
Abstract
Aspergillus fumigatus is a saprotrophic filamentous fungus and also the most prevalent airborne fungal pathogen of humans. Depending on the host’s immune status, the variety of diseases caused by A. fumigatus ranges from allergies in immunocompetent hosts to life-threatening invasive infections in patients with impaired immunity. In contrast to the majority of other Aspergillus species, which are in most cases nonpathogenic, A. fumigatus features an armory of virulence determinants to establish an infection. For example, A. fumigatus is able to evade the human complement system by binding or degrading complement regulators. Furthermore, the fungus interferes with lung epithelial cells, alveolar macrophages, and neutrophil granulocytes to prevent killing by these immune cells. This chapter summarizes the different strategies of A. fumigatus to manipulate the immune response. We also discuss the potential impact of recent advances in immunoproteomics to improve diagnosis and therapy of an A. fumigatus infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fungal pathogens cause a wide range of diseases ranging from allergies and superficial infections to life-threatening invasive mycoses. Often, the outcome of a fungal infection depends on the immune status of the host organism. In particular, individuals with a compromised immune system represent a group at high risk to develop fatal fungal infection. The continuous progress in intensive care, e.g., in chemotherapy and organ or bone marrow transplantation, contributes to the steadily increasing number of patients with impaired immune status [1].
The genus Aspergillus comprises more than 250 species, including on the one hand “good guys” that are industrially used for production of pharmaceuticals, beverages, and food additives; but on the other hand also, several “bad guys” that call for toxin-based crop spoilage or being the causative agent of severe fungal infections. Among the latter group, Aspergillus fumigatus is the number one airborne fungal pathogen of humans. To date, neither reliable diagnostic tools nor effective treatment options are available resulting in unacceptable high mortality rates of patients suffering from invasive fungal infections [2]. Therefore, the identification of new diagnostic markers and the development of novel therapeutics for specific intervention are of great importance. Especially, the characterization of the pathogen’s strategies to defend against attacks of host immune cells is interesting to understand pathogenicity and is important for the identification of potential therapeutic targets.
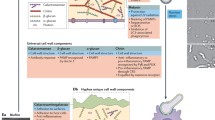
This article focuses on the interaction of A. fumigatus with components of the human immune system. In detail, we discuss the various strategies of this fungus to interfere with lung epithelial cells and phagocytes such as macrophages and neutrophils, and we illustrate how A. fumigatus evades the human complement (Fig. 1). Finally, we will discuss recent advances in immunoproteomics and their impact on target identification and improvement of diagnosis (Fig. 2).

Schematic depiction of the interaction between A. fumigatus and the immune system of the host (for details see text)

Identification of immunoreactive proteins of A. fumigatus by immunoblot analysis of 2D-electrophoresis maps with patient sera
Interaction of A. fumigatus with lung epithelial cells
Conidia of A. fumigatus are comparatively small with a diameter of only 2–3 μm. They are propagated easily through the air and enter the human host via the airway, where they infect the lung tissue and intrude to the lower respiratory system [3]. Epithelial cells of the lung therefore represent the first contact barrier where A. fumigatus interacts with host cells. The alveolus is lined by alveolar epithelial type I and type II cells and especially type II pneumocytes maintaining the alveolar space are confronted to inhaled conidia. In contrast to macrophages or neutrophils, pulmonary epithelial cells represent nonprofessional phagocytes. Upon contact, conidia were shown to strongly adhere to type II pneumocytes of the A549 cell line, which then start to engulf the fungus. Endocytosed conidia are able to survive and reside inside A549 cells [4, 5].
According to the fact that manipulation of host cell apoptosis is an important strategy of many pathogens to establish an infection, it was found that A. fumigatus inhibits apoptosis in different epithelial cell types [6]. Only recently, it was shown that the fungal 1,8-dihydroxynaphthalene (DHN)-melanin is responsible for this effect on the epithelial apoptosis process [7]. DHN-melanin is also essential to prevent phagolysosomal acidification in alveolar epithelial cells to survive intracellularly. A current hypothesis is that since phagocytic activity of epithelial cells is rather low, some conidia might persist within these cells and thereby represent the infectious reservoir after impairment of the host’s immune system [7].
Interestingly, the mechanisms by which A. fumigatus interfere with nonprofessional phagocytes is largely similar to professional phagocytic cells, i.e., macrophages and neutrophils, as described in detail in the following.
Interaction of A. fumigatus with alveolar macrophages
Invading conidia in the lung tissue encounter the resident leucocytes which constitute the first line of the host’s immune defense. Alveolar macrophages are homed just beneath the alveolar surfactant film and represent 90 % of the resident leucocytes in the lung [8]. They are credited with a major contribution to the initial immune response against Aspergillus infections besides neutrophil granulocytes. Alveolar macrophages originate from immigrating blood monocytes or from precursor cells residing in the lung [8]. Patients with reduced numbers of those phagocytes, e.g., due to lymphatic or leukemic malignomes or after stem cell or solid organ transplantation, have increased susceptibility towards aspergillosis [2, 9]. The recognition of conidia by alveolar macrophages leads to phagocytosis and induces the expression of inflammatory chemokines and cytokines such as TNF-α, IL-12, IFN-γ, IL-18, IL-6, IL-1β, MIP-1, MIP-1α, MIP-2, G-CSF, and GM-CSF [10–12]. They recruit further professional phagocytes such as circulating macrophages, neutrophil granulocytes, and dendritic cells (DCs) to the site of infection. The interaction of A. fumigatus with neutrophils is described in more detail below. Dendritic cells are able to phagocytose conidia and even hyphae, and their function in antigen presentation via the major histocompatibility complex class I (MHC I) is important to initiate the adaptive branch of the immune system by the activation of T cell and antibody responses [13, 14].
The fungal surface is composed of α- and β-glucans, chitins, galactomannans, and other polysaccharides. In particular, β-1,3-glucans and galactomannans are highly immunogenic molecules and prominent pathogen-associated patterns (PAMPs) of fungi [15, 16]. However, the rodlet layer composed by the hydrophobic RodA protein and the pigment DHN-melanin on the surface of resting A. fumigatus conidia render the conidia largely inert towards recognition by the immune system [17]. Swelling and germination of conidia are accompanied by a subsequent loss of the protective melanin and hydrophobin layer and thereby lead to an increasing exposure of immunological surface components and enhanced phagocytosis [16, 18]. Pigmentless strains, due to deletion or mutation of the pksP gene encoding the polyketide synthase essential for melanin biosynthesis, display a smooth surface structure and enhanced phagocytosis during co-incubation with macrophages [19–21]. DHN-melanin has been shown to be important for correct cell wall assembly and ascribed a function in the defense against the host’s immune response [22, 23]. Remarkably, a lack of DHN-melanin is accompanied with attenuation of virulence [19, 24].
Different pathogen recognition receptors (PRRs), which are expressed by cells of the innate immune system, are responsible for the detection of specific PAMPs [25]. The DC-specific intracellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN), which is mainly expressed on DCs, binds fungal galactomannans also on macrophages and initiates phagocytosis [26]. Toll-like receptors (TLRs) 2 and 4 have been attributed with immune recognition and modulation of the immune response in Aspergillus infections [27–29], although their actual function in invasive aspergillosis has not been rigorously elucidated yet. TLR2 was ascribed to sense chitin on the fungal surface, and TLR4 presumably detects α-glucans [30, 31]. A contribution of TLR9 to spore detection is discussed [32]. The main route of fungal recognition is the specific binding of the C-type lectin dectin-1 to β-1,3-glucans which are exposed on the fungal surface [11]. Dectin-1 is expressed on macrophages, monocytes, neutrophils, DCs, and a subset of T cells [33].
