Both endogenous type I IFNs, which are derived from immune and tumor cells, and exogenous type I IFNs, which are produced by recombinant technology, trigger signaling cascades by interacting with their cognate transmembrane receptor, the IFN-α/β receptor 1 (IFNAR1)–IFNAR2 heterodimer [8]. Following the interaction of type I IFNs with their receptors, pre-associated tyrosine kinase 2 (TYK2) and Janus kinase 1 (JAK1) are activated, leading to the recruitment and phosphorylation of signal transducer and activator of transcription 1 (STAT1) and STAT2. Subsequently, STAT1 and STAT2 heterodimers are combined with interferon regulatory factor 9 (IRF9) to form a novel complex called IFN-stimulated gene factor 3 (ISGF3) [4, 7,8,9,10,11]. Then, ISGF3 translocates into the nucleus to bind IFN-stimulated response elements (ISREs) for the synthesis of various proteins from interferon-stimulated genes (ISGs) [12,13,14]. In addition to this classical signaling pathway, many “non-classical” pathways exist, such as the mitogen-activated protein kinase (MAPK) pathway, the mammalian target of rapamycin (mTOR) pathway, and the GCD (GCD-GTPases/cyclin-dependent kinases) pathway, which are activated by type I IFNs and trigger the expression of additional ISGs [8, 15]. This diversity of type I IFN-mediated signaling pathways may partly explain the extensive biological functions exhibited by type I IFNs.
After recombinant IFN-α2 was approved by the United States Food and Drug Administration (FDA) as the first human immunotherapeutic for cancer, type I IFNs have been widely used alone and in combination with other immunotherapeutic agents to treat solid and hematologic malignancies [11]. In this review, we outline the cell sources of type I IFNs and their immune regulation in the tumor microenvironment (TME) and discuss how the treatments that exploit type I IFN pathways could potentially be used to enhance the efficacy of immune checkpoint blockade (ICB) treatment in patients with cancer.
Cell sources of type I IFNs in the TME
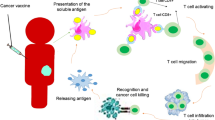
To optimize the clinical manipulation of the type I IFN system for anti-tumor therapy, it is critical to understand which cells conduce to the production of type I IFNs in the TME (Fig. 2). Although it is known that almost all cell types can produce type I IFNs in response to nucleic acids, nucleotides, or synthetic compounds[11], the primary stimulators of type I IFN production and which cells act as the primary IFN-producers in the TME have yet to be conclusively identified.

The production of type I IFNs in the TME. Tumor-derived HMGB1 induces the production of type I IFNs in DCs via the TLR4-MyD88 pathway. Tumor-derived DNA activates the cGAS/STING pathway to drive the expression of type I IFNs through chaperoning HMGB1, autophagosome, exosome, LL37, or CLEC9A into DCs. And manganese increases dsDNA binding to cGAS. Damaged DNA caused by ionizing radiation (IR) in tumor and endothelial cells triggers cGAS/STING signal to produce type I IFNs. Poly I:C induces overexpressed TLR3 to promote IFN-β production via the UNC93B1 signaling pathway in paclitaxel-resistant colon cancer (HCT-8/PTX) cells. Anthracycline induces the secretion of type I IFNs through the TLR3-TICAM1 pathway in tumor cells. TAMs expressing E-FABPs can produce high levels of IFN-β by upregulating LD. Furthermore, tumor-derived DNA enters into CAFs by transcytosis and is distinguished by ZBP1 and DDX41, then activates STING-IRF3 pathway and induces IFN-β expression
Tumor-infiltrating dendritic cells (DCs)
Plasmacytoid dendritic cells (pDCs) are universally acknowledged to be the major producers of type I IFNs against stimulators, such as viruses, endogenous nucleic acids, and synthetic oligoribonucleotides/oligodeoxyribonucleotides [16]; however, it is not known if this paradigm holds for tumors. In solid tumors and tumor-draining lymph nodes, emerging evidence indicates that the invading host cells involved in the production of the majority of cellular IFN-β are tumor-infiltrating CD11b+DCs, and that the binding of tumor-derived DNA to cytosolic DNA receptors serves as the likely trigger for the activation of tumor-infiltrating DCs [17,18,19]. The accumulation of tumor-derived DNA in the cytoplasm is recognized by the receptor cyclic GMP-AMP (cGAMP) synthase (cGAS), which induces the formation of a second messenger, cGAMP [20,21,22]. Subsequently, cGAMP, in combination with the stimulator of interferon genes (STING), an adaptor protein anchored in the endoplasmic reticulum (ER), robustly initiates conformational changes and the translocation of STING from the ER to perinuclear sites, resulting in the aggregation and activation of TANK-binding kinase 1 (TBK1). In turn, activated TBK1 contributes to the phosphorylation of the interferon regulatory factor family, namely IRF3, IRF5, and IRF7, which then translocates to the nucleus and functions together with nuclear factor κB (NF-κB) to induce the abundant secretion of type I IFNs [17, 22, 23]. Recently, researchers found that manganese (Mn), a transition metal, is released from membrane-enclosed organelles upon viral infection and directly bounds to cGAS, which increases the sensitivity of the cGAS-STING pathway for double-stranded DNA (dsDNA) to produce type I IFNs [24]. Moreover, this group found that Mn could promote DC maturation and antigen presentation for antitumor immune responses through the cGAS-STING pathway and type I IFN induction [25]. Additionally, high-mobility group box 1 (HMGB1) proteins derived from dead tumor cells trigger toll-like receptor 4 (TLR4) on DCs to produce type I IFNs through myeloid differentiation factor 88 (MyD88) signaling [26].
However, the underlying mechanism by which tumor cell DNA gains entrance to the cytoplasm of DCs has not yet been established. One possible mechanism of this DNA transfer is through a medium, such as the antimicrobial peptide LL37 [27] and the C-type lectin domain-containing 9A (CLEC9A) receptor [28], that can mediate the uptake of DNA from dying tumor cells. Another possible mechanism is the chaperoning of HMGB1, autophagosome, or exosome uptake to achieve DNA transfer [18]. Gap junctions between tumor cells and DCs might be another mechanism for tumor cell DNA transfer into the cytosol of tumor-infiltrating DCs [29]. In addition, cGAMP in tumor cells, acting as a secreted immunotransmitter, can enter bystander cells through connexin-mediated channels, such as the recently discovered cGAMP transporter SLC19A1, to trigger the cGAMP-STING pathway and cause IFN-β production [30].
Although RNAs have historically been considered the principal stimuli for IFN-α and IFN-β production by activating IRF3 through retinoic acidinducible gene I (RIG-I)-like receptors (RLRs) or toll-like receptors (such as TLR3, TLR7) during antiviral immune responses, tumor-derived RNA is only minimally stimulatory in growing tumors [18].
Tumor cells
Tumor cells are the most abundant components in the TME and are important IFN-β producers after recognizing different pattern recognition receptors (PRRs). For instance, the contribution of overexpressed TLR3 induced by poly I:C to the production of IFN-β via the TLR3-UNC93B1 signaling pathway was observed in paclitaxel-resistant colon cancer (HCT-8/PTX) cells[31]. Anthracycline-dependent production of type I IFNs was also demonstrated to be produced by tumor cells following activation of the TLR3-TICAM1 signaling pathway [32]. B16 murine melanoma cells stimulated with TLR4 agonists, such as lipopolysaccharide (LPS), contributed to IFN-β induction [33]. Furthermore, evidence suggests that the degradation of cytosolic dsDNA induced by radiation is the trigger for type I IFN secretion in tumor cells; the tumor cell-intrinsic activation of type-I IFNs was also confirmed to be dependent on the cGAS-STING pathway [34, 35]. In addition, aside from being produced directly through the cGAS-STING pathway, IFN-β production by tumor cells might occur indirectly through the existing gap junctions, where cGAMP transfers from DCs to the tumor cells, thus inducing the transcription of IFN-β genes [17, 36].
Tumor endothelial cells
Tumor vascular research has mainly focused on tumor endothelial cells. In one experiment, researchers unexpectedly found that tumor endothelial cells, not DCs, are the main IFN-β producers upon spontaneous and enforced STING activation [37]. The possible mechanisms are their higher capacity to produce IFN-β in response to STING activation and their relative abundance in the TME compared with tumor-infiltrating immune cells [37]. In addition, no IFN-α was found in the TME, most likely due to the weak capacity of endothelial cells to produce IFN-α upon STING activation or the relative absence of IRF7, upon which IFN-α expression depends [37, 38]. Similarly, tumor DNA and tumor-derived cGAMP might act as stimuli for IFN-β production in endothelial cells via the signaling cascade involved in the cGAS-STING pathway. However, the mechanism by which they transfer into intracellular endothelial cells remains unclear. As mentioned above, gap junctions, connexin-mediated channels, and phagocytosis are possible mechanisms [39].
Tumor-associated macrophages (TAMs)
Macrophages are important target cells for viral infection and the mainly infiltrated immune cells in many solid TMEs; however, macrophage-produced type I IFNs are rarely observed in the TME. Interestingly, after stimulation with STING agonists, such as ML-RR-S2 CDA and DMXAA, IFN-β expression was observed in many cell types, such as DCs, bone marrow-derived macrophages, T lymphocytes from naive mice, and mouse embryonic fibroblasts, but not in B16 murine melanoma cells [40]. Furthermore, researchers found that TAMs that were sorted from pre-established B16 tumors expressed the highest IFN-β after being treated with STING agonists compared to that of DCs, T cells, and endothelial cells [40]. This study suggests that TAMs might be a type I IFN source in the TME. This is consistent with a previous study which found that TAMs expressing epidermal fatty acid-binding proteins (E-FABP) can produce high levels of IFN-β by upregulating lipid droplet (LD) formation [41].
Cancer-associated fibroblasts (CAFs)
A recent study found that the expression of IFN-β genes can be induced by cytoplasmic DNA sensors sensing aberrant DNA in CAFs. Following transcytosis of tumor cell DNA into CAFs, cytoplasmic DNA sensors (ZBP1 and DDX41) mechanically detect aberrant DNA, which then contributes to the activation of the STING-IRF3 pathway, resulting in the expression of IFN-β and other cytokines [42]. In addition, CAFs isolated from patients with breast cancer are capable of producing IFN-β upon stimulation of DNA fragments released by apoptotic cells [43]. In a recent study, breast CAFs were also shown to produce IFN-β after co-culture with the human breast epithelial cell line MDA-MB-231 [44].
