
Abstract
Astrocytes are the most abundant cell type in the brain. They were long considered only as passive support for neuronal cells. However, recent data have revealed many active roles for these cells both in maintenance of the normal physiological homeostasis in the brain as well as in neurodegeneration and disease. Moreover, human astrocytes have been found to be much more complex than their rodent counterparts, and to date, astrocytes are known to actively participate in a multitude of processes such as neurotransmitter uptake and recycling, gliotransmitter release, neuroenergetics, inflammation, modulation of synaptic activity, ionic balance, maintenance of the blood–brain barrier, and many other crucial functions of the brain. This review focuses on the role of astrocytes in human neurodegenerative disease and the potential of the novel stem cell-based platforms in modeling astrocytic functions in health and in disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Astrocytes are the most abundant non-neuronal cell type in the central nervous system (CNS). They serve numerous critical functions in the brain, including regulation of synapse formation and pruning, as well as regulation of neuroinflammation and lactate and glutamate levels in the brain [1]. Astrocytes play a key role also in several homeostatic and neuroprotective functions and are essential for the integrity of the blood–brain barrier (BBB). Primary astrocyte defects have been implicated to impact neurodegenerative diseases, and both a loss of their normal supportive function as well as a gain of toxic functions are thought to underlie the neuronal death in acute or chronic brain diseases [2, 3]. This review will address the main functions of astrocytes involved in neurodegeneration.
Astrocytes in synaptic defects
Synaptic dysfunction and degeneration are common features in many neurodegenerative diseases, such as Alzheimer’s disease (AD), and amyotrophic lateral sclerosis (ALS). Synaptic defects establish in the early stages of the diseases and synapse loss is often considered a primary pathological feature and not just secondary to the typical neuron loss in these diseases.
Astrocytes are part of the tripartite synapse
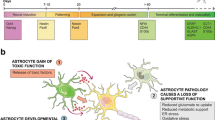
Astrocytes have critical roles in controlling synaptic development, functionality, plasticity, and elimination (reviewed in Ref. [4]). First of all, the presence of astrocytes is required for synapse formation, and glial depletion results in loss of synaptic connections followed by neurodegeneration. Astrocytes secrete synaptogenic factors, including thrombospondins, which regulate the number and formation of synapses [5, 6]. In addition to the secreted signaling molecules, a direct contact of neurons with astrocytes seems to be essential for synapse formation [6]. Second, astrocytes regulate the functionality of synapses in many ways. They contact and ensheath the pre- and post-synaptic endings of neurons in the human brain [7] and communicate with them to form the tripartite synapse [8] (Fig. 1a). Astrocytes fine-tune the strength of synaptic signals by regulating the pre-synaptic vesicle release and post-synaptic receptor composition through secreted molecules [4]. For example, astrocyte-secreted cholesterol has been shown to enhance pre-synaptic glutamatergic signaling [9]. Astrocytes also provide structural support and produce many cell adhesion and extracellular matrix-related molecules (e.g., neuroligins, cadherins, glypicans, and laminins) required for proper synapse formation and function (reviewed in Ref. [10]). In addition, astrocytes and astrocyte-secreted factors play a role in the different aspects of neuronal circuits and synaptic plasticity, including long-term potentiation, long-term depression, and synaptic scaling. Astrocytes are capable of regulating and clearing neurotransmitters in the synaptic cleft, but they also communicate by secreting the so-called gliotransmitters (e.g., GABA, glutamate, d-serine, and ATP) as a result of increased intracellular Ca2+ levels. These gliotransmitters act on various excitatory and inhibitory targets and are known to modulate synaptic transmission and plasticity (reviewed in Ref. [11]). The recent finding that transplantation of human astrocytes significantly improves the cognitive behavior of mice further supports the role of astrocytes in synaptic plasticity and cognition [12]. Finally, astrocytes have been shown to participate in synapse elimination and pruning, and thus refinement of neuronal circuits, through phagocytosis [13]. This process is regulated by two phagocytic receptor pathways, multiple EGF-like domains 10 (MEGF10) and c-mer proto-oncogene tyrosine kinase (MERTK) at least in mice. Taken together, astrocytes are constantly monitoring and regulating synapses in many ways through direct contact and various signaling molecules, and thus, they are likely to play an essential role in synaptic defects linked to neurodegenerative disorders.

a Astrocytes and the pre- and post-synaptic endings of neurons form the tripartite synapse. Astrocytes clear neurotransmitters and secrete gliotransmitters, e.g., GABA, glutamate, d-serine, and ATP [5, 6]. In addition, they provide structural support and produce many cell adhesion and extracellular matrix-related molecules required for proper synaptic function. In AD, APOE4 astrocytes have decreased rate of synapse pruning and turnover in the brain [18]. In AD expression of GABA, the inhibitory gliotransmitter, in reactive astrocytes, is increased, whereas reduced other gliotransmitter release, especially ATP [22,23,24]. b Blood–brain barrier. Astrocytic perivascular endfeet express water channel aquaporin-4 (AQP4) and the ATP-sensitive inward rectifier potassium channel Kir4.1, and transporter proteins such as glucose transporter-1 and P-glycoprotein [37]. Astrocytes support the BBB through the release of several growth factors including vascular endothelial growth factor (VEGF), glial cell line-derived neurotrophic factor (GDNF), basic fibroblast growth factor (bFGF), and angiopoietin 1 (ANG-1) [41]. Astrocytes communicate with each other through gap junction proteins mainly connexin (Cx) 43 and Cx 30. Astrocytic APOE4 promotes BBB disruption in AD. The release of pro-inflammatory cytokines including IL-6, IL-1B, and TNF-A in PD increase neuronal death and rearrange TJ protein expression on ECs. Astrocyte ability to maintain water and potassium homeostasis is reduced in ALS due to increased expression of AQP4 and reduced Kir4.1 [75]. c Astrocytes provide metabolic support to the neurons and maintain neurotransmitter homeostasis, actively responding on neuronal signals [86]. In neurodegenerative disease, these functions are compromised. Astrocytes exposed to amyloid peptides alter glucose uptake and its downstream metabolism in parallel with increased hydroperoxide and glutathione release. AD impacts astrocytic metabolism causing increased glutamate and ATP release, what in turn results in stimulation of microglial activation and neuronal vulnerability [101]. Astrocytic expression of PD-related A53T mutant alpha-synuclein results in down regulation of glutamate transporters and wide-spread gliosis [108]. In ALS, chronic activation of the α2-Na/K ATPase/α-adducin complex in astrocytes has been demonstrated, resulting in biased energy metabolism and acquisition of pro-inflammatory phenotype [131]. d Astrocytes mediate inflammatory effects in neurodegeneration. Astrocytic activity is affected by plethora of cytokines; INF-γ, IL-1β, IL-6, and TNF-α lead to classical activation, whereas IL-10, IL-4, and IL-13 induce alternative activation with decreased ROS and NO production alleviating inflammation [139]. In addition, astrocytes release factors affecting their inflammatory environment. Different cytokine levels are known to be affected in neurodegenerative disorders. In AD, astrocyte associated inflammation has been linked to IL-1β, IL-6, TNF-α, and TGF-β leading to activation of microglial cells [145]. Whereas, levels of IL-1β, TGF-β, and IFNs have been demonstrated to be elevated in post-mortem tissues of ALS patients [161]. Finally, in PD, levels of IL-1β, TNF-α, and IFN- γ are shown to be affected [153, 154]. e Astrocytes and oxidative stress in neurodegenerative diseases. In normal physiological conditions, astrocytes are glycolytic, whereas neurons are more oxidative and produce much more ROS than astrocytes do; however, increased ROS production by astrocytes has been reported in both AD and ALS [187, 196]. On the other hand, astrocytes have much higher antioxidant capacity than neurons and express higher levels of Nrf2, catalase and GSH, than neurons. Oxidative stress induces GSH release from astrocytes, which is degraded, and the cysteine-rich degradation products are taken up by neurons and used for neuronal GSH synthesis. [198,199,200]
Astrocytes are linked to synaptic defects in neurodegenerative diseases
The best evidence for synaptic defects in neurodegenerative diseases comes from AD studies. In AD, synapse dysfunction is one of the most striking early pathological features. There is a progressive loss of synapses during the disease course, and in fact, the extent of synapse loss is the best correlate with cognitive decline [14]. The exact mechanisms behind the synapse loss and synaptic defects still remain unclear, although several theories involving beta-amyloid (Aβ) plaques, complement cascade pathway, and microglia have been proposed [15] [16]. In addition, the importance of astrocytes in AD and synaptic defects is currently strongly emerging and this review will address some of the recent advances supporting it. For example, astrocyte-derived cholesterol has been shown to be a crucial factor in promoting synaptogenesis [9]. Apolipoprotein E (apoE), the major cholesterol carrier in the brain, is mainly synthesized and secreted by astrocytes. APOE polymorphisms are the main genetic determinants for AD with the APOE4 allele being associated with increased AD risk [17]. In mouse studies, APOE4 astrocytes showed decreased rate of synapse pruning and turnover in the brain, leading to the accumulation of senescent, non-functional synapses, when compared to APOE3 astrocytes [18]. This could be linked to the increased synaptic vulnerability in AD. Zhao and colleagues used human iPSC-derived neurons and astrocytes in a transwell-based co-culture system to study the effects of APOE4 astrocytes on control neurons [19]. They found that APOE4 astrocytes were less capable of promoting synapse formation and the expression of synaptic proteins was decreased in comparison with APOE3 astrocytes. Furthermore, APOE4 astrocytes reduced the survival of neurons and eventually led to neurodegeneration. These data support the importance of astrocytes and APOE genotype in synaptic deficiencies and neurodegeneration linked to AD. Another astrocyte-secreted synaptogenic molecule, thrombospondin-1 (TSP-1), has decreased expression level in the brain of AD patients [20]. On the other hand, treatment with TSP-1 was shown to reverse the Aβ-induced synaptic dysfunction in vivo.
Intriguingly, astrocytes were very recently found to engulf and phagocytose dystrophic neurons and pre-synaptic elements associated with amyloid plaques in APP/PS1 mutant AD mice as well as in post-mortem human brains [21]. This result supports the role of astrocytes in synaptic regulation and phagocytosis also in the human brain, although the exact meaning for disease pathogenesis remains to be elucidated. It is hypothesized that the astrocytic clearance of dystrophic neurites and dysfunctional synapses is inadequate in AD, resulting in their accumulation and impaired neural networks. In mice, the rate of synapse pruning decreases upon aging leading to the accumulation of senescent synapses, a possible explanation behind the cognitive decline [4, 13].
Reactive astrocytes in post-mortem AD patient’s brain show markedly increased expression of GABA, the inhibitory gliotransmitter. The enhanced GABA signaling has been linked with memory and learning deficits in mouse models of AD [22]. In addition, a specific type of reactive astrocytes shows diminished potential to promote synaptogenesis and synaptic function, leading to a decreased number of synapses with only weak signals and, finally, neurodegeneration [23]. This kind of reactive astrocytes seems to be abundant in post-mortem AD brain. In mouse primary hippocampal cultures, astrocytes internalized oligomeric tau, which was followed by reduced gliotransmitter release, especially ATP [24]. The reduced ATP release from astrocytes reduced the synaptic transmission of neurons and expression of both pre- and post-synaptic proteins in them.
In other neurodegenerative diseases, there is so far less evidence supporting the role of astrocytes in synaptic defects. In ALS, astrocytes have a well-established role in the degeneration of motor neurons and astrocytes with both sporadic (sALS) and familial (fALS) background seem to have similar effects when studied either in vitro or in vivo [25,26,27,28]. ALS is characterized by synaptic disconnections at the neuromuscular junction leading to axonal degeneration and, finally, death of neurons through either cell-autonomous or non-cell-autonomous mechanisms [29]. When transplanted into the mouse spinal cord, human sALS astrocytes have been shown to lead to reduced pre-synaptic connections and denervation at the neuromuscular junctions [28]. There is also a significant loss and degeneration of synapses in the frontal cortex in ALS, but this has not been clearly linked to astrocytes or astrogliosis in the post-mortem human brain [30].
In synucleinopathies such as Parkinson’ disease (PD) and dementia with Lewy bodies (DLB), a pre-synaptic protein α-synuclein aggregates to form insoluble fibrils. Recently, numerous small deposits of α-synuclein aggregates that are not associated with Lewy bodies were demonstrated at the presynapses in PD DLB cases. Moreover, dendritic spines showed retraction, whereas the presynapses were mostly preserved, suggesting a neurotransmitter deprivation [31]. As α-synuclein released from neuronal cells can be taken up by astrocytes and induce inflammatory response and reduced release of neurotrophic factors, astrocytes may regulate synaptic functions related to pre-synaptic deposition of α-synuclein (Table 1).
In Huntington’s disease patients, reduced glutamate uptake has been detected and this is linked to astrocytes, since glutamate is cleared from the perisynaptic area mostly by astrocytes [32]. In Down’s syndrome, there is reduced level of astrocyte-secreted TSP-1 leading to decreased synaptic density and spine malformations [33]. Taken together, these findings support the importance of astrocytes, astrocyte-secreted gliotransmitters, and other factors in synapse defects and neurodegeneration, although more human studies are needed for confirmation.