After pathogen recognition, phagocytosis is induced. Within the phagocyte, the conidium-containing phagosome fuses with lysosomes to form the phagolysosomes. Killing of the pathogen is observed starting around 6 h after ingestion [34]. Swelling of the conidium and acidification of the phagolysosome is prerequisite for efficient elimination [35]. During the fusion events with lysosomes, hydrolytic enzymes are exchanged. They comprise proteases, most importantly the cathepsins D, B, H, L, and S [36]. Further enzymes are hydrolases such as β-hexosaminidase and β-glucoronidase, lipases, and DNases [37]. Upon acidification of the compartment, these hydrolytic enzymes gain their catalytic activity towards the ingested particle.
A requirement for the biocidal property of the phagolysosome is the acidification of the compartment. The vacuolar ATPase (vATPase), a multiprotein membrane complex, has a major contribution in lowering the intracompartmental pH from pH >6 to pH <4.5 by pumping H+ ions across the membrane in an ATP-dependent reaction [35]. Treatment of macrophages with the vATPase inhibitor bafilomycin A resulted in a complete abolishment of acidification and confirmed the major role of the vATPase in the phagolysosomal acidification [38]. Conidia of the A. fumigatus wild type are localized to a much lower rate in such acidified compartments. Thywissen et al. [39] found the conidial pigment DHN-melanin to be a crucial factor for the inhibition of phagolysosomal acidification. In assays to investigate phagocytosis and intracellular processing of wild-type conidia compared to conidia of the pigmentless pksP mutant, wild-type conidia revealed retarded uptake by macrophages compared to mutant conidia. The intracellular phagolysosomal maturation process, especially fusion events with endosomes and lysosomes, was reported to be similar for both strains. By contrast, a striking difference in the acidification of the phagolysosome was observed. Whereas phagolysosomes that contained pksP mutant conidia acidified to pH below 4.5, the wild-type conidia containing phagolysosomes showed no reduction of pH. Melanin ghosts showed the same effect of reduced acidification, emphasizing the role of DHN-melanin in this mechanism [24, 39]. Other melanins such as DOPA melanin were not able to inhibit phagolysosomal acidification [40]. A contribution of the hydrophobin layer could be excluded, because conidia of a ∆rodA mutant strain did not inhibit acidification of the phagolysosome [17, 24, 39]. Furthermore, DHN-melanin deficiency did not interfere with the formation of rodlet structures on the conidial surface [39].
Melanized conidia of A. fumigatus were shown to inhibit apoptosis by activation of PI 3-kinase/Akt signaling pathway [41]. The internalization of melanized conidia prevented macrophages from cell death when apoptosis was induced via the intrinsic pathway with staurosporine or via the extrinsic pathway with the Fas ligand [41, 42]. Opposed to nonpigmented conidia or hyphae, a sustained PI 3-kinase/Akt signaling was observed in macrophages, epithelial cells, and pneumocytes that resulted in activation of the protein kinase B (PKB) and sustained levels of antiapoptotic proteins of the Bcl family such as Bcl-2 and Mcl-1, reduced cytochrome c release from mitochondria, and consequently reduced apoptosis promoting activation of effector caspases 3, 6, and 7. PI 3-kinase/Akt signaling was demonstrated to further enhance activation of FoxO and NFkB that initiates gene expression and production of inflammatory cytokines [6, 41–43]. The effect was found to be unique for different melanins, including DHN-melanin, DHN-melanin precursors, and DOPA melanin. Volling and co-workers proposed a two-step strategy comprising first the inhibition of acidification of the phagolysosome which then, secondly, resulted in inhibition of apoptosis. The apoptosis inhibition was further attributed to the reactive oxygen intermediates (ROI) quenching effect of melanins: Enhanced ROI signaling usually promotes cytochrome c release but is counteracted by the presence of the pigment on internalized conidia [41]. In contrast, germlings and hyphae of A. fumigatus induce apoptosis via the release of the toxin gliotoxin in a dectin-1 and TLR2-dependent manner to evade immune responses [44, 45]. It was hypothesized that inhibition of apoptosis by A. fumigatus conidia creates a protective intracellular niche for the fungus to evade fungicidal effects of immune cells and to escape the disruption of infected cells by cytotoxic T cells as well as a shuttle for transport to lymph nodes and spleen [24, 46].
Interaction of A. fumigatus with neutrophil granulocytes
Beside macrophages, neutrophil granulocytes are important effector cells of the innate immunity. They represent the most abundant immune cells and belong to the first line of defense against bacterial and fungal infections. Each day, around 5–10 × 1010 neutrophils are produced in the bone marrow [47]. After full differentiation, they are released into the bloodstream where they have a limited lifespan of about 6–8 h. However, without external stimuli, neutrophils can circulate for up to 5 days outside the bone marrow [48]. In the event of infection, a large number of neutrophils is recruited to the affected tissue in response to cytokines such as IL-8, IFN-γ, C5a, and leukotriene B4 [49]. Neutrophils are the key effector cells in the immune response against A. fumigatus. Accordingly, neutropenic patients face the highest risk to develop an invasive aspergillosis [50]. Depletion of neutrophils in mice infected with A. fumigatus conidia induced high mortality rates and hyphae-induced lesions in the lung. In contrast, infected mice with depletion of macrophages were still able to prevent conidial germination resulting in 100 % survival [51].
Neutrophils recognize A. fumigatus at least via dectin-1 and TLRs 2 and 4 [52–54] and possess different mechanisms to kill the fungus. As professional phagocytes, they ingest conidia and germlings very rapidly to kill them by a respiratory burst and degranulation [55, 56]. Neutrophils contain three different types of granules. Primary or azurophilic granules contain myeloperoxidase (MPO), lysozyme, and antimicrobial proteins like cathepsin G, elastase, and proteinase 3. Secondary or specific granules hold lactoferrin, lysozyme, lipocalin, and membrane proteins like flavochrome b558. Tertiary granules contain gelatinase [57, 58]. Upon activation, neutrophils secrete cytokines and chemokines, e.g., CXCL1/2/3, CCL2/3/4, and IL-8 to attract further immune cells [52, 59]. Neutrophils attach to hyphae that are too big to be phagocytosed. By means of degranulation and oxidative and non-oxidative killing mechanisms, fungal hyphae are damaged [60]. Oxidative killing is depending on the assembly of the NADPH oxidase. This enzyme complex produces superoxide anions that are further converted to toxic compounds like H2O2, hydroxyl anions, and hypochlorous acid [61]. Patients suffering from chronic granulomatous deficiency (CGD), an inherited disorder caused by defects in the NADPH oxidase subunit gp91phox, are at high risk for invasive Aspergillus infections. Neutrophils from gp91phox-deficient mice or CGD patients have impaired fungicidal activity in vitro [55, 62]. Co-incubation of A. fumigatus hyphae with human neutrophils led to upregulation of genes encoding catalases and cytochrome c peroxidase [63]. Although previous studies hypothesized that ROI might be involved in killing of A. fumigatus, ROI detoxification mutants of A. fumigatus revealed no difference in virulence in mouse infection models when compared to the A. fumigatus wild type [64–66]. Therefore, it seems highly unlikely that ROI play an essential role in killing of this fungal pathogen.
Neutrophils can also produce reactive nitrogen intermediates (RNI) against microorganisms [67]. RNI can easily diffuse through membranes, nitrosylate proteins, and damage membranes and DNA. RNI can react with ROI to form toxic products like peroxynitrite. RNI also function as signal molecules in the immune response [67]. A. fumigatus employs two systems to detoxify RNI: flavohemoglobins (FhpA and FhpB) and the S-nitrosoglutathione reductase GnoA [68]. However, virulence in a murine model of pulmonary aspergillosis was not dependent on the ability of the fungus to counteract RNI produced by host immune cells [68].
Neutrophils can also use non-oxidative mechanisms to fight against pathogens. One is the discharge of the granule content, designated as degranulation. Defensins exert fungistatic activity and can kill A. fumigatus extracellularly [69, 70]. Zarember et al. [56] showed that lactoferrin can inhibit fungal growth by sequestering free iron ions. Furthermore, serine proteases like the PMN elastase (ELA) contribute to microbial killing [71, 72]. In line, ELA knockout (ELANE) mice exhibited a higher fungal burden indicating that ELA is involved in fungal clearance in vivo. However, in a murine model for invasive aspergillosis, ELANE mice showed survival rates like wild-type mice [73], indicating that the meaning of ELA, if there is any, is more complex.