The effect of type I IFNs on anti-tumor immunity
Ever since type I IFNs were first demonstrated to possess therapeutic anti-tumor potential over 50 years ago [45], an increasing number of researchers have studied the prominent role that type I IFNs play in tumor immune surveillance. It is widely accepted that type I IFNs can affect tumor cell growth, proliferation, differentiation, migration, apoptosis, and other functions via cytotoxic, cytostatic, and antiangiogenic effects [46]. In addition, type I IFNs upregulate major histocompatibility complex class I (MHC I) proteins and enhance tumor-associated antigen (TAA) expression, resulting in both increased recognition and uptake of TAA by antigen-presenting cells (APC) and antigen presentation to cytotoxic T cells (CD8+ T) [3, 9]. However, a recent study found that the activation of type I IFN signaling induced by radiation can help tumor cells avoid CD8+ T cell-mediated killing through the regulation of the serine proteinase inhibitor Serpinb9. This result might suggest a mechanism by which tumor cells develop resistance to antitumor immunity after radiation as well as potential targets for intervention to improve antitumor effects [47]. Thus, type I IFNs may play a dual role in tumor cells for tumor immunity. An overview of the effects of type I IFNs on different target cells in the TME is presented in Fig. 3.

The effects of type I IFNs in the TME. Type I IFNs induce tumor cell apoptosis and inhibit tumor cell proliferation and metastasis. In addition, type I IFNs upregulate TAA and MHC I expression in tumor cells. Type I IFNs are essential for NK cell maturation and activation. Moreover, type I IFNs increase NK cell cytotoxicity. Type I IFNs promote DC differentiation, maturation, and migration into lymph nodes to activate CD8+ T cells. Moreover, type I IFNs increase DC intratumoral accumulation. Type I IFNs reduce Treg infiltration into tumor and Treg proliferation. Type I IFNs inhibit neutrophil infiltration, longevity, and chemokine production. In addition, type I IFNs decrease differentiation and maturation of MDSCs, and block their suppressive activity on CD8+ T cells
Dendritic cells
The predominant role of DCs in antitumor immune responses is to uptake and present TAAs to tumor-specific CD8+ T lymphocytes. In this process, type I IFNs exert multiple effects on DCs, most likely by stimulating DC differentiation and maturation and up-regulating the activity of DCs via cross-presentation of TAAs to CD8+ T cells [3]. Notably, the migration of professional APCs from the tumor site to the lymph nodes is a key prerequisite for immune response initiation. Experiments have shown that type I IFNs are required for DCs to promote their migration toward lymph nodes [9, 19]. Moreover, although it is clear that type I IFNs can induce intratumoral accumulation of CD8α+ DCs, the detailed mechanism by which this occurs remains unclear [19]. Patients with various malignancies, particularly renal cell carcinoma or melanoma, who received DC vaccines combined with or without recombinant IFN-α2, have shown encouraging objective responses or sometimes long-term patient survival [4, 48]. Nevertheless, more preclinical and clinical studies are still needed to confirm the safety and effectiveness of type I IFN-stimulated DC vaccines.
Natural killer cells
Natural killer (NK) cells, which are generally the first line of defense against pathogenic infection and tumors, are dependent on type I IFNs for maturation, activation, and homeostasis in the TME [49, 50]. NK cells are significantly decreased and exert heavily impaired cytotoxic capacity in IFNAR1 and IFNAR2-deficient mice compared with their wild-type counterparts, confirming the essential role of type I IFNs in NK cell activation [51]. In addition, NK cells with TYK2 and STAT1 deficiency, the downstream signaling components of the type I IFN pathway, display impaired cytotoxic function against tumor cells [51]. Moreover, NK cells can be primed by cytokines derived from other immune cells; these immune cells are activated via essential signals provided by type I IFNs. For instance, IL-15, the master cytokine that promotes NK cell maturation, proliferation, and function, can be produced by DCs in the presence of type I IFN [51]. To enhance NK cell cytotoxicity to tumor cells, a human IFN-α gene-modified NK cell line was established. The increased cytolytic activity in the NK cell line showed an upregulation of perforin, granzyme B, and Fas ligand as well as the secretion of cytokines, like tumor necrosis factor-α and IFN-γ, in vitro and in xenograft tumor models [51, 52]. Accordingly, a significant increase of circulating CD56bright NK cells that produced increased levels of IFN-γ has been observed during IFN-α treatment in patients with chronic myeloproliferative neoplasms [53].
Neutrophils
Due to their diversity and plasticity in the TME, neutrophils act as a “double-edged sword” that can mediate both tumor-promoting and antitumor effects [54]. Type I IFNs induce phenotypic and functional changes in neutrophils and tend to restrain tumor progression. Studies have shown that IFN-β interferes with the accumulation of neutrophils by suppressing the expression of the C-X-C motif chemokine receptors, CXCR2 and CXCR4, and by blocking their ligands, CXCL1, CXCL2, or CXCL12, in tumor cells [55, 56]. In addition, neutrophils exhibit decreased longevity in response to endogenous IFN-β. This might be due to the ability of IFN-β to (1) induce high cytotoxic reactive oxygen species (ROS) production by neutrophils [57], (2) modulate the expression of the proapoptotic Bcl-2 protein, Bax, and its anti-apoptotic counterpart, BCL-xL, in neutrophils into a proapoptotic ratio, (3) upregulate the IFN-β-dependent death receptor, Fas, on neutrophils, or (4) decrease the expression of granulocyte colony-stimulating factor, G-CSF, in neutrophils [55, 58]. In addition, type I IFNs have been shown to suppress some pro-angiogenic chemokines, such as vascular endothelial growth factor (VEGF), neutrophil-derived matrix metallopeptidase 9 (MMP9, which degrades the extracellular matrix), and CXCLs (CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, and CXCL8, which are required for direct activation of endothelial cells). This serves as an antiangiogenic mechanism during tumorigenesis [56]. Moreover, although high amounts of ROS are toxic to endothelial cells, IFN-dependent ROS production by neutrophils can also exhibit antiangiogenic properties [55]. In the context of metastatic processes, type I IFNs play a crucial role in elevating the plasma levels of G-CSF and increasing the expression of CXCR2 on neutrophils [58, 59]. In summary, type I IFNs reduce the infiltration, longevity, and chemokine production of neutrophils to mediate antitumor activity [60]. Based on these findings, researchers have analyzed neutrophil characteristics in the human system and found that patients with melanoma undergoing adjuvant type I IFN therapy have lower neutrophil counts and upregulation of co-stimulatory molecules, like ICAM1, compared with the untreated control group [57, 60].
Regulatory T (Treg) cells
Treg cells are dedicated to maintaining immune homeostasis in the host, which limits the antitumor immune response; consequently, they are considered a target for immunotherapy in the TME [61]. The suppressive effect of type I IFNs on Tregs in the TME has been extensively demonstrated. In a CT26 colon cancer model, intratumoral IFN-α gene transduction significantly reduced the frequency of Treg cells. Furthermore, IL-6, a Treg-inhibitory cytokine, was produced by intratumoral CD11c+ cells in response to IFN-α stimulation. In addition, IFN-α-mediated IL-6 leads to the trans-differentiation of Tregs into Th17 cells, which might partly explain the reduction in Tregs [62]. Bacher et al. indicated that IFN-α eliminates the suppressive function of Treg cells through a pathway that involves the stimulation of MEK/ERK-mediated phosphodiesterase 4 (PDE4) activation and the consequent depletion of cyclic AMP (cAMP) [63]. In addition to directly inhibiting Treg proliferation and function, type I IFNs can indirectly limit the recruitment of Treg cells to the TME by blocking CCL22, a Treg-attracting chemokine that is extensively expressed in many tumors and is beneficial to the intratumoral accumulation of Treg cells [64]. Similarly, when CCL17, another Treg-attracting chemokine expressed on CT26 cells, was blocked by IFN-α, tumor-infiltrating Treg cells decreased and CT26-specific CD8+ T cells increased [65]. In human breast cancer, tumor-associated pDCs are highly repressed due to their ability to produce IFN-α. This defect in IFN-α production strongly promotes the infiltration and expansion of Treg cells in the TME and leads to tumor progression and poor survival [66].
Myeloid-derived suppressor cells
Similarly, myeloid-derived suppressor cells (MDSCs), which play a significant role in tumor-associated immunosuppression and are known to hamper successful immunotherapies in tumor-bearing hosts and patients with cancer [67], have been shown to be influenced by type I IFNs. For instance, in a C26 colon carcinoma model, the in vivo short-term application of recombinant IFN-α disturbed the differentiation and maturation of MDSCs and blocked their suppressive function on T cell proliferation [68]. Furthermore, treating tumor-bearing mice with the TLR3 and MDA-5 ligand poly I:C reduced the suppressive activity of MDSCs and induced the production of high amounts of type I IFNs [69]. In addition, a study found that autocrine IFN-α/β from the TME upregulates TRAIL expression on activated T cells, which elicits MDSC apoptosis through the TRAIL–DR5 pathway. Furthermore, they found that using neutralizing IFNAR1 abolishes the type I IFN-induced MDSC apoptosis [70].
Immunotherapies based on type I IFNs
It is widely accepted that type I IFNs have a great impact on both tumor inhibition and stimulating antitumor immune responses, while the systemic administration of type I IFNs is accompanied by many adverse outcomes, including fatigue, nausea, anorexia, flulike symptoms, dizziness, hepatotoxicity, severe depression, leukopenia, and possibly the expression of immunosuppressive enzyme indoleamine 2,3-dioxygenase 1 (IDO1) [71, 72]. Therefore, attempts have been made to target the delivery of type I IFNs to the TME. As an anti-tumor strategy, treatment with type I IFN agonists is usually preferred over the use of recombinant type I IFN (Fig. 4).

Immunotherapies based on type I IFNs. Type I IFN inducers, including TLR agonists, STING agonists, chemotherapeutics and targeted drugs, radiation therapy, oncolytic viruses, and others, are undergoing preclinical and clinical trials
TLR agonists
TLR agonists, including poly A:U, poly I:C, poly I:C plus polylysine (poly(ICLC)), CpG adjuvant, Bacillus Calmette–Guérin (BCG), monophosphoryl lipid A, imiquimod (R837), resiquimod (R848), and motolimod (VTX-2337), are all potent type I IFN inducers [3, 73,74,75,76]. Poly I:C was recently reported to reinforce the potency of cytotoxic chemotherapeutics in paclitaxel-resistant colon tumor cell lines through the TLR3-UNC93B1-IFN-β signaling cascade [31]. In the preclinical studies, researchers mainly focused on the delivery efficiency of TLR agonists into the tumor site; intratumoral injection or nanoparticle-conjugate of TLR7/8 agonists were widely studied to overcome lowering the local drug dose in tumors [77,78,79,80]. The only TLR agonists currently approved for use by the FDA for the treatment of human patients with cancer are BCG, monophosphoryl lipid A, and imiquimod. Trials using poly A:U, poly I:C, or poly(ICLC) have demonstrated clinical benefits in several tumors (NCT00694551/ NCT00058123/NCT01188096) [3], but the use of resiquimod, motolimod, and other TLR7/TLR8 agonists as immunostimulatory agents in patients with cancer have shown disappointing results in recent clinical tests [76, 81, 82].
