
Abstract
Background
As an organelle essential for intracellular energy supply, mitochondria are involved in intracellular metabolism and inflammation, and cell death. The interaction of mitochondria with the NLRP3 inflammasome in the development of lung diseases has been extensively studied. However, the exact mechanism by which mitochondria mediate the activation of the NLRP3 inflammasome and trigger lung disease is still unclear.
Methods
The literatures related to mitochondrial stress, NLRP3 inflammasome and lung diseases were searched in PubMed.
Results
This review aims to provide new insights into the recently discovered mitochondrial regulation of the NLRP3 inflammasome in lung diseases. It also describes the crucial roles of mitochondrial autophagy, long noncoding RNA, micro RNA, altered mitochondrial membrane potential, cell membrane receptors, and ion channels in mitochondrial stress and regulation of the NLRP3 inflammasome, in addition to the reduction of mitochondrial stress by nuclear factor erythroid 2-related factor 2 (Nrf2). The effective components of potential drugs for the treatment of lung diseases under this mechanism are also summarized.
Conclusion
This review provides a resource for the discovery of new therapeutic mechanisms and suggests ideas for the development of new therapeutic drugs, thus promoting the rapid treatment of lung diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
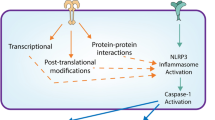
The human innate immune response, as the first line of defense against pathogenic invasion, is initiated by the pattern recognition receptor encoded by germline genes [1]. Pathogenic organisms are identified by pathogen-associated molecular patterns, in which a series of inflammatory responses are triggered to eliminate the associated pathogen or microbial infection to repair damaged tissue [2]. Since they were first proposed in 2002, inflammasomes have received widespread attention from the scientific community, which found that the NLRP3 inflammasome plays an important role as the core of the inflammatory response [3]. NLRP3 inflammasomes comprise complex protein bodies assembled from sensor protein NLRP3, adaptor protein of apoptosis-associated speck-like protein (ASC), and effector protein caspase-1 [4, 5]. Sensor protein NLRP3 is composed of three structural domains: a leucine-rich repeat (LRR) sequence, a central nucleotide-binding NACHT domain with ATPase activity, and an N-terminal pyrin domain (PYD) [6]. When external stimuli attack, sensor protein NLRP3 recruits ASC, forming a PYD–PYD structural domain with the PYD in ASC that results in the aggregation of ASC into ASC specks [7]. ASC specks then recruit the effector protein pro-caspase-1, binding to the caspase recruitment domain (CARD) in pro-caspase-1 to form a CARD-CARD structural domain. Eventually, the NLRP3 inflammasome is formed, which further activates caspase-1 [8, 9] to promote the maturation of interleukin (IL)-1β and IL-18, and shears Gasdermin D (GSDMD). Additionally, the N-terminal domain of GSDMD is then transferred to the cell membrane to form pores, leading to the release of cell contents and pyroptosis [10].
Currently, the NLRP3 inflammasome is understood to be mainly activated by two types of signals: a primary signal and an activation signal [11]. Toll-like receptors (TLRs), cytokine receptors, nucleotide-binding domain and LRR-containing (NLR) ligands, and other factors located on the cell membrane act as the primary signal to activate nuclear factor NF-κB into the nucleus, further promoting expression of NLRP3, pro-IL-18, and pro-IL-1β [12, 13]. Activation signals, such as reactive oxygen species (ROS), lysosomal damage, K+ efflux, Ca2+ inward flow, and ATP, have all been shown to promote the assembly and activation of the NLRP3 inflammasome [14, 15]. The activated inflammasome promotes caspase-1 activation and further promotes the maturation of pro-inflammatory factors and pyroptosis (Fig. 1) [16]. Therefore, aberrant expression of the NLRP3 inflammasome can trigger inflammatory damage in the body. Such aberrant expression has been identified in many types of lung disease, including chronic obstructive pulmonary disease (COPD), lung cancer, acute lung injury (ALI), pulmonary fibrosis, and others (Fig. 2).

Activation and assembly of the NLRP3 inflammasome. NIMA-related kinase 7 (NEK7); Thioredoxin-Interacting Protein (TXNIP)

Mitochondrial stress and the NLRP3 inflammasome mediate lung disease
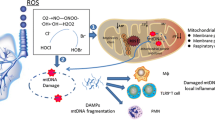
Mitochondria are bilayer membrane organelles that provide energy for various cellular biological activities and are present in most cells, with participatory roles in cell proliferation, differentiation, senescence, apoptosis, and other processes [17]. Mitochondrial energy production requires extensive oxidative phosphorylation. During this process, the electron transport chain is blocked and part of the oxygen undergoes incomplete reduction to produce toxic substances, including superoxide anions, hydrogen peroxide and hydroxyl radicals [18, 19]. These substances are partially converted into ROS, and comprise the primary source of endogenous ROS production [20]. Additionally, compared with nuclear DNA, mitochondrial DNA (mtDNA) is more sensitive to oxidative stress [21]. Damage to mtDNA leads to mitochondrial dysfunction, in which mitochondrial double-stranded RNAs (mt-dsRNAs) are highly expressed and released extracellularly, leading to innate immune activation [22]. In response to stimulation, mitochondria can be induced to divide by mitochondrial fission factor (Mff) and dynamin-related protein 1 (Drp1). Alternatively, cells rely on mitofusin-2 (Mfn2) and optic atrophy 1 (OPA1) to fuse neighboring mitochondria and regulate mitochondrial kinetic balance [23]. In contrast, protein kinase R (PKR), a major sensor of mitochondrial stress-mediated cell death, recognizes most of the mt-dsRNAs [24]. Mt-dsRNAs activate PKR autophosphorylation and inhibit translation, while activating the release of inflammasome and pro-inflammatory factors, disrupting cellular homeostasis. Autophagy not only prevents the activation of PKR but also removes intracytoplasmic mt-dsRNAs, thus reducing the damage to the organism from mitochondrial stress [25]. Additionally, autophagy is involved in regulating dynamic homeostasis and can effectively remove damaged mitochondria from the cell. In damaged mitochondria, PTEN-induced putative kinase 1 (PINK1) accumulates on the outer mitochondrial membrane, marking the mitochondria for removal. After undergoing auto-phosphorylation, PINK1 recruits the E3 ligase Parkin, which ubiquinates mitochondrial outer membrane proteins and triggers the mitophagic process [26], removing excess mtROS, mtDNA and other substances that cause physiological reactions. In summary, in addition to giving the cell energy, mitochondria are crucial in cellular physiological activities. The link between mitochondria and the NLRP3 inflammasome has been widely studied in lung diseases, and it includes mitochondrial ROS (mtROS) and DNA (mtDNA), mitochondrial autophagy, and mitochondrial membrane proteins [27, 28], each of which is associated with activation of the NLRP3 inflammasome. However, it is not yet apparent how mitochondria and the NLRP3 inflammasome function to mediate lung disease onset and progression. Therefore, this review aims to clarify the role of mitochondrial stress and NLRP3 inflammasome in lung diseases to find new strategies for prevention and treatment.