A decade ago, Brinkmann et al. [74] described another extracellular killing mechanisms of neutrophils, the formation of neutrophil extracellular traps (NETs). It was shown in vitro and in mice that neutrophils produce NETs upon contact with A. fumigatus [75]. NETs consist of DNA fibers decorated with histones and antimicrobial proteins. NETosis is a specific form of cell death, at which the cell content is mixed and released into the surrounding. ROI produced by the NADPH oxidase complex are required for NETosis, which can also be induced exogenously by phorbol-12-myristate-13-acetate (PMA), LPS, and IL-8 [76]. In addition to histones, NETs mainly contain the proteins elastase, lactoferrin, cathepsin G, calprotectin, and MPO [77]. The decondensation of chromatin is regulated by neutrophil elastase and MPO [71]. Also, the Raf-MEK-ERK-pathway plays a role in the formation of NETs [78]. Obviously, NETs attach to and attack the pathogen by antimicrobial proteins. Calprotectin has a major antifungal effect, but also defensins, cathelicidin LL37, and histones show antifungal activity [79–81]. Pentraxin 3 (PTX3) is a soluble pattern recognition receptor produced by specific cells but also stored in granula in neutrophils. It is released also during formation of NETs and exhibits anti-Aspergillus activity [79, 82]. Exogenous addition of PTX3 early in infection restored antifungal resistance and restrained the inflammatory response to A. fumigatus [83]. Also, genetic deficiency of PTX3 affects the antifungal capacity of neutrophils and might contribute to the risk of invasive aspergillosis in hematopoietic stem cell transplantation patients [84]. Although NETs effectively kill several bacteria and pathogenic fungi such as C. albicans [74, 80, 81], they only act fungistatically against A. fumigatus [75, 85].
A. fumigatus evades the human complement system
The human complement system comprises approximately 30 serum-derived or membrane-associated proteins to exert its manifold effects on host-pathogen interactions and inflammatory events [86]. These effects aim at the maintenance of tissue homeostasis, resolution of inflammation, and clearance of pathogens, apoptotic cells, or debris. Moreover, the complement system provides a platform for the cross-talk between innate and humoral circuits via the interaction of complement activation products and surface receptors resulting in governing both T and B cell responses [87]. The non-redundant role of the complement system in the onset of invasive aspergillosis has been well documented in DBA/2N mouse models [88]. Complement system deficiencies correlate with a higher mortality rates in C5 knockout mice challenged with A. fumigatus [89, 90].
Complement activation can be triggered by three distinct pathways: the classical, the alternative, and the lectin pathway, all leading to proteolytic cleavage of the central C3 complement factor by C3 convertases and activation of the terminal pathway. Terminal pathway products include C3a and C3b that lead to the formation of terminal complement complex (TCC) and membrane attack complex (MAC) [87].
The complement machinery is induced by both Aspergillus conidia and hyphae. Resting conidia mainly activate complement by the alternative pathway. Conidial germination and hyphal formation, processes accompanied by changes of the cell wall composition, and exposure of surface α- and β-glucans shift complement activation to the classical pathway. Complement activation by all three forms of A. fumigatus features specific kinetics with slowest initiation by resting conidia [91].
Opsonization of pathogens by C3 complement fragments is a key strategy in complement-mediated clearance. Complement opsonization represents a chemotactic compass for immune cells and enhances phagocytosis, ROI production, and killing by alveolar macrophages, monocytes, and neutrophils [87]. Opsonization strongly depends on the availability of putative docking sites for C3 fragments on the fungal surface. A strong hint supporting this hypothesis was provided by two findings. Firstly, DHN-melanin serves as an efficient camouflage tool that reduces the exposure of surface antigens for C3 binding thus masking the fungal spores from efficient complement opsonization. DHN-melanin-deficient conidia bind more C3 fragments on their surface and exhibit reduced virulence in mice [19, 92]. These findings, together with reports of DHN-melanin as a scavenger of ROI and as an inhibitor of phagolysosome acidification in phagocytes, portrait DHN-melanin as a primary tool for A. fumigatus to hijack immune surveillance [46]. Secondly, complement deposition has been shown to be markedly different between virulent and non-virulent Aspergilli with A. fumigatus and A. flavus binding significantly less C3 in comparison to A. glaucus and A. nidulans [90].
The glucan surface cell wall carbohydrate structures can bind mannose-binding lectin (MBL), facilitate activation of the lectin pathway, and lead to C4 deposition in a concentration-dependent manner. MBL can then back up C3 proteolytic cleavage via C2 bypass mechanism disengaging the C3 convertase and thus initiating the alternative pathway. Such a scenario is not a specific hallmark of A. fumigatus alone but is also valid for A. flavus, A. niger, and A. terreus [93]. Crosdale and colleagues [94] highlighted the crucial role of serum MBL levels in the immune clearance of Aspergilli by investigating the correlation between the presence of mutations in the MBL gene promoter region that downregulated serum levels of MBL and the severity of chronic necrotizing pulmonary aspergillosis [94, 95].
Aspergillus-driven complement activation can also occur via the opsonization by PTX3 receptor that interacts either with ficolin-2 [96] via the lectin pathway or associates with C1q from the classical pathway [97]. In agreement with these findings, a recent report linked a single nucleotide gene polymorphism in PTX3 in homozygous haplotype donor individuals to an increased risk of invasive aspergillosis in stem cell recipients of such donor cells [84].
The fine tuning of the regulation of the complement machinery is achieved by the action of complement inhibitors. A. fumigatus sabotages complement activities by acquisition of complement inhibitors such as factor H (FH), factor H-related protein FHL-1, a splicing product of the FH gene, and C4 binding protein (C4bp). Factor H-binding sites were pinpointed to the N-terminal short consensus repeats (SCRs) 1 to 7 and one in the C-terminal SCR 20. FH and C4bp are soluble proteins promoting cleavage of C4b and C3b and accelerating disintegration of assembled C3 convertase. As a functional consequence, complement system is downregulated [98]. Furthermore, A. fumigatus produces complement inhibitor (CI) with yet unresolved structure that interferes with the alternative pathway and C3b-driven phagocytosis and killing [99].
A. fumigatus evades the complement also via secretion of extracellular proteases. For example, Alp1, a serine protease shown to degrade collagen, fibrinogen, and other extracellular matrix proteins, also targets complement components such as C1q, C3, C4, C5, MBL, and factor D [100, 101].
Taken together, A. fumigatus has developed a wide variety of armory to combat complement system activities. Unraveling the tools of such armory contributes to the elucidation of pathogenesis mechanisms and the development of therapeutic approaches for invasive aspergillosis by interference with the human complement system.
Immunoproteomics to identify A. fumigatus antigens
As described in the previous sections, interaction of A. fumigatus with the immune system takes place at several levels during infection of the human host. Next to biochemical and phenotypic single factor analysis and the application of other omics methods like transcriptomics, (immuno-) proteome approaches have been applied to investigate Aspergillus proteins that are likely communicating with host cells and effector proteins of the innate and adaptive immune system during infection. In general, proteomic techniques involve a protein cleanup and the reduction of the sample complexity by single or multi-dimensional electrophoretic and/or chromatographic separation procedures prior or subsequent to the enzymatic protein digestion [102]. The resulting peptide sequences are commonly measured by high-resolution tandem mass spectrometry and finally matched and statistically evaluated by sophisticated protein database search algorithms such as Mascot or Sequest [102].