STING agonists
Since STING stimulation results in type I IFN production, many cyclic dinucleotides (CDNs) have been synthesized to stimulate the STING signal. DMXAA, also known as ASA404 or vadimezan, is a strong STING agonist without significant local or systemic toxicity and was shown to have anti-tumor immunity in mouse models. In a previous study, DMXAA showed anti-angiogenic effects [83], and recent reports found that STING activation by DMXAA reduced bone cancer pain and local tumor burden [84], and promoted CAR T cell trafficking and persistence in breast cancer [85]. Although it had various beneficial effects in preclinical models, DMXAA was ineffective when combined with platinum-based chemotherapy in a phase 3 efficacy trial in human patients with advanced non-small-cell lung cancer (NSCLC) [86, 87]. Other STING agonists include ADU-S100, BMS-986301, GSK3745417, MK-1454, MK-2118, SB11285, and E7766. Among these, ADU-S100 is being tested in combination with ICBs. BMS-986301, GSK3745417, MK-1454, MK-2118, and SB11285 are being tested as standalone immunotherapeutic interventions and/or in combination with ICBs. E7766 is being tested as a standalone immunotherapeutic intervention (NCT04144140) [87].
Since intratumoral administration of CDNs notably induced STING activation, resulting in the cytotoxicity of many cell types in TME and the production of systemic inflammatory cytokines, liposomal nanoparticle-delivered or extracellular vesicle loaded STING agonists have been created. Liposomal cGAMP-NPs induced type I IFN production through STING stimulation and suppressed tumor growth by reprograming the TME. Moreover, liposomal cGAMP-NPs showed a synergistic effect with ICBs in a triple-negative breast cancer (TNBC) mice model [88]. ExoSTING, an engineered extracellular vesicle loaded with CDN, showed an enhanced therapeutic effect compared to free CDN [89]. Furthermore, the first-in-human study of ExoSTING in advanced/metastatic, recurrent, injectable solid tumors has been initiated (NCT04592484). Other STING agonists, including CRD-100 [90] and GSK532 [91], have shown potential anti-tumor immune responses and are under clinical investigation.
In addition to these direct type I IFN agonists, other “indirect” type I IFN agonists, such as chemotherapeutical and targeted drugs, radiation therapy (RT), and oncolytic viruses (OVs), are possible agents that can be used to promote type I IFN signaling.
Chemotherapeutics and targeted drugs
Experiments in mouse models have shown that IFNAR1 expression on tumor cells and not host cells is required for the satisfactory efficacy of anthracycline-based therapy [92, 93]. Some chemotherapeutics, such as anthracyclines, promote the activation of TLR3 in mouse and human tumor cells, leading to the secretion of type I IFNs, which then activate an autocrine or paracrine IFNARdependent circuit that drives the expression of some ISGs [32]. Similarly, anthracyclines and oxaliplatin can induce tumor cell death via the release of HMGB1, which is recognized by TLR4 expressed on DCs and subsequently activates MyD88, resulting in type I IFN production [26]. Trastuzumab, the main treatment for human epidermal growth factor receptor 2 (HER2/ErbB-2)-positive breast cancer, has been proposed to be mechanistically dependent on the release of type I and type II IFNs [92]. Additionally, anti-colony-stimulating factor-1 receptor (anti-CSF-1R), which depletes the majority of F4/80+ TAMs, induces intratumoral type I IFN signaling, sensitizing tumors to cisplatin, thus improving its therapeutic effect [94]. Moreover, studies have suggested that the cisplatin + anti-CSF-1R synergistic therapeutic efficacy against tumors can be further enhanced by targeting neutrophil-dependent immunosuppression, which is critical for an antitumor immune response during cisplatin treatment [94].
Radiation therapy
Local radiotherapy serves as a highly targeted, effective therapy for tumors due to its ability to induce lethal DNA damage and direct cell death and/or senescence. In this context, studies have shown that ablative RT increases intratumoral production of IFN-β, and the antitumor effect of RT is dependent on the production of type I IFNs [95]. Furthermore, a recent study showed that both the cGAS/STING-dependent DNA-sensing pathway and the MAVS-dependent RNA-sensing pathway are crucial for type I IFN signaling induced by radiation alone, or radiation plus ataxia-telangiectasia-mutated (ATM) and Rad3-related protein kinase inhibitor (ATRi). This suggests a mechanism by which radiation induces cell-intrinsic type I IFN signaling pathways and cytosolic nucleic acid-sensing pathways [96]. Additionally, in combination with intratumoral injection of the TLR9 agonist SD-101, RT exhibits a type I IFN-induced antitumor effect in patients (NCT02266147) [97].
Oncolytic virotherapy
Oncolytic virotherapy is a promising cancer therapy in which replicated viruses are used to infect and replicate in growing tumor cells, leading to their lysis. In addition to their direct oncolytic ability, OVs can cause local and contained infections that activate the antitumor immune response via ectopic expression of inflammatory molecules.
T-VEC (Talimogene laherparepvec) was designed from Herpes simplex virus type 1 (HSV-1) to produce granulocyte–macrophage colony-stimulating factor (GM-CSF) and is the first approved OV by the FDA to treat surgically unresectable skin and lymph node lesions in patients with advanced melanoma [98]. Using a single-cell RNA sequence, a recent study reported that T-VEC treatment in patients with primary cutaneous B cell lymphoma induced IFN-α/β signaling and early NK and DC infiltration, which further caused cytotoxic T cell enrichment and a decrease in Treg [99]. Oncolysis by PVSRIPO, the recombinant poliovirus/rhinovirus chimera, releases the proteome of cancer cells and induces type I IFN-dominant responses in DCs, resulting in antitumor immunity [99]. In a phase 1 trial of recurrent glioblastoma, intratumoral delivery of PVSRIPO confirmed the absence of neurovirulent potential and revealed durable radiographic responses [100]. Further trials in breast cancer (NCT03564782) and melanoma (NCT03712358) are being conducted. Furthermore, a recent report found that PVSRIPO-mediated antitumor immunotherapy depended on type-I/III IFN from macrophages and DCs in the TME, but was independent of tumor cell lysis [101]. This study further supports the wide-ranging anti-tumor immune response of OVs in the TME. Local intratumor therapy of B16 melanoma with oncolytic Newcastle Disease Virus (NDV), an avian paramyxovirus with robust type I IFN-inducing and oncolytic properties, promotes lymphocyte infiltration and anti-tumor immunity in distant tumors without distant virus spread [102]. MEDI9253, a recombinant NDV encoding IL-12, has been tested in combination with ICBs in a clinical trial (NCT04613492). Similarly, the therapeutic activity of an IL-12-encoding variant of the Semliki Forest virus (SFV-IL12) strongly relies on a vector-induced IFN-I response that stimulates the IPS-1- and Trif-dependent pathways [103]. Furthermore, Vvax001, an RNA replicon vaccine based on SFV, has been demonstrated to be safe and induced immune responses in all participants with HPV-induced cancers [104]. Moreover, the antitumor immunity of the Maraba rhabdovirus is greatly impaired when IFNAR1 is blocked in mouse tumor models, suggesting the Maraba virus is dependent on the type I IFN-mediated anti-tumor immune response [105]. Moreover, a Maraba-based OV, MAGE-A3, is currently being tested in phase 1/2 studies (NCT02285816/NCT02879760). A recent study suggested that OVs can be co-administered with antigenic peptides in personalized anti-cancer vaccines to target patient-specific mutations [106]. However, even though OVs have been approved by the FDA and achieved certain efficacy in some patients, the resulting increase in type I IFNs can be problematic. NDV-induced type I IFN increases PD-L1 expression in the TME and mediates the resistance to immunotherapy; however, intratumoral NDV in combination with ICBs expands its therapeutic efficacy [107]. Moreover, combining adoptive cell therapy with OVs induced the autoimmune side effect of type I IFN [108] and OVs drove type I IFN to restrict CAR T cell therapy [109]. Thus, combination therapy with OVs needs to be specifically optimized. Importantly, IFNs exhibit antiviral effects, and some tumors that respond well to OV therapy often have IFN pathway defects, as these defects provide an advantage to tumors; however, this attribute makes them particularly vulnerable to viral infections and is, therefore, an intriguing prospect for OV therapy [110].
Similarly, the success of conventional chemotherapeutics, RT, and OVs, to some extent, is dependent on type I IFN signaling. In addition to these therapies, several strategies have been developed to selectively activate type I IFN systems within tumor sites. These include recombinant type I IFNs, type I IFN-expressing cells, and type I IFN-encoding vectors [72]. For instance, the induced pluripotent stem cell (iPSC)-derived proliferating myeloid cell (iPSC-pMC), a gene-modified cell, was engineered to specifically produce IFN-α; its local administration may also promote host XCR1+ DCs to enhance CD8+ T cell activation, resulting in remarkably reproducible and effective antitumor immunity without clear side effects. Furthermore, the combination of IFN-α-iPSC-pMC with ICBs increases perforin+CD8+ T cell accumulation, thereby causing a long-lasting antitumor immune response [111].
Type I IFNs and ICBs
Despite the unprecedented clinical success of ICBs, particularly the PD-1/PD-L1 blockade, only a limited proportion of patients respond well to ICBs employed as a monotherapy treatment owing to the establishment of primary, adaptive, and acquired resistance [112]. Consequently, the development of combination therapies that promote tumor suppression has sparked the interest of researchers. ICB combination partners that have been approved by the FDA have been found to benefit a significant number of patients with cancer after combination therapies [113]. Among the many different combinatorial regimens, many ICB combination partners in development are based on type I IFNs (Table 1–4). Nonetheless, some clinical trials combination type I IFN inducers with ICBs did not acquire robust antitumor effects. Recently, report indicated that sustained type I IFN was observed in anti-PD-1 resistant tumors. PD-L1 and NOS2 expression in both tumor and DCs were induced by the sustained IFN-β transcription, and NOS2 inhibition maintained long-term control of tumors with anti-PD-1 treatment [114]. These studies suggested that the combination type I IFN inducers and ICBs should be used in specific tumor scenarios.