Astrocytes in the maintenance of the blood–brain barrier in neurodegenerative diseases
The blood–brain barrier (BBB) is a semipermeable membrane that separates the circulating blood from the CNS. It regulates delivery of oxygen and important nutrients to the brain through active and passive transport and prevents neurotoxins from entering the brain. It also has a clearance function and removes carbon dioxide and toxic metabolites from the CNS. A well-functioning BBB is essential for maintaining healthy brain tissue, and BBB breakdown or dysfunction is associated with a variety of neurological diseases due to the accumulation of blood-derived neurotoxins in the CNS that causes progressive neurodegeneration [34]. Although the most important components of the BBB are endothelial cells (ECs), astrocytes contribute to BBB stability through their direct contact of perivascular endfeet with ECs and pericytes (Fig. 1b).
Astrocytes through their endfeet establish the link between the endothelial cells and neurons and are important in the development and maintenance of BBB. Astrocytic perivascular endfeet surround ~ 98% of the basal parenchymal membrane of brain microvessels and express molecules which regulate ionic concentrations and protein transporters in the BBB [35, 36]. Among the most abundant ones are water channel aquaporin-4 (AQP4) and the ATP-sensitive inward rectifier potassium channel Kir4.1, as well as transporter proteins such as glucose transporter-1 and P-glycoprotein, indicating the importance of the endfeet polarization for proper functioning of BBB [37]. Interestingly, the location of AQP4 follows the distribution of inwardly rectifying potassium channel 4.1 (Kir4.1) [38], and together, they control water balance and ion homeostasis at the level of BBB and neuronal synapses [39, 40]. Furthermore, astrocytes support the BBB maintenance through the release of several growth factors, including vascular endothelial growth factor (VEGF), glial cell line-derived neurotrophic factor (GDNF), basic fibroblast growth factor (bFGF), and angiopoietin 1 (ANG-1) [41]. The upregulation of ANG-1 leads to enhanced barrier tightness through increased expression of junctional proteins, such as occludin and claudins and inhibition of leukocyte transmigration after TNFα stimulation. The identification of astrocyte-secreted factors suggests that mature astrocytes rather modulate and maintain the barrier function than induce its cerebrovascular integrity, because astrocytes initially appear at the neurovascular unit (NVU) postnatally [42]. Astrocytes regulate the blood flow according to demands of neuronal synaptic activity [43] by elevating Ca2+ levels in the endfeet [44]. Depending on released mediators such as prostaglandin E, nitric oxide, or arachidonic acid from astrocytes, blood vessels in brain constrict or dilate [45, 46]. Astrocytes also produce a cholesterol and phospholipid transporter molecule APOE, which mediates regulatory processes related to brain homeostasis [47]. Adult ApoE knockout mice show increased permeability selectively of cerebral vessels and leakage of serum proteins into the CNS tissue [48]. Whereas APOE3, the most abundant human APOE isoform, and APOE2 mediate physiological BBB tightness, APOE4 promotes BBB disruption, as observed in mutant mice in which mouse Apoe was replaced by human APOE isoforms [49]. Thus, homeostasis of the neurovascular junction is fundamental for cognitive functions, and homeostatic imbalances may be linked to cognitive changes.
Astrocytes in BBB dysfunction
Reactive astrogliosis is a common feature of astrocytes and compromises neuronal survival. This process involves both molecular and morphological changes in astrocytes and highly depends on the events triggering it. Therefore, it may have both beneficial and detrimental effects on surrounding cells [50]. Astrocytes communicate with each other through gap junction proteins, mainly connexin (Cx) 43 and Cx 30. Mice lacking Cx43 and Cx30 expression on astrocyte endfeet manifest a significant loss of AQP4 and β-dystroglycan, a transmembrane receptor anchoring astrocyte endfeet to the perivascular basal lamina. The absence of astroglial connexins weakens the BBB that opens upon increased hydrostatic vascular pressure and shear stress [51].
Alzheimer’s disease
Accumulating data support the importance of vascular astrocytes in AD pathology [52, 53]. Astrocyte endfeet surrounding vascular Aβ deposits showed morphological changes including retraction and swelling, and reduced expression of GLUT1 and lactate transporters in transgenic arctic β-amyloid (arcAβ) mice expressing human amyloid precursor protein (APP) [54]. These changes occur at early stages of the disease and are consistent with NVU uncoupling, suggesting that astrocyte dysfunction contributes to the early behavioral and cognitive impairments. AQP4 deficient mice are unable to clear soluble Aβ efficiently, proposing that this pathway may remove Aβ from the CNS [38]. In humans, the loss of perivascular AQP4 localization was associated with increased Aβ burden and increased AD pathology (Braak stage) [55]. Furthermore, primary human astrocytes obtained from non-demented human brain tissue upregulate the expression of neprilysin and scavenger receptor B1 as a response to Aβ–APOE complexes. This function appears to be defective in astrocytes derived from AD brain [56]. APOE4, which in CNS is mainly secreted by astrocytes, accelerates pericyte loss and microvascular reduction by enhancing pro-inflammatory pathway both in pericytes and ECs [57]. These findings suggest that Aβ clearance of astrocytes is impaired in AD due to astrocyte polarization induced by brain Aβ deposition [58, 59]. Nevertheless, the contribution of Aβ-induced changes to BBB is still unclear.
AD patients’ brains are characterized by high prevalence of degenerated endothelium and impairment of BBB transport system [34, 60]. BBB breakdown in the hippocampus precedes neurodegeneration [61, 62] and follow-up studies in AD patients have confirmed BBB leakage in both gray and white matter regions due to a loss of tight junction barrier function [61, 63, 64]. Moreover, cognitive decline was associated with stronger BBB leakage in patients with an early AD, suggesting that BBB damage might be part of a cascade of pathologic events that eventually leads to dementia [61]. In aging brain, altered AQP4 expression and its redistribution from astrocytic endfoot membranes to non-end-foot membranes compromise Aβ clearance through the glymphatic system and likely contribute to AD pathology [55].
Parkinson’s disease
Initially, it was assumed that the BBB stays unaltered during the development of PD, but several studies with human and animal models have linked BBB dysfunction with the development of PD pathology. In studies with PD patients, the increased permeability of the BBB was associated with decreased expression of P-glycoprotein demonstrated by increased uptake of [11C]-verapamil in the midbrain [65] and frontal white matter regions in PD patients [66]. Taking into account that P-glycoprotein is an active drug transporter, it is possible that the reduction of P-glycoprotein may be associated with accumulation of neurotoxins and aggregated proteins in the PD brain affecting directly dopaminergic neuron survival [67]. In addition to human studies, several toxin-induced animal models, including 6-OHDA treated rats and MPTP-treated mice, have shown a disruption of the BBB that is linked to the loss of dopaminergic neurons [68, 69]. It has also been shown that activation of microglia and astrocytes in PD leads to the release of pro-inflammatory cytokines, including IL-6, IL-1B, and TNFα, which can increase neuronal death and rearrange tight junction protein expression on ECs [70]. Moreover, the loss of signaling interaction between astrocytic perivascular endfeet and ECs leads to hypertrophy and upregulation of GFAP and vimentin in astrocytes triggering reactive astrogliosis and weakens BBB tightness [41].
Amyotrophic lateral sclerosis
In ALS, neurodegeneration of upper and lower motor neurons in the brain and spinal cord is not completely understood. The pathogenesis of the disease is complex with recent evidence showing BBB disturbances in the brain of fALS patients that carry SOD1 mutation and in post-mortem brains of sALS patients [71]. Primary degeneration of ECs and pericytes compromising vascular barrier integrity both in the brain and spinal cord of ALS patients has been described. The BBB disruption could be the main obstacle for efficient drug delivery to the CNS due to the impairment of P-glycoprotein efflux transporter in the brain and spinal cord microvessels [72, 73]. Therefore, restoration of BBB integrity could promote delivery of therapeutics to the CNS as well as help to remove waste products from the brain, providing an improved environment for motor neuron survival. The astrocyte contribution to BBB disruption in ALS has been studied only in rodents. In end-stage SOD1, rats increased expression of AQP4 in the spinal cord gray matter and around blood vessels promoted perivascular edema-linked BBB opening in ALS [74]. As elevated AQP4 was also detected near motor neurons, it is possible that dysfunctional astrocytes contribute to further motor neuron degeneration [75]. In parallel Kir4.1 expression was decreased in the brainstem and cortex of SOD1 rats. Thus, ability of astrocytes to properly maintain water and potassium homeostasis in CNS is reduced, not only affecting the BBB but also impeding motor neuron survival in ALS. In pre-symptomatic SOD1 mutant mice, BBB breakdown has been demonstrated prior to motor neuron degeneration and inflammatory changes in the spinal cord [76]. Whether connexin-mediated changes are directly affecting BBB function in ALS, is not known yet. However, although BBB damage might not be a causative event in ALS, it is still a significant, and possibly critical, pathologic effector for the disease progression.
In vitro models of the BBB
To better understand the role of human astrocytes in BBB functionality in neurodegenerative diseases and to develop new treatment strategies with predictive drug permeability, there is a demand for development of improved BBB models which include astrocytes and other components of human origin. Current in vitro BBB models mostly use brain microvessels and astrocytes of bovine, porcine, or rodent origin [77]. Even though the models are well established, the characteristics and functionality of the BBB in these species differ from that in humans especially in the expression level of important transporters such as P-glycoprotein [78,79,80,81]. The first human BBB models were generated from primary and immortalized human cells [82, 83]. However, the limited availability of primary human cells and lack of barrier tightness in immortalized cells underlines the need for more accessible and relevant cell source for BBB modeling. Recent advances in stem cell technology have created the opportunity to generate cells important for building human in vitro BBB [84, 85]. Stem cell sources have demonstrated a significant advantage over other cell types in respect of human origin, scalability, self-renewal, and potential to generate all BBB cell components from the same patient.
Metabolic dysfunction of astrocytes in neurodegenerative diseases
Astrocytes are actively participating in many metabolic functions of the CNS. Even though for the time being none of the human neurodegenerative diseases is known to originate from the astrocytic cell-autonomous dysfunction, astrocyte–neuronal metabolic coupling plays a central role in the physiological CNS homeostasis (Fig. 1c). The role of astrocytes spans far beyond the participation in a tripartite synapse and astrocytes have an impact on virtually every function classically attributed to neurons. Moreover, astrocytes provide metabolic and structural support to neuronal circuits. A recently discovered ability of astrocytes to regulate the respiratory rhythm-generating neuronal circuits through mechanism involving vesicular release indicates a key role of astrocytes in modulation of CNS activities [86].
The metabolic demand of CNS is very high, comprising approximately ten times over the average for all tissues. The major metabolic consumers here are the functions needed for maintenance of the membrane potential and ion transport across the membranes. On the other hand, the brain contains little substances for energy production and relays mainly on supplies from the circulation [87]. The main energy substrate for the CNS function is glucose, in parallel with ketone bodies which complement energy source upon fasting [88]. Astrocytes are thought to be the main glucose utilizing cells converting it to lactate and pyruvate and creating glycogen deposits. Astrocytes are also capable of ketogenesis, which besides providing an alternative source for energy metabolism may also protect against accumulation of non-esterified fatty acids and prevent formation of pro-apoptotic ceramide [89]. Providing anatomically a link between circulation and neuronal circuits, astrocytes serve as a major regulator of neuronal metabolism. All these activities are coupled and orchestrated with neuronal activity. Thus, astrocytes also support neurons by delivering substrates for energy metabolism, biosynthesis and neurotransmission, as well as take care of waste processing.
Even though neurons and astrocytes have an equal capacity to fully oxidize both glucose and lactate, they demonstrate physiological preferences resulting in astrocytes being mostly glycolytic in contrast to neurons that favor oxidative phosphorylation. There may be several reasons determining such metabolic specialization. First off, compared to astrocytes neurons have higher energy need, which can be met by oxidative metabolism. Second, neurons may favor glucose utilization through pentose phosphate pathway (PPP) to maintain sufficient NADPH levels and antioxidant potential. Upon these circumstances, neuronal lactate oxidation allows a support to sufficient mitochondrial ATP production without increasing glycolytic activity [90].
Astrocytes are indispensable for supporting both GABAergic and glutamatergic neurotransmission in brain. The available pool of these neurotransmitters depends on the glutamine synthesis in astrocytes [91]. In contrast to neurons, astrocytes express pyruvate carboxylase and are able to replenish their citric acid cycle intermediates carboxylating pyruvate to oxaloacetate, what is known as anaplerosis [88] [92].
The major waste produced by neuronal activity are carbon dioxide and ammonia. While carbon dioxide diffuses across BBB, ammonia is taken up by astrocytes and then released to neurons [92]. Ammonia enters astrocytes through potassium channels and transporters and being processed is released as glutamine [93].
In the adult brain, glycogen is deposited in astrocytes [94]. Glycogenolysis and the lactate produced in astrocytes play a critical role in long-term memory formation [95]. It has been demonstrated that pharmacological inhibition of either astrocytic glycogenolysis or monocarboxylate transporter responsible for neuronal lactate uptake impair memory and this impairment can be reversed by administration of lactate or glucose [96]. Thus, compromised astrocyte metabolic functions may result in neuronal dysfunction and lead to the development of neurodegeneration. Here, we will focus on the most relevant metabolic aspects of astrocytes having importance for neurodegenerative diseases.