Besides investigation of the dynamics of the A. fumigatus (wild-type or mutant) proteome, when cultivated under different conditions, e.g., upon confrontation with particular stresses [65, 103–106], Aspergillus cell wall and cell membrane subproteomes are of especial interest in the context of A. fumigatus pathogenicity. The fungal cell wall and the cytoplasmic membrane have a crucial impact on the virulence of the pathogen by constituting the first barriers interacting with the environment and consequently with the host’s immune defense [107]. Although proteins are only minor components of a fungal cell wall which is mainly composed of polysaccharides, they can play a major role in the interaction with the host, especially as antigens/allergens, adhesins, enzymes, or immunomodulators. In A. fumigatus, several conidial surface proteins such as the surface layer protein (hydrophobin) rodlet A, the aspartic protease PEP2, a putative disulfide isomerase, or an extracellular lipase, have been identified upon β-1,3-glucanase treatment of dormant conidia [108]. The presence of an extracellular lipase on the conidial surface may induce cell damage and adherence of the spores within the bronchoalveolar tract during inhalation [108]. In addition, proteins localized within the cytoplasmic lipid bilayer are also crucial for the interaction of the fungus with the environment and, in particular, with the host’s immune system. Ouyang et al. [109] analyzed total membrane preparations of A. fumigatus by SDS-PAGE separation, in-gel digestion, and a subsequent 2D-LC-MS/MS analysis. Thereby, the authors identified 530 membrane-associated proteins of which 17 were integral membrane proteins involved in N-, O-glycosylation, or glycophosphatidylinositol (GPI) anchor biosynthesis. For identification of GPI-anchored proteins, Bruneau et al. [110] released these proteins in a soluble form from the membrane fraction prior to proteome analysis by the combined action of the detergent n-octylglucoside and an endogenous phospholipase C activity.
In addition to analyzing solitary cultures, it has recently become attractive to directly investigate the interplay of Aspergillus with selected host cells by studying the changes in the individual proteomes and the interactome of co-cultures by proteomic (and transcriptomic) procedures [63, 111–113]. These strategies are especially suitable to dissect the interrelationships between host cells and the pathogen during infection. A study focusing on the proteome of human umbilical vein endothelial cells (HUVECs) upon co-incubation with A. fumigatus was recently performed [114]. By applying a mass spectrometry label-free proteomic approach, the molecular mechanism by which A. fumigatus can activate human HUVECs during blood vessel invasion was investigated. Angioinvasion is a key feature of invasive pulmonary aspergillosis (IPA). Endothelial cells act as physiological barriers that facilitate leukocyte migration and local immune response against microbial pathogens by secretion of cytokines/chemokines and other signal molecules. Vascular invasion of A. fumigatus leads to an activated prothrombic phenotype of HUVECs by which leukocyte migration, and an effective immune response is inhibited. A total of 89 proteins were differentially regulated during interaction of HUVECs with A. fumigatus germlings, i.e., 57 proteins were downregulated and 32 were upregulated. Another 409 proteins have been detected that were exclusive to one experimental condition (treatment or control). The group of upregulated proteins or proteins that have been exclusively identified in the interaction of HUVECs with A. fumigatus included particularly proteins with proangiogenic properties, namely, intercellular adhesion molecule-1 (ICAM-1), hepatocyte growth factor (HGF), fibroblast growth factor (FGF), activated leukocyte cell adhesion molecule (ALCAM), and basignin. As a consequence, it has been suggested that the vascular invasion by A. fumigatus activates multiple proteins that are involved in angiogenesis [114].
Another important focus in the field of immunoproteomics is the identification of Aspergillus proteins that serve as allergens and antigens in different Aspergillus-derived diseases by provoking a cellular and humoral immune response. Sensitization is one undesirable effect of the immune system that might arise when in contact with Aspergillus species (mostly A. fumigatus) due to prolonged inhalation and/or colonization of the lung. In particular, individuals suffering from cystic fibrosis or asthma are prone to develop an allergic reaction to Aspergillus, which can manifest itself as allergic pulmonary aspergillosis (ABPA) and exacerbates the health status of these patients [115]. ABPA is proposed to arise as a consequence of an inflammatory response to Aspergillus allergens, and Aspergillus causes bronchial epithelial cell damage that triggers a Th2 hypersensitivity immune response. Therefore, the clinical picture is generally accompanied with an elevated total IgE level and Aspergillus-specific elevated IgE and IgG levels [115]. Such anti-Aspergillus antibody profiles of ABPA patients (and of A. fumigatus sensitized asthmatics) have been analyzed in a number of immunoproteomic studies [116–119]. Gautam et al. [116], for example, studied the IgE immunoreactivity of A. fumigatus proteins by 2D gel electrophoresis followed by immunoblotting of 3-week old culture filtrate proteins with sera from A. fumigatus-sensitized asthmatics and ABPA patients. The authors identified five known and 11 novel antigens by MALDI-TOF mass spectrometry, including an extracellular arabinase, a chitosanase, and a catalase. Based on an enzyme-linked immunosorbent assay (ELISA), Glaser et al. (2009) showed that 94 % of ABPA patients (n = 64) and 46 % of A. fumigatus-sensitized asthmatics (n = 24) had Asp f 34-specific serum IgE [120]. Next to a better understanding of the disease, identified allergens can be used as recombinant proteins in novel ABPA-diagnosing assays that are based on the identification of anti-Aspergillus IgE and/or IgG antibodies in sera of patients. Interestingly, for ABPA, so far, only five recombinant proteins (rAsp f1–f4 and f6) of more than 20 known antigens/allergens are commercially available to proof an allergic immune response against A. fumigatus [121].
Such immunoproteomic analyses of Aspergillus proteins with respect to their interaction with patients’ antibodies have been also extended to cohorts of patients suffering from invasive forms of Aspergillus infections, like IPA. Although the highest risk group of patients suffering from invasive aspergillosis (IA) is strongly immunocompromised, there is a rising incidence of IA in non-immunocompromised but critically ill patients (up to 5.8 %). Shi et al. (2012) tested the immunoreactivity of 2D gel electrophoresis-separated extracellular proteins of A. fumigatus with sera from such patients with proven IA [122]. The authors identified 17 different antigens by application of MALDI-TOF mass spectrometry. The most intense immunoreactivity could be assigned to the secretory gliotoxin oxidase GliT (TR). Antibodies specific to TR have been proposed as a potential biomarker for the serologic diagnosis of IA in non-neutropenic patients that exhibit low serum galactomannan sensitivity [123].
Aside from detection of relevant Aspergillus proteins that have to be expressed during infection to elicit an immune response and their potential as diagnostic markers, the identification of vaccine candidates is an important objective in immunoproteomic approaches [124]. For identification of protective antibodies, Asif et al. (2010), for example, investigated two infected rabbits developing a protective immune response against invasive Aspergillosis by application of a 2D gel electrophoresis immunoblotting approach that has been combined with an identification of the antigenic proteome by LC-MS/MS determination [125]. In total, the authors identified 59 antigens, including proteins related to glycolysis and other primary metabolic pathways, oxidative stress response, and protein folding (heat shock proteins) as potential vaccine candidates.
Conclusions
A. fumigatus has developed a number of immune evasion mechanisms which interfere at the different levels of the infection process with the response of the human host. These include recognition of conidia, modulation of phagocytosis, intracellular processing, NET formation, and complement activation. New techniques and recent advances to analyze changes on the molecular level in both the fungal pathogen and host cells provide exciting possibilities to pin down essential steps in host pathogen interaction. For example, next generation sequencing allows expeditious acquisition of genome information of a multitude of A. fumigatus clinical and environmental isolates. RNAseq and sophisticated LC-MS analyses enable monitoring interaction-induced changes on the transcript level including alternative splicing, RNA editing, and protein modifications on both sides, the pathogen and the host. Last and not the least, automated analysis of digitalized and processed images or movies of the interaction of the pathogen with host cells was recently established [126]. Combined in a systems biology approach [127, 128], all these data will allow to model the interaction of fungal pathogens with different immune cells and to identify regulatory circuits of this interaction with the overall aim to improve diagnosis and to identify novel targets for the development of tailor-made antifungal drugs.