Role of mitochondrial stress and the NLRP3 inflammasome in COPD
Mitochondrial dynamics and the effect of the NLRP3 inflammasome on COPD
COPD has become the third leading cause of death globally, affecting 250 million people and imposing a serious burden on society [29]. The chief features of COPD are irreversible airflow obstruction and airway inflammation, and the core susceptibility factor is oxidative stress [30]. Currently, no effective therapeutic drugs are able to prevent the decline in lung function in COPD over the long term. The main intracellular source of ROS, mitochondrial dysfunction, plays a significant role in the emergence of COPD. Mitochondrial function is regulated by fusion of healthy mitochondrial fragments to generate new mitochondria, and fission to remove damaged mitochondria [31]. Low levels of particulate matter (PM) ≤ 10 µm affect mitochondrial fusion and fission by precisely regulating the protein levels of OPA1 and Drp1. As a result of decreased levels of microtubule-associated protein 1A/1B light chain 3 (LC3)-II and phosphorylated AMP-activated protein kinase (AMPK), the NLRP3 inflammasome is activated, leading to inflammation in the lung [32]. Furthermore, using the cigarette smoke (CS)-induced COPD model, the expression of mitochondrial splitting proteins Mff and Drp1, caspase-1 and NLRP3 was discovered to increase, while the expression of mitochondrial fusion proteins Mfn2 and OPA1 dropped. Furthermore, these phenomena were reversed by pharmacological inhibition or knockdown of transient receptor potential cation subfamily V member 4 (TRPV4), TRPV1, or transient receptor potential cation channel member A1 (TRPA1) [33, 34]. TRPV4, TRPV1, and TRPA1 evidently alleviate CS-induced mitochondrial damage and NLRP3 inflammasome activation, suggesting that these channels are promising targets for new COPD treatments. Additionally, CS-induced oxidative stress on the endoplasmic reticulum has been reported to lead to disruption of endoplasmic reticulum calcium homeostasis. ROS production is then induced in mitochondria, which activates the NLRP3 inflammasome [35]. Furthermore, the same study showed that melatonin can reduce the accumulation of sequestosome 1 (p62/SQSTM1) by upregulating the expression of PINK1, Parkin and the autophagy marker LC3B-II. Autophagic breakdown of the injured mitochondria is induced, which inhibits the formation of mtROS and further decreases the activation of the NLRP3 inflammasome. However, in one study, excessive autophagy worsened the mitochondrial damage and contributed negatively to the injury of human umbilical vein endothelial cells [36]. These results were corroborated using the mitochondrial antioxidant mitoquinone, which was found to reduce mtROS production and excessive autophagy. The preservation of mitochondrial function allowed for restoration of the endothelial barrier function and a decrease in the NF-κB/NLRP3-mediated inflammatory response. Clarification of the specific mechanism of autophagy in COPD caused by CS will require further investigation.
Effect of endogenous antioxidants and the NLRP3 inflammasome on COPD
ROS act primarily as activators in bronchial epithelial cell pyroptosis and acute exacerbation of COPD, activating the NLRP3 inflammasome and exacerbating disease progression [37, 38]. Compared with Nrf2 pathway, nuclear factor erythroid 2-related factor 2 (Nrf2) has a distinct advantage as an endogenous antioxidant pathway for the scavenging of ROS in vivo. Studies have shown that the amelioration of COPD inflammation by either (-)-epicatechin or the lipoxin receptor agonist BML-111 was exerted by upregulating the expression level of Nrf2 and attenuating the ROS/NLRP3/caspase-1 pathway [39, 40]. What is unique about (-)-epicatechin is that it reduces ubiquitination of Nrf2 by promoting tripartite motif-containing 25 (TRIM25)-mediated degradation of Kelch-like ECH-associated protein 1 (Keap1). The exact mechanism of BML-111 is unclear in that mtDNA also often acts as an activator of the NLRP3 inflammasome and is more vulnerable to oxidative damage than nuclear DNA [21]. Giordano et al. [29] found that sublethal amounts of CS not only caused an increase in mitochondrial membrane potential and dysregulation of mitochondrial dynamics, but also mediated the release of mtDNA from extracellular vesicles, triggering increases in NLRP3, pro-inflammatory factors and aging markers in cell cultures. These results were confirmed in the plasma of COPD patients. This study clearly suggested that DNA enzymes as therapeutic agents might be useful as a new therapeutic pathway for COPD patients.
Role of mitochondrial stress and the NLRP3 inflammasome in lung cancer
Impact of smoking and environment on lung cancer
Lung cancer stands out among other cancers because of its high mortality rate [41]. Currently, two types are recognized: small cell lung cancer and non-small cell lung cancer (NSCLC). NSCLC is the more common, accounting for approximately 85% of cases [42], and includes adenocarcinomas, squamous and large cell lung cancers [43]. According to multiple studies, lung cancer is the primary malignancy diagnosed in 11.6% of cases and the leading cause of death from cancer in 18.4% of cases. In high-income countries, lung cancer is 10 times more likely to develop in heavy smokers than in non-smokers, with non-smokers having a 1%–2% lifetime probability of developing lung cancer [44]. By contrast, in low-income countries, the number of lung cancer diagnoses continues to increase because of higher levels of exposure to environmental pollution and secondhand smoke [45, 46]. Currently, early surgical resection is the primary method of treating NSCLC (e.g., lobectomy). For nonsurgical patients, stereotactic radiotherapy and percutaneous thermal ablation procedures are used to treat advanced NSCLC [47]. Despite the fact that treatments for lung cancer have advanced quickly in the past 10 years, clinical outcomes for the disease remain unsatisfactory. Therefore, seeking new therapeutic agents and elucidating their molecular mechanisms against lung cancer are urgently important.
