
Abstract
Complement is the major humoral component of the innate immune system. It recognizes pathogen- and damage-associated molecular patterns, and initiates the immune response in coordination with innate and adaptive immunity. When activated, the complement system unleashes powerful cytotoxic and inflammatory mechanisms, and thus its tight control is crucial to prevent damage to host tissues and allow restoration of immune homeostasis. Factor H is the major soluble inhibitor of complement, where its binding to self markers (i.e., particular glycan structures) prevents complement activation and amplification on host surfaces. Not surprisingly, mutations and polymorphisms that affect recognition of self by factor H are associated with diseases of complement dysregulation, such as age-related macular degeneration and atypical haemolytic uremic syndrome. In addition, pathogens (i.e., non-self) and cancer cells (i.e., altered-self) can hijack factor H to evade the immune response. Here we review recent (and not so recent) literature on the structure and function of factor H, including the emerging roles of this protein in the pathophysiology of infectious diseases and cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
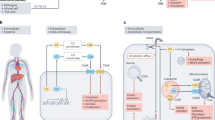
Innate immunity comprises a humoral (macromolecular) and a cellular arm [1, 2]. The complement system is a major component of the former, comprised of a cascade of more than 30 proteins, which is activated via three distinct pathways: alternative (AP), classical (CP) and lectin (LP) [3]; see [4] for complement nomenclature. Activation of the classical and lectin pathways is restricted to the recognition of immune complexes and non-self carbohydrates, respectively, by soluble pattern recognition molecules/receptors (PRMs/PRRs), including the C1q moiety in the CP, and mannose-binding lectin (MBL) and ficolins (both in LP) [3, 5]. Associated serine proteases (i.e., C1r/C1s in CP and mannan-binding lectin-associated serine proteases (MASPs) in LP) then cleave C4 and C2, thus generating C4b and C2a fragments that non-covalently associate to form the CP/LP C3 convertase (C4bC2a). This enzyme complex catalyzes the hydrolysis of C3 to C3a and C3b, where the latter contains a highly reactive thioester that can form a covalent bond with a nucleophile, e.g. on a nearby cell or in the extracellular matrix. Surface-bound C3b can then form a complex with factor B (FB), which is cleaved by factor D to Bb, leading to the formation of the alternative pathway C3 convertase (C3bBb), which is stabilized by factor P (FP; also termed properdin) and catalyzes further hydrolysis of C3. Therefore, as shown in Fig. 1, when the AP C3 convertase forms on an activating surface, lacking an appropriate level of complement inhibitors, the AP can act as an amplification loop for the complement system, regardless of how this is initially triggered (see below and [6]). AP activation also occurs in the absence of a recognition event through a tick over mechanism. Here, C3 undergoes non-enzymatic hydrolysis of the thioester bond to generate C3(H20) [7] that can bind FB, which is itself cleaved by FD to make a fluid-phase C3-convertase (i.e., C3(H20)Bb). This convertase is capable of cleaving further C3 molecules to C3b that can then bind covalently to available surfaces (via an ester bond as described above) and lead to amplification of complement if left unchecked.

The activation, amplification and regulation of the complement system. The complement system can be activated by three different pathways: the alternative pathway (AP), classical pathway (CP) and lectin pathway (LP). While the AP is constitutively active and undergoes a constant tick over, the CP and LP are triggered by antibody- and carbohydrate-mediated recognition mechanisms, respectively. Regardless of how this protein cascade is initiated, it is the AP that amplifies the complement system; e.g. if the AP C3 convertase is formed on an activator surface. All three pathways lead to the conversion of C3 into C3b, which can become covalently attached (via a thioester) to any nearby nucleophile (e.g., a hydroxyl or amine group on a surface); C3 cleavage releases the small protein fragment C3a, a potent anaphylatoxin. The remaining C3b can associate with factor B (FB), which is itself cleaved by factor D (FD). This forms the AP C3 convertase (i.e., C3bBb) where this is stabilized by factor P (FP). If this convertase is not deactivated then any free C3 in the vicinity will be converted into C3b and thus begins a positive feedback cycle, referred to as the amplification loop. If left to run unchecked, this will lead to the opsonization of the target surface (with C3b) and the formation of the AP C5 convertase (i.e., C3bBbC3b), which represents the initiation of the terminal pathway of complement and the subsequent formation of the membrane-attack complex (MAC) that can lead to lysis (e.g., of bacteria). Host cells have a number of cell surface proteins capable of down-regulating the complement cascade that can either dissociate the AP C3 convertase, known as decay acceleration activity, or act as cofactors for the proteolytic cleavage, by factor I (FI), to inactive C3b (iC3b). The soluble C3b-binding protein factor H (FH) can associate with host cells via the recognition of glycan markers of self and thereby down-regulate complement through its decay accelerating and cofactor activities. FH (and the truncated FHL-1 product of the CFH gene) can also recognize self markers on acellular structures such as the extracellular matrix, and in this context FH/FHL-1 are the only negative regulators of the complement AP pathway. Therefore, FH and FHL-1 have a pivotal role in the prevention of complement activation in host tissues. This schematic is modified from [184]
Thus all three pathways converge in the formation of C3 convertases and in the deposition of C3b that opsonizes pathogens (and host cell debris) and promotes their phagocytosis [6]. Moreover, by binding C3b, the C3 convertases can be converted to C5 convertases (i.e. C4bC2aC3b and C3bBbC3b for the CP/LP and AP, respectively) leading to the formation of the membrane-attack complex (MAC, C5b–C9n) that can cause lysis of the target cells (see Fig. 1); the anaphylatoxins C3a and C5a, generated as by-products of convertase formation (i.e. C3 and C5 cleavage), act as potent chemoattractants and activators of immune cells. In addition to these well established effector functions of complement (i.e., being opsonic, lytic and pro-inflammatory), it has become increasingly apparent that this protein cascade also plays key roles in selection, maintenance and activation of B lymphocytes, where C3 and its proteolytic fragments are necessary to elicit a robust antibody response in antigen-primed B cells [8]. Furthermore, an involvement of complement is emerging in T cell immunity, with regard to activation, polarization and survival of T cell subsets [6, 9, 10]. Therefore, the biological activities of complement extend beyond recognition/disposal of invading pathogens and encompass mechanisms in both innate and adaptive immunity, where the complement system has been described as a natural adjuvant and a modulator of the adaptive response [11].
In the light of the pleiotropic functions of the complement system and, most importantly, the destructive potential of its activation (because of the potent amplification ability of the AP), complement requires a number of regulatory mechanisms that allow its tight control to confine it to appropriate pathogenic surfaces, and thereby prevent collateral damage to healthy host tissues [3]. Distinct phases of complement activation are indeed checked by fluid-phase and cell-associated inhibitors so that the final outcome represents an intricate balance between detection and destruction of non-self and minimization of damage to self [3, 6, 12]. In this regard, both C3b and C4b undergo proteolytic processing by factor I (FI), where this enzyme requires, as cofactors, either fluid-phase regulators, i.e. factor H (FH) and C4-binding protein (C4BP) for AP and CP/LP, respectively, or membrane proteins on the cell surface, i.e. complement receptor type 1 (CR1; CD35) and membrane cofactor protein (MCP; CD46). Also, the rate of formation and dissociation (decay) of the C3 (and C5) convertases is controlled by FH, decay-accelerating factor (DAF; CD55), CR1 and C4BP that all serve to reduce the amount of active convertase present; on the other hand, FP stabilizes the C3 and C5 convertases of the AP. In addition, the formation of the membrane-attack complex is inhibited by CD59 (also known as MAC-inhibitory protein) and vitronectin (S protein).
FH is the major regulator of the AP in solution phase, but also has a critical role in the inhibition of complement amplification on cells and in the extracellular matrix of host tissues due to its ability to recognize (and associate with) self surfaces (i.e., via glycan markers) to ensure that these are non-activating (see [13, 14]). In this regard, FH is amongst the most abundant complement components in human blood (with plasma concentrations ranging from 200 to 800 μg/ml; see [15]), where the liver is the major source of FH protein in vivo. However, FH is secreted by additional cell types including monocytes [16], fibroblasts [17], endothelial cells [18], platelets [19], and retinal pigment epithelial cells (RPE) [20, 21], that likely contribute to local titers of the protein in tissues [22]. Furthermore, the complement factor H (CFH) gene undergoes alternative splicing, where a truncated form of FH, termed factor H-like protein 1 (FHL-1) [23], can also be expressed locally, e.g. by RPE cells, as well as by the liver [21, 23]. Thus, systemic and locally secreted FH, most likely, both contribute to the regulation of complement activation in the context of immune surveillance [21, 24, 25].
Given the central role of FH in regulation of complement amplification (Fig. 1), proper functioning of this soluble inhibitor is crucial, as exemplified by human pathologies associated with FH, including those with mutations and polymorphisms in the CFH gene; e.g. typical and atypical hemolytic uremic syndrome (HUS and aHUS, respectively) [13, 26–28], age-related macular degeneration (AMD) [29–31] and dense deposit disease (DDD) [32, 33]. In this review, we discuss the structure and function of FH as a fundamental complement inhibitor and focus on its roles in complement-mediated diseases and the immune evasion strategies adopted by pathogens and cancer cells.
Structural insights into the complement inhibitory activities of factor H
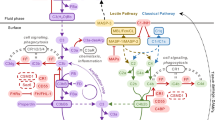
The human FH gene (termed CFH) is located on chromosome 1q32 in the regulators of complement activation (RCA) gene cluster. This encodes an ~155 kDa glycoprotein comprised of 20 contiguous complement control protein (CCP) modules [23, 34] (Fig. 2), also known as short consensus repeats or sushi domains, which occur in many other complement and non-complement proteins [23, 35]; the alternatively spliced transcript, FHL-1 is composed of CCPs 1–7 followed by a unique 4-amino acid sequence. Each CCP consists of ~60 amino acids arranged in a typical β-sandwich fold (containing up to 8 β strands), which approximate to prolate ellipsoids (∼4 nm × ∼2 nm × ∼1.5 nm in size), with the N- and C-termini situated at opposite ends of the longest axis [34]. The 20 CCPs in the FH protein are connected to one another by short, sometimes flexible, linkers (of between three and eight amino acid residues) and arranged in an extended head to tail fashion, which makes the intact protein resemble a pearl necklace. As yet high-resolution 3D structures of the intact FH protein have not been generated, most likely due to its large size, glycosylation and inherent flexibility. Nevertheless, extensive investigations on recombinant constructs spanning individual/multiple CCPs of FH (either alone or in complex with ligands) have been carried out, and NMR and/or X-ray crystal structures are now available for all CCPs apart from 14 and 17 (see [13, 34, 36–41]). Despite a high degree of similarity in the 3D folds for these CCPs, different regions of FH display distinct functional activities (see Fig. 2), where it is now recognized that these originate from both diversity in the sequences of the individual CCPs (and thus differences in their functional surfaces) and from the different intermolecular interactions between neighboring CCPs that dictate their relative orientation [35]. Integration of these high-resolution data with lower-resolution structural information from small-angle X-ray scattering (SAXS) has provided insights into the overall organization/shape of the elongated FH molecule [34], e.g. such that CCPs 8–15 likely adopt a hinge like structure [36, 37, 42]; discussed in more detail below.

Domain structure and ligand-binding activities of factor H and FHL-1. Complement FH is comprised of twenty CCP modules, whereas FHL-1 shares the first seven of these followed by a unique four amino acid sequence (SFTL). The four N-terminal CCPs of FH/FHL-1 (colored blue) confer regulatory activity through mediating C3b and FI binding; C3b (via its C3d region) also binds to the C-terminal CCPs 19–20 of FH. The CCPs 6–8 and/or 19–20 regions (colored yellow) support the interactions with a wide variety of ligands including glycans (GAGs and sialic acid), pentraxins (CRP and PTX3; where the latter binds through the its C- and N-terminal domains, respectively), the lipid peroxidation product MDA, apoptotic/necrotic cells (e.g., via annexin II and DNA), extracellular matrix proteins (e.g., chondroadherin and fibromodulin), ApoE, shiga toxin from enterohaemorrhagic E. coli, and FH-binding proteins from various other bacteria. Although it is presumed that many of the ligands that bind FH via CCPs 6–8 will interact also with FHL-1, most of these have yet to be tested
As highlighted in Fig. 1, FH possesses three complement regulatory activities: the protein (1) inhibits formation of the AP C3 convertase (C3bBb), (2) accelerates the dissociation of this complex, and (3) acts as a cofactor for the serine protease FI. FH does this by interacting directly with C3b, competing with FB for binding to C3b, thus inhibiting assembly of the C3 convertase and facilitating the decay of the already formed complexes (by displacing Bb from C3b). Furthermore, this interaction also makes C3b susceptible to cleavage by FI (i.e., to generate iC3b, an inactive form of C3b that does not bind FB). It is the 4 CCPs at the N-terminal end of the FH protein that mediate these inhibitory functions (see Fig. 2). This is now understood at a molecular level based on the crystal structure of C3b in complex with CCPs 1–4 of FH [41], where this and other structural studies have provided valuable insights into the functional biology of FH [34–40, 42, 43]. Importantly, these CCPs of FH bind to C3b in an extended configuration, with a remarkably long contact interface that covers the whole flank of the C3b protein [41]. This interface on C3b is generated during its transition from C3 (to which FH does not bind), and involves all four FH CCP modules and multiple regions of C3b, including the CUB module and thioester-containing domain (TED). The decay acceleration activity of FH, and inhibition of the formation of the AP C3 convertase, is mainly mediated by the first two N-terminal CCP domains of FH, which recognize a region of C3b that is involved in FB binding, e.g. dislocating Bb from C3b by electrostatic repulsion and via a steric hindrance mechanism. In addition, the interaction of FH with C3b provides a large contact surface for the binding of the serine protease FI bringing it into close proximity to the CUB domain of C3b (i.e., where the cleavages sites are located). Furthermore, given that the CUB domain is proteolytically cleaved when C3b is converted to iC3b, this study [41] also explains why CCPs 1–4 of FH do not bind iC3b [34]. The iC3b fragment generated retains the TED region and, therefore, when it is attached to a cell surface can undergo additional hydrolysis to smaller fragments (e.g., C3dg and C3d) by FI and membrane-bound cofactors (e.g., CR1). Importantly, C3d (essentially corresponding to just the thioester domain) is a ligand of CR2 on B cells, where this interaction is essential for lymphocyte activation and maturation [44].