TLR agonists
Since TLR agonists trigger innate immune cell activation and enhance type I IFNs production in the TME, this is a feasible strategy to synergize with ICBs. As a TLR2/4 agonist, the treatment of BCG combined with ICBs has been regarded as a regimen in clinical trials (Table 1). Interestingly, only anti-PD-1 or anti-PD-L1 antibodies are associated with BCG in these ongoing trials. One trial using BCG followed by ipilimumab treatment in advanced metastatic melanoma reported immune-related adverse events and no evidence of clinical benefits (NCT01838200) [115]. Additionally, these combined trials only studied the effects of BCG in bladder cancer, which is probably due to its FDA approval for bladder cancer. For TLR3 agonists, poly I:C is used to enhance the efficacy of ICBs in preclinical models (Table 1). In an engineered immune cell-poor melanoma mouse model, the targeted activation of the type I IFN system with poly I:C in combination with anti-PD-1 strongly prolonged murine life, thereby suggesting a possible effective strategy to increase the therapeutic efficacy of anti-PD-1/PD-L1 in patients with immune cell-poor melanomas [116]. Similarly, in a study using multiple models of TNBC, poly I:C stimulated PD-L1 expression via type I IFNs, and poly I:C administered in combination with anti-PD-1 treatment was more effective than treatment with anti-PD-1 alone [117]. Moreover, intratumoral administration with BO-112 (nanoplexed poly I:C) reduced MC38, 4T1, and B16-F10 growth in mice, and enhanced the antitumor effect of anti-PD-L1 [118]. Thus, many clinical trials combining TLR3 agonists and ICBs are in progress (Table 1). So far, about 18 TLR7/8 agonists are used in clinical trials for the treatment of cancer and infections [119]. Some of them are combined with ICBs to treat cancers in phase 1/2 (Table 1). BDB001, a TLR 7/8 dual agonist, has been safely administered intravenously to reprogram dendritic cells for antitumor activities [120]. Moreover, intravenously administered BDB001 combined with pembrolizumab has been well-tolerated and resulted in systemic immune activation in a phase 1 dose-escalation trial (NCT03486301) [121]. BDC-1001, a novel TLR 7/8 agonist with HER2 conjugation, has shown immune-mediated antitumor efficacy in preclinical tumor models resistant to anti-HER2 treatments, and its dose escalation by combination with pembrolizumab is ongoing (NCT04278144) [122]. TLR9 agonists have been studied with ICBs in multiple tumor types, including melanoma, lymphoma, head and neck squamous cell carcinoma (HNSCC), and NSCLC (Table 1). Since TLR9 is an intracellular nucleic acid sensor, it has been stimulated with synthetic oligonucleotides (ODN) to activate type I IFN signals. IMO-2125 (Tilsotolimod, commonly referred to as immunomodulatory oligonucleotide (IMO)) is one of the most advanced TLR9 agonists used in clinical trials. In the phase 1/2 trial of PD-1 inhibitor refractory advanced melanoma (NCT02644967), intratumoral IMO-2125 in combination with ipilimumab showed a 71.4% disease control rate and 22.4% overall response rate (ORR) [123]. Based upon these promising results, a phase 3 trial of IMO-2125 in combination with ipilimumab in anti-PD-1 refractory melanoma has been conducted (NCT03445533), but the preliminary results did not meet its primary ORR endpoint [124]. SD-101 is a CpG-ODN with cytidine-phospho-guanosine (CpG) motifs and intratumoral SD-101 overcame PD-1 blockade resistance in mice bearing CT26 tumors [125]. Moreover, SD-101 in combination with pembrolizumab resulted in an ORR of 78% in patients with unresectable or metastatic malignant melanoma (NCT02521870) [126]. However, combining SD-101 with ipilimumab and radiation in patients with recurrent low-grade B cell lymphoma showed as an unpromising therapeutic option (NCT02254772) [127]. Another CpG-ODN TLR9 agonist, CMP-001, enhanced anti-PD-1 therapy in mice-bearing mEERL HNSCC [128] and its combination clinical trial in recurrent or metastatic HNSCC is ongoing (NCT04633278). In addition, a phase 1b study (NCT03084640) of CMP-001 in combination with pembrolizumab had a manageable safety profile and durable response in 25% of patients with metastatic melanoma [129], and further phase 2 trials have been initiated to confirm the efficacy of CMP-001 and nivolumab in advanced melanoma (NCT04401995/NCT04698187/NCT04695977). Furthermore, phase 1/2 trials using CMP-001 and pembrolizumab are ongoing in patients with lymphoma (NCT03983668) and melanoma (NCT02680184/NCT04708418). It is worth noting that the combination of CMP-001 and nivolumab in patients with stage IIIB/C/D melanoma has shown acceptable toxicity and promising efficacy (60% major pathologic response rate (MPR) and 82% 1-year relapse-free survival) [130]. MGN1703, which is DNA-based and essentially different from the CpG-ODN TLR9 agonist, has shown immune activation and anti-tumor efficacy in metastatic solid tumors [131]. However, MGN1703 showed no relevant efficacy in phase 2 trials (NCT02200081) on extensive-stage small-cell lung cancer, even though its favorable safety profile promoted further trials [132]. Further studies found that MGN1703 strengthened the effect of ICBs in preclinical models [133, 134], which supported an ongoing trial combining MGN1703 with ipilimumab in advanced solid tumors (NCT02668770). Thus far, out of all these TLR agonists, TLR9 in combination with ICBs has shown the most encouraging clinical data.
STING agonists
ICBs, particularly anti-PD-1 and anti-CTLA-4, failed to induce antitumor effects in cGAS or STINGdeficient mice when administered alone, which indicated that cGAS-STING signal may need to be screened in patients before STING agonists in combination with ICBs. However, the efficacy of ICBs was substantially increased in tumor models when combined with either the cGAS product, cGAMP, or a synthetic cGAMP analog in cGAS-STING signal sufficient context [37, 135,136,137]. These results suggest that the combination of ICBs with treatments that aim at the cGAS-STING axis could be an effective strategy to overcome immunosuppression and increase patient responsiveness. Therefore, clinical trials combining STING agonists with ICBs are underway [87]. We summarized these clinical trials in Table 2. Reports found that intratumoral low-dose ADU-S100 (MIW815), one of the synthetic CDNs for STING activation, induced local activation of tumor-specific CD8+ T cells for durable anti-tumor immunity and their combination with ICBs resulted in better anti-tumor effects in a poorly immunogenic tumor model [138]. Moreover, intraperitoneal administration of ADU-S100 in colon cancer suppressed aberrant angiogenesis and resulted in an inflamed TME in a type I IFN-dependent manner. Consequently, the combination of ADU-S100 and anti-PD-1 antibody further enhanced the antitumor effect [139]. These preclinical findings supported the clinical trials. In a phase 1b study of PD-1-naive TNBC and PD-1-relapsed/refractory melanoma, the combination of ADU-S100 with spartalizumab was well-tolerated and demonstrated antitumor activity [140]. Furthermore, trials combining ADU-S100 with ipilimumab or pembrolizumab have been initiated (NCT02675439/NCT03937141). The preliminary data of MK-1454, another CDN, alone or in combination with pembrolizumab in patients with advanced solid tumors or lymphomas, resulted in encouraging efficacy and an acceptable safety profile (NCT03010176), which makes us look forward to the final result [141]. In addition, intratumoral MK-1454 in combination with pembrolizumab to treat metastatic or unresectable, recurrent HNSCC is being tested in phase 2 (NCT04220866). In addition, to increase STING-dependent type I IFN production, researchers have engineered CDNs with cancer vaccines to form the STINGVAX. Interestingly, PD-L1 expression in tumor cells from STINGVAX-treated mice was significantly upregulated. Thus, combining PD-1 blockade with STINGVAX increased the anti-tumor efficacy in many tumor models that did not respond to anti-PD-1 alone, which supports the rationale for clinical evaluation of STINGVAX in combination with anti-PD-1, particularly in settings where patients failed to respond to ICB monotherapy [135]. Moreover, using non-pathogenic E coli Nissle, researchers designed SYNB1891, an agonist for targeting STING, to induce anti-tumor immunity and immunological memory [142]. In addition, a phase 1 study of SYNB1891 injection alone or in combination with atezolizumab is being tested in advanced/metastatic solid tumors and lymphoma (NCT04167137). Since the STING signal was discovered after TLR, clinical trials using STING agonists in combination with ICBs are mostly in phase 1 (Table 2).
Radiation therapy
RT has been demonstrated as a promising strategy to induce type I IFN secretion in the TME. RT is a well-established, accessible, and comparatively economical procedure that has been widely used in combination with ICBs in clinical trials, owing to its cytotoxic, and multiple immunomodulatory effects on tumor cells as well as its relatively limited and manageable clinical side effects [143,144,145]. Multiple phase 1/2 clinical trials that combined RT with ICBs have acquired promising results and smoothly implemented phase 3 clinical trials [144]. Among these, durvalumab improved the progression-free survival and overall survival rate in stage III unresectable NSCLLC after chemoradiotherapy (NCT02125461) [146]. In addition, ongoing phase 3 trials are using durvalumab in combination with chemoradiation in patients with locally advanced cervical cancer (NCT03830866) [147] and limited-stage small-cell lung cancer (NCT03703297) [148]. However, ipilimumab had no significant effect in terms of overall survival compared to the placebo after RT in a multicenter, randomized, double-blind, phase 3 trial (NCT00861614) in patients with metastatic castration-resistant prostate cancer [149]. Moreover, in two phase 3 glioblastoma trials (NCT02667587/NCT02617589), nivolumab plus RT did not meet the primary endpoint of overall survival [150]. Despite some disappointing clinical survival benefit results, trials combining RT with ICBs are ongoing in multiple tumors. We summarized the present combination regimens of RT with ICBs in phase 3 trials in Table 3. ICBs may be a feasible option in patients where RT failed, but the disappointing data in some of the phase 3 trials suggested that we need to broaden our understanding of the underlying molecular, cellular, and systemic mechanisms of these combination treatments. Recently, researchers found that RT induced the upregulation of PD-L1 and TGF-β, which can be blocked by bintrafusp alfa, a bifunctional fusion protein targeting both PD-L1 and TGF-β. Moreover, bintrafusp alfa in combination with RT eradicated therapy-resistant tumors [151]. This study indicated that involving a specific target for RT in combination with ICBs may be more effective in specific patients, especially in RT-induced immunosuppressive molecules.