Effect of mitochondrial stress and the NLRP3 inflammasome on smoking- or silica-induced lung cancer
Mitochondria, as important mediators of cellular metabolism, induce apoptosis under a dysregulated state. In cancer cells in particular, ROS often play a regulatory role in inducing apoptosis [48]; however, the exact mechanism in lung cancer cells remains unclear. Studies have shown that CS is highly associated with lung cancer, and that benzo(a)pyrene (BaP) in cigarettes is one of the inducers of lung cancer. This led to the establishment of a lung cancer model induced by BaP, and a study examining treatment with IL-27 and IL-28B [49]. The results showed that IL-27 and IL-28B not only downregulated the expression of the NLRP3 inflammasome but also reduced the aggregation of ROS in lung cancer [50]. The combination downregulated ROS-mediated activation of ERK1/2 and inhibited mast cell aggregation in lung tissue, thus acting as a treatment for lung cancer. The link between silica and lung cancer risk is controversial [51]. A recent study showed that silica-induced DNA damage was not caused by high or low ROS levels, but it was determined by silica-induced NLRP3 phosphorylation and mitochondrial depolarization, which led to DNA damage. Furthermore, silencing NLRP3 with small interfering RNA and CRISPR Cas9 prevented mitochondria depolarization and protected against DNA damage [52]. Further studies revealed that mitochondrial depolarization was critical for triggering DNA damage and was largely dependent on phosphorylation of serine 198 of NLRP3. Nevertheless, Zheng et al. showed that silica could damage DNA through a different mechanism of action [53]. In their study, silica acting on P2X7 induced mitochondrial production of ROS that activated the NLRP3 inflammasome, leading to the accumulation of double-strand breaks (DSBs). However, ataxia-telangiectasia mutated (ATM) activation of autotaxin (ATX) was dependent on the above process, which then triggered lung inflammation and DNA damage, resulting in the emergence of lung cancer. Taken together, these studies showed that both depolarization of mitochondrial membranes and activation of the ROS/NLRP3/ATX axis are closely related to lung carcinogenesis.
Effect of mitochondrial stress and the NLRP3 inflammasome on lung cancer migration
Malignant tumors are unsettling mostly because of their propensity for spreading, which also serves as the main cause of failure of tumor treatments. Therefore, the prevention of malignant tumor spread is often more important than treatment [54]. To examine whether bacterial infection could aggravate the development of lung cancer, Ma et al. [55] induced A549 cells with lipopolysaccharide (LPS) and discovered that LPS encouraged A549 cells to proliferate and migrate. They also found that salidroside, by promoting AMPK protein expression, suppressed NLRP3 inflammasome activation and ROS production, while also reducing the proliferation and migration of LPS-induced A549 cells. Ergosterol peroxide (EP) isolated from marine fungus Phoma sp was also shown to inhibit lung cancer migration [48]. In this study, EP induced A549 autophagy through mitochondrial damage, and this outcome was reversed by the ROS inhibitor N-acetylcysteine. EP also inhibited the activity of the NLRP3 inflammasome, thus reducing the proliferation and migration of lung cancer cells mediated by LPS. Additionally, EP synergized with the inhibition of A549 cell viability by the anti-cancer drug sorafenib. The biggest concern of breast cancer patients after surgery is the spread of the tumor, which is often directed to the more vascular organs; for example, breast cancer spreads to the lungs [56], which then spreads to other organs. Elemene nanoemulsion and hydrophilic As4S4 nanoparticles not only clear ROS, but also prevent NLRP3 inflammasome bodies from activating both in vitro and in vivo, and efficiently prevent breast cancer cells from spreading to the lungs, thus reducing the spread of cancer [57, 58]. Inhibiting the spread of a tumor to the lungs is a priority.
Antioxidant and anti-lung cancer effects of natural drugs on mitochondria and the NLRP3 inflammasome
In recent years, natural drugs, with their good therapeutic effects and few toxic side effects, have gradually come to play an important role in cancer treatment [59]. This is especially true in lung cancer treatment, which is currently limited to drugs with highly toxic side effects and thus in urgent need of new therapeutics. Studies have shown that most drugs activate NF-κB entry into the nucleus by regulating ROS levels in cells, causing the NLRP3 inflammasome to assemble and become active, and then increase caspase-1 protein expression to mediate lung cancer pyroptosis [60,61,62]. However, their mechanisms for regulating intracellular oxidative stress are very different. For example, by inhibiting the expression of glutathione peroxidase 4 (GPX4) and depolarizing mitochondrial membranes, shiitake polysaccharide alters the oxidative state of lung cancer cells [63]. Shiitake polysaccharide also downregulates the long noncoding RNA encoded by the X inactive-specific transcript (LncRNA-XIST) gene, which is highly expressed in lung cancer tissues, thereby promoting the Mn superoxide dismutase (Mn-SOD)-mediated increase in ROS levels and apoptosis [64]. Another natural drug, cucurbitacin B, reportedly exerts its antitumor activities via binding to Toll-like receptor 4 (TLR4) which leads to elevated mtROS levels, accumulation of mitochondrial membrane protein Tom20, and the release of cytoplasmic Ca2+ [15]. In summary, lung cancer development is significantly influenced by the activation of the NLRP3 inflammasome and mitochondrial oxidative stress. According to ongoing studies, the development of future lung cancer treatments will increasingly focus on the NLRP3 inflammasome and mitochondrial oxidative stress as key targets.