The C-terminal two CCPs of FH (CCPs 19–20) have also been shown to be involved in FH’s interaction with the C3b (Fig. 2), where crystal structures of these CCPs in complex with C3d have been determined [39, 40]. These studies show that the binding site on FH mostly involves CCP19 along with the inter-domain linker between CCPs 19–20 (see [13]). Most importantly, from the superimposition of the X-ray structures of one of these structures [39] (where NMR and site directed mutagenesis allowed selection of the physiologically relevant interface) with that for C3b in complex with CCPs 1–4 [41] (described above) it is apparent that CCPs 19–20 recognizes a site on the TED domain that is close to, but distinct from, that occupied by CCPs 1–4. Thus, this strongly suggests that both ends of the FH protein (i.e., CCPs 1–4 and 19–20) can simultaneously bind the same C3b molecule [39].
Based on NMR studies on CCPs 10–11, CCPs 11–12 and CCPs 12–13, and SAXS modeling of CCPs 10–15 and CCPs 8–15 [36, 37, 42], it has been proposed that the middle region of FH (CCPs 8–15) adopts a hinge-like structure, with the CCPs 10–13 in a rigid V-like arrangement. Furthermore, combining these data with a SAXS-derived rod-like structure of CCPs 15–19 [39] and the crystal structures of CCPs 6–8 [45], and the CCPs 1–4 and 19–20 in complex with regions of C3, has allowed the generation of a model of FH where the protein bends back on itself [34]. Thus, this arrangement would provide a mechanism for how the N- and C-terminal ends of FH (i.e., CCPs 1–4 and CCPs 19–20) could bind simultaneously to distinct sites on the same C3b molecule. A key role in such structural organization is assumed to be played by CCP14 that is believed to alternate between poorly structured and compact conformations, depending on intra-molecular interactions (i.e., between neighboring CCPs) and ligand binding (i.e., to C3b associated with a non-activating surface) [37], thus allowing the two C3b binding regions of FH to swing around the central hinge region [34, 37]. Based on this hypothesis, a model has been proposed for the action of FH, where the protein is assumed to preferentially adopt a latent conformation with a low-affinity for C3b, which can switch to a higher-affinity activated structure upon recognition of C3b on self surfaces [34]. This transition has been proposed to be energetically driven by the binding of CCPs 1–4 to C3b and the interaction of CCPs 19–20 and CCPs 6–8 with glycans (such as glycosaminoglycans (GAGs) and sialic acid) on host surfaces, which would compensate for the loss of intra-molecular interactions stabilizing the latent conformation. As noted above, FHL-1 is identical to the full-length FH over the first 7 CCP domains and, therefore, contains the CCP 1–4 region associated with decay accelerating and cofactor activities (see [21]). However, given that FHL-1 lacks the sites for C3b binding and host surface recognition within the C-terminal region of FH (in CCPs 19–20), it is likely to be most effective as an inhibitor of complement activation within the fluid-phase [46].
Interaction of factor H with glycans—discriminating self from non-self
The ability to discriminate between self, non-self and altered-self is perhaps one of the most striking and important properties of the immune system. In this regard, sialic acid-capped glycans and the GAG chains of proteoglycans are abundant on the cell surface and in the extracellular matrix of host tissues, where these structures function as markers of self [14, 31]. By interacting with these surface patterns, it is thought that FH can prevent excessive or inappropriate AP activation/amplification on host tissues; e.g., the interaction with sialic acid increases the affinity of FH for C3b on the mammalian plasma membrane, which enhances the cofactor and decay accelerating activities of FH [13, 47]. On the other hand, microbes (non-self) normally do not express high levels of these glycans and are, therefore, poor binders of FH, thus permitting amplification of the complement AP [48]; moreover the LP is activated via recognition of lipopolysaccharide on Gram-negative bacteria, lipoteichoic acid on Gram-positive bacteria and β-glucan on fungi [5].
As shown in Fig. 2, FH has two distinct glycan binding regions, where CCPs 6–8 have been shown to interact with GAGs alone, whereas CCPs 19–20 bind GAGs and sialylated sugars, i.e. containing Neu5Acα2-3Gal (see [13, 14, 31, 49, 50]). Recently, elegant structural work revealed that when FH is bound to C3b, a major portion of its CCP20 module is exposed and properly positioned (relative to the thioester attached TED region) to interact simultaneously with a sialylated ligand (or potentially GAGs) on the cell surface [13], i.e. supporting formation of a ternary FH-C3b-glycan complex. Furthermore, this study allowed the identification of key amino acid residues in FH CCP20 that bind the glycerol side chain and carboxyl group of Neu5Ac (i.e., the predominant form of sialic acid found in mammalian cells). Interestingly, CCP20 acts as a hot spot for HUS-associated mutations, which indeed modify the Neu5Ac-binding pocket and alter the overall affinity of FH for sialic acid [13], ultimately leading to the defective complement regulation observed in this pathology (see [25, 51]); see further details below.
The CCP 6–8 and CCP 19–20 regions of FH have different specificities for GAGs [49, 50]; the former interacts with heparan sulfate (HS) and dermatan sulfate (DS), while the latter binds to just HS. Furthermore, biochemical and bioimaging studies [49, 50] have revealed that these regions mediate the binding to different (and distinct) HS structures, thereby providing a possible mechanism of tissue specificity; see reviews [14, 31, 52, 53]. Here, it is important to note that GAGs are highly diverse molecules with a huge sequence repertoire (e.g., based on variable sulfation/domain structure [14, 31]); in the case of HS, this is referred to as the heparanome, with differences in HS structure from cell type to cell type and tissue to tissue. For example, in the human eye there is considerable diversity in both GAG structure and in the proteoglycan core proteins to which they attach within different layers of the retina [54, 55]. Because of this, and since many proteins interact with HS, it has been hypothesized that tissue-specific differences in HS structure may act as post codes (or area/ZIP codes) that help regulate protein binding to cell surfaces and to the matrix [31]. For example, FH and FP can both interact with HS but their specificities are distinct such that they do not compete for binding sites; this means that the local HS structure can potentially dictate the relative amounts of these negative and positive regulators of the AP that are associated with a surface, and thus whether complement activation is promoted or inhibited (see [52]).
As noted above the two GAG-binding sites in FH also have different specificities. In this regard, the CCP 19–20 region of FH is solely responsible for the anchoring of this protein to the glomerular basement membrane in the human kidney (partly through HS recognition) [49]; conversely, it is predominantly the CCPs 6–8 region that mediates the recruitment of FH in the human eye by supporting its binding to HS (and DS) on the RPE and Bruch’s membrane, that together form the outer blood-retinal barrier. Why these two regions of FH should be tuned to interacting with different GAG structures is not understood, but it does provide an explanation for why coding changes in CCPs 6–8 and CCPs 19–20 (that affect GAG binding), via mutations/polymorphisms in the CFH gene, are differentially associated as risk factors for AMD and aHUS, which are diseases of the eye and kidney, respectively [31, 49]. Furthermore, how these two glycan-recognition regions co-operate is not well established; utilizing two binding sites simultaneously (that has been termed combinatorial recognition capacity) would be expected to lead to a higher avidity of interaction (see [34, 49]), but what effect this would have on the conformation of the FH protein and its association with C3b has not been determined experimentally. An attractive speculation is that the interaction of CCPs 6–8 with GAGs could position CCP4 of FH proximal to the (non-activator) surface and thereby facilitate the binding of CCPs 1–4 with a C3b molecule attached to this surface via its thioester, while CCPs 9–18 adopt a conformation that allows simultaneous binding of CCP 19–20 with another glycan (i.e., either sialic acid or a GAG) close by on the surface [34]. Structural studies on the CCPs 6–8 region indicate that all three CCP modules are likely involved in HS binding, where there is a central GAG-binding surface in CCP7 and additional binding sites in CCP6 and in the linker between CCPs 7 and 8 [45, 53]; biophysical data indicate that these sites together may allow the binding of extended HS sequences, e.g. of 24 saccharides in length. Given that FHL-1 lacks CCP8 (and CCPs thereafter) and has a unique SFTL sequence attached to the C-terminus of CCP7 (Fig. 2) it is possible that its specificity may differ somewhat from the full-length protein; it is known that FHL-1 can bind to heparin (a highly sulfated form of HS), but as yet the characterization of its GAG-binding properties have not been reported [21].
The importance of the GAG-binding specificity to FH’s function in self recognition is evidenced by the Y402H polymorphism that changes a tyrosine to histidine at amino acid 402 within CCP7 altering the sulfation pattern recognized in HS [45, 50, 56]; the 402H variant of FH requires more highly sulfated HS for binding, whereas the 402Y form of the protein recognizes a broader range of HS structures. It is unusual that a single amino acid change has such a pronounced effect on HS binding but structural studies (involving NMR analysis and crystallography) indicate that the two FH variants likely use a different mode of interaction (see [45, 53]); evidence suggests that the histidine-402 can interact directly with a sulfate group on HS, whereas when a tyrosine is in this sequence position such an interaction is not possible. The 402H and 402Y variants of FH have differential ability to interact with HS within the human Bruch’s membrane, where the former binds less well presumably because the HS sequences it recognizes are rare compared to those bound by the 402Y form of the protein [53]. This is an important finding since the Y402H polymorphism has been found to be a major risk factor for AMD, a disease where dysregulation of the complement AP has been strongly implicated and where the pathology develops in the central part of the retina (the macula) at the interface of the Bruch’s membrane and RPE (see [30]); the restricted binding specificity of the disease variant of FH has been hypothesized to lead to its impaired association with the Bruch’s membrane, and because this extracellular matrix doesn’t have any other inhibitors of the AP, this would allow inappropriate complement activation driving disease progression [50, 53]. Moreover, recent work has shown that there is a substantial reduction in the amount of HS in human Bruch’s membrane with age (and a small, but significant, reduction in the level of HS sulfation) that likely explains the larger difference in the relative binding activities of the 402H and 402Y variants in old versus young tissues [57]; interestingly, similar changes in HS level and composition are not seen in a closely positioned region of the eye (i.e., the neurosensory retina). Thus, it can be envisaged that tissue-specific (and even more localized) changes in the amount and/or composition of glycans with age or disease would have a profound effect on the ability of FH to recognize markers of self and, thereby, affect its regulatory functions (see [14, 30, 31]). In addition to AMD, this is of relevance to a number of pathologies where there is dysregulation of the complement AP, including aHUS and DDD; see below for further detailed discussion.
Other ligands of factor H
Matrix proteins
The extracellular matrix contributes to all aspects of tissue/organ function, dictating the tensile strength and turgor of the tissue and directing cell phenotype and function both through the communication of mechanical cues and by acting as a reservoir and gate keeper for the myriad of extracellular signaling molecules. A remarkable example of matrix is that of articular cartilage, since it accounts for ~95% of the tissue volume; the cartilage matrix is composed of collagens and other fibril-forming proteins that provide tensile strength and elasticity as well as proteoglycans that hydrate the tissue (and thus give rise to its load-bearing properties) and also contribute to formation of the fibrillar network [58]. Importantly, the matrix has been found to play a major role in the regulation of both the adaptive and the innate immune system [59] and, as already described above, the glycomatrix (i.e., GAGs and sialic acid) serve to prevent inappropriate complement activation on host tissues [14]. However, in inflammatory diseases the dysregulated synthesis of matrix components and matrix breakdown not only leads to the activation of further inflammation via toll-like receptors (i.e. responding to these damage-associated molecular patterns [59]), but also can influence the complement system in a multitude of ways (see [60]). For instance, many components of the cartilage matrix have been implicated in the modulation of the complement system, e.g. in the context of rheumatoid arthritis, where several small leucine-rich proteoglycans (SLRPs) appear to play a role [60–63]; these matrix molecules, hidden in the cartilage matrix in healthy tissues, are released into the synovial fluid (e.g., as fragments) where they can interact with components of complement. In this regard, three SLRPs (fibromodulin, osteoadherin and chondroadherin) have been shown to be potent activators of the CP, via the selective recruitment of C1q [61, 62]. However, these proteins can also bind FH (and C4BP), where these interactions have been proposed to limit complement activation to the early steps of the CP [61–63]; interestingly, it is the CCP 6–8 region of FH that mediates the interaction with fibromodulin and chondroadherin, where in both cases the Y402H polymorphism reduces the strength of binding [61, 64]. The NC4 domain of type IX collagen, which is proteolytically released from cartilage during articular joint disease, can directly inhibit complement activation via its association with C3, C4 and C9, where for example the latter inhibits C9 polymerization and the formation of the membrane-attack complex [65]; it can also bind to FH and enhance FH’s cofactor activity for the FI-mediated breakdown of C3b. Furthermore, the NC4 fragment can also interact with fibromodulin and osteoadherin and inhibit their binding to C1q, illustrating the highly complex role of the matrix in the regulation of complement.