Oncolytic virotherapy
Currently, few OVs have been officially approved for use in clinical settings. However, combinations of various OVs with ICBs are currently being tested in clinical trials, including HSV-1/2, Adenovirus (Ad), Vaccinia virus (VV), Coxsackievirus; Polio/rhinovirus, Maraba virus (MRB), Vesicular stomatitis virus (VSV), and Reovirus (Table 4). T-VEC has gained a durable response in melanoma [152] and its combination with ipilimumab in unresected melanoma has shown greater antitumor activity without additional safety concerns compared to monotherapy (NCT01740297) [153]. In addition to its combination with anti-CTLA-4, T-VEC combined with pembrolizumab increased IFN-γ and CD8+ T cells in patients with advanced melanoma [154]. For most trials using T-VEC plus ICBs, phase 1 has completed and phase 2 will likely soon be initiated (Table 4). Moreover, the combination of T-VEC with pembrolizumab has shown promising ORR and CR rates in advanced melanoma (NCT02263508) [155]. However, in a multicenter phase 1b study, the combination of T-VEC and pembrolizumab has shown a tolerable safety profile in recurrent or metastatic HNSCC, but its efficacy was similar to that of pembrolizumab monotherapy [156]. Thus, this combination regimen will not be continued to phase 3. HF10, another HSV-1-derived OV, is well-tolerated and has continued viral antitumor activity in refractory and superficial cancers [157]. Furthermore, the combination of HF10 and ipilimumab showed favorable profiles in two phase 2 studies of patients with unresectable or metastatic melanoma (NCT02272855/NCT03153085) [158, 159]. Other HSV-1-derived OVs in combination with ICBs are being tested in phase 1/2 trials (Table 4). In preclinical studies, both NDV and Maraba-based OVs induced increased PD-L1 expression in melanoma [107] and breast cancer [105]. Therefore, the combination of OVs with ICBs is a promising strategy to overcome PD-L1-induced immunotherapy resistance. Zamarin et al. showed that localized oncolytic NDV in combination with systemic anti-CTLA-4 blockade can eradicate tumors in B16 melanoma by producing curative immune responses that require CD8+ T cells, NK cells, and type I IFNs [102]. Therefore, MEDI9253 (NDV human IL-12) in combination with durvalumab is being used in an ongoing phase 1 trial (NCT04613492). Ad is another large class of OVs, and many of them are combined with ICBs in phase 1/2 trials (Table 4). ONCOS-102, an Ad-based OV that expresses GM-CSF, in combination with pembrolizumab reduced tumor volume in a humanized A2058 melanoma mouse model that did not benefit from pembrolizumab monotherapy [160]. Moreover, a phase 1 trial of ONCOS-102 plus pembrolizumab has shown a 33% ORR in advanced or unresectable melanoma progressing after PD-1 blockade (NCT03003676) [161]. These promising data promoted the use of other Ads in combination with ICBs in trials (Table 4). MRB-based OVs are used in ongoing trials as well, such as MG1-MAGEA3 and MG1-E6E7. To expand more tumor-associated antigens in the TME, two trials used MRB and Ad-based OVs (MG1-MAGEA3 and Ad-MAGEA3; MG1-E6E7 and Ad-E6E7) in combination with ICBs for NSCLC or HPV-associated cancers (NCT02879760/ NCT03618953). The use of Coxsackievirus A21 (CVA21), a naturally occurring OV, in combination with ICBs in a phase 1 trial has shown a well-tolerated and durable response (NCT02307149/NCT02565992) [162, 163]. Moreover, phase 2 trials using CVA21 in combination with pembrolizumab are recruiting participants (Table 4). Recently, researchers found that monotherapy with Measles virus-based neutrophil-activating protein or anti-PD-1 treatment has shown a modest survival benefit in Measles virus-resistant syngeneic glioblastoma models, and that combination treatment had a synergic effect [164]. Other OVs, such as PVSRIPO, VSV-IFNβ-NIS, Pelareorep (Reovirus), and OH2 (HSV-2), in combination with ICBs are in trials as well. In general, the use of OVs plus ICBs is a promising regimen, especially T-VEC plus pembrolizumab in patients with melanoma, which is currently in phase 3.
Others
Currently, many additional curative combinatorial agents targeting the type I IFN signaling axis are being developed. For instance, based on the characteristic of Mn to induce type I IFN production, the combination of Mn and anti-PD-1 antibody has been used in a phase 1 study of patients with advanced metastatic solid tumors (NCT03991559), which showed manageable safety and promising efficacy [25]. In addition, several phase 2 studies using a combined regimen of Mn and anti-PD-1 antibody in advanced solid tumors or lymphoma are ongoing (NCT04004234/ NCT03989310/NCT03989336/NCT04873440). The nucleoside analog 6-thio-2’-deoxyguanosine (6-thio-dG), which induces damage to telomeric DNA in telomerase-expressing tumor cells to initiate the host cytosolic DNA-sensing type I IFNs/STING pathway, was reported to overcome ICB resistance in advanced tumors and was used in combination with ICBs in tumor models [165]. Moreover, oral delivery of live Lactobacillus rhamnosus GG (LGG), one of the most well-characterized and used probiotics, synergizes with anti-PD-1 to augment antitumor immunity. Combination therapy of LGG and anti-PD-1 mechanically increases tumor-infiltrating DCs and IFN-β induction through the cGAS-STING-IRF7 cascade [166].
Conclusions and perspectives
Type I IFNs were first described for their strong antiviral properties, and mounting evidence confirmed their anti-tumor activity. To date, IFN-α and IFN-β have achieved some beneficial results in patients with cancer, and emerging clinical data support the key role of type I IFN inducers in combination with ICBs. These antitumor effects originate from type I IFNs produced by tumor cells or immune effector cells in response to pathogenic molecular stimuli. In turn, type I IFNs exert anti-tumor functions by directly influencing tumor cell progression and indirectly modulating anti-tumor immune cells in multiple ways. Nevertheless, large gaps in our understanding of the mechanism by which substances and signals are transferred between tumor cells, type I IFN, and immune cells remain. Thus, further elucidating the reciprocal interactions among tumor cells, type I IFN, and immune cells will be of interest for future studies. Furthermore, the cell population of type I IFN-produced cells, such as DC and endothelial cells, is limited in the TME; thus, the baseline of type I IFNs is lower. This prompts researchers to find the regulatory mechanism of type I IFNs. A more in-depth appreciation of the molecular signaling cascades and the series of genes activated by type I IFNs will provide multiple opportunities for enhancing IFN-I-based tumor therapy.
To overcome the lower baseline level, many type I IFN inducers were used to treat tumors in preclinical models and clinical trials. However, its anti-tumor effect is unsatisfactory in certain circumstances, which may be due to recombinant type I IFNs causing systemic side effects or certain type I IFN inducers causing the increased production of immunosuppressive molecules, such as ROS from neutrophil, PD-L1 in tumor cells, and IDO in immune cells. In addition, the specific tumor background should be considered when performing the therapeutic schedule. However, intratumoral injection or nanoparticle-packaged type I IFN inducers have been used in trials to reduce these side effects. Furthermore, to neutralize immunosuppressive molecules, type I IFN inducers in combination with ICB, IDO, and other specific inhibitors may overcome this resistance.
Many clinical trials that combine type I IFN inducers with ICBs are in progress. Some final results of phase 3 studies did not meet the primary endpoints; thus, the essential mechanism of resistance caused by ICB or type I IFN inducer monotherapy needs to be clarified. These studies also suggest that not all patients are suitable for combination therapy of type I IFN inducers and ICBs. The addition of type I IFN signaling as an efficacy prediction factor of ICB may promote the precisive application of type I IFN inducers and ICBs in future. Therefore, we look forward to improved clinical effects in these ongoing trials.
Data availability
Not applicable.
Abbreviations
- ICB:
-
Immune checkpoint blockade
- IFNs:
-
Interferons
- IFNAR1:
-
IFN-α/β receptor 1
- IFNAR2:
-
IFN-α/β receptor 2
- TYK2:
-
Tyrosine kinase 2
- JAK1:
-
Janus kinase 1
- STAT1:
-
Signal transducer and activator of transcription 1
- STAT2:
-
Signal transducer and activator of transcription 2
- IRF9:
-
Interferon regulatory factor 9
- ISGF3:
-
IFN-stimulated gene factor 3
- ISREs:
-
IFN-stimulated response elements
- ISGs:
-
Interferon-stimulated genes
- MAPK:
-
Mitogen-activated protein kinase
- mTOR:
-
Mammalian target of rapamycin
- GCD:
-
GCD-GTPases/cyclin-dependent kinases
- FDA:
-
Food and Drug Administration
- TME:
-
Tumor microenvironment
- DCs:
-
Dendritic cells
- pDCs:
-
Plasmacytoid dendritic cells
- CD:
-
Cluster of differentiation
- GMP:
-
Guanosine monophosphate
- AMP:
-
Adenosine monophosphate
- cGAMP:
-
Cyclic GMP–AMP
- cGAS:
-
CGAMP synthase
- STING:
-
Stimulator of interferon genes
- ER:
-
Endoplasmic reticulum
- TBK1:
-
TANK-binding kinase 1
- IRF:
-
Interferon regulatory factor
- NF-κB:
-
Nuclear factor-kappa B
- Mn:
-
Manganese
- dsDNA:
-
Double-stranded DNA
- HMGB1:
-
High-mobility group box 1
- TLR:
-
Toll-like receptor
- MyD88:
-
Myeloid differentiation factor 88
- CLEC9A:
-
C-type lectin domain-containing 9A
- SLC19A1:
-
Solute Carrier Family 19 Member 1
- RIG-I:
-
Retinoic acidinducible gene I
- RLRs:
-
RIG-I-like receptors
- PRRs:
-
Pattern recognition receptors
- UNC93B1:
-
Unc-93 homolog B1
- PTX:
-
Paclitaxel
- TICAM1:
-
Toll-like receptor adaptor molecule 1
- LPS:
-
Lipopolysaccharide
- TAMs:
-
Tumor-associated macrophages
- E-FABP:
-
Epidermal fatty acid-binding proteins
- LD:
-
Lipid droplet
- CAFs:
-
Cancer-associated fibroblasts
- ZBP1:
-
Z-DNA binding protein 1
- DDX41:
-
DEAD-Box Helicase 41
- MHC I:
-
Major histocompatibility complex class I
- TAA:
-
Tumor-associated antigen
- APC:
-
Antigen-presenting cells
- NK:
-
Natural killer
- IL:
-
Interleukin
- CXCR:
-
C-X-C chemokine receptor
- CXCL:
-
C-X-C chemokine ligand
- ROS:
-
Reactive oxygen species
- Bcl-2:
-
B cell lymphoma-2
- BCL-xL:
-
B cell lymphoma-extra-large
- G-CSF:
-
Granulocyte colony-stimulating factor
- VEGF:
-
Vascular endothelial growth factor
- MMP9:
-
Matrix metallopeptidase 9
- Treg:
-
Regulatory T cells
- MEK:
-
Mitogen-activated protein kinase kinase
- ERK:
-
Extracellular signal-regulated kinase
- PDE4:
-
Phosphodiesterase 4
- cAMP:
-
Cyclic AMP
- CCL:
-
C–C chemokine ligand
- MDSCs:
-
Myeloid-derived suppressor cells
- TRAIL:
-
Tumor necrosis factor (TNF)-related apoptosis-inducing ligand
- DR5:
-
Death receptor 5
- IDO1:
-
Indoleamine 2,3-dioxygenase 1
- Poly A:U:
-
Polyadenylic–polyuridylic acid
- Poly(ICLC):
-
Poly I:C plus polylysine
- CpG:
-
Cytidine-phospho-guanosine
- BCG:
-
Bacillus Calmette–Guérin
- CDNs:
-
Cyclic dinucleotides
- CAR T cell:
-
Chimeric antigen receptor T cell
- NSCLC:
-
Non-small-cell lung cancer
- cGAMP-NPs:
-
CGAMP nanoparticles
- RT:
-
Radiation therapy
- OVs:
-
Oncolytic viruses
- HER2/ErbB-2:
-
Human epidermal growth factor receptor 2
- Anti-CSF-1R:
-
Anti-colony-stimulating factor-1 receptor
- ATM:
-
Ataxia-telangiectasia-mutated
- ATRi:
-
ATM and Rad3-related protein kinase inhibitor
- T-VEC:
-
Talimogene laherparepvec
- HSV:
-
Herpes simplex virus
- GM-CSF:
-
Granulocyte–macrophage colony-stimulating factor
- NDV:
-
Newcastle disease virus
- IPS-1:
-
Interferon-beta promoter stimulator 1
- Trif:
-
Toll/IL-1R domain-containing adaptor inducing IFN-β factor
- iPSC:
-
Induced pluripotent stem cell
- iPSC-pMC:
-
IPSC-derived proliferating myeloid cell
- XCR1:
-
X-C Motif Chemokine Receptor 1
- PD-1:
-
Programmed cell death 1
- PD-L1:
-
Programmed death ligand 1
- ODN:
-
Oligonucleotides
- IMO:
-
Immunomodulatory oligonucleotide
- ORR:
-
Objective response rate
- HNSCC:
-
Head and neck squamous cell carcinoma
- MPR:
-
Major pathologic response rate
- CTLA-4:
-
Cytotoxic lymphocyte-associated antigen-4
- TNBC:
-
Triple-negative breast cancer
- TGF-β:
-
Transforming growth factor beta
- Ad:
-
Adenovirus
- VV:
-
Vaccinia virus
- MRB:
-
Maraba virus
- VSV:
-
Vesicular stomatitis virus
- CR:
-
Complete response
- CVA21:
-
Coxsackievirus A21
- 6-thio-dG:
-
6-Thio-20-deoxyguanosine
- LGG:
-
Lactobacillus rhamnosus GG
- IMRT:
-
Intensity-modulated radiation therapy
- IGRT:
-
Image guided radiation therapy
- EBRT:
-
External beam radiotherapy
- SBRT:
-
Stereotactic body radiotherapy
- 3D-CRT:
-
3-Dimensional conformal radiation therapy
- SABR:
-
Stereotactic ablative radiotherapy
- PCI:
-
Prophylactic cranial irradiation
References
Isaacs A, Lindenmann J (1957) Virus interference. I. the interferon. Proc R Soc Lond B Biol Sci 147:258–267
Isaacs A, Lindenmann J, Valentine RC (1957) Virus interference. II. Some properties of interferon. Proc R Soc Lond B Biol Sci 147:268–273
Parker BS, Rautela J, Hertzog PJ (2016) Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer 16:131–144
Sprooten J, Agostinis P, Garg AD (2019) Type I interferons and dendritic cells in cancer immunotherapy. Int Rev Cell Mol Biol 348:217–262
Pestka S, Krause CD, Walter MR (2004) Interferons, interferon-like cytokines, and their receptors. Immunol Rev 202:8–32
Oritani K, Kanakura Y (2005) IFN-zeta/ limitin: a member of type I IFN with mild lympho-myelosuppression. J Cell Mol Med 9:244–254
Lazear HM, Schoggins JW, Diamond MS (2019) Shared and distinct functions of type I and type III interferons. Immunity 50:907–923
Platanias LC (2005) Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5:375–386
Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A et al (2011) Direct effects of type I interferons on cells of the immune system. Clin Cancer Res 17:2619–2627
Stark GR, Darnell JE Jr (2012) The JAK-STAT pathway at twenty. Immunity 36:503–514
Borden EC (2019) Interferons alpha and beta in cancer: therapeutic opportunities from new insights. Nat Rev Drug Discov 18:219–234
Levy DE, Kessler DS, Pine R, Darnell JE Jr (1989) Cytoplasmic activation of ISGF3, the positive regulator of interferon-alpha-stimulated transcription, reconstituted in vitro. Genes Dev 3:1362–1371
Kessler DS, Veals SA, Fu XY, Levy DE (1990) Interferon-alpha regulates nuclear translocation and DNA-binding affinity of ISGF3, a multimeric transcriptional activator. Genes Dev 4:1753–1765
Platanitis E, Demiroz D, Schneller A et al (2019) A molecular switch from STAT2-IRF9 to ISGF3 underlies interferon-induced gene transcription. Nat Commun 10:2921
Saleiro D, Platanias LC (2019) Interferon signaling in cancer Non-canonical pathways and control of intracellular immune checkpoints. Semin Immunol 43:101299
Swiecki M, Colonna M (2015) The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol 15:471–485
Andzinski L, Spanier J, Kasnitz N et al (2016) Growing tumors induce a local STING dependent Type I IFN response in dendritic cells. Int J Cancer 139:1350–1357
Woo SR, Fuertes MB, Corrales L et al (2014) STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41:830–842
Fuertes MB, Kacha AK, Kline J et al (2011) Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med 208:2005–2016
Wu J, Sun L, Chen X et al (2013) Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339:826–830
Gao P, Ascano M, Wu Y et al (2013) Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153:1094–1107
Zhang X, Bai XC, Chen ZJ (2020) Structures and mechanisms in the cGAS-STING innate immunity pathway. Immunity 53:43–53
Corrales L, McWhirter SM, Dubensky TW Jr, Gajewski TF (2016) The host STING pathway at the interface of cancer and immunity. J Clin Invest 126:2404–2411
Wang C, Guan Y, Lv M et al (2018) Manganese increases the sensitivity of the cGAS-STING pathway for double-stranded DNA and Is required for the host defense against DNA viruses. Immunity 48:675–687 (e677)
Lv M, Chen M, Zhang R et al (2020) Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res 30:966–979
Apetoh L, Ghiringhelli F, Tesniere A et al (2007) Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 13:1050–1059
Lande R, Gregorio J, Facchinetti V et al (2007) Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449:564–569
Sancho D, Joffre OP, Keller AM et al (2009) Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458:899–903
Saccheri F, Pozzi C, Avogadri F et al (2010) Bacteria-induced gap junctions in tumors favor antigen cross-presentation and antitumor immunity. Sci Transl Med 2:44ra57
Luteijn RD, Zaver SA, Gowen BG et al (2019) SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 573:434–438
Zhao J, Xue Y, Pan Y et al (2019) Toll-like receptor 3 agonist poly I: C reinforces the potency of cytotoxic chemotherapy via the TLR3-UNC93B1-IFN-beta signaling axis in paclitaxel-resistant colon cancer. J Cell Physiol 234:7051–7061
Sistigu A, Yamazaki T, Vacchelli E et al (2014) Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 20:1301–1309
Nunez NG, Andreani V, Crespo MI et al (2012) IFNbeta produced by TLR4-activated tumor cells is involved in improving the antitumoral immune response. Cancer Res 72:592–603
Deng L, Liang H, Xu M et al (2014) STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41:843–852
Vanpouille-Box C, Alard A, Aryankalayil MJ et al (2017) DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 8:15618
Ablasser A, Schmid-Burgk JL, Hemmerling I et al (2013) Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503:530–534
Demaria O, De Gassart A, Coso S et al (2015) STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A 112:15408–15413
Ma F, Li B, Yu Y et al (2015) Positive feedback regulation of type I interferon by the interferon-stimulated gene STING. EMBO Rep 16:202–212
Ehnfors J, Kost-Alimova M, Persson NL et al (2009) Horizontal transfer of tumor DNA to endothelial cells in vivo. Cell Death Differ 16:749–757
Corrales L, Glickman LH, McWhirter SM et al (2015) Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep 11:1018–1030
Zhang Y, Sun Y, Rao E et al (2014) Fatty acid-binding protein E-FABP restricts tumor growth by promoting IFN-beta responses in tumor-associated macrophages. Cancer Res 74:2986–2998
Arwert EN, Milford EL, Rullan A et al (2020) STING and IRF3 in stromal fibroblasts enable sensing of genomic stress in cancer cells to undermine oncolytic viral therapy. Nat Cell Biol 22:758–766
Yoon N, Park MS, Shigemoto T et al (2016) Activated human mesenchymal stem/stromal cells suppress metastatic features of MDA-MB-231 cells by secreting IFN-beta. Cell Death Dis 7:e2191
Broad RV, Jones SJ, Teske MC et al (2021) Inhibition of interferon-signalling halts cancer-associated fibroblast-dependent protection of breast cancer cells from chemotherapy. Br J Cancer 124:1110–1120
Gresser I, Bourali C, Levy JP et al (1969) Increased survival in mice inoculated with tumor cells and treated with interferon preparations. Proc Natl Acad Sci U S A 63:51–57
Zhou L, Zhang Y, Wang Y et al (2020) A dual role of type I interferons in antitumor immunity. Adv Biosyst 4:e1900237
Chen J, Cao Y, Markelc B et al (2019) Type I IFN protects cancer cells from CD8+ T cell–mediated cytotoxicity after radiation. J Clin Investig 129:4224–4238
Butterfield LH, Vujanovic L, Santos PM et al (2019) Multiple antigen-engineered DC vaccines with or without IFNalpha to promote antitumor immunity in melanoma. J Immunother Cancer 7:113
Swann JB, Hayakawa Y, Zerafa N et al (2007) Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J Immunol 178:7540–7549
Marcus A, Mao AJ, Lensink-Vasan M et al (2018) Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity 49:754–763 (e754)
Muller L, Aigner P, Stoiber D (2017) Type I Interferons and natural killer cell regulation in cancer. Front Immunol 8:304
Jiang W, Zhang C, Tian Z, Zhang J (2013) hIFN-alpha gene modification augments human natural killer cell line anti-human hepatocellular carcinoma function. Gene Ther 20:1062–1069