Mitochondrial stress and role of the NLRP3 inflammasome in acute lung injury
Oxidative stress and acute lung injury
The COVID-19 pandemic has been an unprecedented catastrophe for the world. COVID-19 mainly causes pulmonary complications such as pneumonia. Severe pneumonia can rapidly develop into acute respiratory distress syndrome or ALI [65]. However, the drugs currently used to treat ALI, which include vasodilators, ketoconazole and antioxidant therapy [66], are not specific or efficacious for ALI, despite some advancements in the field [67]. Therapeutics that counter oxidative stress and promote anti-inflammatory responses are expected to be a breakthrough in the treatment of ALI.
Amelioration of ALI by inhibiting mtROS and NLRP3 inflammasome activation
Mitochondria, as a major source of endogenous oxidative stress in cells, occupy a significant position in the body’s response to oxidative stress and inflammation [68]. Studies have shown that renal ischemia–reperfusion-induced ALI alters mitochondrial membrane potential as well as mitochondrial morphology [69]. NaHS and dexmedetomidine effectively reduced inflammatory factor levels by inhibiting NLRP3 inflammasome activation, which prevented mitochondrial damage, morphological and structural alterations, and mitochondrial dysfunction, thereby inhibiting mtROS production [70, 71]. Furthermore, cases of ALI caused by acid or bacterial inhalation were shown to exhibit elevated levels of mtROS and reduced aconitase activity as a superoxide anion-sensitive enzyme [72]. The administration of Mito-TEMPO revealed that NLRP3 inflammasome activation was inhibited and expression of IL-1β, TNF-α and IL-6 was downregulated [73]. These results suggest that mtROS cause ALI by precisely mediating the NLRP3 inflammasome; hence, targeted inhibition of mtROS may be one key to treating ALI. Endogenous antioxidants are also critical for the treatment of ALI. Melatonin reduces the generation of mtROS by activating the Nrf2/HO-1 pathway and increasing the expression of Mn-SOD and NAD(P)H:quinone oxidoreductase 1 (NQO1), which prevents the release of inflammatory factors and reverses the upregulation of NLRP3 and caspase-1 brought on by LPS. These effects of melatonin were also shown to reduce the occurrence of pyroptosis [74]. Additionally, calycosin [75] has been found to raise SOD and glutathione (GSH) levels in lung tissue while suppressing NLRP3 inflammasome activation and preventing the interaction of NLRP3, ASC and caspase-1. Notably, all of these effects are mediated through the regulation of mtROS, indicating that the NLRP3 inflammasome is inextricably activated by mtROS. Consequently, recent research on mitochondrial oxidative stress in ALI has been relatively shallow and mainly focused on pharmacological antioxidant studies. For example, drugs that treat or protect against ALI by inhibiting the ROS-mediated TXNIP/NLRP3 [76,77,78], TLR4/TRAF6 [79] and TRPM2/Ca2+/NLRP3 axes, or by activating the Nrf2/HO-1/NQO1 axis [80,81,82], reduce intracellular ROS levels to further decrease NLRP3 activation. Medications that suppress the NLRP3 inflammasome pathway via reduction of oxidative stress (Table 1) are characterized by mainly natural plant components. With further research, they will hopefully become a new approach to preventing or effectively treating ALI.
Potential mitochondrial targets and improvement of ALI via the NLRP3 inflammasome
Autophagy selectively clears damaged mitochondria, which negatively regulates mtROS production and inflammasome activation [83]. The regulation of autophagy in LPS-induced ALI was found to be worsened by a faulty FUNDC1 gene, which encodes a mitochondrial outer membrane protein, as shown by significant elevations in ROS, NLRP3, caspase-1 and inflammatory markers, along with significant inhibition of autophagy in lung tissue [84]. Sestrin2 (sesn2) showed the same phenomenon: knockdown of sesn2 led to upregulated mtROS expression levels and inhibition of mitochondrial autophagy [85]. Thus, lung cell pyroptosis appears to result from excessive promotion of NLRP3 inflammasome activation. These studies also demonstrated that FUNDC1 and Sestrin2 mediate mitochondrial autophagy and are vital for the modulation of mtROS, which might represent a viable therapy for ALI. Additionally, ROS cause elevated levels of lipid peroxides, possibly leading to iron-dependent death, which is mainly characterized by increased membrane density and rupture, and mitochondrial atrophy [86]. Expression of the iron death biomarker GPX4 was decreased, while expression of prostaglandin endoperoxidase synthase 2 (PTGS2) was increased, in ALI [87, 88]. Furthermore, mitochondrial acetaldehyde dehydrogenase 2 (ALDH2) expression inhibited iron-dependent death and pyroptosis, exerting a protective effect against ALI [89]. Clearly, iron-dependent cell death is involved in the ALI process. Sirtuin-3 (SIRT3), a mitochondrial NAD + deacetylase, plays a key role in inflammation regulation and lung injury. Pro-inflammatory factors are major players in ALI and are important treatment targets [67]. PKR acts as a stressor for mt-dsRNAs, its activation significantly increases the expression of IL-1β and high mobility group box-1 protein (HMGB1) [90], and it interacts with NLRP3 to activate the NLRP3 inflammasome [91]. These functions suggest inhibition of PKR expression as a potential strategy for the treatment of ALI. SIRT3 deficiency disrupted mitochondrial redox homeostasis, activating the NLRP3 inflammasome and leading to inflammation. While administration of the SIRT3 activator viniferin can attenuate ALI in wild-type mice, it is not effective in SIRT3-deficient mice [92]. Therefore, PKR has a role in the development of ALI, either by mediating mitochondrial autophagy or regulating the expression of PTGS2, SIRT3, or PKR, and it is expected to be a key target in the treatment of ALI.
Role of mitochondrial stress and the NLRP3 inflammasome in pulmonary fibrosis
Pathogenic characteristics of pulmonary fibrosis
Pulmonary fibrosis manifests as fibroblast proliferation and massive extracellular matrix aggregation, with inflammatory injury and tissue structural damage [102]. Because the etiology is absent in the vast majority of patients with pulmonary fibrosis, the most prevalent form of which is idiopathic pulmonary fibrosis, the condition is known as idiopathic interstitial pneumonia. Currently, there is no effective treatment and a high mortality rate for idiopathic pulmonary fibrosis [103]. We must urgently elucidate the pathophysiologic mechanisms and seek effective therapeutic drugs.