References
Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC (2012) Hidden killers: human fungal infections. Sci Transl Med 4(165):3004404
Brakhage AA (2005) Systemic fungal infections caused by Aspergillus species: epidemiology, infection process and virulence determinants. Curr Drug Targets 6(8):875–886
Brakhage AA, Langfelder K (2002) Menacing mold: the molecular biology of Aspergillus fumigatus. Annu Rev Microbiol 56:433–455
Wasylnka JA, Moore MM (2003) Aspergillus fumigatus conidia survive and germinate in acidic organelles of A549 epithelial cells. J Cell Sci 116(Pt 8):1579–1587
Paris S, Boisvieux-Ulrich E, Crestani B, Houcine O, Taramelli D, Lombardi L, Latge JP (1997) Internalization of Aspergillus fumigatus conidia by epithelial and endothelial cells. Infect Immun 65(4):1510–1514
Berkova N, Lair-Fulleringer S, Femenia F, Huet D, Wagner MC, Gorna K, Tournier F, Ibrahim-Granet O, Guillot J, Chermette R, Boireau P, Latge JP (2006) Aspergillus fumigatus conidia inhibit tumour necrosis factor- or staurosporine-induced apoptosis in epithelial cells. Int Immunol 18(1):139–150
Amin S, Thywissen A, Heinekamp T, Saluz HP, Brakhage AA (2014) Melanin dependent survival of Aspergillus fumigatus conidia in lung epithelial cells. Int J Med Microbiol IJMM 304(5–6):626–636. doi:10.1016/j.ijmm.2014.04.009
Hasenberg M, Behnsen J, Krappmann S, Brakhage A, Gunzer M (2011) Phagocyte responses towards Aspergillus fumigatus. Int J Med Microbiol IJMM 301(5):436–444. doi:10.1016/j.ijmm.2011.04.012
Latge JP (1999) Aspergillus fumigatus and aspergillosis. Clin Microbiol Rev 12(2):310–350
Balloy V, Huerre M, Latge JP, Chignard M (2005) Differences in patterns of infection and inflammation for corticosteroid treatment and chemotherapy in experimental invasive pulmonary aspergillosis. Infect Immun 73(1):494–503. doi:10.1128/IAI. 73.1.494-503.2005
Steele C, Rapaka RR, Metz A, Pop SM, Williams DL, Gordon S, Kolls JK, Brown GD (2005) The beta-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog 1(4):e42. doi:10.1371/journal.ppat.0010042
Gersuk GM, Underhill DM, Zhu L, Marr KA (2006) Dectin-1 and TLRs permit macrophages to distinguish between different Aspergillus fumigatus cellular states. J Immunol 176(6):3717–3724
Ramirez-Ortiz ZG, Means TK (2012) The role of dendritic cells in the innate recognition of pathogenic fungi (A. fumigatus, C. neoformans and C. albicans). Virulence 3(7):635–646. doi:10.4161/viru.22295
Cramer RA, Rivera A, Hohl TM (2011) Immune responses against Aspergillus fumigatus: what have we learned? Curr Opin Infect Dis 24(4):315–322. doi:10.1097/QCO.0b013e328348b159
Medzhitov R, Janeway C Jr (2000) Innate immunity. N Engl J Med 343(5):338–344. doi:10.1056/NEJM200008033430506
Luther K, Torosantucci A, Brakhage AA, Heesemann J, Ebel F (2007) Phagocytosis of Aspergillus fumigatus conidia by murine macrophages involves recognition by the dectin-1 beta-glucan receptor and Toll-like receptor 2. Cell Microbiol 9(2):368–381
Aimanianda V, Bayry J, Bozza S, Kniemeyer O, Perruccio K, Elluru SR, Clavaud C, Paris S, Brakhage AA, Kaveri SV, Romani L, Latge JP (2009) Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature 460(7259):1117–1121. doi:10.1038/nature08264
Hohl TM, Van Epps HL, Rivera A, Morgan LA, Chen PL, Feldmesser M, Pamer EG (2005) Aspergillus fumigatus triggers inflammatory responses by stage-specific beta-glucan display. PLoS Pathog 1(3):18
Langfelder K, Jahn B, Gehringer H, Schmidt A, Wanner G, Brakhage AA (1998) Identification of a polyketide synthase gene (pksP) of Aspergillus fumigatus involved in conidial pigment biosynthesis and virulence. Med Microbiol Immunol 187(2):79–89
Jahn B, Koch A, Schmidt A, Wanner G, Gehringer H, Bhakdi S, Brakhage AA (1997) Isolation and characterization of a pigmentless-conidium mutant of Aspergillus fumigatus with altered conidial surface and reduced virulence. Infect Immun 65(12):5110–5117
Jahn B, Langfelder K, Schneider U, Schindel C, Brakhage AA (2002) PKSP-dependent reduction of phagolysosome fusion and intracellular kill of Aspergillus fumigatus conidia by human monocyte-derived macrophages. Cell Microbiol 4(12):793–803
Pihet M, Vandeputte P, Tronchin G, Renier G, Saulnier P, Georgeault S, Mallet R, Chabasse D, Symoens F, Bouchara JP (2009) Melanin is an essential component for the integrity of the cell wall of Aspergillus fumigatus conidia. BMC Microbiol 9:177. doi:10.1186/1471-2180-9-177
Bayry J, Beaussart A, Dufrene YF, Sharma M, Bansal K, Kniemeyer O, Aimanianda V, Brakhage AA, Kaveri SV, Kwon-Chung KJ, Latge JP, Beauvais A (2014) Surface structure characterization of Aspergillus fumigatus conidia mutated in the melanin synthesis pathway and their human cellular immune response. Infect Immun 82(8):3141–3153
Heinekamp T, Thywissen A, Macheleidt J, Keller S, Valiante V, Brakhage AA (2012) Aspergillus fumigatus melanins: interference with the host endocytosis pathway and impact on virulence. Front Microbiol 3:440. doi:10.3389/fmicb.2012.00440
Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S (2005) Macrophage receptors and immune recognition. Annu Rev Immunol 23:901–944. doi:10.1146/annurev.immunol.23.021704.115816
Serrano-Gomez D, Dominguez-Soto A, Ancochea J, Jimenez-Heffernan JA, Leal JA, Corbi AL (2004) Dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin mediates binding and internalization of Aspergillus fumigatus conidia by dendritic cells and macrophages. J Immunol 173(9):5635–5643
Meier A, Kirschning CJ, Nikolaus T, Wagner H, Heesemann J, Ebel F (2003) Toll-like receptor (TLR) 2 and TLR4 are essential for Aspergillus-induced activation of murine macrophages. Cell Microbiol 5(8):561–570
Netea MG, Warris A, Van der Meer JW, Fenton MJ, Verver-Janssen TJ, Jacobs LE, Andresen T, Verweij PE, Kullberg BJ (2003) Aspergillus fumigatus evades immune recognition during germination through loss of toll-like receptor-4-mediated signal transduction. J Infect Dis 188(2):320–326. doi:10.1086/376456
Chai LY, Vonk AG, Kullberg BJ, Verweij PE, Verschueren I, van der Meer JW, Joosten LA, Latge JP, Netea MG (2011) Aspergillus fumigatus cell wall components differentially modulate host TLR2 and TLR4 responses. Microbes Infect Inst Pasteur 13(2):151–159. doi:10.1016/j.micinf.2010.10.005
Levitz SM (2010) Innate recognition of fungal cell walls. PLoS Pathog 6(4):e1000758. doi:10.1371/journal.ppat.1000758
Bellocchio S, Montagnoli C, Bozza S, Gaziano R, Rossi G, Mambula SS, Vecchi A, Mantovani A, Levitz SM, Romani L (2004) The contribution of the Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J Immunol 172(5):3059–3069
Ramaprakash H, Ito T, Standiford TJ, Kunkel SL, Hogaboam CM (2009) Toll-like receptor 9 modulates immune responses to Aspergillus fumigatus conidia in immunodeficient and allergic mice. Infect Immun 77(1):108–119. doi:10.1128/IAI. 00998-08
Willment JA, Marshall AS, Reid DM, Williams DL, Wong SY, Gordon S, Brown GD (2005) The human beta-glucan receptor is widely expressed and functionally equivalent to murine Dectin-1 on primary cells. Eur J Immunol 35(5):1539–1547. doi:10.1002/eji.200425725