Alzheimer’s disease
Astrocytic metabolic dysfunction has been suggested be a triggering factor for amyloid beta (Aβ) deposition [97]. While amyloid deposition has been mostly seen as a consequence of reduced Aβ clearance, increased production of amyloidogenic species may take place through metabolic regulation of beta and gamma secretase levels [98, 99], or aberrant post-translational modification of amyloid precursor protein APP [100]. It has been demonstrated that exposure of cultured astrocytes to amyloid peptides alters their glucose uptake and its downstream metabolism in parallel with increased hydroperoxide and glutathione release [101]. The pro-inflammatory environment typically present in AD brain may alter astrocytic metabolism causing increased glutamate and ATP release through connexin 43 hemichannels, which in turn could result in microglial activation and neuronal vulnerability [102, 103].
Parkinson’s disease and Huntington’s disease
The degenerating brain areas in PD, such as SN, contain relatively less astrocytes than other brain areas, making dopaminergic neurons potentially more vulnerable to the metabolic and energy disturbances [104]. In line with this assumption, it has been demonstrated that glucose deprivation promotes α-synuclein aggregation [105], whereas increased lactate supply inhibits it [106], providing a rationale for involvement of astrocytic metabolic dysfunction in PD pathogenesis. Moreover, astrocytes isolated from PD animal models expressing mutant α-synuclein or deficient for Parkin showed functional and morphological deficits of mitochondria, which was linked to astrocytes’ inability to support neuronal differentiation [107]. In addition, expression of PD-related A53T mutant α-synuclein in astrocytes was found to result in downregulation of glutamate transporters and wide-spread gliosis [108]. In the context of PD treatment, astrocytes have been shown to convert l-DOPA to dopamine [109], and in the striatum, astrocytes serve for l-DOPA storage [110]. It is, therefore, likely that metabolic and energy disturbances in astrocytes contribute to not only development of PD, but also in deteriorating treatment responses.
Several lines of research indicate astrocytic contribution also in HD. In a mouse model of HD, a deficit of astrocytic GABA release through GAT-3 has been thought to cause reduced tonic inhibition neuronal output in the striatum [111].
Furthermore, in HD mouse models astrocytes have shown increased Ca2+-dependent exocytotic glutamate release which correlates with elevated biosynthesis of glutamate due to upregulation of astrocytic pyruvate carboxylase [112].
Amyotrophic lateral sclerosis
Both non-cell-autonomous and cell-autonomous mechanisms are implicated in glial cell contribution to ALS [113,114,115], and astrocytes appear to be major determinants of ALS progression. Animal studies indicate a toxic gain of function in ALS astrocytes, which may converge into impaired supportive capacity of astrocytes to motoneurons [26, 116]. Subsequently, the initial astrocyte cell-autonomous dysfunction may lead to the non-cell-autonomous neuronal toxicity [117, 118].
Application of metabolomics, transcriptomics, and lipidomics on ALS models indicates significant dysregulation of a number of metabolic pathways in ALS astrocytes [119,120,121,122,123,124,125]. Metabolomics studies on models composed from motoneurons and astrocytes expressing mutant human SOD1 revealed disruption of the TCA cycle and glutamate metabolism under oxidative stress induced by menadione [126]. Furthermore, dysregulation of purine, pyrimidine, lysine, and glycerophospholipid metabolism pathways was demonstrated in ALS astrocytes co-cultured with motoneurons. In this model, glutamate exposure caused significant alterations in the astrocyte metabolic fingerprint and associated with decreased lactate levels by affecting lactate efflux from astrocytes [127]. Moreover, these findings correlate with the data from CSF analyses of human ALS patients [120]. Importantly, ALS in human patients has been associated with hypermetabolism, possibly involving aberrant astrocytic mitochondrial metabolism [128], which is present already at the pre-symptomatic state in SOD1 mutant mouse models and may thus have a key role of in early disease pathogenesis [129].
Analysis of ALS models expressing mutant SOD1 reveals prominent astrocytic alterations linked to metabolic dysregulation due to impairment of the astrocyte lactate transport and pro-nerve growth factor-p75 receptor signaling [129]. In addition, aberrant activation of metabotropic receptor 5 mGluR5 has been demonstrated in ALS astrocytes causing persistent elevation of intracellular calcium [130]. More recently, a specific chronic activation of the α2-Na/K ATPase/α-adducin complex in ALS astrocytes has been demonstrated, resulting in biased energy metabolism and acquisition of pro-inflammatory phenotype [131].
It has been demonstrated that astrocytes also potentially regulate neuronal proteostasis by affecting autophagic degradation pathways. The ability of conditioned medium from ALS-patient iPSC-derived astrocytes to modulate the autophagy pathway neuronal cells has been linked to the decreased expression of LC3-II and concomitant increase in the expression of SOD1 [132].
Overall, the current research demonstrates that astrocytic non-cell-autonomous and cell-autonomous mechanisms including metabolic dysfunction and related changes in gene expression play a central role in pathogenesis of neurodegenerative diseases, and identification of molecular targets in astrocytes can serve for designing therapy that could prevent or slow down the disease progression.
Astrocytes in the inflammation associated with neurodegeneration
Although the exact mechanisms of neurodegeneration are not known, recent data have identified the inflammatory processes tightly linked to multiple neurodegenerative pathways [133]. Regardless of the immune reaction within the CNS, it always involves astrocytes and microglia [134], the resident immune cells of the CNS. Under physiological conditions, astrocytes are key cells for the maintenance of homeostatic balance [135]. However, under pathological inflammatory state, they may produce neurotoxic factors that amplify the disease state, indicating the persistence of inflammatory stimuli or failure in normal resolution mechanisms [136]. This can result in the production of neurotoxic mediators, such as cytokines and interleukins [137], which are associated with several neurodegenerative diseases such as AD, PD, and ALS. The inflammation associated with these diseases is not typically the initiating factor of neurodegeneration. However, the emerging evidence on the sustained inflammatory response associated with the contribution of astrocytes in disease progression suggests contributory role for neuroinflammation in neuronal dysfunction and death.
Traditionally, increased GFAP expression has been considered as a hallmark of astrocyte activation in neurodegenerative disorders [138]. In addition, increased levels of pro-inflammatory cytokines such as INF-γ, IL-1β, IL-6, and TNFα are known to result in classical astrocyte activation, leading to increased activation of NF-κB pathway, production of ROS and NO, and further release of IL-1β, IL-6, and TNFα. On the contrary, augmented levels of IL-4 and IL-13 induce an alternative activation with increased secretion of IL-4 and decreased production of ROS and NO (Fig. 1d). However, astrocyte deactivation, including reduced immune surveillance and pro-inflammatory signaling, is mediated through high levels of IL-10 and TGF-β [139].
Alzheimer’s disease
Neuroinflammation is a major contributor to AD, driven by the activation of microglia and astrocytes. A recent study of CSF markers from several hundred individuals with mild cognitive impairment and patients with AD dementia revealed increased levels of YKL-40, ICAM-1, VCAM-1, IL-15, and Flt-1 during the pre-clinical, prodromal, and dementia stages of AD [140]. There was a correlation of these biomarkers with increased CSF levels of total tau, especially for YKL-40, a glycoprotein secreted by microglia and astrocytes. Interestingly, high astrocytic expression of IL-15 has also been shown to promote tissue damage in multiple sclerosis and following brain ischemia [141].
Aβ, the main pathological hallmark of AD, activates astrocytes to release chemokines and cytokines, and in an interaction with the increased levels of pro-inflammatory cytokines even increases the production of Aβ in neurons and astrocytes [142]. In fact, hypertrophic reactive astrocytes are strongly associated with Aβ pathology, as they accumulate around Aβ plaques, and are seen [143]. Especially, the pro-inflammatory A1 phenotype of astrocytes is seen in post-mortem tissue from AD patients. While Aβ has been demonstrated to induce a pro-inflammatory profile, and even astrogliosis in AD, it is not clear whether it is Aβ that induces this phenotype [23] [144].
In AD, neuroinflammation is characterized by the accumulation of cytokines such as IL-1β, IL-6, TNF-α, or TGF-β, which can, through activation of β- and γ-secretase activity, further contribute to cerebral amyloid deposition, augmentation of APP expression, Aβ formation, and finally to recruitment and activation of microglial cells [145].
Pro-inflammatory astrocyte activation appears to be linked to the release of synaptotoxic factors and loss of glutamate regulation. Dysregulation of Ca2+ homeostasis is thought to play the central role in these events. Due to the complexity of the Ca2+ balancing mechanisms, any malfunction in the regulatory checkpoints may contribute to the overall pathological alterations in neurodegenerative diseases. The Ca2+/calmodulin-dependent protein phosphatase, calcineurin has been suggested to provide a l link between Ca2+ dysregulation and astrocyte activation [146]. Calcineurin is highly expressed by subsets of activated astrocytes in both humans and animal models, resulting in the consecutive NFAT activation and contributing to the AD neuropathology [147].
Parkinson’s disease
One of the key aspects in PD pathophysiology is neuroinflammation, including presence of reactive astrocytes, in the substantia nigra pars compacta [148]. This is thought to be a downstream response to the death of dopaminergic neurons, even though there is also increasing amount of evidence, suggesting that astrocytes have an initiating role in PD pathophysiology [149].
Several genes implicated in PD pathology, including SNCA, DJ-1, PLA2G6, ATP13A2, LRRK2, PINK, and Parkin, have been demonstrated to play a role in the origination and regulation of astrocyte activation in response to inflammatory stimuli [149]. Both α-synuclein and DJ-1 have been shown to affect the astrocytic TLR4 signaling [150,151,152]. Furthermore, if looking at the secreted pro-inflammatory mediators, Parkin expression is known to be modified by IL-1β and TNF-α stimulation [153] and DJ-1 has been shown to regulate astrocyte activation by IFN-γ [154]. Finally, functional ATP13A2 expression prevents NLRP3 inflammasome activation [155].
Interestingly, a recent study demonstrated that post-mortem brain samples from PD patients display increased astrocytic senescence. Furthermore, paraquat, a known chemical to cause parkinsonism, induced astrocytic senescence and senescence-associated secretory phenotype with robust secretion of numerous inflammatory cytokines, chemokines, growth factors, and proteases both in in vitro and in in vivo settings [156]. Another recent report studying transplantation of neural progenitors in PD utilized co-transplantation of rat astrocytes and showed increased anti-inflammatory as well as decreased pro-inflammatory profile associated with behavioral improvement in the animals [235]. This further emphasizes the dual role of astrocytes in the neurodegeneration as astrocytes can either support the neurons or cause their degeneration.
Amyotrophic lateral sclerosis
ALS is considered to be a non-cell-autonomous disease with substantial involvement of astrocytes [157]. Post-mortem studies of ALS patients have revealed increased GFAP expression in the motor cortex, motor nuclei of the brain stem, the corticospinal tract, and the ventral horn of the spinal tract [158]. Furthermore, astrocytic production of pro-inflammatory mediators, such as type 1 interferons, TGF-β1, and prostaglandin D2, has been demonstrated to be upregulated in ALS-patient post-mortem tissues and in ALS models based on embryonic stem cell-derived human astrocytes [113, 159, 160]. In addition, the astrocytic NLRP3 inflammasome expression has been shown to be increased, leading to augmented IL-1β production in the post-mortem tissues of sALS patients [161]. Most recently, the advances in the patient neuroimaging have allowed direct visualization of neuroinflammation in ALS. Although in the post-mortem tissues from ALS patients, astrocytosis is seen in parts of the corticospinal tract other than pons [162], PET imaging with [11C]-DED, a commonly used marker of activated astrocytes, was able to demonstrate increased uptake rate in pons and white matter [163], which may reflect corticospinal tract astrocytosis. Finally, a strong astrocytic A1-phenotype has been discovered in the motor cortex of ALS patients, further highlighting the pro-inflammatory phenotype of astrocytes in the areas affected by ALS [23].
Huntington’s disease
Increased susceptibility to pro-inflammatory stimulation has been demonstrated in several mouse models of HD [164].
The enhanced inflammatory response in the brain was observed only in models with expression of mHTT including glial cells. The studies on primary astrocytes revealed increased IkB kinase activity, resulting in prolongation of NF-kB activation and upregulating pro-inflammatory factors, particularly TNF-α. Importantly, enhanced activation NF-kB-p65 was observed in astrocytes of patients and mice with HD [164]. Further attempts to block soluble TNF-α signaling using a dominant negative inhibitor XPro1595 caused decrease of TNF-α in the cortex and striatum, resulting in increased neuronal density and improved motor function. In addition, diminished amount of mutant HTT aggregates, and decreased gliosis was evident in brains of R6/2 mice [165].
Astrocytes and oxidative stress in neurodegenerative diseases
Brain metabolism uses vast amounts of oxygen, and thus also produces free oxygen radicals that may damage the cells. At the same time, brain has relatively low antioxidant defense mechanisms and high incidence of peroxidation substrates, such as enrichment of unsaturated fatty acids and redox active transition metals [166]. Thus, the brain is one of the most susceptible tissues for oxidative damage. Oxidative stress plays an important role in various neurodegenerative disorders, in which age is one of the main risk factors and cumulative oxidative stress is considered as one of the mechanisms driving the age-related changes [167].
The level of oxidative stress in tissues is dependent on the balance between two separate mechanisms: 1) production of ROS (reactive oxygen species) and 2) the ability of antioxidant defense mechanisms to scavenge ROS. Yet, ROS are not only deleterious, but also essential signaling molecules needed for many adaptive processes and during development [168, 169]. Oxidative stress in the brain is thus not only related to the level of oxidative damage but also to the intriguing redox signaling, which can, for example, regulate growth of neural progenitor cells [170] and control axonal pathfinding and regeneration [171].