Effect of mitochondrial autophagy and the NLRP3 inflammasome on pulmonary fibrosis
In pulmonary fibrosis, the abnormal expression of mtROS induces mtDNA damage, which in turn activates macrophages and fibroblasts [104]. Therefore, ROS production and mtDNA damage are considered to be main features of pulmonary fibrosis. Sakai et al. [105] developed the cytoplasmic ·OH and mitochondrial ·OH scavengers TA293 and MitoTA293 to study this phenomenon. They demonstrated that cytoplasmic ·OH depleted intracellular GSH content, which triggered a decrease in mitochondrial GSH content and further induced H2O2 production in mitochondria, damaging the mtDNA and activating the NLRP3 inflammasome. In contrast, ·OH in mitochondria induced cell division and scavenged mtDNA in response to oxidative damage, consequently preventing the NLRP3 inflammasome from being activated and slowing the development of pulmonary fibrosis. Mn-SOD is a mitochondrial detoxifying agent that protects mitochondria from oxidative damage [106]. Anakinra, a recombinant IL-1 receptor antagonist, extended the lifespan of Mn-SOD by encouraging Mn-SOD binding with ubiquitin-specific protease 36 (USP36) and constitutive photomorphogenic 9 (COP9) signalosomes, thereby preventing oxidative damage to mitochondria and the generation of ROS, and avoiding the activation of the NLRP3 inflammasome [107].
Exosomal micro-RNA (miRNA) Let-7 has also been reported to control the expression of leptin-like oxidized low-density lipoprotein receptor-1 (LOX1) in lung epithelial cells, which in turn efficiently inhibits the formation of mtROS and DNA damage. Activation of the NLRP3 inflammasome and the process of pulmonary fibrosis were also inhibited [108]. However, more research is required to determine whether LOX1 may be a useful target for the therapy of pulmonary fibrosis. Pulmonary fibrosis occurs in response to apoptosis of damaged lung epithelial cells, which are subsequently phagocytosed and aggregated by macrophages [109]. Additionally, patients who experience exacerbations of acute idiopathic pulmonary fibrosis exacerbations also exhibit this phenomenon. Changes in both radiation-apoptotic lung cancer cells and microbiota were found that could activate the NLRP3 inflammasome by mediating mtROS; however, the former required cathepsin B involvement and the latter was associated with AIM2 inflammasome [48, 110]. Autophagy, similar to apoptosis, is a very important biological phenomenon in cells and is essential for the development of pulmonary fibrosis. Following its induction of redox imbalance, Ang II was shown to activate autophagy, which effectively reduced the quantity of ROS in cells, exacerbating pulmonary fibrosis [111]. Furthermore, the activation of autophagy was mainly dependent on the upregulation of NADPH oxidase-4 (NOX4) and its mediation of ROS/AMPK pathway activation, which decreased the accumulation of ROS [112]. Meanwhile, Ang II induced P62/SQSTM1 to degrade ubiquitinated ASC containing CARD, thereby inhibiting NLRP3 inflammasome activation and attenuating pulmonary fibrosis.
Effects of mitochondrial stress and the NLRP3 inflammasome on PM-induced pulmonary fibrosis
Although PM2.5 is found at a low level in the air, it has received widespread attention because it contains a variety of substances that are harmful to humans [113] and has unique and complex physicochemical properties. By conditioning the physicochemical properties of PM2.5, metal ions, polycyclic aromatic hydrocarbons and ROS in PM2.5 were found to encourage the activation of the NLRP3 inflammasome and generated mtROS [62]. Intranasal instillation of PM2.5 drops led to upregulation of NOX4 protein expression, increased levels of mtROS levels, and activation of the NLRP3 inflammasome in lung tissue after 3 weeks, and a fibrotic phenotype in the lung tissue after 9 weeks [112]. These results were attributed to mitochondrial damage and autophagy [114], and activation of the TGFβ1-NOX4-NLRP3 pathway. However, PM2.5 is not the only airborne PM capable of triggering pulmonary fibrosis. Carbon nanotubes, and silica and carbon black nanoparticles are all involved in affecting mitochondrial membrane potential as well as ROS production [115, 116], but they activate the NLRP3 inflammasome in different ways. For example, carbon black nanoparticles regulate NLRP3 inflammasome activation through miRNA-96, targeting FOXO3a to trigger pulmonary fibrosis [117]. To develop animal models to research the prevention and management of pulmonary fibrosis, understanding the disease processes affected by such particles is essential, which is expected to be a challenge in overcoming lung diseases caused by environmental pollution.
Mitochondrial stress and role of the NLRP3 inflammasome in lung infection and other lung diseases
Mitochondrial stress and the NLRP3 inflammasome ameliorate viral pneumonia
Influenza virus is a highly infectious virus of the human respiratory tract, and numerous inactivated vaccines and antiviral medications have been created following considerable research [118]. However, human health remains at serious risk due to the high genetic variability and emergence of drug-resistant strains of the virus [119]. Therefore, studies on the pathogenic mechanism of influenza viruses and screening for antiviral drugs should continue. Mitochondrial dynamics have been shown to be closely related to the pathogenesis of influenza viruses [120]. In the swine influenza virus, increased expression of the receptor-interacting protein kinase 1 (RIPK1) resulted in phosphorylation of a serine at position 579 in Drp1, promoted mitochondrial division and caused activation of the NLRP3 inflammasome [121]. However, to treat influenza pneumonia, berberine was shown to block the activation of the NLRP3 inflammasome and promote mitochondrial autophagy through the Bcl-2/adenovirus E1B-19-kDa interacting protein 3 (BNIP3) pathway [122]. Additionally, inhibition of the calcium receptor stromal interaction molecule 1 (STIM1) reversed the lung injury caused by miRNA-233 and ROS upregulation induced by the influenza virus [123]. Moreover, PKR, as a key to the body's defense against viral infection [124], recognizes viral dsRNA, mediates the activation of MAPK and NF-κB signaling, and promotes the release of inflammatory factors [125, 126]. The adenovirus-5 virus-associated RNAi inhibits the activation of the NLRP3 inflammasome precisely by targeting PKR and inhibiting PKR auto-phosphorylation, while blocking PKR–ASC interaction [127]. Reduning combined with ribavirin also attenuates the pathological injury caused by severe influenza pneumonia, reportedly impacting the inhibition of NLRP3 activation, mostly through lowering ROS levels [128]. Therefore, NLRP3 inflammasome activation and modulation of the mitochondrial kinetic balance may be crucial components of future strategies to prevent or treat viral lung illnesses.