Philippe B, Ibrahim-Granet O, Prevost MC, Gougerot-Pocidalo MA, Sanchez Perez M, Van der Meeren A, Latge JP (2003) Killing of Aspergillus fumigatus by alveolar macrophages is mediated by reactive oxidant intermediates. Infect Immun 71(6):3034–3042
Ibrahim-Granet O, Philippe B, Boleti H, Boisvieux-Ulrich E, Grenet D, Stern M, Latge JP (2003) Phagocytosis and intracellular fate of Aspergillus fumigatus conidia in alveolar macrophages. Infect Immun 71(2):891–903
Claus V, Jahraus A, Tjelle T, Berg T, Kirschke H, Faulstich H, Griffiths G (1998) Lysosomal enzyme trafficking between phagosomes, endosomes, and lysosomes in J774 macrophages. Enrichment of cathepsin H in early endosomes. J Biol Chem 273(16):9842–9851
Garin J, Diez R, Kieffer S, Dermine JF, Duclos S, Gagnon E, Sadoul R, Rondeau C, Desjardins M (2001) The phagosome proteome: insight into phagosome functions. J Cell Biol 152(1):165–180
Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y (1991) Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 266(26):17707–17712
Thywissen A, Heinekamp T, Dahse HM, Schmaler-Ripcke J, Nietzsche S, Zipfel PF, Brakhage AA (2011) Conidial dihydroxynaphthalene melanin of the human pathogenic fungus Aspergillus fumigatus interferes with the host endocytosis pathway. Front Microbiol 2(96)
Levitz SM, Nong SH, Seetoo KF, Harrison TS, Speizer RA, Simons ER (1999) Cryptococcus neoformans resides in an acidic phagolysosome of human macrophages. Infect Immun 67(2):885–890
Volling K, Thywissen A, Brakhage AA, Saluz HP (2011) Phagocytosis of melanized Aspergillus conidia by macrophages exerts cytoprotective effects by sustained PI3K/Akt signalling. Cell Microbiol 13(8):1130–1148. doi:10.1111/j.1462-5822.2011.01605.x
Volling K, Brakhage AA, Saluz HP (2007) Apoptosis inhibition of alveolar macrophages upon interaction with conidia of Aspergillus fumigatus. FEMS Microbiol Lett 275(2):250–254. doi:10.1111/j.1574-6968.2007.00883.x
Femenia F, Huet D, Lair-Fulleringer S, Wagner MC, Sarfati J, Shingarova L, Guillot J, Boireau P, Chermette R, Berkova N (2009) Effects of conidia of various Aspergillus species on apoptosis of human pneumocytes and bronchial epithelial cells. Mycopathologia 167(5):249–262. doi:10.1007/s11046-008-9175-3
Stanzani M, Orciuolo E, Lewis R, Kontoyiannis DP, Martins SL, St John LS, Komanduri KV (2005) Aspergillus fumigatus suppresses the human cellular immune response via gliotoxin-mediated apoptosis of monocytes. Blood 105(6):2258–2265. doi:10.1182/blood-2004-09-3421
Pardo J, Urban C, Galvez EM, Ekert PG, Muller U, Kwon-Chung J, Lobigs M, Mullbacher A, Wallich R, Borner C, Simon MM (2006) The mitochondrial protein Bak is pivotal for gliotoxin-induced apoptosis and a critical host factor of Aspergillus fumigatus virulence in mice. J Cell Biol 174(4):509–519. doi:10.1083/jcb.200604044
Brakhage AA, Bruns S, Thywissen A, Zipfel PF, Behnsen J (2010) Interaction of phagocytes with filamentous fungi. Curr Opin Microbiol 13(4):409–415
Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER (2010) Neutrophil kinetics in health and disease. Trends Immunol 31(8):318–324. doi:10.1016/j.it.2010.05.006
Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JAM, Tesselaar K, Koenderman L (2010) In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 116(4):625–627. doi:10.1182/blood-2010-01-259028
Geering B, Stoeckle C, Conus S, Simon HU (2013) Living and dying for inflammation: neutrophils, eosinophils, basophils. Trends Immunol 34(8):398–409. doi:10.1016/j.it.2013.04.002
Chignard M (2009) In: Latgé J-P, Steinbach WJ (eds) Aspergillus fumigatus and aspergillosis. ASM Press, Washington DC, pp 229–238
Mircescu MM, Lipuma L, van Rooijen N, Pamer EG, Hohl TM (2009) Essential role for neutrophils but not alveolar macrophages at early time points following Aspergillus fumigatus infection. J Infect Dis 200(4):647–656. doi:10.1086/600380
Braedel S, Radsak M, Einsele H, Latge JP, Michan A, Loeffler J, Haddad Z, Grigoleit U, Schild H, Hebart H (2004) Aspergillus fumigatus antigens activate innate immune cells via toll-like receptors 2 and 4. Br J Haematol 125(3):392–399. doi:10.1111/j.1365-2141.2004.04922.x
Kennedy AD, Willment JA, Dorward DW, Williams DL, Brown GD, DeLeo FR (2007) Dectin-1 promotes fungicidal activity of human neutrophils. Eur J Immunol 37(2):467–478. doi:10.1002/eji.200636653
Werner JL, Metz AE, Horn D, Schoeb TR, Hewitt MM, Schwiebert LM, Faro-Trindade I, Brown GD, Steele C (2009) Requisite role for the dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J Immunol 182(8):4938–4946. doi:10.4049/jimmunol.0804250
Park SJ, Mehrad B (2009) Innate immunity to Aspergillus species. Clin Microbiol Rev 22(4):535–551. doi:10.1128/CMR. 00014-09
Zarember KA, Sugui JA, Chang YC, Kwon-Chung KJ, Gallin JI (2007) Human polymorphonuclear leukocytes inhibit Aspergillus fumigatus conidial growth by lactoferrin-mediated iron depletion. J Immunol 178(10):6367–6373
Borregaard N (2010) Neutrophils, from marrow to microbes. Immunity 33(5):657–670. doi:10.1016/j.immuni.2010.11.011
Segal AW (2005) How neutrophils kill microbes. Annu Rev Immunol 23:197–223. doi:10.1146/annurev.immunol.23.021704.115653
Scapini P, Lapinet-Vera JA, Gasperini S, Calzetti F, Bazzoni F, Cassatella MA (2000) The neutrophil as a cellular source of chemokines. Immunol Rev 177:195–203
Boyle KB, Stephens LR, Hawkins PT (2012) Activation of the neutrophil NADPH oxidase by Aspergillus fumigatus. Ann N Y Acad Sci 1273:68–73. doi:10.1111/j.1749-6632.2012.06821.x
Diamond RD, Clark RA (1982) Damage to Aspergillus fumigatus and Rhizopus oryzae hyphae by oxidative and nonoxidative microbicidal products of human neutrophils in vitro. Infect Immun 38(2):487–495
Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC (1995) Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet 9(2):202–209. doi:10.1038/ng0295-202
Sugui JA, Kim HS, Zarember KA, Chang YC, Gallin JI, Nierman WC, Kwon-Chung KJ (2008) Genes differentially expressed in conidia and hyphae of Aspergillus fumigatus upon exposure to human neutrophils. PLoS One 3(7):e2655. doi:10.1371/journal.pone.0002655
Lamarre C, Ibrahim-Granet O, Du C, Calderone R, Latge JP (2007) Characterization of the SKN7 ortholog of Aspergillus fumigatus. Fungal Genet Biol: FG & B 44(7):682–690. doi:10.1016/j.fgb.2007.01.009
Lessing F, Kniemeyer O, Wozniok I, Loeffler J, Kurzai O, Haertl A, Brakhage AA (2007) The Aspergillus fumigatus transcriptional regulator AfYap1 represents the major regulator for defense against reactive oxygen intermediates but is dispensable for pathogenicity in an intranasal mouse infection model. Eukaryot Cell 6(12):2290–2302. doi:10.1128/EC.00267-07
Lambou K, Lamarre C, Beau R, Dufour N, Latge JP (2010) Functional analysis of the superoxide dismutase family in Aspergillus fumigatus. Mol Microbiol 75(4):910–923
Bogdan C (2011) Reactive oxygen and reactive nitrogen intermediates in the immune system. Immunology of infectious diseases. ASM Press, Washington, DC