The production of ROS is directly linked to the metabolic activity of the cells, and specifically, the mitochondrial respiratory activity as mitochondria is the main source of ROS [172, 173]. Apart from mitochondria, also peroxisomes produce ROS as byproducts of their metabolic processes such as fatty acid oxidation [174]. Mitochondrial ROS are produced as a consequence of normal respiratory chain (RC) activity. While the normal respiratory function leads to relatively low ROS generation, RC defects may increase ROS production significantly (reviewed in Ref. [116]).
As mitochondria are major producers of ROS, they are also well equipped with defense mechanisms for ROS scavenging (reviewed in Ref. [118]).
Oxidative stress in astrocytes
Astrocytes are glycolytic in their metabolism, to the extent that they survive without active respiratory chain [175]; thus, neurons have been considered as the major ROS producers in the brain. However, recent reports have shown that astrocytes can contribute to the redox status of the brain both directly by producing ROS and indirectly by affecting neuronal metabolism. Neurons rely on astrocytes for lactate supply and diminished lactate supply from astrocytes is likely to alter the oxidative microenvironment in the brain by increasing neuronal respiratory activity and reducing neurovascular coupling [176]. In addition, upon inflammation ROS is produced by both activated microglia and astrocytes to serve in the immune response and to trigger signaling cascades [177, 178]. Several antioxidant response pathway genes are regulated by the transcription factor Nrf2, which binds to the antioxidant response element (ARE) present in these genes. In normal physiological conditions, Keap1 targets Nrf2 for ubiquitin-mediated degradation; however, in oxidative conditions, Keap1 is not able to bind Nrf2 leading to accumulation of Nrf2 in the nucleus and induction of ARE-containing genes [179, 180]. This Nrf2–ARE pathway is particularly weak in neurons, whereas astrocytes both show strong response to Nrf2 activation and have higher expression of ARE-containing genes than neurons (Fig. 1e) [181]. Isolated neurons are known to possess very limited antioxidant defenses, including low catalase and GSH levels, both regulated by Nrf2, and Nrf2 deficiency does not affect their expression [182]. Astrocytes on the other hand are much more resistant to oxidative insults. They have high capacity for production and storage of GSH and oxidative stress increases GSH release from astrocytes. The GSH released from astrocytes is degraded and the cysteine-rich products are taken up by neurons to be used as building blocks for neuronal GSH synthesis in a process regulated by Nrf2 [183, 184]. Nrf2 deficiency also increases astrocyte vulnerability to oxidative stress [182].
Despite their low antioxidant defenses but high mitochondrial respiratory activity and consequently relatively high ROS production, post-mitotic neurons survive and remain functional for decades. This is thought to be achieved through antioxidant support from surrounding astrocytes [185].
Alzheimer’s disease
Aβ plays a key role in oxidative stress induction in AD and astrocytes seem to be involved in this. Aβ exposure in neuron–astrocyte co-cultures leads to a loss of mitochondrial membrane potential and increased ROS production in astrocytes but not in neurons [186]. The increase in ROS is due to the activation of astrocytic NADPH oxidase, as inhibition of it blocks the ROS production and protects neurons [186]. The increased ROS due to activated NADPH oxidase further activates PARP, which leads to decreased NAD levels and metabolic failure in astrocytes further leading to neuronal death [187]. These data from rodent models strongly suggest that astrocytes have a central role in oxidative stress induction in AD. Human data are still scarce; however, our recent report from PSEN1 mutant patient astrocytes showed that also human AD patient astrocytes manifest Aβ deposits and altered metabolism with increased ROS production and these result in reduced neuronal support [188].
Oxidative stress directly affects Ca2+-homeostasis [189], which is one of the hallmarks of astrocytes and impaired in AD. In astrocytes, increased ROS production together with Ca2+ leakage from ER leads to opening of the mitochondrial permeability transition pore and transient depolarization of mitochondria, resulting in loading of cellular Ca2+ from ER to mitochondria [190]. The transient events were not detrimental; however, they eventually lead to complete loss of membrane potential and necrotic cell death and further increase in ROS production [190].
Astrocytic activation of transcription factor Nrf2 results in upregulation of antioxidant responses spreading to surrounded neurons [191, 192]. It has been demonstrated that lentiviral Nrf2 gene transfer causes a robust reduction in astrocytic activation and induction of Nrf2 target gene heme oxygenase 1 [193]. Finally, activation of the Nrf2 pathway by small molecules has been shown to confer protection in several neurodegenerative disorders, including AD and PD [194], and studies using mice with astrocyte-specific overexpression of Nrf2 prove that this neuronal protection is mediated by astrocytes, further signaling the importance of astrocytes in oxidative stress and neurodegeneration [184, 195].
Amyotrophic lateral sclerosis
RC dysfunction in an SOD1 mutant rat model for ALS has been shown to lead to increased ROS production by astrocytes and motor neuron loss, which can be prevented by treating the astrocytes with antioxidants, suggesting a role for astrocytes in induction of oxidative stress also in ALS [196].
NF-κB transcription factors are central in inflammation and activated mainly by pro-inflammatory stimulation, which is present in many neurodegenerative diseases. However, also ROS has a strong role in regulating NF-κB signaling, and again, certain NF-κB-regulated genes are involved in regulating cellular ROS levels [197]. The most direct way for NF-κB to regulate cellular ROS levels is through activating antioxidant genes such as SOD2, SOD1, and thioredoxins [198,199,200]. However, as inflammation stimulates ROS production, NF-κB can activate also genes that lead to increased ROS. These include NADPH oxidase and inducible nitric oxide synthase (iNOS) [201, 202].
How ROS regulates NF-κB signaling in astrocytes is more complex. First of all, ROS has been reported to stimulate NF-κB in the cytoplasm, but inhibit it in the nucleus and many of the interactions of ROS and NF-κB are cell type and even astrocyte-specific [203]. Oxidation of cysteines is the most well characterized way that ROS affects NF-κB pathway. The initial reactions to sulfenic acid are usually reversible by antioxidants, but further oxidation leads to irreversible changes [203]. ROS can inhibit NF-κB DNA binding by direct oxidation of a cysteine of p50 [204]. The same cysteine can also be s-nitrosylated by NO produced by iNOS, which is activated by NF-κB, thus creating a negative feedback loop [205]. NF-κB activation may also further induce COX-2 and phospholipase A2 expression and promote inflammation [206].
Modelling astrocytic dysfunction in human iPSC platforms
The majority of the current human data from neurodegenerative diseases originates from post-mortem samples, representing thus only the end stage of the disease. Novel methodologies have enabled the generation of comprehensive iPSC cell libraries [207], which will in the future be an invaluable model resource for studying neurodegeneration. For example, iPSC-based studies have shown aberrant morphological changes in AD astrocytes [208], as well as APOE-related neurotrophic and synaptic disturbances [19]. Our recent results further confirm the central role of astrocytes in AD and synaptic defects. We studied the consequences of mutant PSEN1 ΔE9 in patient iPSC-derived astrocytes and how it could be linked to AD pathology [188]. Our PSEN1 ΔE9 astrocytes demonstrated increased Aβ production and oxidative stress while showing reduced lactate production. Furthermore, the PSEN1 ΔE9 astrocytes influenced the Ca2+ signaling in healthy neurons. Both the glutamate and GABA-induced Ca2+ responses were reduced in neurons cultured with AD astrocytes in comparison with when the neurons were cultured with isogenic control astrocytes, suggesting that synaptic abnormalities indeed occur early in the disease course via astrocyte dysfunction. In addition, we were able to demonstrate that PSEN1 ΔE9 mutant astrocytes have altered cytokine secretion after pro-inflammatory stimulation [188].
In addition, the novel stem cell-based BBB models are very promising [209,210,211]. They have already resulted in advances in HD studies, showing that hiPSC-derived microvascular ECs had intrinsic abnormalities in angiogenesis and barrier properties [212]. In another recent study, a human BBB model based on hiPSCs from a patient carrying a mutation in transporter MCT8 revealed a decreased transport of l-triiodothyronine affecting neuronal function [213]. Studies to model BBB in AD, stroke, and traumatic brain injury are being developed [214]. hiPSC-based BBB models hold great promises for disease modeling and drug development and could provide new insights into how vascular dysfunction contributes to neurodegenerative diseases.
New iPSC techniques have also been exploited in ALS studies. Studies on ALS astrocytes of SOD1 mice and sALS patients as well in human ALS iPSC-derived astrocytes revealed elevated levels of connexin 43, which were associated with increased intracellular Ca2+ levels that regulate motor neuron survival [215]. One of the key roles of astrocytes is to support motoneurons, and importantly, a recent study discovered a neuronal help-me signal, which seems to be disrupted in ALS-patient-derived iPSC astrocytes and to lead to impaired STAT3 activation [118]. In this study, cell-autonomous mechanisms were found to diminish EphB1-ephrin-B1-mediated reverse signaling and STAT3 activation in ALS astrocytes [118]. The deficit was observed to cause a failure in maintenance of neuroprotective status that is normally induced by neuronal signals upon injury in healthy controls. Moreover, the detailed analysis of ALS astrocytes revealed an increased expression of PHLDA3, a regulator of Akt phosphorylation previously linked with cell death and degeneration in response to oxidative stress [216]. Thus, astrocytes in ALS and potentially in other neurodegenerative diseases may display loss-of-function type changes that could be valuable in future development of more efficient therapies.
The iPSC-based models revealing a central role of astrocyte dysfunctions in pathogenesis of neurodegenerative diseases makes them attractive target for drug discovery. Due to complexity of pathophysiological mechanisms, pre-clinical models that adequately reflect this are indispensable for the development of novel pharmacological treatments [217]. Currently, a number of in vitro models clearly indicate a pivotal role of astrocytes modulating neuronal responses, including their ability to survive environmental stress.
An important aspect on the drug discovery is the safety issues, particularly neurotoxicity in humans [218]. The induction of seizures is among the main adverse effects causing a failure of clinical trials. In a study, where hiPSC- derived neurons were co-cultured with astrocytes, activation of pathways related to AMPA and NMDA receptors, neuronal polarity, and axon guidance was demonstrated [219]. These results indicate that hiPSC-derived astrocytes promote neuronal susceptibility to convulsant drugs and that co-culture of hiPSC-derived neurons with hiPSC-derived astrocytes allows for a pre-clinical risk assessment of drug-induced seizures.
Astrocytes differentiated from human ESCs and iPSC have been used to screen compound libraries aiming at identification of molecules that could counteract oxidative stress, a common denominator in neurodegeneration [220]. Here, in a study by Ishii et al., a total of 4100 FDA approved drugs and compounds were screened and 9 of the 22 hits were validated in astrocytes differentiated from human iPSC. The protective mechanism proposed for the validated hits was implicating activation of antioxidant response though induction of Nrf2.
Transplantation of glial progenitor cells and astrocytes
Animal models are used extensively to validate therapeutic targets, but confirmation of cellular and molecular mechanisms in human cells in vivo could increase the probability of successful target validation and drug development. Human glial chimeras may provide useful models for assessing the role of glia targets in human cognitive processing. The ability to construct the mice with glia generated from human ESCs and iPSCs allows the establishment of human glial chimeric mice with glia from defined disease genotypes and phenotypes. The capability promises to significantly advance our understanding of the contribution of glia to the pathogenesis of a wide variety of neurological disorders, many of which have simply been assumed to be entirely neuronal by origin.
Chimeric glial brains can be generated in vivo by a neonatal transplantation of human glial progenitors (A2B5+/PSA–NCAM cells obtained from 17- to 22-week-old human fetuses). Maturated recipient’s brains show large numbers and high proportions of both human glial progenitors and astrocytes, which retain specific hominid features despite of integration with the host astroglia and formation of gap junctions. The chimeric mice show increased performance in a number of behavioral tests such as maze navigation and fear conditioning [12], indicating the special features of human astrocytes and the value of such chimeric mouse models.
When human glial progenitor cells are transplanted into congenitally hypomyelinated shiverer mice, which have deficits in myelin generation and die by 18–21 weeks of age, the lethal phenotype is rescued and neurological performance improved [221]. The follow-up of 1 year after transplantation revealed massive expansion of human glia, outcompeting host cells [222].
Thus, the models based on disease-specific iPSC-derived human glial chimeras may provide an opportunity for better understanding of astrocytic dysfunction in a broad range of neurodegenerative and neuropsychiatric disorders [223].
A chimeric model comprising neonatal engraftment of human glial progenitor cells derived from mutant huntingtin transduced fetal hGPCs into immunodeficient mice has been established [224]. In these animals, striatal neurons are hyperexcitable and they display worse motor performance than controls. In contrast, striatal transplantation of normal glia into R6/2 HD mice counteracts the disease phenotype by restoration of interstitial potassium homeostasis, improving electrophysiological and behavioral test results, slowing down disease progression and extending the survival.
Recently, a human glia chimeric model was established by transplanting astroglia progenitors differentiated from iPSC lines of childhood-onset schizophrenic subjects [225]. These glial chimeras developed abnormal astrocytic morphology and hypomyelination compared to glial chimeras generated with cells derived from control iPSC lines. An impairment in the expression of differentiation-associated genes was evident and glial progenitors of schizophrenic subjects and corresponding chimeric mice demonstrated a broad range of behavioral and sleep abnormalities. Even though schizophrenia is not a neurodegenerative disease, this study shows further evidence for the importance of astrocytes in brain diseases.