Mitochondrial stress and the NLRP3 inflammasome ameliorate asthma
Asthma, a common lung disease, has pathogenic and therapeutic mechanisms closely related to mitochondrial dynamics and NLRP3 inflammasome activation. Among the pathogenic mechanisms, ozone, as a potent oxidizing gas, is strongly linked to the emergence of a number of lung diseases [129], including airway inflammation with bronchial hyper-responsiveness. Acute ozone exposure was found to lead to upregulated expression of mitochondrial complex II and IV, and induced elevated expression of mitochondrial Drp1 and Mff, decreased Mfn2, and upregulated mtROS levels in lung tissue [130]. Thus, an imbalance in mitochondrial dynamics may be critical in triggering bronchial hyper-responsiveness. Controlling the production of asthmatic mucus can alleviate asthma [131]. Specifically, the aryl hydrocarbon receptor (AhR) effectively inhibits asthmatic mucus production by inhibiting NLRP3 inflammasome activation and reducing IL-1β expression via decreased mtROS levels [132], thereby avoiding the IL-1β-induced overexpression of mucin 5AC that leads to asthmatic mucus production and asthma exacerbation. One study revealed that patients with severe asthma exhibit mitochondrial malfunction and ROS aggregation [133] manifested by inhibition of the autophagy controller transcription factor EB (TFEB) signaling pathway. This inhibition results in diminished mitochondrial autophagy and the inability to clear damaged mitochondria, which activates the NLRP3 signaling pathway and exacerbates asthma. Therefore, the regulation of mitochondrial oxidative stress is crucial for the prevention and treatment of asthma.
Others
According to a growing body of research, oxidative stress and inflammasome activation are linked to increased lung inflammation [134,135,136]. Indeed, modulation of this pathway and inhibition of inflammasome activation have become effective therapeutic approaches (Table 2) in lung conditions, such as radiation pneumonia [137], pulmonary ischemia–reperfusion injury [138, 139], and chronic intermittent hypoxia [140]. Carbon monoxide releasing molecule-2 (CORM-2) was shown to not only decrease the expression of TLR2 and TLR4 but also restrain NADPH oxidase activity, thereby reducing mtROS production and exerting a protective effect against PM-induced lung inflammation [141]. In another example, resveratrol protected against nickel-induced pneumonia by reducing the accumulation of ROS [142]. The important role of G protein-coupled receptor 43 (GPR43) in lung injury has also been confirmed at the molecular level, with upregulated GPR43 gene expression shown to inhibit NLRP3 inflammasome activation through the PPAR-gamma/Nox1/EBP50/p47phox axis, and thereby treat inflammatory responses to mitochondrial injury [143]. The role of oxidative stress with NLRP3 has also been confirmed in bacterial pneumonia. Furthermore, fungal β-glucan, Glaesserella parasuis and Aspergillus fumigatus were shown to induce inflammation in the lungs by regulating ROS levels and activating inflammasomes [144, 145]. The latter could be directly inhibited by allicin in terms of reduced spore activity and amelioration of lung inflammation by regulating expression of NLRP3 and caspase-1, and inhibiting excessive autophagy [146]. This study demonstrated how crucial the NLRP3 inflammasome and mitochondrial oxidative stress are to the inflammatory response, and we predict that continued research in this area will ultimately overcome the therapeutic challenges of lung disease.
Conclusion
Mitochondria are intracellular multifunctional organelles that are crucial targets for the management of oxidative stress, inflammatory response, cancer and more. Mitochondrial dysfunction leads to a series of stress responses (Fig. 3), including alterations in mitochondrial membrane potential, Mn-SOD content, ALDH activity, Tom20 expression and mitochondrial dynamics. These changes serve as activation signals for the NLRP3 inflammasome, mediating mtROS and mtDNA with mt-dsRNAs, and altering the histopathological state of the lung. Therefore, removing damaged mitochondria and reducing the stress response triggered by mitochondrial dysfunction are two effective approaches to improving lung diseases. Currently, the main therapeutic approaches in lung disease can be divided into five areas (Fig. 4): (1) prevention of mitochondrial dysfunction by blocking ion channels or cell membrane proteins to diminish the stimulation of external factors; (2) inhibition of mtROS production by reducing intracellular Ca2+ disorder and decreasing mitochondrial depolarization; (3) alteration of intracellular mitochondrial dynamics, or increased mitochondrial autophagy to remove damaged mitochondria, to prevent ROS accumulation; (4) promotion of endogenous antioxidant pathways or prolongation of the duration of action of antioxidant proteins to reduce ROS accumulation and inhibit activation of the NLRP3 inflammasome; and (5) regulation of noncoding RNA expression, inhibition of mtDNA damage or mtROS production, or direct blockade of NLRP3 inflammasome assembly. Mitochondrial stress from an altered cellular environment is mainly manifested by mtROS production and mtDNA damage. Strategies to control mtROS generation and prevent NLRP3 inflammasome activation will undoubtedly become the focus of future treatments for lung disease.

Therapeutic mechanisms of mitochondrial stress and NLRP3 inflammasome activation in lung disease. Mitochondrial membrane potential (MMP)

Major treatment approaches for lung diseases
Data availability
Not available.
References
Liu Z, Wu C, Pan Y, Liu H, Wang X, Yang Y, Gu M, Zhang Y, Wang X. NDR2 promotes the antiviral immune response via facilitating TRIM25-mediated RIG-I activation in macrophages. Sci Adv. 2019;5(2):eaav0163. https://doi.org/10.1126/sciadv.aav0163.
Wu L, Yan C, Czader M, Foreman O, Blum JS, Kapur R, Du H. Inhibition of PPARγ in myeloid-lineage cells induces systemic inflammation, immunosuppression, and tumorigenesis. Blood. 2012;119(1):115–26. https://doi.org/10.1182/blood-2011-06-363093.