Lapp K, Vödisch M, Kroll K, Strassburger M, Kniemeyer O, Heinekamp T, Brakhage AA (2014) Characterization of the Aspergillus fumigatus detoxification systems for reactive nitrogen intermediates and their impact on virulence. Front Microbiol 5. doi:10.3389/fmicb.2014.00469
Auvynet C, Rosenstein Y (2009) Multifunctional host defense peptides: antimicrobial peptides, the small yet big players in innate and adaptive immunity. FEBS J 276(22):6497–6508. doi:10.1111/j.1742-4658.2009.07360.x
Levitz SM, Selsted ME, Ganz T, Lehrer RI, Diamond RD (1986) In vitro killing of spores and hyphae of Aspergillus fumigatus and Rhizopus oryzae by rabbit neutrophil cationic peptides and bronchoalveolar macrophages. J Infect Dis 154(3):483–489
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A (2010) Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 191(3):677–691. doi:10.1083/jcb.201006052
Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW (2002) Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 416(6878):291–297. doi:10.1038/416291a
Prufer S, Weber M, Stein P, Bosmann M, Stassen M, Kreft A, Schild H, Radsak MP (2014) Oxidative burst and neutrophil elastase contribute to clearance of Aspergillus fumigatus pneumonia in mice. Immunobiology 219(2):87–96. doi:10.1016/j.imbio.2013.08.010
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303(5663):1532–1535. doi:10.1126/science.1092385
Bruns S, Kniemeyer O, Hasenberg M, Aimanianda V, Nietzsche S, Thywissen A, Jeron A, Latge JP, Brakhage AA, Gunzer M (2010) Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog 6(4):1000873
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176(2):231–241. doi:10.1083/jcb.200606027
Brinkmann V, Zychlinsky A (2012) Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol 198(5):773–783
Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, Waldmann H (2011) Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol 7(2):75–77
Jaillon S, Peri G, Delneste Y, Frémaux I, Doni A, Moalli F, Garlanda C, Romani L, Gascan H, Bellocchio S, Bozza S, Cassatella MA, Jeannin P, Mantovani A (2007) The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J Exp Med 204(4):793–804. doi:10.1084/jem.20061301
Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A (2009) Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog 5(10):e1000639. doi:10.1371/journal.ppat.1000639
von Köckritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, Medina E (2008) Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 111(6):3070–3080. doi:10.1182/blood-2007-07-104018
Garlanda C, Hirsch E, Bozza S, Salustri A, De Acetis M, Nota R, Maccagno A, Riva F, Bottazzi B, Peri G, Doni A, Vago L, Botto M, De Santis R, Carminati P, Siracusa G, Altruda F, Vecchi A, Romani L, Mantovani A (2002) Non-redundant role of the long pentraxin PTX3 in anti-fungal innate immune response. Nature 420(6912):182–186. doi:10.1038/nature01195
D’Angelo C, De Luca A, Zelante T, Bonifazi P, Moretti S, Giovannini G, Iannitti RG, Zagarella S, Bozza S, Campo S, Salvatori G, Romani L (2009) Exogenous pentraxin 3 restores antifungal resistance and restrains inflammation in murine chronic granulomatous disease. J Immunol 183(7):4609–4618. doi:10.4049/jimmunol.0900345
Cunha C, Aversa F, Lacerda JF, Busca A, Kurzai O, Grube M, Loffler J, Maertens JA, Bell AS, Inforzato A, Barbati E, Almeida B, Santos e Sousa P, Barbui A, Potenza L, Caira M, Rodrigues F, Salvatori G, Pagano L, Luppi M, Mantovani A, Velardi A, Romani L, Carvalho A (2014) Genetic PTX3 deficiency and aspergillosis in stem-cell transplantation. N Engl J Med 370(5):421–432. doi:10.1056/NEJMoa1211161
McCormick A, Heesemann L, Wagener J, Marcos V, Hartl D, Loeffler J, Heesemann J, Ebel F (2010) NETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes Infect Inst Pasteur 12(12–13):928–936. doi:10.1016/j.micinf.2010.06.009
Zipfel PF, Skerka C (2009) Complement regulators and inhibitory proteins. Nat Rev Immunol 9(10):729–740
Ricklin D, Hajishengallis G, Yang K, Lambris JD (2010) Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11(9):785–797
Hector RF, Yee E, Collins MS (1990) Use of DBA/2N mice in models of systemic candidiasis and pulmonary and systemic aspergillosis. Infect Immun 58(5):1476–1478
Svirshchevskaya EV, Shevchenko MA, Huet D, Femenia F, Latge JP, Boireau P, Berkova NP (2009) Susceptibility of mice to invasive aspergillosis correlates with delayed cell influx into the lungs. Int J Immunogenet 36(5):289–299
Henwick S, Hetherington SV, Patrick CC (1993) Complement binding to Aspergillus conidia correlates with pathogenicity. J Lab Clin Med 122(1):27–35
Kozel TR, Wilson MA, Farrell TP, Levitz SM (1989) Activation of C3 and binding to Aspergillus fumigatus conidia and hyphae. Infect Immun 57(11):3412–3417
Tsai HF, Chang YC, Washburn RG, Wheeler MH, Kwon-Chung KJ (1998) The developmentally regulated alb1 gene of Aspergillus fumigatus: its role in modulation of conidial morphology and virulence. J Bacteriol 180(12):3031–3038
Neth O, Jack DL, Dodds AW, Holzel H, Klein NJ, Turner MW (2000) Mannose-binding lectin binds to a range of clinically relevant microorganisms and promotes complement deposition. Infect Immun 68(2):688–693
Crosdale DJ, Poulton KV, Ollier WE, Thomson W, Denning DW (2001) Mannose-binding lectin gene polymorphisms as a susceptibility factor for chronic necrotizing pulmonary aspergillosis. J Infect Dis 184(5):653–656
Kaur S, Gupta VK, Thiel S, Sarma PU, Madan T (2007) Protective role of mannan-binding lectin in a murine model of invasive pulmonary aspergillosis. Clin Exp Immunol 148(2):382–389
Ma YJ, Doni A, Hummelshoj T, Honore C, Bastone A, Mantovani A, Thielens NM, Garred P (2009) Synergy between ficolin-2 and pentraxin 3 boosts innate immune recognition and complement deposition. J Biol Chem 284(41):28263–28275
Moalli F, Doni A, Deban L, Zelante T, Zagarella S, Bottazzi B, Romani L, Mantovani A, Garlanda C (2010) Role of complement and Fcγ receptors in the protective activity of the long pentraxin PTX3 against Aspergillus fumigatus. Blood 116(24):5170–5180
Behnsen J, Hartmann A, Schmaler J, Gehrke A, Brakhage AA, Zipfel PF (2008) The opportunistic human pathogenic fungus Aspergillus fumigatus evades the host complement system. Infect Immun 76(2):820–827
Washburn RG, DeHart DJ, Agwu DE, Bryant-Varela BJ, Julian NC (1990) Aspergillus fumigatus complement inhibitor: production, characterization, and purification by hydrophobic interaction and thin-layer chromatography. Infect Immun 58(11):3508–3515
Rambach G, Dum D, Mohsenipour I, Hagleitner M, Wurzner R, Lass-Florl C, Speth C (2010) Secretion of a fungal protease represents a complement evasion mechanism in cerebral aspergillosis. Mol Immunol 47(7–8):1438–1449
Behnsen J, Lessing F, Schindler S, Wartenberg D, Jacobsen ID, Thoen M, Zipfel PF, Brakhage AA (2010) Secreted Aspergillus fumigatus protease Alp1 degrades human complement proteins C3, C4, and C5. Infect Immun 78(8):3585–3594