Apoptotic and necrotic cells
To maintain tissue homeostasis, old and damaged cells undergo apoptosis and are replaced by new ones [66]. In the apoptotic process several changes occur in the morphology of the nuclear and plasma membrane that lead to recognition and phagocytosis of apoptotic cells by professional phagocytes; this includes several roles for complement, including opsonization (e.g., with C1q and C3b) and lysis of apoptotic cells [67, 68]. Rapid and efficient removal of apoptotic cells is clearly important to make space for living cells and to maintain tissue homeostasis; especially, since uncleared dying cells can undergo secondary necrosis, with the release of their intracellular contents, where this has been linked to autoimmune diseases (see [69]). During apoptosis the activation of caspases leads to the release of soluble chemo-attractants that can guide phagocytes towards apoptotic cells, where the discrimination of dying from live cells involves exposure of molecules on the cell surface and changes in charge and glycosylation patterns [70–73]. Importantly, FH can bind to molecules that become exposed on cells during apoptosis (and necrosis), including DNA, histones and annexin II (and possibly the altered glycocalyx), where it is believed that this serves to minimize the downstream pro-inflammatory effects of complement activation and the release of autoantigens that would trigger autoimmunity [62, 64, 68, 69]; FH binding may also compensate for the loss of some membrane-bound inhibitors of complement. In this regard, blocking of FH binding to apoptotic cells leads to increased opsonization with C3b and MAC-mediated lysis [69]. Three regions of the FH protein (CCPs 6–8, 8–15 and 19–20) have been implicated in the binding to apoptotic cells (whereas only CCPs 6–8 and 19–20 are involved in the association of FH with necrotic cells), where all of these regions mediate the binding to histones, the CCP 6–8 region mediates the interaction with annexin II and both the CCP 6–8 and CCP 19–20 regions bind DNA; the Y402H polymorphism affects the binding of FH to annexin II and DNA, impairing the former and enhancing the latter [64, 68]. Interestingly, FH did not bind to either GAGs or sialic acid on apoptotic cells revealing that FH uses a different mechanism of action compared to that described above for the recognition of healthy host cells.
As discussed in the section below, FH also interacts with pentraxins, e.g. C-reactive protein (CRP) and pentraxin 3 (PTX3), which are soluble pattern recognition molecules (PRMs) that recognize apoptotic and necrotic cells and recruit FH on to their surface [74–77].
Pentraxins
Pentraxins, which can be subdivided into short and long forms, are characterized by a cyclic multimeric structure, and members of this family share a 200-amino acid pentraxin domain that is highly conserved in evolution [1, 78–80]. CRP and serum amyloid P component (SAP), which are pentameric and considered to be the prototypic short pentraxins, are both produced in the liver during inflammation (e.g. in response to IL-6); they play key roles in innate immune and tissue homeostasis, where they help remove cellular debris and matrix components [1, 80, 81]. PTX3 is a long pentraxin that was identified as an IL-1-inducible gene in endothelial cells and as a TNF-stimulated gene in fibroblasts [82]. PTX3 is produced locally (e.g. during inflammation) by various cell types, including vascular, stromal and innate immune cells [83]. While the short pentraxins CRP and SAP are composed primarily of a pentraxin domain, PTX3 has an additional ~160 residue domain at its N-terminal end [80, 84]. Both short and long pentraxins interact with components of the three complement pathways, including FH [74, 76] and C4BP [85, 86].
PTX3, which is an octamer composed of eight identical protomers [87], enhances the binding of FH to apoptotic cells, and thus the deposition of iC3b (see [76]). The binding of PTX3 to FH occurs through CCP7 and CCPs 19–20 (see Fig. 2), where these regions interact with the C-terminal and N-terminal domains of PTX3, respectively [76]. In this case, the Y402H polymorphism was reported to have no effect [76], however, FHL-1 is able to bind [88]. Interestingly, the glycosylation status of PTX3’s C-terminal domain has been shown to affect its interaction with FH, suggesting that tissue specific differences in this post-translational modification might play a role in tuning PTX3 activity [76, 89]. Furthermore, mutations in CCP 20, associated with increased risk for aHUS, have been reported to affect the interaction with PTX3, which has allowed the identification of a putative binding site on this terminal CCP of FH [88].
CRP cooperates with the complement system to remove apoptotic/necrotic cells [74, 90]. Indeed, it can activate both CP and LP by interacting with C1q and ficolins, respectively; this promotes complement-mediated opsonization and enhanced phagocytosis of target cells [91, 92]. In particular, CRP can bind to Fcγ receptors on leukocytes and thereby directly activate phagocytosis [93]. The interaction of FH with CRP [75, 94] has been shown to play important roles at sites of tissue damage and local inflammation, for example, in the atherosclerotic plaque [94]; the binding is believed to be mediated primarily by CCP7 with other sites in CCPs 8–11 (see [64, 95]) and CCPs 19–20 [75, 77]. Complement activation in the arterial intima can be initiated by components of both CP (i.e., C1q) and AP (i.e., C3b) that have been deposited on activating surfaces, such as immune complexes, CRP and oxidized LDL particles. When bound to CRP and cell surface glycans, FH down-regulates complement and protects the endothelium from MAC-mediated damage. Importantly, the FH Y402H polymorphism markedly reduces CRP binding capacity [64, 95], and could, therefore, lead to aberrant regulation of the complement cascade, e.g. in the context of AMD (see below). In other studies it has been proposed that it is the monomeric (pro-inflammatory) rather than pentameric (anti-inflammatory) form of CRP that interacts with FH and, in this way, limits complement activation, increases phagocytosis of apoptotic cells and reduces the inflammatory response by macrophages [77]. Interestingly, it has been observed that the 402H variant of FH has reduced binding to monomeric CRP as compared to the 402Y allele, and, therefore, in this context, may have an impaired anti-inflammatory activity [64, 96, 97].
Cellular receptors
FH can bind several cell types and its functions go beyond the canonical role of a complement inhibitor [98]. For example, Avery et al. first identified complement receptor 3 (CR3, also known as CD11b/CD18 or integrin αMβ2) as a FH receptor on human neutrophils [99], where CR3 engagement by FH and FHL-1 modulates cell activation and function [100]; interestingly, recent data show that while FH can support neutrophil recruitment, it may also play an anti-inflammatory role, reducing tissue damage by influencing the formation of neutrophil extracellular traps [101]. Available data indicate that FH likely also binds CR4, possibly triggering a similar cellular response, e.g. on macrophages and dendritic cells that (unlike neutrophils) express significant amounts of CR4. Also new roles for the FH-CR3 complex have been proposed based on the observation that, when FH is bound to the yeast form of C. albicans, neutrophils strongly adhere and exhibit increased phagocytosis of this opportunistic fungal pathogen [99]. Therefore, in a sharp contrast to its role as a complement inhibitor, FH might promote the physical contact between neutrophils and certain pathogens, thereby increasing antimicrobial activity. Conversely, it has also been found that the FH-CR3 interaction can facilitate the entry of certain pathogens (i.e., Streptococcus pneumoniae and Neisseria gonorrhoeae) into host cells [102, 103].
In addition to CR3 and CR4, other membrane-bound molecules have been proposed as putative FH receptors, such as integrins (e.g. αIIbβ3) and L-selectin on human neutrophils, B lymphocytes, monocytes, and platelets [104–108]; as discussed below, thrombomodulin, a cell-surface thrombin-binding protein and regulator of the coagulation system, is also a ligand for FH on endothelial cells [109, 110]. Moreover, it has been observed that FH binds human B lymphocytes, thus inducing calcium-dependent FI release [105] and triggering blastogenesis [111]. Also, a chemotactic effect on monocytes has been reported for both intact [112] and thrombin-cleaved FH [113]. These additional interactions appear to mediate different functions to those involved in host recognition and complement regulation, ranging from the control of cell adhesion and migration to cytokine induction and a potential direct modulatory role on B cells. In this regard, FH might have a direct anti-inflammatory and tolerogenic activity towards infiltrated leukocytes, as suggested by the recent observation that monocyte-derived dendritic cells that have been treated with FH have lower expression of maturation markers and costimulatory molecules, decreased production of pro-inflammatory Th1-cytokines (i.e., IL-12, TNF, IFN-γ, IL-6, and IL-8), and preferential production of immunomodulatory mediators (i.e., IL-10 and TGF-β) [114].
Miscellaneous ligands
FH has been identified as a binding partner for adrenomedullin (AM) (see [115, 116]), a regulatory peptide expressed in many tissues and cell types (including monocytes, macrophage, lymphocytes and granulocytes, and that is particularly abundant in the vascular endothelium), which has important physiological roles encompassing effects on vasodilatation, bronchodilatation, regulation of hormone secretion, control of apoptosis, angiogenesis and antimicrobial action [117, 118]. FH, studied under the name of AM-binding protein-1 [115, 116], has been found to enhance AM-mediated induction of cAMP in fibroblasts, augment the AM-mediated growth of a cancer cell line, and suppress the bactericidal capability of AM on E. coli. These activities are thought to be due to a stabilization effect of FH on AM, where FH inhibits the cleavage of AM by its candidate protease matrix metalloprotease 2 (see [116]) and co-administration of AM and FH reduces liver and kidney damage as well as systemic inflammation in a rat model of haemorrhagic shock [115]. Given the prominent roles of AM in blood pressure homeostasis, these data strongly encourage the targeting of AM with FH or FH-derived peptides/fragments as a pharmacological approach in the therapy of haemorrhagic shock and sepsis. In this regard, FH has been reported to interact with AM through a number of regions, where a high affinity site may be present in CCPs 15–20; see the excellent review by Sim et al. [116] for further discussion.
Aberrant accumulation of prion protein (PrP) is recognized as an important process in a number of neurodegenerative transmissible spongiform encephalopathies such as Creutzfeldt-Jakob disease in humans and bovine spongiform encephalopathy in cattle (see [119]). The complement system might play a role in the pathogenesis of these conditions, given that C1q, C3b and MAC are present in the PrP plaques of human brains with prion disease [120]; moreover, oligomers of human PrP can activate both the AP and CP. However, complement activation is restricted to its early stages (i.e., MAC formation initiated by PrP is at a low level in comparison to the deposition of C3b and C4b), where this has been ascribed to the ability of PrP to establish additional interactions with FH and C4BP [119]. Thus, complement might represent a target for neurodegenerative diseases associated with prions.
Additional ligands of FH include markers of oxidative stress, such as malondialdehyde (MDA) and malondialdehyde acetaldehyde (MAA) [121–123], and serum apolipoprotein E (ApoE) [124]. For example, FH is a major MDA-binding protein that interacts via CCPs 7 and 20 (see Fig. 2), where the Y402H polymorphism leads to substantially impaired binding [123]. Through its interaction with MDA, FH has been shown to bind to apoptotic and necrotic cells, inactivate complement (via its cofactor activity) on MDA-labeled surfaces, inhibit the uptake of MDA-modified proteins by macrophages and neutralize the pro-inflammatory effects of MDA in leukocytes and stromal cells, e.g. inhibiting the production of IL-8 in RPE cells. Thus, FH’s recognition of products of lipid peroxidation might help prevent excessive complement activation at sites of oxidative tissue damage and inflammation, e.g. in the context of chronic inflammatory diseases [125]. FH can also interact with ApoE, via CCPs 5–7 (see Fig. 2), on high density lipoprotein (HDL) particles in plasma and can thereby regulate/prevent complement activation [124].
Factor H and immune evasion
Microbes have evolved several strategies to escape the immune response allowing their spread in the host [48, 126]. Given that the complement system is at the forefront of the immunological battle against invading microorganisms, it is not surprising that a number of its components have become the targets of major evasion mechanisms. For example, some microbes express proteolytic enzymes that inactivate C3b on their own surface or contain glycophosphatidylinositol-anchored complement inhibitors [127]. An additional way for pathogens to escape complement attack is by recruiting (i.e., hijacking) complement inhibitors from the host, such as FH (and C4BP) [128–130]. Indeed, some pathogens mimic a host surface by capturing these soluble complement regulators or, alternatively, target and inactivate complement inhibitors with secreted proteins/proteases [48, 131]; once a given pathogen has recruited complement inhibitors on to its own surface, it is likely protected from the complement attack as if it were self. There are several examples of bacteria that can acquire FH via the presentation of specific binding proteins. Amongst these are the factor H binding protein (fHbp) of Neisseria meningitidis [132], the outer surface protein E [133] and CspA [134] of Borrelia burgdorferi, Sbi (the staphylococcal binder of IgG) from Staphylococcus aureus [135] and the pneumococcal surface protein C of Streptococcus pneumoniae [129, 136, 137]. Despite the fact that these organisms are evolutionary distinct from each other and the FH ligands they express are structurally different, they all share a related binding site on FH that is localized on a common face of CCP20 [126]; these interactions enhance the binding of FH CCPs 19–20 to C3b, thus leading to a stable ternary complex between FH, C3b and the microbial protein, with increased co-factor activity (with conversion of C3b to iC3b). Thus, this super-evasion site, resulting from convergent evolution, provides further compelling evidence for the role of FH as a key regulator of the immune system.
An additional evasion site has been identified in the CCP7 domain, which mediates the recruitment of FH onto the surface of Streptococcus pyogenes, via the M protein, the main virulence factor of group A streptococcus (see [53, 138]). Interestingly, the Y402H polymorphism leads to reduced binding of FH to this organism and its increased phagocytosis in blood. These observations suggest that the presence of the 402H allotype might be protective against infections from group A streptococcus, which can be fatal in childhood, and might partly explain the high frequency of Y402H in some populations, e.g. ~30% of European-descended individuals carry the 402H allele [53]. The CCP 6–7 region (in addition to CCP20) has been described as a major binding site for fHbp of Neisseria meningitidis, however, the Y402H polymorphism does not affect this interaction [139]. Interestingly, the high affinity of fHbp for FH suggests that N. meningitidis could rapidly sequester FH from plasma, depleting its levels in circulation and allowing C3 turnover, a process that might contribute to the dramatic haemorrhagic rash seen in meningococcal sepsis. Other bacteria, for instance Pseudomonas aeruginosa, express sialic acid on their surface [140], where this is used to mimic a self-glycan pattern and recruit FH from the host’s blood, thereby escaping the complement-mediated immune response [141]. It has recently been suggested that the binding of FH to ApoE on HDL (via its CCP 5–7 region) provides an explanation for why it is beneficial for bacteria to bind to this part of FH [124]; i.e. by binding FH in this way bacteria are able to mimic plasma HDL increasing their survival in blood.