Zhang X-N, Yu Z-L, Chen J-Y, Li X-Y, Wang Z-P, Wu M, Liu L-T. The crosstalk between NLRP3 inflammasome and gut microbiome in atherosclerosis. Pharmacol Res. 2022;181:106289. https://doi.org/10.1016/j.phrs.2022.106289.
Zhang X, Wang R, Hu D, Sun X, Fujioka H, Lundberg K, Chan ER, Wang Q, Xu R, Flanagan ME, Pieper AA, Qi X. Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer’s disease. Sci Adv. 2020;6(49):eabb8680. https://doi.org/10.1126/sciadv.abb8680.
Qiao C-Y, Li Y, Shang Y, Jiang M, Liu J, Zhan Z-Y, Ye H, Lin Y-C, Jiao J-Y, Sun R-H, Zhang Z-H, Piao M-H, Wu Y-L, Nan J-X, Lian L-H. Management of gout-associated MSU crystals-induced NLRP3 inflammasome activation by procyanidin B2: targeting IL-1β and cathepsin B in macrophages. Inflammopharmacology. 2020;28(6):1481–93. https://doi.org/10.1007/s10787-020-00758-8.
Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20(13):3328. https://doi.org/10.3390/ijms20133328.
Yu S-H, Sun X, Kim M-K, Akther M, Han J-H, Kim T-Y, Jiang J, Kang T-B, Lee K-H. Chrysanthemum indicum extract inhibits NLRP3 and AIM2 inflammasome activation via regulating ASC phosphorylation. J Ethnopharmacol. 2019;239:111917. https://doi.org/10.1016/j.jep.2019.111917.
Paik S, Kim JK, Silwal P, Sasakawa C, Jo E-K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol. 2021;18(5):1141–60. https://doi.org/10.1038/s41423-021-00670-3.
Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol. 2021;18(9):2114–27. https://doi.org/10.1038/s41423-021-00740-6.
He W-T, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang Z-H, Zhong C-Q, Han J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25(12):1285–98. https://doi.org/10.1038/cr.2015.139.
Han X, Sun S, Sun Y, Song Q, Zhu J, Song N, Chen M, Sun T, Xia M, Ding J, Lu M, Yao H, Hu G. Small molecule-driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: implications for Parkinson disease. Autophagy. 2019;15(11):1860–81. https://doi.org/10.1080/15548627.2019.1596481.
Lang T, Lee JPW, Elgass K, Pinar AA, Tate MD, Aitken EH, Fan H, Creed SJ, Deen NS, Traore DAK, Mueller I, Stanisic D, Baiwog FS, Skene C, Wilce MCJ, Mansell A, Morand EF, Harris J. Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat Commun. 2018;9(1):2223. https://doi.org/10.1038/s41467-018-04581-2.
Xu X, Jin L, Jiang T, Lu Y, Aosai F, Piao H-N, Xu G-H, Jin C-H, Jin X-J, Ma J, Piao L-X. Ginsenoside Rh2 attenuates microglial activation against toxoplasmic encephalitis via TLR4/NF-κB signaling pathway. J Ginseng Res. 2020;44(5):704–16. https://doi.org/10.1016/j.jgr.2019.06.002.
Childers GM, Perry CA, Blachut B, Martin N, Bortner CD, Sieber S, Li J-L, Fessler MB, Harry GJ. Assessing the association of mitochondrial function and inflammasome activation in murine macrophages exposed to select mitotoxic tri-organotin compounds. Environ Health Perspect. 2021;129(4):47015. https://doi.org/10.1289/ehp8314.
Yuan R, Zhao W, Wang Q-Q, He J, Han S, Gao H, Feng Y, Yang S. Cucurbitacin B inhibits non-small cell lung cancer in vivo and in vitro by triggering TLR4/NLRP3/GSDMD-dependent pyroptosis. Pharmacol Res. 2021;170:105748. https://doi.org/10.1016/j.phrs.2021.105748.
Li Q, Cao Y, Dang C, Han B, Han R, Ma H, Hao J, Wang L. Inhibition of double-strand DNA-sensing cGAS ameliorates brain injury after ischemic stroke. EMBO Mol Med. 2020;12(4):e11002. https://doi.org/10.15252/emmm.201911002.
Ren T-B, Zhang Q-L, Su D, Zhang X-X, Yuan L, Zhang X-B. Detection of analytes in mitochondria without interference from other sites based on an innovative ratiometric fluorophore. Chem Sci. 2018;9(24):5461–6. https://doi.org/10.1039/c8sc01673a.
Wang Y, Tang B, Long L, Luo P, Xiang W, Li X, Wang H, Jiang Q, Tan X, Luo S, Li H, Wang Z, Chen Z, Leng Y, Jiang Z, Wang Y, Ma L, Wang R, Zeng C, Liu Z, Wang Y, Miao H, Shi C. Improvement of obesity-associated disorders by a small-molecule drug targeting mitochondria of adipose tissue macrophages. Nat Commun. 2021;12(1):102. https://doi.org/10.1038/s41467-020-20315-9.
Perales-Clemente E, Cook AN, Evans JM, Roellinger S, Secreto F, Emmanuele V, Oglesbee D, Mootha VK, Hirano M, Schon EA, Terzic A, Nelson TJ. Natural underlying mtDNA heteroplasmy as a potential source of intra-person hiPSC variability. EMBO J. 2016;35(18):1979–90. https://doi.org/10.15252/embj.201694892.
Redman LM, Smith SR, Burton JH, Martin CK, Il’yasova D, Ravussin E. Metabolic slowing and reduced oxidative damage with sustained caloric restriction support the rate of living and oxidative damage theories of aging. Cell Metab. 2018;27(4):805-815.e804. https://doi.org/10.1016/j.cmet.2018.02.019.
Sanchez-Guerra M, Peng C, Trevisi L, Cardenas A, Wilson A, Osorio-Yáñez C, Niedzwiecki MM, Zhong J, Svensson K, Acevedo MT, Solano-Gonzalez M, Amarasiriwardena CJ, Estrada-Gutierrez G, Brennan KJM, Schnaas L, Just AC, Laue HE, Wright RJ, Téllez-Rojo MM, Wright RO, Baccarelli AA. Altered cord blood mitochondrial DNA content and pregnancy lead exposure in the PROGRESS cohort. Environ Int. 2019;125:437–44. https://doi.org/10.1016/j.envint.2019.01.077.