The demonstrated non-cell-autonomous mechanisms in ALS provide a rationale for developing therapeutic approaches based on transplantation of glial progenitors. In a recent study human neural progenitor, cells engineered to produce GDNF were transplanted into the G93A SOD1 ALS rat cortex [226]. The transplanted cells maturated to astrocytes and released GDNF. The protection of motoneurons, delayed disease onset, and extended survival by transplanted GDNF producing progenitors were evident.
Spinal neural progenitor cells derived from healthy fALS and sALS iPSCs have been transplanted into cervical spinal cord of adult SCID mice [28]. Nine months later differentiated human astrocytes were found in large areas of the spinal cord, replacing host astrocytes and forming contacts with neurons. Importantly, both fALS and sALS cell transplantations caused reduction in motoneuron numbers and remaining motoneurons showed reduced inputs from inhibitory neurons and exhibited disorganized neurofilaments and aggregated ubiquitin. In addition, the cellular pathology in the spinal cord was accompanied with motor deficits. These results indicate that both fALS and sALS iPSC-derived astrocytes are capable to induce ALS-like pathological phenotype [227]. Importantly, transplantation into wt and ALS spinal cord showed no gross differences in the engraftment or gene expression in the differentiated human astrocytes, suggesting that the phenotype of transplanted glial progenitors is independent from the neurodegenerative environment supporting thus clinical transplantations into ALS patients [228].
Furthermore, HD patient-specific iPSCs were differentiated into neural progenitor cells and functional neurons in vitro [229]. When transplanted in vivo, they gave rise to neurons in the adult mouse brain. In parallel, astrocytes derived from HD iPSCs showed increased cytoplasmic vacuolation. Altogether, these results support feasibility to recapitulate HD phenotype under basal conditions what has been demonstrated in primary cells from HD patients.
In studies, where transplantation of glial-rich neural progenitors derived from human iPSCs was found to be beneficial, the potential therapeutic mechanism was assumed take place via activation of AKT signaling [230]. In one study, where primary glial-restricted precursor cells were transplanted focally into cervical spinal cord of a transgenic ALS rat, concomitant reduction of gliosis, extended survival, attenuated motor neuron loss and improved motor functions was observed to be partially mediated by astrocyte glutamate transporter GLT1 [231].
Human fetal-derived neural progenitor cells have been shown to survive well and mainly differentiate into astrocytes when transplanted into aging rat spinal cord [232]. Interestingly, differentiation into astrocytes was considerably more efficient in aged rats (500 and 600 days) than when using younger animals (100 and 200 days), possibly because aged environment may have more factors that stimulate astrocyte maturation compared to young adults. The differentiated astrocytes of the transplantation prevented the motoneuron loss that is associated with aging, even though improved motor performance was not evident.
One approach to obtain healthy, potentially therapeutic astrocytes into the CNS is injection of human mesenchymal stem cells (MSCs) into the cerebrospinal fluid. When symptomatic transgenic ALS rats received such injection, MSCs infiltrated into the nervous parenchyma and migrated substantially into the ventral gray matter [233]. Importantly, the injected MSCs efficiently differentiated into astrocytes, reduced motoneuron loss, and prolonged survival. The beneficial effect of human MSC-derived astrocytes was possibly mediated by reduced inflammation, since the neuroprotection correlated with the inhibition of microglial proliferation and reduction of COX-2 and NOX-2 expressions.
Transplantation of precursor cell-derived astrocytes has also been studies in a rat model of PD. The astrocytes were generated in vitro by directed differentiation of glial precursors. The transplantation into striatum of the 6-hydroxydopamine lesioned rat model restored HT expression rescued GABAergic interneurons and improved behavioral recovery [234]. It is thought that transplanted cells produced beneficial trophic factors and antioxidants. Further studies have explored cografting astrocytes, derived from the midbrain and demonstrated that they remarkably enhanced neural progenitor cell differentiation into maturated DA neurons and promoted DA neuron engraftment in PD rats for at least 6 months after transplantation [235].
A number of studies have investigated astrocyte transplantation in AD models. When astrocytes isolated from adult and neonatal mice were transplanted into the hippocampi of AD model APdE9 mice most of them were found near Aβ deposits and internalized human Aβ immunoreactive material [236]. In the follow-up study, a significant reduction in Aβ burden was observed after 2 months in parallel colocalization with transplanted astrocytes, and the remaining Aβ deposits showed fragmentation [237]. The mechanism demonstrated was associated with astrocytic expression of Aβ degrading proteases such as neprilysin (NEP), angiotensin-converting enzyme-1 (ACE-1), and endothelin-converting enzyme-2 (ECE-2).
Another approach by autologous transplantation of glia cells isolated from intestine—enteric glial cells (EGCs) has demonstrated an improvement in neuropathology and restoration of cognitive deficits in of a rat model of Aβ-induced neurodegeneration [238].
Conclusions
Astrocytes have been shown to take active part in nearly all CNS functions and participate both in initiation and progression of neurodegenerative disease. As human astrocytes are much more complex than their rodent counterparts, human models are needed to be able to better understand the role of astrocytes in human neurological disease. Moreover, there is strong evidence, indicating that activation of microglia precedes the pro-inflammatory activation of astrocytes in rodents. Considering that inflammation is involved in aging and most of the brain diseases, models including both human astrocytes and human microglia that interact with human neurons are going to further reveal the multiple functions of human astrocytes. Thus, the novel human stem cell-based models are indispensable to discover pathological mechanism and for development of therapeutic strategies.
Abbreviations
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- APP:
-
Amyloid precursor protein
- ARE:
-
Antioxidant response element
- BBB:
-
Blood–brain barrier
- CNS:
-
Central nervous system
- EC:
-
Endothelial cells
- ES:
-
Embryonic stem
- GDNF:
-
Glial cell line-derived neurotrophic factor
- MERTK:
-
Mer proto-oncogene tyrosine kinase
- MS:
-
Multiple sclerosis
- PD:
-
Parkinson’s disease
- PET:
-
Positron emission topography
- PPP:
-
Pentose phosphate pathway
- RC:
-
Respiratory chain
- RET:
-
Reverse electron transport
- ROS:
-
Reactive oxygen species
- VEGF:
-
Vascular endothelial growth factor
References
Oberheim NA, Takano T, Han X et al (2009) Uniquely hominid features of adult human astrocytes. J Neurosci 29:3276–3287. https://doi.org/10.1523/JNEUROSCI.4707-08.2009
Phatnani H, Maniatis T (2015) Astrocytes in neurodegenerative disease: table 1. Cold Spring Harb Perspect Biol 7:a020628. https://doi.org/10.1101/cshperspect.a020628
Karkkainen V, Magga J, Koistinaho J, Malm T (2012) Brain environment and Alzheimer’s disease mutations affect the survival, migration and differentiation of neural progenitor cells. Curr Alzheimer Res 9:1030–1042. https://doi.org/10.2174/156720512803569028
Chung WS, Allen NJ, Eroglu C (2015) Astrocytes control synapse formation, function, and elimination. Cold Spring Harb Perspect Biol 7:a020370
Christopherson KS, Ullian EM, Stokes CCA et al (2005) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120:421–433. https://doi.org/10.1016/j.cell.2004.12.020
Barker AJ, Koch SM, Reed J et al (2008) Developmental control of synaptic receptivity. J Neurosci 28:8150–8160. https://doi.org/10.1523/JNEUROSCI.1744-08.2008
Witcher MR, Park YD, Lee MR et al (2010) Three-dimensional relationships between perisynaptic astroglia and human hippocampal synapses. Glia 58:572–587
Araque A, Parpura V, Sanzgiri RP, Haydon PG (1999) Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22:208–215
Mauch DH, Nagler K, Schumacher S et al (2001) CNS synaptogenesis promoted by glia-derived cholesterol. Science 294:1354–1357
Hillen AEJ, Burbach JPH, Hol EM (2018) Cell adhesion and matricellular support by astrocytes of the tripartite synapse. Prog Neurobiol 82:30047–30048
Araque A, Carmignoto G, Haydon PG et al (2014) Gliotransmitters travel in time and space. Neuron 81:728–739. https://doi.org/10.1016/j.neuron.2014.02.007
Han X, Chen M, Wang F et al (2013) Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell. https://doi.org/10.1016/j.stem.2012.12.015
Chung WS, Clarke LE, Wang GX et al (2013) Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504:394–400
Terry RD, Masliah E, Salmon DP et al (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580
Hong S, Beja-Glasser VF, Nfonoyim BM et al (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–716. https://doi.org/10.1126/science.aad8373
Forner S, Baglietto-Vargas D, Martini AC et al (2017) Synaptic impairment in Alzheimer’s disease: a dysregulated symphony. Trends Neurosci 40:347–357. https://doi.org/10.1016/j.tins.2017.04.002
Liu C-C, Kanekiyo T, Xu H et al (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9:106–118. https://doi.org/10.1038/nrneurol.2012.263
Chung WS, Verghese PB, Chakraborty C et al (2016) Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc Natl Acad Sci USA 113:10186–10191
Zhao J, Davis MD, Martens YA et al (2017) APOE ε4/ε4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum Mol Genet 26:2690–2700. https://doi.org/10.1093/hmg/ddx155
Son SM, Nam DW, Cha M-Y et al (2015) Thrombospondin-1 prevents amyloid beta-mediated synaptic pathology in Alzheimer’s disease. Neurobiol Aging 36:3214–3227. https://doi.org/10.1016/j.neurobiolaging.2015.09.005
Gomez-Arboledas A, Davila JC, Sanchez-Mejias E et al (2018) Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 66:637–653
Jo S, Yarishkin O, Hwang YJ et al (2014) GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med 20:886–896
Liddelow SA, Guttenplan KA, Clarke LE et al (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. https://doi.org/10.1038/nature21029
Piacentini R, Puma DDL et al (2017) Reduced gliotransmitter release from astrocytes mediates tau-induced synaptic dysfunction in cultured hippocampal neurons. Glia 65:1302–1316
Haidet-Phillips AM, Hester ME, Miranda CJ et al (2011) Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 29:824–828. https://doi.org/10.1038/nbt.1957
Hall CE, Yao Z, Choi M et al (2017) Progressive motor neuron pathology and the role of astrocytes in a human stem cell model of VCP-related ALS. Cell Rep 19:1739–1749. https://doi.org/10.1016/j.celrep.2017.05.024
Meyer K, Ferraiuolo L, Miranda CJ et al (2014) Direct Convers patient fibroblasts Demonstr non-cell Auton Toxic astrocytes to Mot neurons Fam sporadic ALS. Proc Natl Acad Sci USA 111:829–832
Qian K, Huang H, Peterson A et al (2017) Sporadic ALS astrocytes induce neuronal degeneration in vivo. Stem Cell Reports 8:843–855. https://doi.org/10.1016/J.STEMCR.2017.03.003
Arbour D, Vande Velde C, Robitaille R (2017) New perspectives on amyotrophic lateral sclerosis: the role of glial cells at the neuromuscular junction. J Physiol 595:647–661. https://doi.org/10.1113/JP270213
Henstridge CM, Sideris DI, Carroll E et al (2018) Synapse loss in the prefrontal cortex is associated with cognitive decline in amyotrophic lateral sclerosis. Acta Neuropathol 135:213–226
Schulz-Schaeffer WJ (2010) The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol 120:131–143. https://doi.org/10.1007/s00401-010-0711-0
Hassel B, Tessler S, Faull RLM, Emson PC (2008) Glutamate uptake is reduced in prefrontal cortex in Huntington’s disease. Neurochem Res 33:232–237. https://doi.org/10.1007/s11064-007-9463-1
Garcia O, Torres M, Helguera P et al (2010) A role for thrombospondin-1 deficits in astrocyte-mediated spine and synaptic pathology in Down’s syndrome. PLoS One 5:e14200. https://doi.org/10.1371/journal.pone.0014200
Zlokovic BV (2008) The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron 57:178–201. https://doi.org/10.1016/j.neuron.2008.01.003