Similar evasion strategies have been developed by fungi. For example, the opportunistic fungus Aspergillus fumigatus has acquired the ability to recruit both FH and C4BP onto its cell wall [128], however, in this case the microbial ligands are unknown [142]. As noted above, some pathogens can escape the immune response by synthesizing and secreting soluble proteases that can target selected complement components. This is the case for Candida albicans whose aspartic protease 2 inactivates FH and the macrophage FH-receptors CR3 and CR4 [131]. In this context, it appears that the selective degradation of FH by the microbe-encoded protease helps circumvent the pro-phagocytic activity of FH (see above).
Although not directly related to the recognition of microorganisms, a noticeable example of FH-mediated immune evasion is represented by mosquitoes, which are important vectors in the transmission of many human diseases (e.g., malaria, dengue, yellow fever etc.) [143]. In this regard, human FH from a blood meal has been found to bind the luminal surface of the mosquito’s midgut and, via its cofactor activity, prevent complement activation on these surfaces, allowing the mosquito to tolerate the ingestion of large quantities of human blood. Interestingly, the merozoites from Plasmodium falciparum (i.e., the invasive form of the malarial parasite released on erythrocyte rupture) have been shown to evade the complement system by capturing FH and FHL-1 from serum (via the Pf92 protein) and down-regulating the AP [130].
The discovery of complement-evasion strategies employed by microorganisms, as described above, has led to the development of complement vaccines [48, 144]. Potential targets of these are the ligands of complement inhibitors made by bacteria; such that, if binding is prevented then complement activation would be expected to be promoted on the bacterial surface leading to its phagocytosis or lysis [48]. For example, recent progress has been made towards the development of a vaccine for N. meningitidis by targeting the factor H-binding protein fHbp [145]. Alternatively, engineered forms of FH might be used to elicit rapid and robust complement responses against specific pathogens. This is the approach that is forming the basis of a novel strategy for the treatment of the sexually transmitted infection gonorrhea, where the infectious agent Neisseria gonorrhoeae has developed resistance to almost every conventional antibiotic [146–148]. This bacteria escapes complement activation by interacting via its factor H binding protein with FH at sites within CCPs 6–7 and CCPs 18–20 [146]. Based on this, a chimeric protein comprising CCPs 18–20 of human FH fused to IgG Fc was designed, where key amino acids involved in FH binding to host cells were mutated while retaining its binding to gonococci [147]; this construct activated the CP and promoted a strong complement-dependent bactericidal activity. Recent work has incorporated a mutation into the CCP 19–20 region of the fusion protein that abolishes its binding to erythrocytes while retaining the interaction with N. gonorrhoeae, which promotes complement-mediated killing of the bacteria but without causing lysis of red blood cells [148]; in vivo testing in a mouse model of gonococcal infection has provided evidence that this has potential as a therapeutic against multidrug-resistant Neisseria gonorrhoeae.
Factor H-associated diseases
FH plays a prominent role in the pathogenesis of several diseases in which genetic variations in the CFH gene are associated with modifying risk and/or where complement dysregulation is central to their etiology; these include typical and atypical HUS [27, 149, 150], AMD [30] and DDD [32, 33]. Furthermore, FH has long been regarded as a target for immune evasion by cancer cells [151], in a similar way to pathogens that hijack it to protect themselves from a complement-mediated response [126]. However, this paradigm has been recently challenged by the observation that reduced or impaired recruitment of FH to the tumor site can cause local inflammation, which in turn sustains rather than prevents cancerogenesis [152]; see further discussion below.
Hemolytic uremic syndrome
HUS is the most common cause of acute renal failure amongst children and its clinical symptoms are often a combination of hemolytic anemia, thrombocytopenia and acute renal failure [153]. This disease is classified as either typical or atypical and referred to as HUS and aHUS, respectively. HUS is characterized by bloody diarrhea caused by verotoxin/shiga toxin producing enterohaemorrhagic E. coli (EHEC) [27]. The pathogenesis of this form of HUS is still poorly understood, however, it has been reported that shiga toxin causes damage to endothelial and epithelial cells by inhibiting protein synthesis and inducing apoptosis [154]. Importantly, this toxin activates complement, mostly via AP in the fluid-phase, and delays the cofactor activity of FH on the cell surface, thus exacerbating complement-dependent tissue damage [155]. Shiga toxin interacts with FH at sites within CCPs 6–8 (and CCPs 18–20) where the 402Y variant of FH had somewhat higher binding activity compared to the 402H form, such that individuals carrying the former allele might be more vulnerable to HUS. Furthermore, a direct interaction between shiga toxin and FHL-1 has been described, indicating that this truncated variant of FH might also have a role in the pathogenesis of EHEC-associated HUS [26].
aHUS is not caused by an infection, but instead is directly associated with FH mutations that impair recognition of self surfaces [13, 156]. As described above, the C-terminal CCP20 domain of FH is known to mediate interactions with glycan markers on certain host cells (e.g., endothelial cells) and in the matrix, and in this way provides protection from autologous complement activation [49]. Most aHUS-associated FH mutations map to this region of the protein (see [13]), and have been described to impair cell binding and cell surface protection from complement attack, thus contributing to endothelial injury and microvascular thrombus formation, with major clinical manifestations at the level of the central nervous system and kidney [40, 156]; indeed, recent work has revealed that certain aHUS mutations impair the recognition of sialic acid by FH on endothelial cells, erythrocytes and platelets, indicating that sialic acid (rather than HS) is critical for FH-mediated complement regulation on these cell types [157]. Furthermore, many patients with aHUS have FH autoantibodies that are directed against the C-terminal domain (i.e. CCP20), thus inhibiting the binding of FH to host surfaces [158–160].
In addition to FH, mutations in other genes have been reported to be associated with aHUS, including C3, FB, FI, MCP and thrombomodulin [156, 161, 162]. Thrombomodulin is a cell surface FH-binding protein that enhances FH cofactor activity probably through stabilization of the FH-C3b complex on endothelial cells [110], where aHUS-associated mutations in thrombomodulin interfere with the FH interaction [109]. Thus, thrombomodulin, in addition to its classical role as an inhibitor of the coagulation system, also provides an alternative mechanism by which FH can protect endothelial surfaces.
Dense deposit disease (DDD)
Dense deposit disease, formerly known as membranoproliferative glomerulonephritis type II, is a renal disorder characterized by unchecked AP activation in the plasma and high levels of C3 deposition on the glomeruli, with severe impairment of the kidney’s draining functions [32, 33]; as its name suggests electron-dense intramembranous deposits form within the glomerular basement membrane, a specialized extracellular matrix of the kidney. Several genes have been associated with this pathology, and most of them code for components of the complement system, including FH [28, 33]. DDD-associated mutations in the CFH gene mainly affect FH protein folding and secretion, and its cofactor activity [163, 164]; for example, a mutation causing the replacement of a cysteine with a tyrosine at position 431 (in CCP7) induces protein aggregation [163], whereas mutations in CCP4 lead to impaired cofactor activity [41]. DDD (and aHUS) can also be caused by total deficiencies in FH production, where the defective control of AP activation leads to C3b deposition in kidney glomeruli and a secondary deficiency in C3 (due to its uncontrolled turnover) (see [165]). Furthermore, autoantibodies against FH can also be associated with DDD where these block formation of the C3b/FH complex, providing another mechanism for uncontrolled complement activation [166, 167].
Age-related macular degeneration (AMD)
Age-related macular degeneration is the most common form of vision loss in the developed world. Indeed, it is predicted that by the year 2020, 196 million people worldwide will suffer from some form of the disease [168]. The condition manifests itself in the central part of the retina, called the macula, resulting ultimately in the complete loss of central vision. Late-stage (advanced) AMD is divided into two forms: wet and dry. In the former, there is growth of new (leaky) blood vessels from the choroid across the RPE and Bruch’s membrane into the retina and in the latter there is loss of cells and tissue structure in the choroid, RPE and neurosensory retina [30, 169]. Late-stage AMD is usually preceded by the formation of deposits called drusen, which contain complement activation products, that form on/in the Bruch’s membrane; as noted above this is an extracellular matrix that along with the RPE forms the outer blood-retinal barrier, allowing the transport of nutrients and waste products into and out of the retina while preventing the migration of leukocytes to this immune privileged site.
AMD is a multifactorial disease where there is a large genetic component, along with age, diet, and smoking all being additional risk factors [170]. A large number of genetic variants that alter the risk for developing advanced AMD have been identified through numerous genome-wide association studies (see [171, 172]); importantly, the majority of the strongest associations reside in genes of the complement system [172, 173]. Of these, the Y402H polymorphism in CFH is considered to represent the greatest (single) genetic risk factor of developing AMD [29, 170]. As described in detail already, this polymorphism affects the coding sequence of CCP7 in the full-length FH protein and FHL-1, since they are both produced by alternative splicing of the CFH gene; the altered specificity for GAGs (e.g., in the human Bruch’s membrane) as well as changes to other binding functions (e.g., CRP and apoptotic/necrotic cells) are of likely relevance to the pathology of AMD, where local (chronic) inflammation, driven by complement dysregulation, is thought to be of central importance [30, 31, 174].
In the case of FHL-1, which has been shown recently to be the predominant CFH gene product associated with Bruch’s membrane [21], CCP7 represents the only GAG-binding domain for the protein. Therefore, it is likely that FHL-1 is more susceptible to alterations in GAG recognition conferred by the Y402H polymorphism (although no data are yet published on this). Indeed, Bruch’s membrane from human donor eyes [175] failed to retain as much of the AMD-associated 402H variant of FHL-1 compared to the 402Y form when the proteins were individually diffused across this ECM [21]. The finding that the Y402H polymorphism has no effect on systemic levels or activity of FH/FHL-1 [176] is consistent with AMD being a disease of local complement dysregulation, e.g. at the interface of the RPE and Bruch’s membrane, which is where the pathology initially develops.
FH and cancer
There is increasing evidence that complement plays a role in cancerogenesis and in the tumor microenvironment [177]. Membrane-associated inhibitors of the complement system, such as DAF, MCP, CR1 and CD59, are highly expressed on certain tumors, which likely help cancer cells to escape complement activation. Similar strategies are adopted by tumor cells that express and release high amounts of FH, contributing to reduced complement activity in the tumor microenvironment [178]; in these cases, FH has indeed been proposed as a new cancer marker [179]. Interestingly, cancer cells also display an increased amount of sialic acid, and sometimes GAGs, on their surfaces, where this altered self (or super self) is believed to be able to recruit FH and provide protection against complement-mediated lysis (see [14]). Interestingly, sera from patients with early stage, non-metastatic, non-small cell lung cancer were found to contain autoantibodies against FH, where these recognize a conformationally distinct (reduced) form of the protein that is believed to be displayed only on the surface of tumor cells [180]; the autoantibodies interact with CCP 19 and inhibit the binding of FH to lung carcinoma cells, thereby promoting the deposition of C3b and increasing complement mediated tumor cell lysis. Furthermore, a recombinant anti-FH autoantibody has been demonstrated to activate complement on tumor cells, leading to the release of anaphylatoxins that induce complement-dependent cytotoxicity and inhibit tumor growth in vivo [181]. These recent findings provide an exciting opportunity to develop a novel immunotherapeutic strategy to treat cancer by targeting FH.
According to the traditional paradigm, complement is good, in that it recognizes, when not deceived, the cancer cell, and either directly (via MAC-mediated lysis) or indirectly (via complement-dependent cell toxicity) kills or disposes of it. New ideas, however, are emerging that challenge this and point to the complement system as a factor in the tumor’s ability to promote inflammation [152]. For example, in a mouse model of chemically induced carcinogenesis, the genetic deficiency of PTX3 (a ligand of FH; see above) causes an increase in both growth and size of the tumor lesions; this has been ascribed to defective recruitment of FH onto cancer cells in the PTX3 −/− mice. In fact, higher levels of C3 and C5a have been described in tumors from the PTX3-deficient animals, suggesting that complement regulation in the absence of this long pentraxin is subverted. These data indicate that the complement system might promote cancer-related inflammation, and in this way support rather than oppose cancerogenesis and tumor growth [152] (see Fig. 3). An active role of complement in tumor growth and metastasis has been proposed in other studies. For example, upon complement activation, the anaphylatoxins C3a and, in particular, C5a initiate and modulate the inflammatory response by mediating chemotaxis, inflammation and generation of reactive oxygen species (ROS) [182]. The resulting microenvironment appears to favor cancerogenesis as well as tumor immune escape and progression [183]. Thus, FH appears to have opposing roles in cancer and whether it promotes an unfavorable environment for cancer through its anti-inflammatory properties (complement assistance) and/or is hijacked by cancer cells to provide immune evasion (complement resistance) likely depends on the type/stage of cancer involved; clearly there is further work needed to more fully understand FH’s exact role in cancer immunology.

A dual role for FH in cancer. FH is expressed or recruited by cancer cells and this leads to reduced deposition of C3 and less production of the anaphylatoxin C5a. Inhibition of complement attack (complement resistance) can favor tumor survival and promote tumor growth (tumor promotion). However, in certain types of cancer that are inherently promoted and sustained by inflammation, the anti-inflammatory microenvironment generated by FH (complement assistance) might be unfavorable to tumor growth and progression (tumor suppression)
Concluding remarks
In this review we discussed the structure/function relationships of complement FH, with a focus on the emerging roles of this protein in the pathophysiology of infectious diseases, inflammatory conditions and cancer. FH is the major soluble inhibitor of the AP C3 convertase, an enzyme complex that is at the very center of the complement cascade. Therefore, alterations of FH protein structure, due to mutations and polymorphisms, likely lead to aberrant functional outcomes. This is the case for FH mutations associated with aHUS, AMD and DDD all diseases of local complement dysfunction where a loss of appropriate self-recognition, e.g. within the extracellular matrix, appears to be a common factor. Also, FH is a preferential target for complement evasion by a number of pathogens. This has paved the way towards complement vaccines, which are being designed and developed based on FH microbial ligands, and novel FH-based therapeutics. Finally, a dual role is emerging for FH in cancer, where it can either be hijacked by cancer cells to avoid the complement attack, or can be used to dampen cancer-related inflammation.