Riley CH, Hansen M, Brimnes MK et al (2015) Expansion of circulating CD56bright natural killer cells in patients with JAK2-positive chronic myeloproliferative neoplasms during treatment with interferon-alpha. Eur J Haematol 94:227–234
Jaillon S, Ponzetta A, Di Mitri D et al (2020) Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer 20:485–503
Andzinski L, Wu CF, Lienenklaus S et al (2015) Delayed apoptosis of tumor associated neutrophils in the absence of endogenous IFN-beta. Int J Cancer 136:572–583
Jablonska J, Wu CF, Andzinski L et al (2014) CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-beta. Int J Cancer 134:1346–1358
Andzinski L, Kasnitz N, Stahnke S et al (2016) Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int J Cancer 138:1982–1993
Jablonska J, Leschner S, Westphal K et al (2010) Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest 120:1151–1164
Wu CF, Andzinski L, Kasnitz N et al (2015) The lack of type I interferon induces neutrophil-mediated pre-metastatic niche formation in the mouse lung. Int J Cancer 137:837–847
Pylaeva E, Lang S, Jablonska J (2016) The essential role of type I interferons in differentiation and activation of tumor-associated neutrophils. Front Immunol 7:629
Sharabi A, Tsokos MG, Ding Y et al (2018) Regulatory T cells in the treatment of disease. Nat Rev Drug Discov 17:823–844
Hashimoto H, Ueda R, Narumi K et al (2014) Type I IFN gene delivery suppresses regulatory T cells within tumors. Cancer Gene Ther 21:532–541
Bacher N, Raker V, Hofmann C et al (2013) Interferon-alpha suppresses cAMP to disarm human regulatory T cells. Cancer Res 73:5647–5656
Anz D, Rapp M, Eiber S et al (2015) Suppression of Intratumoral CCL22 by type I Interferon inhibits migration of regulatory T cells and blocks cancer progression. Can Res 75:4483–4493
Hirata A, Hashimoto H, Shibasaki C et al (2019) Intratumoral IFN-alpha gene delivery reduces tumor-infiltrating regulatory T cells through the downregulation of tumor CCL17 expression. Cancer Gene Ther 26:334–343
Sisirak V, Faget J, Gobert M et al (2012) Impaired IFN-alpha production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res 72:5188–5197
Li K, Shi H, Zhang B et al (2021) Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther 6:362
Zoglmeier C, Bauer H, Noerenberg D et al (2011) CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin Cancer Res 17:1765–1775
Field AK, Tytell AA, Lampson GP, Hilleman MR (1967) Inducers of interferon and host resistance. II. Multistranded synthetic polynucleotide complexes. Proc Natl Acad Sci U S A 58:1004–1010
Chen J, Sun HW, Yang YY et al (2021) Reprogramming immunosuppressive myeloid cells by activated T cells promotes the response to anti-PD-1 therapy in colorectal cancer. Signal Transduct Target Ther 6:4
Huang L, Li L, Lemos H et al (2013) Cutting edge: DNA sensing via the STING adaptor in myeloid dendritic cells induces potent tolerogenic responses. J Immunol 191:3509–3513
Zitvogel L, Galluzzi L, Kepp O et al (2015) Type I interferons in anticancer immunity. Nat Rev Immunol 15:405–414
Drobits B, Holcmann M, Amberg N et al (2012) Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells. J Clin Invest 122:575–585
Adamus T, Kortylewski M (2018) The revival of CpG oligonucleotide-based cancer immunotherapies. Contemp Oncol (Pozn) 22:56–60
Aranda F, Vacchelli E, Obrist F et al (2014) Trial watch: toll-like receptor agonists in oncological indications. Oncoimmunology 3:e29179
Frega G, Wu Q, Le Naour J et al (2020) Trial watch: experimental TLR7/TLR8 agonists for oncological indications. Oncoimmunology 9:1796002
Sagiv-Barfi I, Czerwinski DK, Levy S et al (2018) Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med 10:4eaan488
Nuhn L, De Koker S, Van Lint S et al (2018) Nanoparticle-conjugate TLR7/8 agonist localized immunotherapy provokes safe antitumoral responses. Adv Mater 30:e1803397
Kim SY, Kim S, Kim JE et al (2019) Lyophilizable and multifaceted toll-like receptor 7/8 agonist-loaded nanoemulsion for the reprogramming of tumor microenvironments and enhanced cancer immunotherapy. ACS Nano 13:12671–12686
Perry JL, Tian S, Sengottuvel N et al (2020) Pulmonary delivery of nanoparticle-bound toll-like receptor 9 agonist for the treatment of metastatic lung cancer. ACS Nano 14:7200–7215
Vollmer J, Krieg AM (2009) Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv Drug Deliv Rev 61:195–204
Brody JD, Ai WZ, Czerwinski DK et al (2010) In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol 28:4324–4332
Tozer GM, Kanthou C, Baguley BC (2005) Disrupting tumour blood vessels. Nat Rev Cancer 5:423–435
Wang K, Donnelly CR, Jiang C et al (2021) STING suppresses bone cancer pain via immune and neuronal modulation. Nat Commun 12:4558
Xu N, Palmer DC, Robeson AC et al (2021) STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J Exp Med 218:e20200844
Lara PN Jr, Douillard JY, Nakagawa K et al (2011) Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J Clin Oncol 29:2965–2971
Le Naour J, Zitvogel L, Galluzzi L et al (2020) Trial watch: STING agonists in cancer therapy. Oncoimmunology 9:1777624
Cheng N, Watkins-Schulz R, Junkins RD et al (2018) A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1-insensitive models of triple-negative breast cancer. JCI Insight 3:e120638
Jang SC, Economides KD, Moniz RJ et al (2021) ExoSTING, an extracellular vesicle loaded with STING agonists, promotes tumor immune surveillance. Commun Biol 4:497
Banerjee M, Middya S, Basu S et al (2018) Abstract B43: Novel small-molecule human STING agonists generate robust Type I interferon responses in tumors. Cancer Immunol Res 6:B43
Yang J, Adam M, Clemens J et al (2018) Abstract 5554: Preclinical characterization of GSK532, a novel STING agonist with potent anti-tumor activity. Cancer Res 78:5554
Stagg J, Loi S, Divisekera U et al (2011) Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc Natl Acad Sci U S A 108:7142–7147
Yang X, Zhang X, Fu ML et al (2014) Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell 25:37–48
Salvagno C, Ciampricotti M, Tuit S et al (2019) Therapeutic targeting of macrophages enhances chemotherapy efficacy by unleashing type I interferon response. Nat Cell Biol 21:511–521
Burnette BC, Liang H, Lee Y et al (2011) The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res 71:2488–2496
Feng X, Tubbs A, Zhang C et al (2020) ATR inhibition potentiates ionizing radiation-induced interferon response via cytosolic nucleic acid-sensing pathways. EMBO J 39:e104036
Frank MJ, Reagan PM, Bartlett NL et al (2018) In situ vaccination with a TLR9 agonist and local low-dose radiation induces systemic responses in untreated indolent lymphoma. Cancer Discov 8:1258–1269
Poh A (2016) First oncolytic viral therapy for melanoma. Cancer Discov 6:6
Ramelyte E, Tastanova A, Balazs Z et al (2021) Oncolytic virotherapy-mediated anti-tumor response: a single-cell perspective. Cancer Cell 39:394–406 (e394)
Desjardins A, Gromeier M, Herndon JE 2nd et al (2018) Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med 379:150–161
Brown MC, Mosaheb MM, Mohme M et al (2021) Viral infection of cells within the tumor microenvironment mediates antitumor immunotherapy via selective TBK1-IRF3 signaling. Nat Commun 12:1858
Zamarin D, Holmgaard RB, Subudhi SK et al (2014) Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med 6:226ra232
Melero I, Quetglas JI, Reboredo M et al (2015) Strict requirement for vector-induced type I interferon in efficacious antitumor responses to virally encoded IL12. Cancer Res 75:497–507
Komdeur FL, Singh A, van de Wall S et al (2021) First-in-human phase I clinical trial of an SFV-based RNA replicon cancer vaccine against HPV-induced cancers. Mol Ther 29:611–625
Bourgeois-Daigneault MC, Roy DG, Aitken AS et al (2018) Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci Transl Med 10:eaao1641
Roy DG, Geoffroy K, Marguerie M et al (2021) Adjuvant oncolytic virotherapy for personalized anti-cancer vaccination. Nat Commun 12:2626
Zamarin D, Ricca JM, Sadekova S et al (2018) PD-L1 in tumor microenvironment mediates resistance to oncolytic immunotherapy. J Clin Invest 128:1413–1428
Walsh SR, Bastin D, Chen L et al (2019) Type I IFN blockade uncouples immunotherapy-induced antitumor immunity and autoimmune toxicity. J Clin Invest 129:518–530
Evgin L, Huff AL, Wongthida P et al (2020) Oncolytic virus-derived type I interferon restricts CAR T cell therapy. Nat Commun 11:3187
Fend L, Yamazaki T, Remy C et al (2017) Immune checkpoint blockade, immunogenic chemotherapy or IFN-alpha blockade boost the local and abscopal effects of oncolytic virotherapy. Cancer Res 77:4146–4157
Tsuchiya N, Zhang R, Iwama T et al (2019) Type I interferon delivery by iPSC-derived myeloid cells elicits antitumor Immunity via XCR1(+) dendritic cells. Cell Rep 29:162–175 (e169)
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A (2017) Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168:707–723
Meric-Bernstam F, Larkin J, Tabernero J, Bonini C (2021) Enhancing anti-tumour efficacy with immunotherapy combinations. The Lancet 397:1010–1022
Jacquelot N, Yamazaki T, Roberti MP et al (2019) Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res 29:846–861
Da Gama DJ, Parakh S, Andrews MC et al (2018) Autoantibodies may predict immune-related toxicity: results from a phase I study of intralesional bacillus calmette-guerin followed by ipilimumab in patients with advanced metastatic melanoma. Front Immunol 9:411
Bald T, Landsberg J, Lopez-Ramos D et al (2014) Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov 4:674–687
Brockwell NK, Owen KL, Zanker D et al (2017) Neoadjuvant interferons: critical for effective PD-1-based immunotherapy in TNBC. Cancer Immunol Res 5:871–884
Aznar MA, Planelles L, Perez-Olivares M et al (2019) Immunotherapeutic effects of intratumoral nanoplexed poly I:C. J Immunother Cancer 7:116
Bhagchandani S, Johnson JA, Irvine DJ (2021) Evolution of Toll-like receptor 7/8 agonist therapeutics and their delivery approaches: from antiviral formulations to vaccine adjuvants. Adv Drug Deliv Rev 175:113803