Haseloff RF, Blasig IE, Bauer HC, Bauer H (2005) In search of the astrocytic factor(s) modulating blood–brain barrier functions in brain capillary endothelial cells in vitro. Cell Mol Neurobiol 25:25–39
Obermeier B, Daneman R, Ransohoff RM (2013) Development, maintenance and disruption of the blood–brain barrier. Nat Med 19:1584–1596. https://doi.org/10.1038/nm.3407
Abbott NJ, Rönnbäck L, Hansson E (2006) Astrocyte-endothelial interactions at the blood–brain barrier. Nat Rev Neurosci 7:41–53. https://doi.org/10.1038/nrn1824
Iliff JJ, Wang M, Liao Y et al (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med 4:147ra111. https://doi.org/10.1126/scitranslmed.3003748
Nielsen S, Nagelhus EA, Amiry-Moghaddam M et al (1997) Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci Off J Soc Neurosci 17:171–180
Newman EA, Frambach DA, Odette LL (1984) Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science 225:1174–1175
Alvarez JI, Katayama T, Prat A (2013) Glial influence on the blood brain barrier. Glia 61:1939–1958. https://doi.org/10.1002/glia.22575
Daneman R, Zhou L, Kebede AA, Barres BA (2010) Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 468:562–566. https://doi.org/10.1038/nature09513
Koehler RC, Gebremedhin D, Harder DR (2006) Role of astrocytes in cerebrovascular regulation. J Appl Physiol (Bethesda, Md 1985) 100:307–317. https://doi.org/10.1152/japplphysiol.00938.2005
Bazargani N, Attwell D (2016) Astrocyte calcium signaling: the third wave. Nat Neurosci 19:182–189. https://doi.org/10.1038/nn.4201
Metea MR, Newman EA (2006) Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J Neurosci Off J Soc Neurosci 26:2862–2870. https://doi.org/10.1523/JNEUROSCI.4048-05.2006
Zonta M, Angulo MC, Gobbo S et al (2003) Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci 6:43–50. https://doi.org/10.1038/nn980
Gee JR, Keller JN (2005) Astrocytes: regulation of brain homeostasis via apolipoprotein E. Int J Biochem Cell Biol 37:1145–1150. https://doi.org/10.1016/j.biocel.2004.10.004
Hafezi-Moghadam A, Thomas KL, Wagner DD (2007) ApoE deficiency leads to a progressive age-dependent blood–brain barrier leakage. Am J Physiol Cell Physiol 292:C1256–C1262. https://doi.org/10.1152/ajpcell.00563.2005
Bell RD, Winkler EA, Singh I et al (2012) Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485:512–516. https://doi.org/10.1038/nature11087
Hamby ME, Sofroniew MV (2010) Reactive astrocytes as therapeutic targets for CNS disorders. Neurother J Am Soc Exp Neurother 7:494–506. https://doi.org/10.1016/j.nurt.2010.07.003
Ezan P, André P, Cisternino S et al (2012) Deletion of astroglial connexins weakens the blood–brain barrier. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 32:1457–1467. https://doi.org/10.1038/jcbfm.2012.45
Birch AM (2014) The contribution of astrocytes to Alzheimer’s disease. Biochem Soc Trans 42:1316–1320. https://doi.org/10.1042/BST20140171
Vincent AJ, Gasperini R, Foa L, Small DH (2010) Astrocytes in Alzheimer’s disease: emerging roles in calcium dysregulation and synaptic plasticity. J Alzheimer’s Dis JAD 22:699–714. https://doi.org/10.3233/JAD-2010-101089
Merlini M, Meyer EP, Ulmann-Schuler A, Nitsch RM (2011) Vascular β-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAβ mice. Acta Neuropathol 122:293–311. https://doi.org/10.1007/s00401-011-0834-y
Zeppenfeld DM, Simon M, Haswell JD et al (2017) Association of perivascular localization of aquaporin-4 with cognition and alzheimer disease in aging brains. JAMA Neurol 74:91–99. https://doi.org/10.1001/jamaneurol.2016.4370
Mulder SD, Veerhuis R, Blankenstein MA, Nielsen HM (2012) The effect of amyloid associated proteins on the expression of genes involved in amyloid-β clearance by adult human astrocytes. Exp Neurol 233:373–379. https://doi.org/10.1016/j.expneurol.2011.11.001
Halliday MR, Rege SV, Ma Q et al (2016) Accelerated pericyte degeneration and blood–brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab 36:216–227. https://doi.org/10.1038/jcbfm.2015.44
Yang J, Lunde LK, Nuntagij P et al (2011) Loss of astrocyte polarization in the tg-ArcSwe mouse model of Alzheimer’s disease. J Alzheimer’s Dis JAD 27:711–722. https://doi.org/10.3233/JAD-2011-110725
Yang W, Wu Q, Yuan C et al (2012) Aquaporin-4 mediates astrocyte response to β-amyloid. Mol Cell Neurosci 49:406–414. https://doi.org/10.1016/j.mcn.2012.02.002
Weiss N, Miller F, Cazaubon S, Couraud P-O (2009) The blood–brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta Biomembr 1788:842–857. https://doi.org/10.1016/j.bbamem.2008.10.022
van de Haar HJ, Burgmans S, Jansen JFA et al (2016) Blood–brain barrier leakage in patients with early Alzheimer disease. Radiology 281:527–535. https://doi.org/10.1148/radiol.2016152244
Montagne A, Barnes SR, Sweeney MD et al (2015) Blood–brain barrier breakdown in the aging human hippocampus. Neuron 85:296–302. https://doi.org/10.1016/j.neuron.2014.12.032
Montagne A, Nation DA, Pa J et al (2016) Brain imaging of neurovascular dysfunction in Alzheimer’s disease. Acta Neuropathol 131:687–707. https://doi.org/10.1007/s00401-016-1570-0
van de Haar HJ, Jansen JFA, van Osch MJP et al (2016) Neurovascular unit impairment in early Alzheimer’s disease measured with magnetic resonance imaging. Neurobiol Aging 45:190–196. https://doi.org/10.1016/j.neurobiolaging.2016.06.006
Kortekaas R, Leenders KL, van Oostrom JCH et al (2005) Blood–brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol 57:176–179. https://doi.org/10.1002/ana.20369
Bartels AL, Willemsen ATM, Kortekaas R et al (2008) Decreased blood–brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J Neural Transm (Vienna, Austria 1996) 115:1001–1009. https://doi.org/10.1007/s00702-008-0030-y
Bartels AL (2011) Blood–brain barrier P-glycoprotein function in neurodegenerative disease. Curr Pharm Des 17:2771–2777
Carvey PM, Zhao CH, Hendey B et al (2005) 6-Hydroxydopamine-induced alterations in blood–brain barrier permeability. Eur J Neurosci 22:1158–1168. https://doi.org/10.1111/j.1460-9568.2005.04281.x
Chen X, Lan X, Roche I et al (2008) Caffeine protects against MPTP-induced blood–brain barrier dysfunction in mouse striatum. J Neurochem 107:1147–1157. https://doi.org/10.1111/j.1471-4159.2008.05697.x
Wong D, Dorovini-Zis K, Vincent SR (2004) Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood–brain barrier. Exp Neurol 190:446–455. https://doi.org/10.1016/j.expneurol.2004.08.008
Garbuzova-Davis S, Sanberg PR (2014) Blood-CNS barrier impairment in ALS patients versus an animal model. Front Cell Neurosci. https://doi.org/10.3389/fncel.2014.00021
Milane A, Fernandez C, Dupuis L et al (2010) P-glycoprotein expression and function are increased in an animal model of amyotrophic lateral sclerosis. Neurosci Lett 472:166–170. https://doi.org/10.1016/j.neulet.2010.01.078
Jablonski MR, Jacob DA, Campos C et al (2012) Selective increase of two ABC drug efflux transporters at the blood-spinal cord barrier suggests induced pharmacoresistance in ALS. Neurobiol Dis 47:194–200. https://doi.org/10.1016/j.nbd.2012.03.040
Nicaise C, Mitrecic D, Demetter P et al (2009) Impaired blood–brain and blood-spinal cord barriers in mutant SOD1-linked ALS rat. Brain Res 1301:152–162. https://doi.org/10.1016/j.brainres.2009.09.018
Nicaise C, Soyfoo MS, Delporte C, Pochet R (2010) Aquaporin-4 as a potential marker of BBB disruption in ALS models. Amyotroph Lateral Scler 11:253–254. https://doi.org/10.3109/17482960902803457
Zhong Z, Deane R, Ali Z et al (2008) ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci 11:420–422. https://doi.org/10.1038/nn2073
Deli MA, Abrahám CS, Kataoka Y, Niwa M (2005) Permeability studies on in vitro blood–brain barrier models: physiology, pathology, and pharmacology. Cell Mol Neurobiol 25:59–127
Aday S, Cecchelli R, Hallier-Vanuxeem D et al (2016) Stem cell-based human blood–brain barrier models for drug discovery and delivery. Trends Biotechnol 34:382–393. https://doi.org/10.1016/j.tibtech.2016.01.001
Hoshi Y, Uchida Y, Tachikawa M et al (2013) Quantitative atlas of blood–brain barrier transporters, receptors, and tight junction proteins in rats and common marmoset. J Pharm Sci 102:3343–3355. https://doi.org/10.1002/jps.23575
Warren MS, Zerangue N, Woodford K et al (2009) Comparative gene expression profiles of ABC transporters in brain microvessel endothelial cells and brain in five species including human. Pharmacol Res 59:404–413. https://doi.org/10.1016/j.phrs.2009.02.007
Syvänen S, Lindhe O, Palner M et al (2009) Species differences in blood–brain barrier transport of three positron emission tomography radioligands with emphasis on P-glycoprotein transport. Drug Metab Dispos 37:635–643. https://doi.org/10.1124/dmd.108.024745
Weksler BB, Subileau EA, Perrière N et al (2005) Blood–brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J 19:1872–1874. https://doi.org/10.1096/fj.04-3458fje
Sano Y, Shimizu F, Abe M et al (2010) Establishment of a new conditionally immortalized human brain microvascular endothelial cell line retaining an in vivo blood–brain barrier function. J Cell Physiol 225:519–528. https://doi.org/10.1002/jcp.22232
Lippmann ES, Azarin SM, Kay JE et al (2012) Derivation of blood–brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol 30:783–791. https://doi.org/10.1038/nbt.2247
Lippmann ES, Al-Ahmad A, Palecek SP, Shusta EV (2013) Modeling the blood–brain barrier using stem cell sources. Fluids Barriers CNS 10:2. https://doi.org/10.1186/2045-8118-10-2
Sheikhbahaei S, Turovsky EA, Hosford PS et al (2018) Astrocytes modulate brainstem respiratory rhythm-generating circuits and determine exercise capacity. Nat Commun 9:370. https://doi.org/10.1038/s41467-017-02723-6
Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21:1133–1145. https://doi.org/10.1097/00004647-200110000-00001
Magistretti PJ, Allaman I (2016) Brain energy and metabolism. In: Pfaff D, Volkow N (eds) Neuroscience in the 21st century. Springer, New York, pp 1879–1909
Guzmán M, Blázquez C (2004) Ketone body synthesis in the brain: possible neuroprotective effects. Prostaglandins Leukot Essent Fat Acids 70:287–292. https://doi.org/10.1016/j.plefa.2003.05.001
Bolaños JP, Almeida A, Moncada S (2010) Glycolysis: a bioenergetic or a survival pathway? Trends Biochem Sci 35:145–149. https://doi.org/10.1016/j.tibs.2009.10.006
Schousboe A, Scafidi S, Bak LK, et al (2014) Glutamate metabolism in the brain focusing on astrocytes. Adv Neurobiol 11:13–30
Weber B, Barros LF (2015) The astrocyte: powerhouse and recycling center. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a020396
Kelly T, Rose CR (2010) Ammonium influx pathways into astrocytes and neurones of hippocampal slices. J Neurochem 115:1123–1136. https://doi.org/10.1111/j.1471-4159.2010.07009.x
Brown AM, Ransom BR (2007) Astrocyte glycogen and brain energy metabolism. Glia 55:1263–1271. https://doi.org/10.1002/glia.20557
Alberini CM, Cruz E, Descalzi G et al (2017) Astrocyte glycogen and lactate: new insights into learning and memory mechanisms. Glia. https://doi.org/10.1002/glia.23250
Newman LA, Korol DL, Gold PE (2011) Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One 6:e28427. https://doi.org/10.1371/journal.pone.0028427
Yan L-J, Xiao M, Chen R, Cai Z (2013) Metabolic dysfunction of astrocyte: an initiating factor in beta-amyloid pathology? Aging Neurodegener 1:7–14
Nadler Y, Alexandrovich A, Grigoriadis N et al (2008) Increased expression of the γ-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia 56:552–567. https://doi.org/10.1002/glia.20638
Zhang J, Zhou W, Qiao H (2011) Bioenergetic homeostasis decides neuroprotection or neurotoxicity induced by volatile anesthetics: a uniform mechanism of dual effects. Med Hypotheses 77:223–229. https://doi.org/10.1016/j.mehy.2011.04.018
Perdivara I, Petrovich R, Allinquant B et al (2009) Elucidation of O-glycosylation structures of the beta-amyloid precursor protein by liquid chromatography-mass spectrometry using electron transfer dissociation and collision induced dissociation. J Proteome Res 8:631–642. https://doi.org/10.1021/pr800758g
Allaman I, Gavillet M, Bélanger M et al (2010) Amyloid–beta aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J Neurosci 30:3326–3338. https://doi.org/10.1523/JNEUROSCI.5098-09.2010
Orellana JA, Shoji KF, Abudara V et al (2011) Amyloid -induced death in neurons involves glial and neuronal hemichannels. J Neurosci 31:4962–4977. https://doi.org/10.1523/JNEUROSCI.6417-10.2011