References
Bottazzi B, Doni A, Garlanda C, Mantovani A (2010) An integrated view of humoral innate immunity: pentraxins as a paradigm. Annu Rev Immunol 28:157–183. doi:10.1146/annurev-immunol-030409-101305
Iwasaki A, Medzhitov R (2010) Regulation of adaptive immunity by the innate immune system. Science 327(5963):291–295. doi:10.1126/science.1183021
Ricklin D, Reis ES, Lambris JD (2016) Complement in disease: a defence system turning offensive. Nat Rev Nephrol 12(7):383–401. doi:10.1038/nrneph.2016.70
Kemper C, Pangburn MK, Fishelson Z (2014) Complement nomenclature 2014. Mol Immunol 61(2):56–58. doi:10.1016/j.molimm.2014.07.004
Medzhitov R (2008) Origin and physiological roles of inflammation. Nature 454(7203):428–435. doi:10.1038/nature07201
Kemper C, Atkinson JP (2007) T-cell regulation: with complements from innate immunity. Nat Rev Immunol 7(1):9–18. doi:10.1038/nri1994
Chen ZA, Pellarin R, Fischer L, Sali A, Nilges M, Barlow PN, Rappsilber J (2016) Structure of complement C3(H2O) revealed by quantitative cross-linking/mass spectrometry and modelling. Mol Cell Proteomics. doi:10.1074/mcp.M115.056473
Carroll MC (2004) The complement system in B cell regulation. Mol Immunol 41(2–3):141–146. doi:10.1016/j.molimm.2004.03.017
Dunkelberger JR, Song WC (2010) Role and mechanism of action of complement in regulating T cell immunity. Mol Immunol 47(13):2176–2186. doi:10.1016/j.molimm.2010.05.008
Kwan WH, van der Touw W, Heeger PS (2012) Complement regulation of T cell immunity. Immunol Res 54(1–3):247–253. doi:10.1007/s12026-012-8327-1
Dunkelberger JR, Song WC (2010) Complement and its role in innate and adaptive immune responses. Cell Res 20(1):34–50. doi:10.1038/cr.2009.139
Ricklin D, Hajishengallis G, Yang K, Lambris JD (2010) Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11(9):785–797. doi:10.1038/ni.1923
Blaum BS, Hannan JP, Herbert AP, Kavanagh D, Uhrin D, Stehle T (2015) Structural basis for sialic acid-mediated self-recognition by complement factor H. Nat Chem Biol 11(1):77–82. doi:10.1038/nchembio.1696
Langford-Smith A, Day AJ, Bishop PN, Clark SJ (2015) Complementing the sugar code: role of GAGs and sialic acid in complement regulation. Front Immunol 6:25. doi:10.3389/fimmu.2015.00025
Perkins SJ, Nan R, Li K, Khan S, Miller A (2012) Complement factor H-ligand interactions: self-association, multivalency and dissociation constants. Immunobiology 217(2):281–297. doi:10.1016/j.imbio.2011.10.003
Whaley K (1980) Biosynthesis of the complement components and the regulatory proteins of the alternative complement pathway by human peripheral blood monocytes. J Exp Med 151(3):501–516
Katz Y, Strunk RC (1988) Synthesis and regulation of complement protein factor H in human skin fibroblasts. J Immunol 141(2):559–563
Brooimans RA, van der Ark AA, Buurman WA, van Es LA, Daha MR (1990) Differential regulation of complement factor H and C3 production in human umbilical vein endothelial cells by IFN-gamma and IL-1. J Immunol 144(10):3835–3840
Devine DV, Rosse WF (1987) Regulation of the activity of platelet-bound C3 convertase of the alternative pathway of complement by platelet factor H. Proc Natl Acad Sci USA 84(16):5873–5877
Chen M, Forrester JV, Xu H (2007) Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp Eye Res 84(4):635–645. doi:10.1016/j.exer.2006.11.015
Clark SJ, Schmidt CQ, White AM, Hakobyan S, Morgan BP, Bishop PN (2014) Identification of factor H-like protein 1 as the predominant complement regulator in Bruch’s membrane: implications for age-related macular degeneration. J Immunol 193(10):4962–4970. doi:10.4049/jimmunol.1401613
Clark SJ, Bishop PN (2015) Role of factor H and related proteins in regulating complement activation in the macula, and relevance to age-related macular degeneration. J Clin Med 4(1):18–31. doi:10.3390/jcm4010018
Ripoche J, Day AJ, Harris TJ, Sim RB (1988) The complete amino acid sequence of human complement factor H. Biochem J 249(2):593–602
de Paula PF, Barbosa JE, Junior PR, Ferriani VP, Latorre MR, Nudelman V, Isaac L (2003) Ontogeny of complement regulatory proteins—concentrations of factor h, factor I, c4b-binding protein, properdin and vitronectin in healthy children of different ages and in adults. Scand J Immunol 58(5):572–577
Meri S (2016) Self-nonself discrimination by the complement system. FEBS Lett. doi:10.1002/1873-3468.12284
Poolpol K, Orth-Holler D, Speth C, Zipfel PF, Skerka C, de Cordoba SR, Brockmeyer J, Bielaszewska M, Wurzner R (2014) Interaction of Shiga toxin 2 with complement regulators of the factor H protein family. Mol Immunol 58(1):77–84. doi:10.1016/j.molimm.2013.11.009
Reis ES, Falcao DA, Isaac L (2006) Clinical aspects and molecular basis of primary deficiencies of complement component C3 and its regulatory proteins factor I and factor H. Scand J Immunol 63(3):155–168. doi:10.1111/j.1365-3083.2006.01729.x
Ying L, Katz Y, Schlesinger M, Carmi R, Shalev H, Haider N, Beck G, Sheffield VC, Landau D (1999) Complement factor H gene mutation associated with autosomal recessive atypical hemolytic uremic syndrome. Am J Hum Genet 65(6):1538–1546. doi:10.1086/302673
Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R (2005) A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA 102(20):7227–7232. doi:10.1073/pnas.0501536102
McHarg S, Clark SJ, Day AJ, Bishop PN (2015) Age-related macular degeneration and the role of the complement system. Mol Immunol 67(1):43–50. doi:10.1016/j.molimm.2015.02.032
Langford-Smith A, Keenan TD, Clark SJ, Bishop PN, Day AJ (2014) The role of complement in age-related macular degeneration: heparan sulphate, a ZIP code for complement factor H? J Innate Immun 6(4):407–416. doi:10.1159/000356513
Smith RJ, Alexander J, Barlow PN, Botto M, Cassavant TL, Cook HT, de Cordoba SR, Hageman GS, Jokiranta TS, Kimberling WJ, Lambris JD, Lanning LD, Levidiotis V, Licht C, Lutz HU, Meri S, Pickering MC, Quigg RJ, Rops AL, Salant DJ, Sethi S, Thurman JM, Tully HF, Tully SP, van der Vlag J, Walker PD, Wurzner R, Zipfel PF (2007) New approaches to the treatment of dense deposit disease. J Am Soc Nephrol 18(9):2447–2456. doi:10.1681/ASN.2007030356
Barbour TD, Pickering MC, Terence Cook H (2013) Dense deposit disease and C3 glomerulopathy. Semin Nephrol 33(6):493–507. doi:10.1016/j.semnephrol.2013.08.002
Makou E, Herbert AP, Barlow PN (2013) Functional anatomy of complement factor H. Biochemistry 52(23):3949–3962. doi:10.1021/bi4003452
Makou E, Herbert AP, Barlow PN (2015) Creating functional sophistication from simple protein building blocks, exemplified by factor H and the regulators of complement activation. Biochem Soc Trans 43(5):812–818. doi:10.1042/BST20150074
Makou E, Mertens HD, Maciejewski M, Soares DC, Matis I, Schmidt CQ, Herbert AP, Svergun DI, Barlow PN (2012) Solution structure of CCP modules 10-12 illuminates functional architecture of the complement regulator, factor H. J Mol Biol 424(5):295–312. doi:10.1016/j.jmb.2012.09.013
Schmidt CQ, Herbert AP, Mertens HD, Guariento M, Soares DC, Uhrin D, Rowe AJ, Svergun DI, Barlow PN (2010) The central portion of factor H (modules 10-15) is compact and contains a structurally deviant CCP module. J Mol Biol 395(1):105–122. doi:10.1016/j.jmb.2009.10.010
Morgan HP, Mertens HD, Guariento M, Schmidt CQ, Soares DC, Svergun DI, Herbert AP, Barlow PN, Hannan JP (2012) Structural analysis of the C-terminal region (modules 18–20) of complement regulator factor H (FH). PLoS One 7(2):e32187. doi:10.1371/journal.pone.0032187
Morgan HP, Schmidt CQ, Guariento M, Blaum BS, Gillespie D, Herbert AP, Kavanagh D, Mertens HD, Svergun DI, Johansson CM, Uhrin D, Barlow PN, Hannan JP (2011) Structural basis for engagement by complement factor H of C3b on a self surface. Nat Struct Mol Biol 18(4):463–470. doi:10.1038/nsmb.2018
Kajander T, Lehtinen MJ, Hyvarinen S, Bhattacharjee A, Leung E, Isenman DE, Meri S, Goldman A, Jokiranta TS (2011) Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc Natl Acad Sci USA 108(7):2897–2902. doi:10.1073/pnas.1017087108
Wu J, Wu YQ, Ricklin D, Janssen BJ, Lambris JD, Gros P (2009) Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat Immunol 10(7):728–733. doi:10.1038/ni.1755
Schmidt CQ, Herbert AP, Kavanagh D, Gandy C, Fenton CJ, Blaum BS, Lyon M, Uhrin D, Barlow PN (2008) A new map of glycosaminoglycan and C3b binding sites on factor H. J Immunol 181(4):2610–2619
Alcorlo M, Lopez-Perrote A, Delgado S, Yebenes H, Subias M, Rodriguez-Gallego C, Rodriguez de Cordoba S, Llorca O (2015) Structural insights on complement activation. FEBS J 282(20):3883–3891. doi:10.1111/febs.13399
De Groot AS, Ross TM, Levitz L, Messitt TJ, Tassone R, Boyle CM, Vincelli AJ, Moise L, Martin W, Knopf PM (2015) C3d adjuvant effects are mediated through the activation of C3d-specific autoreactive T cells. Immunol Cell Biol 93(2):189–197. doi:10.1038/icb.2014.89
Prosser BE, Johnson S, Roversi P, Herbert AP, Blaum BS, Tyrrell J, Jowitt TA, Clark SJ, Tarelli E, Uhrin D, Barlow PN, Sim RB, Day AJ, Lea SM (2007) Structural basis for complement factor H linked age-related macular degeneration. J Exp Med 204(10):2277–2283. doi:10.1084/jem.20071069
Skerka C, Lauer N, Weinberger AA, Keilhauer CN, Suhnel J, Smith R, Schlotzer-Schrehardt U, Fritsche L, Heinen S, Hartmann A, Weber BH, Zipfel PF (2007) Defective complement control of factor H (Y402H) and FHL-1 in age-related macular degeneration. Mol Immunol 44(13):3398–3406. doi:10.1016/j.molimm.2007.02.012
Pangburn MK, Pangburn KL, Koistinen V, Meri S, Sharma AK (2000) Molecular mechanisms of target recognition in an innate immune system: interactions among factor H, C3b, and target in the alternative pathway of human complement. J Immunol 164(9):4742–4751
Meri S, Jordens M, Jarva H (2008) Microbial complement inhibitors as vaccines. Vaccine 26(Suppl 8):I113–I117
Clark SJ, Ridge LA, Herbert AP, Hakobyan S, Mulloy B, Lennon R, Wurzner R, Morgan BP, Uhrin D, Bishop PN, Day AJ (2013) Tissue-specific host recognition by complement factor H is mediated by differential activities of its glycosaminoglycan-binding regions. J Immunol 190(5):2049–2057. doi:10.4049/jimmunol.1201751
Clark SJ, Perveen R, Hakobyan S, Morgan BP, Sim RB, Bishop PN, Day AJ (2010) Impaired binding of the age-related macular degeneration-associated complement factor H 402H allotype to Bruch’s membrane in human retina. J Biol Chem 285(39):30192–30202. doi:10.1074/jbc.M110.103986
Ferreira VP, Herbert AP, Cortes C, McKee KA, Blaum BS, Esswein ST, Uhrin D, Barlow PN, Pangburn MK, Kavanagh D (2009) The binding of factor H to a complex of physiological polyanions and C3b on cells is impaired in atypical hemolytic uremic syndrome. J Immunol 182(11):7009–7018. doi:10.4049/jimmunol.0804031
Clark SJ, Bishop PN, Day AJ (2013) The proteoglycan glycomatrix: a sugar microenvironment essential for complement regulation. Front Immunol 4:412. doi:10.3389/fimmu.2013.00412
Clark SJ, Bishop PN, Day AJ (2010) Complement factor H and age-related macular degeneration: the role of glycosaminoglycan recognition in disease pathology. Biochem Soc Trans 38(5):1342–1348. doi:10.1042/BST0381342
Clark SJ, Keenan TD, Fielder HL, Collinson LJ, Holley RJ, Merry CL, van Kuppevelt TH, Day AJ, Bishop PN (2011) Mapping the differential distribution of glycosaminoglycans in the adult human retina, choroid, and sclera. Invest Ophthalmol Vis Sci 52(9):6511–6521. doi:10.1167/iovs.11-7909