Patel M, Rasco D, Johnson M et al (2020) 324 BDB001, a Toll-Like receptor 7 and 8 (TLR7/8) agonist, can be safely administered intravenously and shows clinical responses in advanced solid tumors. J Immunother Cancer 8:A199
Patel MR, Tolcher AW, Rasco DW et al (2021) BDB001, an intravenously administered toll-like receptor 7 and 8 (TLR7/8) agonist, in combination with pembrolizumab in advanced solid tumors: phase 1 safety and efficacy results. J Clin Oncol 39:2512–2512
Sharma M, Carvajal RD, Hanna GJ et al (2021) Preliminary results from a phase 1/2 study of BDC-1001, a novel HER2 targeting TLR7/8 immune-stimulating antibody conjugate (ISAC), in patients (pts) with advanced HER2-expressing solid tumors. J Clin Oncol 39:2549–2549
Haymaker C, Andtbacka RHI, Johnson DB et al (2020) 1083MO Final results from ILLUMINATE-204, a phase I/II trial of intratumoral tilsotolimod in combination with ipilimumab in PD-1 inhibitor refractory advanced melanoma. Ann Oncol 31:S736
Butler MO, Robert C, Negrier S et al (2019) ILLUMINATE 301: A randomized phase 3 study of tilsotolimod in combination with ipilimumab compared with ipilimumab alone in patients with advanced melanoma following progression on or after anti-PD-1 therapy. J Clin Oncol 37:TPS9599–TPS9599
Wang S, Campos J, Gallotta M et al (2016) Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc Natl Acad Sci U S A 113:E7240–E7249
Ribas A, Medina T, Kummar S et al (2018) SD-101 in Combination with pembrolizumab in advanced melanoma: results of a phase Ib, multicenter study. Cancer Discov 8:1250–1257
Smith M, Garcia-Martinez E, Pitter MR et al (2018) Trial Watch: Toll-like receptor agonists in cancer immunotherapy. Oncoimmunology 7:e1526250
Cheng Y, Lemke-Miltner CD, Wongpattaraworakul W et al (2020) In situ immunization of a TLR9 agonist virus-like particle enhances anti-PD1 therapy. J Immunother Cancer 8:e000940
Ribas A, Medina T, Kirkwood JM et al (2021) Overcoming PD-1 blockade resistance with CpG-A toll-like receptor 9 agonist vidutolimod in patients with metastatic melanoma. Cancer Discov. https://doi.org/10.1158/2159-8290
Davar D, Karunamurthy A, Hartman D et al (2020) 303 Phase II trial of neoadjuvant nivolumab (Nivo) and intra-tumoral (IT) CMP-001 in high-risk resectable melanoma (Neo-C-Nivo): final results. J Immunother Cancer 8:A185–A186
Weihrauch MR, Richly H, von Bergwelt-Baildon MS et al (2015) Phase I clinical study of the toll-like receptor 9 agonist MGN1703 in patients with metastatic solid tumours. Eur J Cancer 51:146–156
Thomas M, Ponce-Aix S, Navarro A et al (2018) Immunotherapeutic maintenance treatment with toll-like receptor 9 agonist lefitolimod in patients with extensive-stage small-cell lung cancer: results from the exploratory, controlled, randomized, international phase II IMPULSE study. Ann Oncol 29:2076–2084
Kapp K, Volz B, Oswald D et al (2019) Beneficial modulation of the tumor microenvironment and generation of anti-tumor responses by TLR9 agonist lefitolimod alone and in combination with checkpoint inhibitors. Oncoimmunology 8:e1659096
Reilley MJ, Morrow B, Ager CR et al (2019) TLR9 activation cooperates with T cell checkpoint blockade to regress poorly immunogenic melanoma. J Immunother Cancer 7:323
Fu J, Kanne DB, Leong M et al (2015) STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med 7:283ra252-283ra252
Wang H, Hu S, Chen X et al (2017) cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A 114:1637–1642
Harding SM, Benci JL, Irianto J et al (2017) Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548:466–470
Sivick KE, Desbien AL, Glickman LH et al (2018) Magnitude of therapeutic STING activation determines CD8(+) T cell-mediated anti-tumor immunity. Cell Rep 25:3074–3085 (e3075)
Lee SJ, Yang H, Kim WR et al (2021) STING activation normalizes the intraperitoneal vascular-immune microenvironment and suppresses peritoneal carcinomatosis of colon cancer. J Immunother Cancer 9:e002195
Meric-Bernstam F, Sandhu SK, Hamid O et al (2019) Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. J Clin Oncol 37:2507–2507
Harrington KJ, Brody J, Ingham M et al (2018) LBA15Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann Oncol 30:v557
Leventhal DS, Sokolovska A, Li N et al (2020) Immunotherapy with engineered bacteria by targeting the STING pathway for anti-tumor immunity. Nat Commun 11:2739
Lhuillier C, Vanpouille-Box C, Galluzzi L et al (2018) Emerging biomarkers for the combination of radiotherapy and immune checkpoint blockers. Semin Cancer Biol 52:125–134
Kang J, Demaria S, Formenti S (2016) Current clinical trials testing the combination of immunotherapy with radiotherapy. J Immunother Cancer 4:51
Vacchelli E, Bloy N, Aranda F et al (2016) Trial watch: immunotherapy plus radiation therapy for oncological indications. Oncoimmunology 5:e1214790
Hui R, Ozguroglu M, Villegas A et al (2019) Patient-reported outcomes with durvalumab after chemoradiotherapy in stage III, unresectable non-small-cell lung cancer (PACIFIC): a randomised, controlled, phase 3 study. Lancet Oncol 20:1670–1680
Mayadev J, Nunes AT, Li M et al (2020) CALLA: Efficacy and safety of concurrent and adjuvant durvalumab with chemoradiotherapy versus chemoradiotherapy alone in women with locally advanced cervical cancer: a phase III, randomized, double-blind, multicenter study. Int J Gynecol Cancer 30:1065–1070
Senan S, Okamoto I, Lee GW et al (2020) Design and rationale for a phase III, randomized, placebo-controlled trial of durvalumab with or without tremelimumab after concurrent chemoradiotherapy for patients With limited-stage small-cell lung cancer: the ADRIATIC study. Clin Lung Cancer 21:e84–e88
Kwon ED, Drake CG, Scher HI et al (2014) Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol 15:700–712
Chan HY, Choi J, Jackson C, Lim M (2021) Combination immunotherapy strategies for glioblastoma. J Neurooncol 151:375–391
Lan Y, Moustafa M, Knoll M et al (2021) Simultaneous targeting of TGF-beta/PD-L1 synergizes with radiotherapy by reprogramming the tumor microenvironment to overcome immune evasion. Cancer Cell 39:1388–1403 (e1310)
Andtbacka RH, Kaufman HL, Collichio F et al (2015) Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol 33:2780–2788
Puzanov I, Milhem MM, Minor D et al (2016) Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol 34:2619–2626
Ribas A, Dummer R, Puzanov I et al (2017) Oncolytic virotherapy promotes intratumoral T cell Infiltration and improves Anti-PD-1 immunotherapy. Cell 170:1109–1119 (e1110)
Long GV, Dummer R, Ribas A et al (2016) Efficacy analysis of MASTERKEY-265 phase 1b study of talimogene laherparepvec (T-VEC) and pembrolizumab (pembro) for unresectable stage IIIB-IV melanoma. J Clin Oncol 34:9568–9568
Harrington KJ, Kong A, Mach N et al (2020) Talimogene laherparepvec and pembrolizumab in recurrent or metastatic squamous cell carcinoma of the head and neck (MASTERKEY-232): a multicenter, phase 1b study. Clin Cancer Res 26:5153–5161
Ferris RL, Gross ND, Nemunaitis JJ et al (2014) Phase I trial of intratumoral therapy using HF10, an oncolytic HSV-1, demonstrates safety in HSV+/HSV- patients with refractory and superficial cancers. J Clin Oncol 32:6082–6082
Andtbacka RHI, Ross MI, Agarwala SS et al (2017) Final results of a phase II multicenter trial of HF10, a replication-competent HSV-1 oncolytic virus, and ipilimumab combination treatment in patients with stage IIIB-IV unresectable or metastatic melanoma. J Clin Oncol 35:9510–9510
Yokota K, Isei T, Uhara H et al (2019) Final results from phase II of combination with canerpaturev (formerly HF10), an oncolytic viral immunotherapy, and ipilimumab in unresectable or metastatic melanoma in second-or later line treatment. Ann Oncol 30:v533–v563
Kuryk L, Moller AW, Jaderberg M (2019) Combination of immunogenic oncolytic adenovirus ONCOS-102 with anti-PD-1 pembrolizumab exhibits synergistic antitumor effect in humanized A2058 melanoma huNOG mouse model. Oncoimmunology 8:e1532763
Shoushtari AN, Olszanski AJ, Nyakas M et al (2021) A pilot study of engineered adenovirus ONCOS-102 in combination with pembrolizumab (pembro) in checkpoint inhibitor refractory advanced or unresectable melanoma. Ann Oncol 32:S867–S905
Curti B, Richards J, Hallmeyer S et al (2017) Abstract CT114: The MITCI (Phase 1b) study: A novelimmunotherapy combination of intralesional Coxsackievirus A21 and systemic ipilimumab in advanced melanoma patients with or without previous immune checkpoint therapy treatment. Cancer Res 77:CT114
Silk AW, Kaufman H, Gabrail N et al (2017) Abstract CT026: Phase 1b study of intratumoral Coxsackievirus A21 (CVA21) and systemic pembrolizumab in advanced melanoma patients: Interim results of the CAPRA clinical trial. Cancer Res 77:CT026
Panagioti E, Kurokawa C, Viker K et al (2021) Immunostimulatory bacterial antigen-armed oncolytic measles virotherapy significantly increases the potency of anti-PD1 checkpoint therapy. J Clin Invest 131:e141614
Mender I, Zhang A, Ren Z et al (2020) Telomere Stress Potentiates STING-Dependent Anti-tumor Immunity. Cancer Cell 38:400–411 (e406)
Si W, Liang H, Bugno J et al (2021) Lactobacillus rhamnosus GG induces cGAS/STING- dependent type I interferon and improves response to immune checkpoint blockade. Gut 71:521–533
Acknowledgements
The figures are created with Biorender.com.
Funding
This work was supported by the National Natural Science Foundation of China (82003018 to D.C.; 81925030 to B.Z.).
Author information
Authors and Affiliations
Contributions
D.C and B.Z: conceived the manuscript. RY and DC: wrote the manuscript, prepared the figures and tables, and contributed to the discussion. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
All authors agreed to publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, R., Zhu, B. & Chen, D. Type I interferon-mediated tumor immunity and its role in immunotherapy. Cell. Mol. Life Sci. 79, 191 (2022). https://doi.org/10.1007/s00018-022-04219-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-022-04219-z