Orellana JA, Froger N, Ezan P et al (2011) ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J Neurochem 118:826–840. https://doi.org/10.1111/j.1471-4159.2011.07210.x
Mena MA, García de Yébenes J (2008) Glial Cells as Players in Parkinsonism: the “Good”, the “Bad”, and the “Mysterious” Glia. Neurosci 14:544–560. https://doi.org/10.1177/1073858408322839
Bellucci A, Collo G, Sarnico I et al (2008) Alpha-synuclein aggregation and cell death triggered by energy deprivation and dopamine overload are counteracted by D2/D3 receptor activation. J Neurochem 106:560–577. https://doi.org/10.1111/j.1471-4159.2008.05406.x
Jiang P, Gan M, Ebrahim AS et al (2013) Adenosine monophosphate-activated protein kinase overactivation leads to accumulation of α-synuclein oligomers and decrease of neurites. Neurobiol Aging 34:1504–1515. https://doi.org/10.1016/j.neurobiolaging.2012.11.001
Schmidt S, Linnartz B, Mendritzki S et al (2011) Genetic mouse models for Parkinson’s disease display severe pathology in glial cell mitochondria. Hum Mol Genet 20:1197–1211. https://doi.org/10.1093/hmg/ddq564
Gu X-L, Long C-X, Sun L et al (2010) Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol Brain 3:12. https://doi.org/10.1186/1756-6606-3-12
Mena MA, de Bernardo S, Casarejos MJ et al (2002) The role of astroglia on the survival of dopamine neurons. Mol Neurobiol 25:245–264. https://doi.org/10.1385/MN:25:3:245
Asanuma M, Miyazaki I, Murakami S et al (2014) Striatal astrocytes act as a reservoir for l-DOPA. PLoS One 9:e106362. https://doi.org/10.1371/journal.pone.0106362
Wójtowicz AM, Dvorzhak A, Semtner M, Grantyn R (2013) Reduced tonic inhibition in striatal output neurons from Huntington mice due to loss of astrocytic GABA release through GAT-3. Front Neural Circuits 7:188. https://doi.org/10.3389/fncir.2013.00188
Lee W, Reyes RC, Gottipati MK et al (2013) Enhanced Ca(2+)-dependent glutamate release from astrocytes of the BACHD Huntington’s disease mouse model. Neurobiol Dis 58:192–199. https://doi.org/10.1016/j.nbd.2013.06.002
Di Giorgio FP, Carrasco MA, Siao MC et al (2007) Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci 10:608–614. https://doi.org/10.1038/nn1885
Nagai M, Re DB, Nagata T et al (2007) Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci 10:615–622. https://doi.org/10.1038/nn1876
Philips T, Rothstein JD (2014) Glial cells in amyotrophic lateral sclerosis. Exp Neurol 262(Pt B):111–120. https://doi.org/10.1016/j.expneurol.2014.05.015
Boillée S, Vande Velde C, Cleveland DW (2006) ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52:39–59. https://doi.org/10.1016/j.neuron.2006.09.018
Serio A, Bilican B, Barmada SJ et al (2013) Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc Natl Acad Sci USA 110:4697–4702. https://doi.org/10.1073/pnas.1300398110
Tyzack GE, Hall CE, Sibley CR et al (2017) A neuroprotective astrocyte state is induced by neuronal signal EphB1 but fails in ALS models. Nat Commun 8:1164. https://doi.org/10.1038/s41467-017-01283-z
Blasco H, Corcia P, Pradat P-F et al (2013) Metabolomics in cerebrospinal fluid of patients with amyotrophic lateral sclerosis: an untargeted approach via high-resolution mass spectrometry. J Proteome Res 12:3746–3754. https://doi.org/10.1021/pr400376e
Blasco H, Patin F, Madji Hounoum B et al (2016) Metabolomics in amyotrophic lateral sclerosis: how far can it take us? Eur J Neurol 23:447–454
Blasco H, Nadal-Desbarats L, Pradat P-F et al (2014) Untargeted 1H-NMR metabolomics in CSF: toward a diagnostic biomarker for motor neuron disease. Neurology 82:1167–1174. https://doi.org/10.1212/WNL.0000000000000274
Patin F, Corcia P, Vourc’h P et al (2017) Omics to explore amyotrophic lateral sclerosis evolution: the central role of arginine and proline metabolism. Mol Neurobiol 54:5361–5374. https://doi.org/10.1007/s12035-016-0078-x
Patin F, Baranek T, Vourc’h P et al (2016) Combined metabolomics and transcriptomics approaches to assess the IL-6 blockade as a therapeutic of ALS: deleterious alteration of lipid metabolism. Neurotherapeutics 13:905–917. https://doi.org/10.1007/s13311-016-0461-3
Blasco H, Veyrat-Durebex C, Bocca C et al (2017) Lipidomics reveals cerebrospinal-fluid signatures of ALS. Sci Rep 7:17652. https://doi.org/10.1038/s41598-017-17389-9
Veyrat-Durebex C, Bocca C, Chupin S et al (2018) Metabolomics and lipidomics profiling of a combined mitochondrial plus endoplasmic reticulum fraction of human fibroblasts: a robust tool for clinical studies. J Proteome Res 17:745–750. https://doi.org/10.1021/acs.jproteome.7b00637
Veyrat-Durebex C, Corcia P, Piver E et al (2016) Disruption of TCA cycle and glutamate metabolism identified by metabolomics in an in vitro model of amyotrophic lateral sclerosis. Mol Neurobiol 53:6910–6924. https://doi.org/10.1007/s12035-015-9567-6
Madji Hounoum B, Mavel S, Coque E et al (2017) Wildtype motoneurons, ALS-linked SOD1 mutation and glutamate profoundly modify astrocyte metabolism and lactate shuttling. Glia 65:592–605. https://doi.org/10.1002/glia.23114
Cistaro A, Valentini MC, Chiò A et al (2011) Brain hypermetabolism in amyotrophic lateral sclerosis: a FDG PET study in ALS of spinal and bulbar onset. Eur J Nucl Med Mol Imaging 39:251–259. https://doi.org/10.1007/s00259-011-1979-6
Ferraiuolo L, Higginbottom A, Heath PR et al (2011) Dysregulation of astrocyte–motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 134:2627–2641. https://doi.org/10.1093/brain/awr193
Martorana F, Brambilla L, Valori CF et al (2012) The BH4 domain of Bcl-XL rescues astrocyte degeneration in amyotrophic lateral sclerosis by modulating intracellular calcium signals. Hum Mol Genet 21:826–840. https://doi.org/10.1093/hmg/ddr513
Gallardo G, Barowski J, Ravits J et al (2014) An α2-Na/K ATPase/α-adducin complex in astrocytes triggers non-cell autonomous neurodegeneration. Nat Neurosci 17:1710–1719. https://doi.org/10.1038/nn.3853
Madill M, McDonagh K, Ma J et al (2017) Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol Brain 10:22. https://doi.org/10.1186/s13041-017-0300-4
Chen WW, Zhang X, Huang WJ (2016) Role of neuroinflammation in neurodegenerative diseases (review). Mol Med Rep 13:3391–3396. https://doi.org/10.3892/mmr.2016.4948
Perry VH, Teeling J (2013) Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol 35:601–612. https://doi.org/10.1007/s00281-013-0382-8
Vasile F, Dossi E, Rouach N (2017) Human astrocytes: structure and functions in the healthy brain. Brain Struct Funct 222:2017–2029. https://doi.org/10.1007/s00429-017-1383-5
Colombo E, Farina C (2016) Astrocytes: key regulators of neuroinflammation. Trends Immunol 37:608–620. https://doi.org/10.1016/j.it.2016.06.006
Glass CK, Saijo K, Winner B et al (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140:918–934. https://doi.org/10.1016/j.cell.2010.02.016
Millington C, Sonego S, Karunaweera N et al (2014) Chronic neuroinflammation in Alzheimer’s disease: new perspectives on animal models and promising candidate drugs. Biomed Res Int 2014:309129. https://doi.org/10.1155/2014/309129
Sofroniew MV (2014) Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neurosci A Rev J Bring Neurobiol Neurol Psychiatry 20:160–172. https://doi.org/10.1177/1073858413504466
Janelidze S, Mattsson N, Stomrud E et al (2018) CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology. https://doi.org/10.1212/wnl.0000000000006082
Li M, Li Z, Yao Y et al (2017) Astrocyte-derived interleukin-15 exacerbates ischemic brain injury via propagation of cellular immunity. Proc Natl Acad Sci USA 114:E396–E405. https://doi.org/10.1073/pnas.1612930114
González-Reyes RE, Nava-Mesa MO, Vargas-Sánchez K et al (2017) Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front Mol Neurosci. https://doi.org/10.3389/fnmol.2017.00427
Medeiros R, LaFerla FM (2013) Astrocytes: conductors of the Alzheimer disease neuroinflammatory symphony. Exp Neurol 239:133–138. https://doi.org/10.1016/j.expneurol.2012.10.007
Batarseh YS, Duong Q-V, Mousa YM et al (2016) Amyloid-β and astrocytes interplay in amyloid-β related disorders. Int J Mol Sci. https://doi.org/10.3390/ijms17030338
Esler WP, Wolfe MS (2001) A portrait of Alzheimer secretases—new features and familiar faces. Science 293:1449–1454. https://doi.org/10.1126/science.1064638
Sompol P, Norris CM (2018) Ca2 + , astrocyte activation and calcineurin/NFAT signaling in age-related neurodegenerative diseases. Front Aging Neurosci 10:199. https://doi.org/10.3389/fnagi.2018.00199
Sompol P, Furman JL, Pleiss MM et al (2017) Calcineurin/NFAT signaling in activated astrocytes drives network hyperexcitability in Aβ-bearing mice. J Neurosci 37:6132–6148. https://doi.org/10.1523/JNEUROSCI.0877-17.2017
Miklossy J, Doudet DD, Schwab C et al (2006) Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp Neurol 197:275–283. https://doi.org/10.1016/j.expneurol.2005.10.034
Booth HDE, Hirst WD, Wade-Martins R (2017) The Role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci 40:358–370. https://doi.org/10.1016/j.tins.2017.04.001
Rannikko EH, Weber SS, Kahle PJ (2015) Exogenous α-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci 16:57. https://doi.org/10.1186/s12868-015-0192-0
Fellner L, Irschick R, Schanda K et al (2013) Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 61:349–360. https://doi.org/10.1002/glia.22437
Kim KS, Kim JS, Park J-Y et al (2013) DJ-1 Associates with lipid rafts by palmitoylation and regulates lipid rafts-dependent endocytosis in astrocytes. Hum Mol Genet 22:4805–4817. https://doi.org/10.1093/hmg/ddt332
Khasnavis S, Pahan K (2014) Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson’s disease. J Neuroimmune Pharmacol 9:569–581. https://doi.org/10.1007/s11481-014-9552-2
Kim J, Choi D, Jeong H et al (2013) DJ-1 facilitates the interaction between STAT1 and its phosphatase, SHP-1, in brain microglia and astrocytes: a novel anti-inflammatory function of DJ-1. Neurobiol Dis 60:1–10. https://doi.org/10.1016/j.nbd.2013.08.007
Qiao C, Yin N, Gu H-Y et al (2016) Atp13a2 deficiency aggravates astrocyte-mediated neuroinflammation via NLRP3 inflammasome activation. CNS Neurosci Ther 22:451–460. https://doi.org/10.1111/cns.12514
Chinta SJ, Woods G, Demaria M et al (2018) Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep 22:930–940. https://doi.org/10.1016/j.celrep.2017.12.092
Ilieva H, Polymenidou M, Cleveland DW (2009) Non–cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 187:761–772. https://doi.org/10.1083/jcb.200908164
Kawamata T, Akiyama H, Yamada T, McGeer PL (1992) Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol 140:691–707
Wang R, Yang B, Zhang D (2011) Activation of interferon signaling pathways in spinal cord astrocytes from an ALS mouse model. Glia 59:946–958. https://doi.org/10.1002/glia.21167
Endo F, Komine O, Fujimori-Tonou N et al (2015) Astrocyte-derived TGF-β1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep 11:592–604. https://doi.org/10.1016/j.celrep.2015.03.053
Johann S, Heitzer M, Kanagaratnam M et al (2015) NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 63:2260–2273. https://doi.org/10.1002/glia.22891
Schiffer D, Cordera S, Cavalla P, Migheli A (1996) Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J Neurol Sci 139(Suppl):27–33
Johansson A, Engler H, Blomquist G et al (2007) Evidence for astrocytosis in ALS demonstrated by [11C](L)-deprenyl-D2 PET. J Neurol Sci 255:17–22. https://doi.org/10.1016/j.jns.2007.01.057
Hsiao H-Y, Chen Y-C, Chen H-M et al (2013) A critical role of astrocyte-mediated nuclear factor-κB-dependent inflammation in Huntington’s disease. Hum Mol Genet 22:1826–1842. https://doi.org/10.1093/hmg/ddt036
Hsiao H-Y, Chiu F-L, Chen C-M et al (2014) Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum Mol Genet 23:4328–4344. https://doi.org/10.1093/hmg/ddu151
Cobley JN, Fiorello ML, Bailey DM (2018) 13 reasons why the brain is susceptible to oxidative stress. Redox Biol 15:490–503
Gandhi S, Abramov AY (2012) Mechanism of oxidative stress in neurodegeneration. Oxid Med Cell Longev 2012:42801
Sena LA, Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48:158–167
Holmstrom KM, Finkel T (2014) Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 15:411–421
Dickinson BC, Peltier J, Stone D et al (2011) Nox2 redox signaling maintains essential cell populations in the brain. Nat Chem Biol 7:106–112
Gauron C, Meda F, Dupont E et al (2016) Hydrogen peroxide controls axon pathfinding during zebrafish development. Dev Biol 414:133–141
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13
Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552:335–344