Keenan TD, Clark SJ, Unwin RD, Ridge LA, Day AJ, Bishop PN (2012) Mapping the differential distribution of proteoglycan core proteins in the adult human retina, choroid, and sclera. Invest Ophthalmol Vis Sci 53(12):7528–7538. doi:10.1167/iovs.12-10797
Clark SJ, Higman VA, Mulloy B, Perkins SJ, Lea SM, Sim RB, Day AJ (2006) His-384 allotypic variant of factor H associated with age-related macular degeneration has different heparin binding properties from the non-disease-associated form. J Biol Chem 281(34):24713–24720. doi:10.1074/jbc.M605083200
Keenan TD, Pickford CE, Holley RJ, Clark SJ, Lin W, Dowsey AW, Merry CL, Day AJ, Bishop PN (2014) Age-dependent changes in heparan sulfate in human Bruch’s membrane: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci 55(8):5370–5379. doi:10.1167/iovs.14-14126
Heinegard D (2009) Proteoglycans and more—from molecules to biology. Int J Exp Pathol 90(6):575–586. doi:10.1111/j.1365-2613.2009.00695.x
Sorokin L (2010) The impact of the extracellular matrix on inflammation. Nat Rev Immunol 10(10):712–723. doi:10.1038/nri2852
Happonen KE, Heinegard D, Saxne T, Blom AM (2012) Interactions of the complement system with molecules of extracellular matrix: relevance for joint diseases. Immunobiology 217(11):1088–1096. doi:10.1016/j.imbio.2012.07.013
Sjoberg AP, Manderson GA, Morgelin M, Day AJ, Heinegard D, Blom AM (2009) Short leucine-rich glycoproteins of the extracellular matrix display diverse patterns of complement interaction and activation. Mol Immunol 46(5):830–839. doi:10.1016/j.molimm.2008.09.018
Sjoberg A, Onnerfjord P, Morgelin M, Heinegard D, Blom AM (2005) The extracellular matrix and inflammation: fibromodulin activates the classical pathway of complement by directly binding C1q. J Biol Chem 280(37):32301–32308. doi:10.1074/jbc.M504828200
Happonen KE, Sjoberg AP, Morgelin M, Heinegard D, Blom AM (2009) Complement inhibitor C4b-binding protein interacts directly with small glycoproteins of the extracellular matrix. J Immunol 182(3):1518–1525
Sjoberg AP, Trouw LA, Clark SJ, Sjolander J, Heinegard D, Sim RB, Day AJ, Blom AM (2007) The factor H variant associated with age-related macular degeneration (His-384) and the non-disease-associated form bind differentially to C-reactive protein, fibromodulin, DNA, and necrotic cells. J Biol Chem 282(15):10894–10900. doi:10.1074/jbc.M610256200
Kalchishkova N, Furst CM, Heinegard D, Blom AM (2011) NC4 Domain of cartilage-specific collagen IX inhibits complement directly due to attenuation of membrane attack formation and indirectly through binding and enhancing activity of complement inhibitors C4B-binding protein and factor H. J Biol Chem 286(32):27915–27926. doi:10.1074/jbc.M111.242834
Kiraz Y, Adan A, Kartal Yandim M, Baran Y (2016) Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. doi:10.1007/s13277-016-5035-9
Fishelson Z, Attali G, Mevorach D (2001) Complement and apoptosis. Mol Immunol 38(2–3):207–219
Leffler J, Herbert AP, Norstrom E, Schmidt CQ, Barlow PN, Blom AM, Martin M (2010) Annexin-II, DNA, and histones serve as factor H ligands on the surface of apoptotic cells. J Biol Chem 285(6):3766–3776. doi:10.1074/jbc.M109.045427
Trouw LA, Bengtsson AA, Gelderman KA, Dahlback B, Sturfelt G, Blom AM (2007) C4b-binding protein and factor H compensate for the loss of membrane-bound complement inhibitors to protect apoptotic cells against excessive complement attack. J Biol Chem 282(39):28540–28548. doi:10.1074/jbc.M704354200
Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS (2009) Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461(7261):282–286. doi:10.1038/nature08296
Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, Barbour SE, Milstien S, Spiegel S (2008) Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J 22(8):2629–2638. doi:10.1096/fj.08-107169
Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, Melville L, Melrose LA, Ogden CA, Nibbs R, Graham G, Combadiere C, Gregory CD (2008) CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 112(13):5026–5036. doi:10.1182/blood-2008-06-162404
Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S (2003) Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 113(6):717–730
Mold C, Gewurz H, Du Clos TW (1999) Regulation of complement activation by C-reactive protein. Immunopharmacology 42(1–3):23–30
Okemefuna AI, Nan R, Miller A, Gor J, Perkins SJ (2010) Complement factor H binds at two independent sites to C-reactive protein in acute phase concentrations. J Biol Chem 285(2):1053–1065. doi:10.1074/jbc.M109.044529
Deban L, Jarva H, Lehtinen MJ, Bottazzi B, Bastone A, Doni A, Jokiranta TS, Mantovani A, Meri S (2008) Binding of the long pentraxin PTX3 to factor H: interacting domains and function in the regulation of complement activation. J Immunol 181(12):8433–8440
Mihlan M, Stippa S, Jozsi M, Zipfel PF (2009) Monomeric CRP contributes to complement control in fluid phase and on cellular surfaces and increases phagocytosis by recruiting factor H. Cell Death Differ 16(12):1630–1640. doi:10.1038/cdd.2009.103
Bottazzi B, Inforzato A, Messa M, Barbagallo M, Magrini E, Garlanda C, Mantovani A (2016) The pentraxins PTX3 and SAP in innate immunity, regulation of inflammation and tissue remodelling. J Hepatol 64(6):1416–1427. doi:10.1016/j.jhep.2016.02.029
Garlanda C, Maina V, Cotena A, Moalli F (2009) The soluble pattern recognition receptor pentraxin-3 in innate immunity, inflammation and fertility. J Reprod Immunol 83(1–2):128–133. doi:10.1016/j.jri.2009.05.006
Inforzato A, Doni A, Barajon I, Leone R, Garlanda C, Bottazzi B, Mantovani A (2013) PTX3 as a paradigm for the interaction of pentraxins with the complement system. Semin Immunol 25(1):79–85. doi:10.1016/j.smim.2013.05.002
Inforzato A, Bottazzi B, Garlanda C, Valentino S, Mantovani A (2012) Pentraxins in humoral innate immunity. Adv Exp Med Biol 946:1–20. doi:10.1007/978-1-4614-0106-3_1
Breviario F, d’Aniello EM, Golay J, Peri G, Bottazzi B, Bairoch A, Saccone S, Marzella R, Predazzi V, Rocchi M et al (1992) Interleukin-1-inducible genes in endothelial cells. Cloning of a new gene related to C-reactive protein and serum amyloid P component. J Biol Chem 267(31):22190–22197
Mantovani A, Valentino S, Gentile S, Inforzato A, Bottazzi B, Garlanda C (2013) The long pentraxin PTX3: a paradigm for humoral pattern recognition molecules. Ann N Y Acad Sci 1285:1–14. doi:10.1111/nyas.12043
Daigo K, Inforzato A, Barajon I, Garlanda C, Bottazzi B, Meri S, Mantovani A (2016) Pentraxins in the activation and regulation of innate immunity. Immunol Rev 274(1):202–217. doi:10.1111/imr.12476
Sjoberg AP, Trouw LA, McGrath FD, Hack CE, Blom AM (2006) Regulation of complement activation by C-reactive protein: targeting of the inhibitory activity of C4b-binding protein. J Immunol 176(12):7612–7620
Braunschweig A, Jozsi M (2011) Human pentraxin 3 binds to the complement regulator C4b-binding protein. PLoS One 6(8):e23991. doi:10.1371/journal.pone.0023991
Inforzato A, Baldock C, Jowitt TA, Holmes DF, Lindstedt R, Marcellini M, Rivieccio V, Briggs DC, Kadler KE, Verdoliva A, Bottazzi B, Mantovani A, Salvatori G, Day AJ (2010) The angiogenic inhibitor long pentraxin PTX3 forms an asymmetric octamer with two binding sites for FGF2. J Biol Chem 285(23):17681–17692. doi:10.1074/jbc.M109.085639
Kopp A, Strobel S, Tortajada A, Rodriguez de Cordoba S, Sanchez-Corral P, Prohaszka Z, Lopez-Trascasa M, Jozsi M (2012) Atypical hemolytic uremic syndrome-associated variants and autoantibodies impair binding of factor h and factor h-related protein 1 to pentraxin 3. J Immunol 189(4):1858–1867. doi:10.4049/jimmunol.1200357
Inforzato A, Reading PC, Barbati E, Bottazzi B, Garlanda C, Mantovani A (2012) The “sweet” side of a long pentraxin: how glycosylation affects PTX3 functions in innate immunity and inflammation. Front Immunol 3:407. doi:10.3389/fimmu.2012.00407
Black S, Kushner I, Samols D (2004) C-reactive Protein. J Biol Chem 279(47):48487–48490. doi:10.1074/jbc.R400025200
Volanakis JE, Kaplan MH (1974) Interaction of C-reactive protein complexes with the complement system. II. Consumption of guinea pig complement by CRP complexes: requirement for human C1q. J Immunol 113(1):9–17
Ng PM, Le Saux A, Lee CM, Tan NS, Lu J, Thiel S, Ho B, Ding JL (2007) C-reactive protein collaborates with plasma lectins to boost immune response against bacteria. EMBO J 26(14):3431–3440. doi:10.1038/sj.emboj.7601762
Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, Sun PD (2008) Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature 456(7224):989–992. doi:10.1038/nature07468
Jarva H, Jokiranta TS, Hellwage J, Zipfel PF, Meri S (1999) Regulation of complement activation by C-reactive protein: targeting the complement inhibitory activity of factor H by an interaction with short consensus repeat domains 7 and 8–11. J Immunol 163(7):3957–3962
Laine M, Jarva H, Seitsonen S, Haapasalo K, Lehtinen MJ, Lindeman N, Anderson DH, Johnson PT, Jarvela I, Jokiranta TS, Hageman GS, Immonen I, Meri S (2007) Y402H polymorphism of complement factor H affects binding affinity to C-reactive protein. J Immunol 178(6):3831–3836
Lauer N, Mihlan M, Hartmann A, Schlotzer-Schrehardt U, Keilhauer C, Scholl HP, Charbel Issa P, Holz F, Weber BH, Skerka C, Zipfel PF (2011) Complement regulation at necrotic cell lesions is impaired by the age-related macular degeneration-associated factor-H His402 risk variant. J Immunol 187(8):4374–4383. doi:10.4049/jimmunol.1002488
Molins B, Fuentes-Prior P, Adan A, Anton R, Arostegui JI, Yague J, Dick AD (2016) Complement factor H binding of monomeric C-reactive protein downregulates proinflammatory activity and is impaired with at risk polymorphic CFH variants. Sci Rep 6:22889. doi:10.1038/srep22889
Kopp A, Hebecker M, Svobodova E, Jozsi M (2012) Factor H: a complement regulator in health and disease, and a mediator of cellular interactions. Biomolecules 2(1):46–75. doi:10.3390/biom2010046
Avery VM, Gordon DL (1993) Characterization of factor H binding to human polymorphonuclear leukocytes. J Immunol 151(10):5545–5553
Losse J, Zipfel PF, Jozsi M (2010) Factor H and factor H-related protein 1 bind to human neutrophils via complement receptor 3, mediate attachment to Candida albicans, and enhance neutrophil antimicrobial activity. J Immunol 184(2):912–921. doi:10.4049/jimmunol.0901702
Schneider AE, Sandor N, Karpati E, Jozsi M (2016) Complement factor H modulates the activation of human neutrophil granulocytes and the generation of neutrophil extracellular traps. Mol Immunol 72:37–48. doi:10.1016/j.molimm.2016.02.011
Agarwal V, Asmat TM, Luo S, Jensch I, Zipfel PF, Hammerschmidt S (2010) Complement regulator Factor H mediates a two-step uptake of Streptococcus pneumoniae by human cells. J Biol Chem 285(30):23486–23495. doi:10.1074/jbc.M110.142703
Agarwal S, Ram S, Ngampasutadol J, Gulati S, Zipfel PF, Rice PA (2010) Factor H facilitates adherence of Neisseria gonorrhoeae to complement receptor 3 on eukaryotic cells. J Immunol 185(7):4344–4353. doi:10.4049/jimmunol.0904191
Malhotra R, Ward M, Sim RB, Bird MI (1999) Identification of human complement Factor H as a ligand for l-selectin. Biochem J 341(Pt 1):61–69
Lambris JD, Dobson NJ, Ross GD (1980) Release of endogenous C3b inactivator from lymphocytes in response to triggering membrane receptors for beta 1H globulin. J Exp Med 152(6):1625–1644