Kniemeyer O (2011) Proteomics of eukaryotic microorganisms: the medically and biotechnologically important fungal genus Aspergillus. Proteomics 11(15):3232–3243
Albrecht D, Guthke R, Brakhage AA, Kniemeyer O (2010) Integrative analysis of the heat shock response in Aspergillus fumigatus. BMC Genomics 11(32):1471–2164
Barker BM, Kroll K, Vodisch M, Mazurie A, Kniemeyer O, Cramer RA (2012) Transcriptomic and proteomic analyses of the Aspergillus fumigatus hypoxia response using an oxygen-controlled fermenter. BMC Genomics 13(62):1471–2164
Bruns S, Seidler M, Albrecht D, Salvenmoser S, Remme N, Hertweck C, Brakhage AA, Kniemeyer O, Muller FM (2010) Functional genomic profiling of Aspergillus fumigatus biofilm reveals enhanced production of the mycotoxin gliotoxin. Proteomics 10(17):3097–3107
Vodisch M, Scherlach K, Winkler R, Hertweck C, Braun HP, Roth M, Haas H, Werner ER, Brakhage AA, Kniemeyer O (2011) Analysis of the Aspergillus fumigatus proteome reveals metabolic changes and the activation of the pseurotin A biosynthesis gene cluster in response to hypoxia. J Proteome Res 10(5):2508–2524
Kniemeyer O, Schmidt AD, Vodisch M, Wartenberg D, Brakhage AA (2011) Identification of virulence determinants of the human pathogenic fungi Aspergillus fumigatus and Candida albicans by proteomics. Int J Med Microbiol IJMM 301(5):368–377
Asif AR, Oellerich M, Amstrong VW, Riemenschneider B, Monod M, Reichard U (2006) Proteome of conidial surface associated proteins of Aspergillus fumigatus reflecting potential vaccine candidates and allergens. J Proteome Res 5(4):954–962
Ouyang H, Luo Y, Zhang L, Li Y, Jin C (2010) Proteome analysis of Aspergillus fumigatus total membrane proteins identifies proteins associated with the glycoconjugates and cell wall biosynthesis using 2D LC-MS/MS. Mol Biotechnol 44(3):177–189
Bruneau JM, Magnin T, Tagat E, Legrand R, Bernard M, Diaquin M, Fudali C, Latge JP (2001) Proteome analysis of Aspergillus fumigatus identifies glycosylphosphatidylinositol-anchored proteins associated to the cell wall biosynthesis. Electrophoresis 22(13):2812–2823
Fekkar A, Balloy V, Pionneau C, Marinach-Patrice C, Chignard M, Mazier D (2012) Secretome of human bronchial epithelial cells in response to the fungal pathogen Aspergillus fumigatus analyzed by differential in-gel electrophoresis. J Infect Dis 205(7):1163–1172
Morton CO, Bouzani M, Loeffler J, Rogers TR (2012) Direct interaction studies between Aspergillus fumigatus and human immune cells; what have we learned about pathogenicity and host immunity? Front Microbiol 3(413)
McDonagh A, Fedorova ND, Crabtree J, Yu Y, Kim S, Chen D, Loss O, Cairns T, Goldman G, Armstrong-James D, Haynes K, Haas H, Schrettl M, May G, Nierman WC, Bignell E (2008) Sub-telomere directed gene expression during initiation of invasive aspergillosis. PLoS Pathog 4(9):1000154
Curty N, Kubitschek-Barreira PH, Neves GW, Gomes D, Pizzatti L, Abdelhay E, Souza GH, Lopes-Bezerra LM (2014) Discovering the infectome of human endothelial cells challenged with Aspergillus fumigatus applying a mass spectrometry label-free approach. J Proteomics 97:126–140
Knutsen AP, Slavin RG (2011) Allergic bronchopulmonary aspergillosis in asthma and cystic fibrosis. Clin Dev Immunol 843763(10):5
Gautam P, Sundaram CS, Madan T, Gade WN, Shah A, Sirdeshmukh R, Sarma PU (2007) Identification of novel allergens of Aspergillus fumigatus using immunoproteomics approach. Clin Exp Allergy 37(8):1239–1249
Singh B, Oellerich M, Kumar R, Kumar M, Bhadoria DP, Reichard U, Gupta VK, Sharma GL, Asif AR (2010) Immuno-reactive molecules identified from the secreted proteome of Aspergillus fumigatus. J Proteome Res 9(11):5517–5529
Chow LP, Liu SL, Yu CJ, Liao HK, Tsai JJ, Tang TK (2000) Identification and expression of an allergen Asp f 13 from Aspergillus fumigatus and epitope mapping using human IgE antibodies and rabbit polyclonal antibodies. Biochem J 2:423–431
Gautam P, Madan T, Gade WN, Sarma PU (2006) Immunoproteomic analysis of secretory proteins of Aspergillus fumigatus with specific IGE immunoreactivity. Indian J Clin Biochem 21(2):12–19
Glaser AG, Kirsch AI, Zeller S, Menz G, Rhyner C, Crameri R (2009) Molecular and immunological characterization of Asp f 34, a novel major cell wall allergen of Aspergillus fumigatus. Allergy 64(8):1144–1151
Singh B, Singh S, Asif AR, Oellerich M, Sharma GL (2014) Allergic Aspergillosis and the antigens of Aspergillus fumigatus. Curr Protein Pept Sci 12:12
Shi LN, Li FQ, Huang M, Lu JF, Kong XX, Wang SQ, Shao HF (2012) Immunoproteomics based identification of thioredoxin reductase GliT and novel Aspergillus fumigatus antigens for serologic diagnosis of invasive aspergillosis. BMC Microbiol 12(11):1471–2180
Shi LN, Li FQ, Lu JF, Kong XX, Wang SQ, Huang M, Shao HF, Shao SH (2012) Antibody specific to thioredoxin reductase as a new biomarker for serodiagnosis of invasive aspergillosis in non-neutropenic patients. Clin Chim Acta 413(9–10):938–943
Reichard U, Herrmann S, Asif AR (2014) Vaccination approaches against opportunistic fungal infections caused by Aspergillus fumigatus. Curr Protein Pept Sci 12:12
Asif AR, Oellerich M, Amstrong VW, Gross U, Reichard U (2010) Analysis of the cellular Aspergillus fumigatus proteome that reacts with sera from rabbits developing an acquired immunity after experimental aspergillosis. Electrophoresis 31(12):1947–1958
Mech F, Thywissen A, Guthke R, Brakhage AA, Figge MT (2011) Automated image analysis of the host-pathogen interaction between phagocytes and Aspergillus fumigatus. PLoS One 6(5):0019591
Albrecht D, Kniemeyer O, Mech F, Gunzer M, Brakhage A, Guthke R (2011) On the way toward systems biology of Aspergillus fumigatus infection. Int J Med Microbiol IJMM 301(5):453–459
Horn F, Heinekamp T, Kniemeyer O, Pollmacher J, Valiante V, Brakhage AA (2012) Systems biology of fungal infection. Front Microbiol 3(108)
Acknowledgments
This work was supported by the Graduate School of Excellence “Jena School for Microbial Communication (JSMC)” funded by the Deutsche Forschungsgemeinschaft (DFG) (www.jsmc.uni-jena.de), the AspBIOmics project (BMBF FKZ 0315900 B) within the coordination action program European Research Area Network PathoGenoMics, and the DFG-funded CRC/Transregio 124 “Human-pathogenic fungi and their human host—Networks of interaction—FungiNet” (www.funginet.de).
Ethical standards
All experiments were in compliance with the German law and were approved by the responsible Federal State authority and ethics committee.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
This article is a contribution to the special issue on Immunopathology of Fungal Diseases - Guest Editor: Jean-Paul Latge
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Heinekamp, T., Schmidt, H., Lapp, K. et al. Interference of Aspergillus fumigatus with the immune response. Semin Immunopathol 37, 141–152 (2015). https://doi.org/10.1007/s00281-014-0465-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-014-0465-1