Sandalio LM, Rodriguez-Serrano M, Romero-Puertas MC, del Rio LA (2013) Role of peroxisomes as a source of reactive oxygen species (ROS) signaling molecules. Subcell Biochem 69:231–255
Supplie LM, Duking T, Campbell G et al (2017) Respiration-Deficient Astrocytes Survive As Glycolytic Cells In Vivo. J Neurosci 37:4231–4242
Belanger M, Allaman I, Magistretti PJ (2011) Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14:724–738
Lull ME, Block ML (2010) Microglial activation and chronic neurodegeneration. Neurotherapeutics 7:354–365
Sheng WS, Hu S, Feng A, Rock RB (2013) Reactive oxygen species from human astrocytes induced functional impairment and oxidative damage. Neurochem Res 38:2148–2159
Ma Q (2013) Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53:401–426
Suzuki T, Motohashi H, Yamamoto M (2013) Toward clinical application of the Keap1–Nrf2 pathway. Trends Pharmacol Sci 34:340–346
Baxter PS, Hardingham GE (2016) Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic Biol Med 100:147–152
Bell KF, Al-Mubarak B, Martel MA et al (2015) Neuronal development is promoted by weakened intrinsic antioxidant defences due to epigenetic repression of Nrf. Nat Commun 6:7066
Hirrlinger J, Schulz JB, Dringen R (2002) Glutathione release from cultured brain cells: multidrug resistance protein 1 mediates the release of GSH from rat astroglial cells. J Neurosci Res 69:318–326
Vargas MR, Johnson DA, Sirkis DW et al (2008) Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci 28:13574–13581
Vargas MR, Johnson JA (2009) The Nrf2–ARE cytoprotective pathway in astrocytes. Expert Rev Mol Med 11:e17
Abramov AY, Canevari L, Duchen MR (2004) Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci 24:565–575
Abeti R, Abramov AY, Duchen MR (2011) Beta-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain 134:1658–1672
Oksanen M, Petersen AJ, Naumenko N et al (2017) PSEN1 mutant iPSC-derived model reveals severe astrocyte pathology in Alzheimer’s disease. Stem Cell Rep 9:1885–1897. https://doi.org/10.1016/j.stemcr.2017.10.016
Lin DT, Wu J, Holstein D et al (2007) Ca2 + signaling, mitochondria and sensitivity to oxidative stress in aging astrocytes. Neurobiol Aging 28:99–111
Jacobson J, Duchen MR (2002) Mitochondrial oxidative stress and cell death in astrocytes—requirement for stored Ca2+ and sustained opening of the permeability transition pore. J Cell Sci 115:1175–1188
Kärkkäinen V, Pomeshchik Y, Savchenko E et al (2014) Nrf2 regulates neurogenesis and protects neural progenitor cells against Aβ toxicity. Stem Cells 32:1904–1916. https://doi.org/10.1002/stem.1666
Pomeshchik Y, Kidin I, Savchenko E et al (2014) Does Nrf2 gene transfer facilitate recovery after contusion spinal cord injury? Antioxid Redox Signal 20:1313–1323. https://doi.org/10.1089/ars.2013.5453
Kanninen K, Heikkinen R, Malm T et al (2009) Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci 106:16505–16510. https://doi.org/10.1073/pnas.0908397106
Johnson DA, Johnson JA (2015) Nrf2–a therapeutic target for the treatment of neurodegenerative diseases. Free Radic Biol Med 88:253–267
Chen PC, Vargas MR, Pani AK et al (2009) Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: critical role for the astrocyte. Proc Natl Acad Sci USA 106:2933–2938
Cassina P, Cassina A, Pehar M et al (2008) Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci 28:4115–4122
Morgan MJ, Liu ZG (2011) Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res 21:103–115
Jones PL, Ping D, Boss JM (1997) Tumor necrosis factor alpha and interleukin-1beta regulate the murine manganese superoxide dismutase gene through a complex intronic enhancer involving C/EBP-beta and NF-kappaB. Mol Cell Biol 17:6970–6981
Kairisalo M, Korhonen L, Blomgren K, Lindholm D (2007) X-linked inhibitor of apoptosis protein increases mitochondrial antioxidants through NF-kappaB activation. Biochem Biophys Res Commun 364:138–144
Rojo AI, Salinas M, Martin D et al (2004) Regulation of Cu/Zn-superoxide dismutase expression via the phosphatidylinositol 3 kinase/Akt pathway and nuclear factor-kappaB. J Neurosci 24:7324–7334
Anrather J, Racchumi G, Iadecola C (2006) NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem 281:5657–5667
Kolyada AY, Savikovsky N, Madias NE (1996) Transcriptional regulation of the human iNOS gene in vascular-smooth-muscle cells and macrophages: evidence for tissue specificity. Biochem Biophys Res Commun 220:600–605
Kabe Y, Ando K, Hirao S et al (2005) Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal 7:395–403
Toledano MB, Leonard WJ (1991) Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc Natl Acad Sci USA 88:4328–4332
Matthews JR, Botting CH, Panico M et al (1996) Inhibition of NF-kappaB DNA binding by nitric oxide. Nucleic Acids Res 24:2236–2242
Hsieh HL, Yang CM (2013) Role of redox signaling in neuroinflammation and neurodegenerative diseases. Biomed Res Int 2013:3
Holmqvist S, Lehtonen Š, Chumarina M et al (2016) Creation of a library of induced pluripotent stem cells from Parkinsonian patients. NPJ Park Dis 2:16009. https://doi.org/10.1038/npjparkd.2016.9
Jones VC, Atkinson-Dell R, Verkhratsky A, Mohamet L (2017) Aberrant iPSC-derived human astrocytes in Alzheimer’s disease. Cell Death Dis 8:e2696. https://doi.org/10.1038/cddis.2017.89
Ribecco-Lutkiewicz M, Sodja C, Haukenfrers J et al (2018) A novel human induced pluripotent stem cell blood–brain barrier model: applicability to study antibody-triggered receptor-mediated transcytosis. Sci Rep 8:1873. https://doi.org/10.1038/s41598-018-19522-8
Appelt-Menzel A, Cubukova A, Günther K et al (2017) Establishment of a human blood–brain barrier co-culture model mimicking the neurovascular unit using induced pluri- and multipotent stem cells. Stem Cell Reports 8:894–906. https://doi.org/10.1016/j.stemcr.2017.02.021
Canfield SG, Stebbins MJ, Morales BS et al (2017) An isogenic blood–brain barrier model comprising brain endothelial cells, astrocytes, and neurons derived from human induced pluripotent stem cells. J Neurochem 140:874–888. https://doi.org/10.1111/jnc.13923
Lim RG, Quan C, Reyes-Ortiz AM et al (2017) Huntington’s disease iPSC-derived brain microvascular endothelial cells reveal WNT-mediated angiogenic and blood–brain barrier deficits. Cell Rep 19:1365–1377. https://doi.org/10.1016/j.celrep.2017.04.021
Vatine GD, Al-Ahmad A, Barriga BK et al (2017) Modeling psychomotor retardation using iPSCs from MCT8-deficient patients indicates a prominent role for the blood–brain barrier. Cell Stem Cell 20:831.e5–843.e5. https://doi.org/10.1016/j.stem.2017.04.002
Neuhaus W (2017) Human induced pluripotent stem cell basedin vitromodels of the blood–brain barrier: the future standard? Neural Regen Res 12:1607–1609. https://doi.org/10.4103/1673-5374.217326
Almad AA, Doreswamy A, Gross SK et al (2016) Connexin 43 in astrocytes contributes to motor neuron toxicity in amyotrophic lateral sclerosis. Glia 64:1154–1169. https://doi.org/10.1002/glia.22989
Takikawa M, Ohki R (2017) A vicious partnership between AKT and PHLDA3 to facilitate neuroendocrine tumors. Cancer Sci 108:1101–1108. https://doi.org/10.1111/cas.13235
Gonzalez DM, Gregory J, Brennand KJ (2017) The importance of non-neuronal cell types in hiPSC-based disease modeling and drug screening. Front cell Dev Biol 5:117. https://doi.org/10.3389/fcell.2017.00117
Olson H, Betton G, Robinson D et al (2000) Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul Toxicol Pharmacol 32:56–67. https://doi.org/10.1006/RTPH.2000.1399
Ishii MN, Yamamoto K, Shoji M et al (2017) Human induced pluripotent stem cell (hiPSC)-derived neurons respond to convulsant drugs when co-cultured with hiPSC-derived astrocytes. Toxicology 389:130–138. https://doi.org/10.1016/J.TOX.2017.06.010
Thorne N, Malik N, Shah S et al (2016) High-throughput phenotypic screening of human astrocytes to identify compounds that protect against oxidative stress. Stem Cells Transl Med 5:613–627. https://doi.org/10.5966/sctm.2015-0170
Windrem MS, Schanz SJ, Guo M et al (2008) Neonatal chimerization with human glial progenitor cells can both remyelinate and rescue the otherwise lethally hypomyelinated shiverer mouse. Cell Stem Cell 2:553–565. https://doi.org/10.1016/j.stem.2008.03.020
Windrem MS, Schanz SJ, Morrow C et al (2014) A competitive advantage by neonatally engrafted human glial progenitors yields mice whose brains are chimeric for human glia. J Neurosci 34:16153–16161. https://doi.org/10.1523/JNEUROSCI.1510-14.2014
Goldman SA, Nedergaard M, Windrem MS (2015) Modeling cognition and disease using human glial chimeric mice. Glia 63:1483–1493. https://doi.org/10.1002/glia.22862
Benraiss A, Wang S, Herrlinger S et al (2016) Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat Commun 7:11758. https://doi.org/10.1038/ncomms11758
Windrem MS, Osipovitch M, Liu Z et al (2017) Human iPSC glial mouse chimeras reveal glial contributions to schizophrenia. Cell Stem Cell 21:195.e6–208.e6. https://doi.org/10.1016/j.stem.2017.06.012
Thomsen GM, Avalos P, Ma AA et al (2018) Transplantation of neural progenitor cells expressing glial cell line-derived neurotrophic factor into the motor cortex as a strategy to treat amyotrophic lateral sclerosis. Stem Cells. https://doi.org/10.1002/stem.2825
Chen H, Qian K, Chen W et al (2015) Human-derived neural progenitors functionally replace astrocytes in adult mice. J Clin Invest 125:1033–1042. https://doi.org/10.1172/JCI69097
Haidet-Phillips AM, Doreswamy A, Gross SK et al (2015) Human glial progenitor engraftment and gene expression is independent of the ALS environment. Exp Neurol 264:188–199. https://doi.org/10.1016/j.expneurol.2014.12.011
Juopperi TA, Kim W, Chiang C-H et al (2012) Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol Brain 5:17. https://doi.org/10.1186/1756-6606-5-17
Kondo T, Funayama M, Tsukita K et al (2014) Focal transplantation of human iPSC-derived glial-rich neural progenitors improves lifespan of ALS mice. Stem cell reports 3:242–249. https://doi.org/10.1016/j.stemcr.2014.05.017
Lepore AC, Rauck B, Dejea C et al (2008) Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat Neurosci 11:1294–1301. https://doi.org/10.1038/nn.2210
Das MM, Avalos P, Suezaki P et al (2016) Human neural progenitors differentiate into astrocytes and protect motor neurons in aging rats. Exp Neurol 280:41–49. https://doi.org/10.1016/J.EXPNEUROL.2016.03.023
Boucherie C, Schäfer S, Lavand’homme P et al (2009) Chimerization of astroglial population in the lumbar spinal cord after mesenchymal stem cell transplantation prolongs survival in a rat model of amyotrophic lateral sclerosis. J Neurosci Res 87:2034–2046. https://doi.org/10.1002/jnr.22038
Proschel C, Stripay JL, Shih C-H et al (2014) Delayed transplantation of precursor cell-derived astrocytes provides multiple benefits in a rat model of Parkinsons. EMBO Mol Med 6:504–518. https://doi.org/10.1002/emmm.201302878
Song J-J, Oh S-M, Kwon O-C et al (2018) Cografting astrocytes improves cell therapeutic outcomes in a Parkinson’s disease model. J Clin Invest 128:463–482. https://doi.org/10.1172/JCI93924
Pihlaja R, Koistinaho J, Malm T et al (2008) Transplanted astrocytes internalize deposited β-amyloid peptides in a transgenic mouse model of Alzheimer’s disease. Glia 56:154–163. https://doi.org/10.1002/glia.20599
Pihlaja R, Koistinaho J, Kauppinen R et al (2011) Multiple cellular and molecular mechanisms Are involved in human Aβ clearance by transplanted adult astrocytes. Glia 59:1643–1657. https://doi.org/10.1002/glia.21212
Esposito G, Sarnelli G, Capoccia E et al (2016) Autologous transplantation of intestine-isolated glia cells improves neuropathology and restores cognitive deficits in β amyloid-induced neurodegeneration. Sci Rep 6:22605. https://doi.org/10.1038/srep22605
Schöll M, Carter SF, Westman E et al (2015) Early astrocytosis in autosomal dominant Alzheimer’s disease measured in vivo by multi-tracer positron emission tomography. Sci Rep 5:16404. https://doi.org/10.1038/srep16404
Acknowledgements
Open access funding provided by University of Helsinki including Helsinki University Central Hospital.
Funding
Funding was provided by Academy of Finland (278343).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Oksanen, M., Lehtonen, S., Jaronen, M. et al. Astrocyte alterations in neurodegenerative pathologies and their modeling in human induced pluripotent stem cell platforms. Cell. Mol. Life Sci. 76, 2739–2760 (2019). https://doi.org/10.1007/s00018-019-03111-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03111-7