Kang YH, Urban BC, Sim RB, Kishore U (2012) Human complement Factor H modulates C1q-mediated phagocytosis of apoptotic cells. Immunobiology 217(4):455–464. doi:10.1016/j.imbio.2011.10.008
Vaziri-Sani F, Hellwage J, Zipfel PF, Sjoholm AG, Iancu R, Karpman D (2005) Factor H binds to washed human platelets. J Thromb Haemost 3(1):154–162. doi:10.1111/j.1538-7836.2004.01010.x
Mnjoyan Z, Li J, Afshar-Kharghan V (2008) Factor H binds to platelet integrin alphaIIbbeta3. Platelets 19(7):512–519. doi:10.1080/09537100802238494
Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, Del-Favero J, Plaisance S, Claes B, Lambrechts D, Zoja C, Remuzzi G, Conway EM (2009) Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med 361(4):345–357. doi:10.1056/NEJMoa0810739
Heurich M, Preston RJ, O’Donnell VB, Morgan BP, Collins PW (2016) Thrombomodulin enhances complement regulation through strong affinity interactions with factor H and C3b-Factor H complex. Thromb Res 145:84–92. doi:10.1016/j.thromres.2016.07.017
Hammann KP, Raile A, Schmitt M, Mussel HH, Peters H, Scheiner O, Dierich MP (1981) beta 1H stimulates mouse-spleen B lymphocytes as demonstrated by increased thymidine incorporation and formation of B cell blasts. Immunobiology 160(3–4):289–301
Nabil K, Rihn B, Jaurand MC, Vignaud JM, Ripoche J, Martinet Y, Martinet N (1997) Identification of human complement factor H as a chemotactic protein for monocytes. Biochem J 326(Pt 2):377–383
Ohtsuka H, Imamura T, Matsushita M, Tanase S, Okada H, Ogawa M, Kambara T (1993) Thrombin generates monocyte chemotactic activity from complement factor H. Immunology 80(1):140–145
Olivar R, Luque A, Cardenas-Brito S, Naranjo-Gomez M, Blom AM, Borras FE, Rodriguez de Cordoba S, Zipfel PF, Aran JM (2016) The complement inhibitor factor H generates an anti-inflammatory and tolerogenic state in monocyte-derived dendritic cells. J Immunol 196(10):4274–4290. doi:10.4049/jimmunol.1500455
Qiang X, Wu R, Ji Y, Zhou M, Wang P (2008) Purification and characterization of human adrenomedullin binding protein-1. Mol Med 14(7–8):443–450. doi:10.2119/2008-00015.Qiang
Sim RB, Ferluga J, Al-Rashidi H, Abbow H, Schwaeble W, Kishore U (2015) Complement factor H in its alternative identity as adrenomedullin-binding protein 1. Mol Immunol 68(1):45–48. doi:10.1016/j.molimm.2015.06.006
Hinson JP, Kapas S, Smith DM (2000) Adrenomedullin, a multifunctional regulatory peptide. Endocr Rev 21(2):138–167. doi:10.1210/edrv.21.2.0396
Zudaire E, Portal-Nunez S, Cuttitta F (2006) The central role of adrenomedullin in host defense. J Leukoc Biol 80(2):237–244. doi:10.1189/jlb.0206123
Sjoberg AP, Nystrom S, Hammarstrom P, Blom AM (2008) Native, amyloid fibrils and beta-oligomers of the C-terminal domain of human prion protein display differential activation of complement and bind C1q, factor H and C4b-binding protein directly. Mol Immunol 45(11):3213–3221. doi:10.1016/j.molimm.2008.02.023
Kovacs GG, Gasque P, Strobel T, Lindeck-Pozza E, Strohschneider M, Ironside JW, Budka H, Guentchev M (2004) Complement activation in human prion disease. Neurobiol Dis 15(1):21–28
Hyvarinen S, Uchida K, Varjosalo M, Jokela R, Jokiranta TS (2014) Recognition of malondialdehyde-modified proteins by the C terminus of complement factor H is mediated via the polyanion binding site and impaired by mutations found in atypical hemolytic uremic syndrome. J Biol Chem 289(7):4295–4306. doi:10.1074/jbc.M113.527416
Shaw PX, Zhang L, Zhang M, Du H, Zhao L, Lee C, Grob S, Lim SL, Hughes G, Lee J, Bedell M, Nelson MH, Lu F, Krupa M, Luo J, Ouyang H, Tu Z, Su Z, Zhu J, Wei X, Feng Z, Duan Y, Yang Z, Ferreyra H, Bartsch DU, Kozak I, Zhang L, Lin F, Sun H, Feng H, Zhang K (2012) Complement factor H genotypes impact risk of age-related macular degeneration by interaction with oxidized phospholipids. Proc Natl Acad Sci USA 109(34):13757–13762. doi:10.1073/pnas.1121309109
Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HP, Charbel Issa P, Cano M, Brandstatter H, Tsimikas S, Skerka C, Superti-Furga G, Handa JT, Zipfel PF, Witztum JL, Binder CJ (2011) Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 478(7367):76–81. doi:10.1038/nature10449
Haapasalo K, van Kessel K, Nissila E, Metso J, Johansson T, Miettinen S, Varjosalo M, Kirveskari J, Kuusela P, Chroni A, Jauhiainen M, van Strijp J, Jokiranta TS (2015) Complement factor H binds to human serum apolipoprotein E and mediates complement regulation on high density lipoprotein particles. J Biol Chem 290(48):28977–28987. doi:10.1074/jbc.M115.669226
Sjoberg AP, Trouw LA, Blom AM (2009) Complement activation and inhibition: a delicate balance. Trends Immunol 30(2):83–90. doi:10.1016/j.it.2008.11.003
Meri T, Amdahl H, Lehtinen MJ, Hyvarinen S, McDowell JV, Bhattacharjee A, Meri S, Marconi R, Goldman A, Jokiranta TS (2013) Microbes bind complement inhibitor factor H via a common site. PLoS Pathog 9(4):e1003308. doi:10.1371/journal.ppat.1003308
Serruto D, Rappuoli R, Scarselli M, Gros P, van Strijp JA (2010) Molecular mechanisms of complement evasion: learning from staphylococci and meningococci. Nat Rev Microbiol 8(6):393–399. doi:10.1038/nrmicro2366
Vogl G, Lesiak I, Jensen DB, Perkhofer S, Eck R, Speth C, Lass-Florl C, Zipfel PF, Blom AM, Dierich MP, Wurzner R (2008) Immune evasion by acquisition of complement inhibitors: the mould Aspergillus binds both factor H and C4b binding protein. Mol Immunol 45(5):1485–1493. doi:10.1016/j.molimm.2007.08.011
Herbert AP, Makou E, Chen ZA, Kerr H, Richards A, Rappsilber J, Barlow PN (2015) Complement evasion mediated by enhancement of captured factor H: implications for protection of self-surfaces from complement. J Immunol 195(10):4986–4998. doi:10.4049/jimmunol.1501388
Kennedy AT, Schmidt CQ, Thompson JK, Weiss GE, Taechalertpaisarn T, Gilson PR, Barlow PN, Crabb BS, Cowman AF, Tham WH (2016) Recruitment of factor H as a novel complement evasion strategy for blood-stage plasmodium falciparum infection. J Immunol 196(3):1239–1248. doi:10.4049/jimmunol.1501581
Svoboda E, Schneider AE, Sandor N, Lermann U, Staib P, Kremlitzka M, Bajtay Z, Barz D, Erdei A, Jozsi M (2015) Secreted aspartic protease 2 of Candida albicans inactivates factor H and the macrophage factor H-receptors CR3 (CD11b/CD18) and CR4 (CD11c/CD18). Immunol Lett 168(1):13–21. doi:10.1016/j.imlet.2015.08.009
Pizza M, Donnelly J, Rappuoli R (2008) Factor H-binding protein, a unique meningococcal vaccine antigen. Vaccine 26(Suppl 8):I46–I48
Hellwage J, Meri T, Heikkila T, Alitalo A, Panelius J, Lahdenne P, Seppala IJ, Meri S (2001) The complement regulator factor H binds to the surface protein OspE of Borrelia burgdorferi. J Biol Chem 276(11):):8427–8435. doi:10.1074/jbc.M007994200
Kraiczy P, Hartmann K, Hellwage J, Skerka C, Kirschfink M, Brade V, Zipfel PF, Wallich R, Stevenson B (2004) Immunological characterization of the complement regulator factor H-binding CRASP and Erp proteins of Borrelia burgdorferi. Int J Med Microbiol 293(Suppl 37):152–157
Haupt K, Reuter M, van den Elsen J, Burman J, Halbich S, Richter J, Skerka C, Zipfel PF (2008) The Staphylococcus aureus protein Sbi acts as a complement inhibitor and forms a tripartite complex with host complement Factor H and C3b. PLoS Pathog 4(12):e1000250. doi:10.1371/journal.ppat.1000250
Janulczyk R, Iannelli F, Sjoholm AG, Pozzi G, Bjorck L (2000) Hic, a novel surface protein of Streptococcus pneumoniae that interferes with complement function. J Biol Chem 275(47):37257–37263. doi:10.1074/jbc.M004572200
Melin M, Di Paolo E, Tikkanen L, Jarva H, Neyt C, Kayhty H, Meri S, Poolman J, Vakevainen M (2010) Interaction of pneumococcal histidine triad proteins with human complement. Infect Immun 78(5):2089–2098. doi:10.1128/IAI.00811-09
Haapasalo K, Jarva H, Siljander T, Tewodros W, Vuopio-Varkila J, Jokiranta TS (2008) Complement factor H allotype 402H is associated with increased C3b opsonization and phagocytosis of Streptococcus pyogenes. Mol Microbiol 70(3):583–594. doi:10.1111/j.1365-2958.2008.06347.x
Schneider MC, Prosser BE, Caesar JJ, Kugelberg E, Li S, Zhang Q, Quoraishi S, Lovett JE, Deane JE, Sim RB, Roversi P, Johnson S, Tang CM, Lea SM (2009) Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature 458(7240):890–893. doi:10.1038/nature07769
Vimr ER, Kalivoda KA, Deszo EL, Steenbergen SM (2004) Diversity of microbial sialic acid metabolism. Microbiol Mol Biol Rev 68(1):132–153
Khatua B, Ghoshal A, Bhattacharya K, Mandal C, Saha B, Crocker PR (2010) Sialic acids acquired by Pseudomonas aeruginosa are involved in reduced complement deposition and siglec mediated host-cell recognition. FEBS Lett 584(3):555–561. doi:10.1016/j.febslet.2009.11.087
Heinekamp T, Schmidt H, Lapp K, Pahtz V, Shopova I, Koster-Eiserfunke N, Kruger T, Kniemeyer O, Brakhage AA (2015) Interference of Aspergillus fumigatus with the immune response. Semin Immunopathol 37(2):141–152. doi:10.1007/s00281-014-0465-1
Khattab A, Barroso M, Miettinen T, Meri S (2015) Anopheles midgut epithelium evades human complement activity by capturing factor H from the blood meal. PLoS Negl Trop Dis 9(2):e0003513. doi:10.1371/journal.pntd.0003513
Seib KL, Scarselli M, Comanducci M, Toneatto D, Masignani V (2015) Neisseria meningitidis factor H-binding protein fHbp: a key virulence factor and vaccine antigen. Expert Rev Vaccines 14(6):841–859. doi:10.1586/14760584.2015.1016915
Rossi R, Konar M, Beernink PT (2016) Meningococcal factor H binding protein vaccine antigens with increased thermal stability and decreased binding of human factor H. Infect Immun 84(6):1735–1742. doi:10.1128/IAI.01491-15
Blom AM, Hallstrom T, Riesbeck K (2009) Complement evasion strategies of pathogens-acquisition of inhibitors and beyond. Mol Immunol 46(14):2808–2817. doi:10.1016/j.molimm.2009.04.025
Shaughnessy J, Ram S, Bhattacharjee A, Pedrosa J, Tran C, Horvath G, Monks B, Visintin A, Jokiranta TS, Rice PA (2011) Molecular characterization of the interaction between sialylated Neisseria gonorrhoeae and factor H. J Biol Chem 286(25):22235–22242. doi:10.1074/jbc.M111.225516
Shaughnessy J, Gulati S, Agarwal S, Unemo M, Ohnishi M, Su XH, Monks BG, Visintin A, Madico G, Lewis LA, Golenbock DT, Reed GW, Rice PA, Ram S (2016) A novel factor H-Fc chimeric immunotherapeutic molecule against Neisseria gonorrhoeae. J Immunol 196(4):1732–1740. doi:10.4049/jimmunol.1500292
Sanchez-Corral P, Perez-Caballero D, Huarte O, Simckes AM, Goicoechea E, Lopez-Trascasa M, de Cordoba SR (2002) Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome. Am J Hum Genet 71(6):1285–1295. doi:10.1086/344515
Francis NJ, McNicholas B, Awan A, Waldron M, Reddan D, Sadlier D, Kavanagh D, Strain L, Marchbank KJ, Harris CL, Goodship TH (2012) A novel hybrid CFH/CFHR3 gene generated by a microhomology-mediated deletion in familial atypical hemolytic uremic syndrome. Blood 119(2):591–601. doi:10.1182/blood-2011-03-339903
Gorter A, Meri S (1999) Immune evasion of tumor cells using membrane-bound complement regulatory proteins. Immunol Today 20(12):576–582
Bonavita E, Gentile S, Rubino M, Maina V, Papait R, Kunderfranco P, Greco C, Feruglio F, Molgora M, Laface I, Tartari S, Doni A, Pasqualini F, Barbati E, Basso G, Galdiero MR, Nebuloni M, Roncalli M, Colombo P, Laghi L, Lambris JD, Jaillon S, Garlanda C, Mantovani A (2015) PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell 160(4):700–714. doi:10.1016/j.cell.2015.01.004
Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, Kavanagh D, Noris M, Pickering M, Sanchez-Corral P, Skerka C, Zipfel P, Smith RJ (2015) Atypical aHUS: state of the art. Mol Immunol 67(1):31–42. doi:10.1016/j.molimm.2015.03.246
Ray PE, Liu XH (2001) Pathogenesis of Shiga toxin-induced hemolytic uremic syndrome. Pediatr Nephrol 16(10):823–839
Orth D, Khan AB, Naim A, Grif K, Brockmeyer J, Karch H, Joannidis M, Clark SJ, Day AJ, Fidanzi S, Stoiber H, Dierich MP, Zimmerhackl LB, Wurzner R (2009) Shiga toxin activates complement and binds factor H: evidence for an active role of complement in hemolytic uremic syndrome. J Immunol 182(10):6394–6400. doi:10.4049/jimmunol.0900151
Berger BE (2016) The alternative pathway of complement and the evolving clinical-pathophysiological spectrum of atypical hemolytic uremic syndrome. Am J Med Sci 352(2):177–190. doi:10.1016/j.amjms.2016.05.003
Hyvarinen S, Meri S, Jokiranta TS (2016) Disturbed sialic acid recognition on endothelial cells and platelets in complement attack causes atypical hemolytic uremic syndrome. Blood 127(22):2701–2710. doi:10.1182/blood-2015-11-680009
Dragon-Durey MA, Loirat C, Cloarec S, Macher MA, Blouin J, Nivet H, Weiss L, Fridman WH, Fremeaux-Bacchi V (2005) Anti-factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol 16(2):555–563. doi:10.1681/ASN.2004050380
Jozsi M, Strobel S, Dahse HM, Liu WS, Hoyer PF, Oppermann M, Skerka C, Zipfel PF (2007) Anti factor H autoantibodies block C-terminal recognition function of factor H in hemolytic uremic syndrome. Blood 110(5):1516–1518. doi:10.1182/blood-2007-02-071472
Bhattacharjee A, Reuter S, Trojnar E, Kolodziejczyk R, Seeberger H, Hyvarinen S, Uzonyi B, Szilagyi A, Prohaszka Z, Goldman A, Jozsi M, Jokiranta TS (2015) The major autoantibody epitope on factor H in atypical hemolytic uremic syndrome is structurally different from its homologous site in factor H-related protein 1, supporting a novel model for induction of autoimmunity in this disease. J Biol Chem 290(15):9500–9510. doi:10.1074/jbc.M114.630871
Warwicker P, Donne RL, Goodship JA, Goodship TH, Howie AJ, Kumararatne DS, Thompson RA, Taylor CM (1999) Familial relapsing haemolytic uraemic syndrome and complement factor H deficiency. Nephrol Dial Transplant 14(5):1229–1233
Rodriguez de Cordoba S, Hidalgo MS, Pinto S, Tortajada A (2014) Genetics of atypical hemolytic uremic syndrome (aHUS). Semin Thromb Hemost 40(4):422–430. doi:10.1055/s-0034-1375296
Montes T, Goicoechea de Jorge E, Ramos R, Goma M, Pujol O, Sanchez-Corral P, Rodriguez de Cordoba S (2008) Genetic deficiency of complement factor H in a patient with age-related macular degeneration and membranoproliferative glomerulonephritis. Mol Immunol 45(10):2897–2904. doi:10.1016/j.molimm.2008.01.027
Licht C, Heinen S, Jozsi M, Loschmann I, Saunders RE, Perkins SJ, Waldherr R, Skerka C, Kirschfink M, Hoppe B, Zipfel PF (2006) Deletion of Lys224 in regulatory domain 4 of Factor H reveals a novel pathomechanism for dense deposit disease (MPGN II). Kidney Int 70(1):42–50. doi:10.1038/sj.ki.5000269
Fakhouri F, de Jorge EG, Brune F, Azam P, Cook HT, Pickering MC (2010) Treatment with human complement factor H rapidly reverses renal complement deposition in factor H-deficient mice. Kidney Int 78(3):279–286. doi:10.1038/ki.2010.132
Goodship TH, Pappworth IY, Toth T, Denton M, Houlberg K, McCormick F, Warland D, Moore I, Hunze EM, Staniforth SJ, Hayes C, Cavalcante DP, Kavanagh D, Strain L, Herbert AP, Schmidt CQ, Barlow PN, Harris CL, Marchbank KJ (2012) Factor H autoantibodies in membranoproliferative glomerulonephritis. Mol Immunol 52(3–4):200–206. doi:10.1016/j.molimm.2012.05.009
Nozal PSS, Ibernon M, Lopez D, Sanchez-Corral P, Rodrıguez de Cordoba S, Jozsi M, Lopez-Trascasa M (2012) Anti-factor H antibody affecting factor H cofactor activity in a patient with dense deposit disease. Clin Kidney J 5:133–136. doi:10.1093/ckj/sfs002
Wong WL, Su X, Li X, Cheung CM, Klein R, Cheng CY, Wong TY (2014) Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Global Health 2(2):e106–e116. doi:10.1016/S2214-109X(13)70145-1
Ferris FL 3rd, Wilkinson CP, Bird A, Chakravarthy U, Chew E, Csaky K, Sadda SR (2013) Clinical classification of age-related macular degeneration. Ophthalmology 120(4):844–851. doi:10.1016/j.ophtha.2012.10.036
Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, Zack DJ, Arakawa S, Cipriani V, Ripke S, Igo RP, Jr., Buitendijk GH, Sim X, Weeks DE, Guymer RH, Merriam JE, Francis PJ, Hannum G, Agarwal A, Armbrecht AM, Audo I, Aung T, Barile GR, Benchaboune M, Bird AC, Bishop PN, Branham KE, Brooks M, Brucker AJ, Cade WH, Cain MS, Campochiaro PA, Chan CC, Cheng CY, Chew EY, Chin KA, Chowers I, Clayton DG, Cojocaru R, Conley YP, Cornes BK, Daly MJ, Dhillon B, Edwards AO, Evangelou E, Fagerness J, Ferreyra HA, Friedman JS, Geirsdottir A, George RJ, Gieger C, Gupta N, Hagstrom SA, Harding SP, Haritoglou C, Heckenlively JR, Holz FG, Hughes G, Ioannidis JP, Ishibashi T, Joseph P, Jun G, Kamatani Y, Katsanis N, C NK, Khan JC, Kim IK, Kiyohara Y, Klein BE, Klein R, Kovach JL, Kozak I, Lee CJ, Lee KE, Lichtner P, Lotery AJ, Meitinger T, Mitchell P, Mohand-Said S, Moore AT, Morgan DJ, Morrison MA, Myers CE, Naj AC, Nakamura Y, Okada Y, Orlin A, Ortube MC, Othman MI, Pappas C, Park KH, Pauer GJ, Peachey NS, Poch O, Priya RR, Reynolds R, Richardson AJ, Ripp R, Rudolph G, Ryu E, Sahel JA, Schaumberg DA, Scholl HP, Schwartz SG, Scott WK, Shahid H, Sigurdsson H, Silvestri G, Sivakumaran TA, Smith RT, Sobrin L, Souied EH, Stambolian DE, Stefansson H, Sturgill-Short GM, Takahashi A, Tosakulwong N, Truitt BJ, Tsironi EE, Uitterlinden AG, van Duijn CM, Vijaya L, Vingerling JR, Vithana EN, Webster AR, Wichmann HE, Winkler TW, Wong TY, Wright AF, Zelenika D, Zhang M, Zhao L, Zhang K, Klein ML, Hageman GS, Lathrop GM, Stefansson K, Allikmets R, Baird PN, Gorin MB, Wang JJ, Klaver CC, Seddon JM, Pericak-Vance MA, Iyengar SK, Yates JR, Swaroop A, Weber BH, Kubo M, Deangelis MM, Leveillard T, Thorsteinsdottir U, Haines JL, Farrer LA, Heid IM, Abecasis GR (2013) Seven new loci associated with age-related macular degeneration. Nat Genet 45(4):433–439, 439e431–e432. doi:10.1038/ng.2578
Black JR, Clark SJ (2016) Age-related macular degeneration: genome-wide association studies to translation. Genet Med 18(4):283–289. doi:10.1038/gim.2015.70
Fritsche LG, Igl W, Bailey JN, Grassmann F, Sengupta S, Bragg-Gresham JL, Burdon KP, Hebbring SJ, Wen C, Gorski M, Kim IK, Cho D, Zack D, Souied E, Scholl HP, Bala E, Lee KE, Hunter DJ, Sardell RJ, Mitchell P, Merriam JE, Cipriani V, Hoffman JD, Schick T, Lechanteur YT, Guymer RH, Johnson MP, Jiang Y, Stanton CM, Buitendijk GH, Zhan X, Kwong AM, Boleda A, Brooks M, Gieser L, Ratnapriya R, Branham KE, Foerster JR, Heckenlively JR, Othman MI, Vote BJ, Liang HH, Souzeau E, McAllister IL, Isaacs T, Hall J, Lake S, Mackey DA, Constable IJ, Craig JE, Kitchner TE, Yang Z, Su Z, Luo H, Chen D, Ouyang H, Flagg K, Lin D, Mao G, Ferreyra H, Stark K, von Strachwitz CN, Wolf A, Brandl C, Rudolph G, Olden M, Morrison MA, Morgan DJ, Schu M, Ahn J, Silvestri G, Tsironi EE, Park KH, Farrer LA, Orlin A, Brucker A, Li M, Curcio CA, Mohand-Said S, Sahel JA, Audo I, Benchaboune M, Cree AJ, Rennie CA, Goverdhan SV, Grunin M, Hagbi-Levi S, Campochiaro P, Katsanis N, Holz FG, Blond F, Blanche H, Deleuze JF, Igo RP Jr, Truitt B, Peachey NS, Meuer SM, Myers CE, Moore EL, Klein R, Hauser MA, Postel EA, Courtenay MD, Schwartz SG, Kovach JL, Scott WK, Liew G, Tan AG, Gopinath B, Merriam JC, Smith RT, Khan JC, Shahid H, Moore AT, McGrath JA, Laux R, Brantley MA Jr, Agarwal A, Ersoy L, Caramoy A, Langmann T, Saksens NT, de Jong EK, Hoyng CB, Cain MS, Richardson AJ, Martin TM, Blangero J, Weeks DE, Dhillon B, van Duijn CM, Doheny KF, Romm J, Klaver CC, Hayward C, Gorin MB, Klein ML, Baird PN, den Hollander AI, Fauser S, Yates JR, Allikmets R, Wang JJ, Schaumberg DA, Klein BE, Hagstrom SA, Chowers I, Lotery AJ, Leveillard T, Zhang K, Brilliant MH, Hewitt AW, Swaroop A, Chew EY, Pericak-Vance MA, DeAngelis M, Stambolian D, Haines JL, Iyengar SK, Weber BH, Abecasis GR, Heid IM (2016) A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet 48(2):134–143. doi:10.1038/ng.3448
Schramm EC, Clark SJ, Triebwasser MP, Raychaudhuri S, Seddon JM, Atkinson JP (2014) Genetic variants in the complement system predisposing to age-related macular degeneration: a review. Mol Immunol 61(2):118–125. doi:10.1016/j.molimm.2014.06.032
Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, Hauser MA, Rickman CB, Bok D, Hageman GS, Johnson LV (2010) The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retinal Eye Res 29(2):95–112. doi:10.1016/j.preteyeres.2009.11.003
McHarg S, Brace N, Bishop PN, Clark SJ (2015) Enrichment of Bruch’s membrane from human donor eyes. J Vis Exp. doi:10.3791/53382
Hecker LA, Edwards AO, Ryu E, Tosakulwong N, Baratz KH, Brown WL, Charbel Issa P, Scholl HP, Pollok-Kopp B, Schmid-Kubista KE, Bailey KR, Oppermann M (2010) Genetic control of the alternative pathway of complement in humans and age-related macular degeneration. Hum Mol Genet 19(1):209–215. doi:10.1093/hmg/ddp472
Mamidi S, Hone S, Kirschfink M (2015) The complement system in cancer: ambivalence between tumour destruction and promotion. Immunobiology. doi:10.1016/j.imbio.2015.11.008
Wilczek E, Rzepko R, Nowis D, Legat M, Golab J, Glab M, Gorlewicz A, Konopacki F, Mazurkiewicz M, Sladowski D, Gornicka B, Wasiutynski A, Wilczynski GM (2008) The possible role of factor H in colon cancer resistance to complement attack. Int J Cancer 122(9):2030–2037. doi:10.1002/ijc.23238
Ferreira VP, Pangburn MK, Cortes C (2010) Complement control protein factor H: the good, the bad, and the inadequate. Mol Immunol 47(13):2187–2197. doi:10.1016/j.molimm.2010.05.007
Campa MJ, Gottlin EB, Bushey RT, Patz EF Jr (2015) Complement factor H antibodies from lung cancer patients induce complement-dependent lysis of tumor cells, suggesting a novel immunotherapeutic strategy. Cancer Immunol Res 3(12):1325–1332. doi:10.1158/2326-6066.CIR-15-0122
Bushey RT, Moody MA, Nicely NL, Haynes BF, Alam SM, Keir ST, Bentley RC, Roy Choudhury K, Gottlin EB, Campa MJ, Liao HX, Patz EF Jr (2016) A therapeutic antibody for cancer, derived from single human B cells. Cell Rep 15(7):1505–1513. doi:10.1016/j.celrep.2016.04.038
Zhou W (2012) The new face of anaphylatoxins in immune regulation. Immunobiology 217(2):225–234. doi:10.1016/j.imbio.2011.07.016
Corrales L, Ajona D, Rafail S, Lasarte JJ, Riezu-Boj JI, Lambris JD, Rouzaut A, Pajares MJ, Montuenga LM, Pio R (2012) Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol 189(9):4674–4683. doi:10.4049/jimmunol.1201654
Kavanagh D, Goodship T (2010) Genetics and complement in atypical HUS. Pediatr Nephrol 25(12):2431–2442. doi:10.1007/s00467-010-1555-5
Acknowledgements
AI is recipient of a Young Investigator Grant from Ministero della Salute (GR-2011-02349539), which also funds RP’s Ph.D. studentship. AJD and SJC would like to thank Fight for Sight, the Macular Society and the Medical Research Council (UK) for their funding of studies into complement and age-related macular degeneration.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Parente, R., Clark, S.J., Inforzato, A. et al. Complement factor H in host defense and immune evasion. Cell. Mol. Life Sci. 74, 1605–1624 (2017). https://doi.org/10.1007/s00018-016-2418-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2418-4