Yoon J, Lee M, Ali AA, Oh YR, Choi YS, Kim S, Lee N, Jang SG, Park S, Chung JH, Kwok SK, Hyon JY, Cha S, Lee YJ, Im SG, Kim Y. Mitochondrial double-stranded RNAs as a pivotal mediator in the pathogenesis of Sjӧgren’s syndrome. Mol Ther Nucleic Acids. 2022;30:257–69. https://doi.org/10.1016/j.omtn.2022.09.020.
Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X, Huang K, Xie Z, Zou M-H. Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation. 2017;136(23):2248–66. https://doi.org/10.1161/circulationaha.117.030235.
Kim Y, Park J, Kim S, Kim M, Kang MG, Kwak C, Kang M, Kim B, Rhee HW, Kim VN. PKR senses nuclear and mitochondrial signals by interacting with endogenous double-stranded RNAs. Mol Cell. 2018;71(6):1051-1063.e1056. https://doi.org/10.1016/j.molcel.2018.07.029.
Kim S, Lee K, Choi YS, Ku J, Kim H, Kharbash R, Yoon J, Lee YS, Kim JH, Lee YJ, Kim Y. Mitochondrial double-stranded RNAs govern the stress response in chondrocytes to promote osteoarthritis development. Cell Rep. 2022;40(6):111178. https://doi.org/10.1016/j.celrep.2022.111178.
Vaillant-Beuchot L, Mary A, Pardossi-Piquard R, Bourgeois A, Lauritzen I, Eysert F, Kinoshita PF, Cazareth J, Badot C, Fragaki K, Bussiere R, Martin C, Mary R, Bauer C, Pagnotta S, Paquis-Flucklinger V, Buée-Scherrer V, Buée L, Lacas-Gervais S, Checler F, Chami M. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer’s disease models and human brains. Acta Neuropathol. 2021;141(1):39–65. https://doi.org/10.1007/s00401-020-02234-7.
Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, McGeough MD, Ellisman MH, Seki E, Gustafsson AB, Hoffman HM, Diaz-Meco MT, Moscat J, Karin M. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164(5):896–910. https://doi.org/10.1016/j.cell.2015.12.057.
Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin X-J, Wong J, Ding S, Seki E, Schnabl B, Hevener AL, Greenberg HB, Kisseleva T, Karin M. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560(7717):198–203. https://doi.org/10.1038/s41586-018-0372-z.
Giordano L, Gregory AD, Verdaguer MP, Ware SA, Harvey H, DeVallance E, Brzoska T, Sundd P, Zhang Y, Sciurba FC, Shapiro SD, Kaufman BA. Extracellular release of mitochondrial DNA: triggered by cigarette smoke and detected in COPD. Cells. 2022;11(3):369. https://doi.org/10.3390/cells11030369.
Kim H-T, Yin W, Jin Y-J, Panza P, Gunawan F, Grohmann B, Buettner C, Sokol AM, Preussner J, Guenther S, Kostin S, Ruppert C, Bhagwat AM, Ma X, Graumann J, Looso M, Guenther A, Adelstein RS, Offermanns S, Stainier DYR. Myh10 deficiency leads to defective extracellular matrix remodeling and pulmonary disease. Nat Commun. 2018;9(1):4600. https://doi.org/10.1038/s41467-018-06833-7.
Schulman JJ, Szczesniak LM, Bunker EN, Nelson HA, Roe MW, Wagner LE, Yule DI, Wojcikiewicz RJH. Bok regulates mitochondrial fusion and morphology. Cell Death Differ. 2019;26(12):2682–94. https://doi.org/10.1038/s41418-019-0327-4.
Chan YL, Wang B, Chen H, Ho KF, Cao J, Hai G, Jalaludin B, Herbert C, Thomas PS, Saad S, Oliver BGG. Pulmonary inflammation induced by low-dose particulate matter exposure in mice. Am J Physiol Lung Cell Mol Physiol. 2019;317(3):L424–30. https://doi.org/10.1152/ajplung.00232.2019.
Wang M, Zhang Y, Xu M, Zhang H, Chen Y, Chung KF, Adcock IM, Li F. Roles of TRPA1 and TRPV1 in cigarette smoke -induced airway epithelial cell injury model. Free Radic Biol Med. 2019;134:229–38. https://doi.org/10.1016/j.freeradbiomed.2019.01.004.
Rao Y, Gai X, Xiong J, Le Y, Sun Y. Transient receptor potential cation channel subfamily V member 4 mediates pyroptosis in chronic obstructive pulmonary disease. Front Physiol. 2021;12:783891. https://doi.org/10.3389/fphys.2021.783891.
Mahalanobish S, Dutta S, Saha S, Sil PC. Melatonin induced suppression of ER stress and mitochondrial dysfunction inhibited NLRP3 inflammasome activation in COPD mice. Food Chem Toxicol. 2020;144:111588. https://doi.org/10.1016/j.fct.2020.111588.
Chen S, Wang Y, Zhang H, Chen R, Lv F, Li Z, Jiang T, Lin D, Zhang H, Yang L, Kong X. The antioxidant MitoQ protects against CSE-induced endothelial barrier injury and inflammation by inhibiting ROS and autophagy in human umbilical vein endothelial cells. Int J Biol Sci. 2019;15(7):1440–51. https://doi.org/10.7150/ijbs.30193.
Uh S-T, Koo SM, Kim Y, Kim K, Park S, Jang AS, Kim D, Kim YH, Park C-S. The activation of NLRP3-inflammsome by stimulation of diesel exhaust particles in lung tissues from emphysema model and RAW 264.7 cell line. Korean J Intern Med. 2017;32(5):865–74. https://doi.org/10.3904/kjim.2016.033.
Zhang M-Y, Jiang Y-X, Yang Y-C, Liu J-Y, Huo C, Ji X-L, Qu Y-Q. Cigarette smoke extract induces pyroptosis in human bronchial epithelial cells through the ROS/NLRP3/caspase-1 pathway. Life Sci. 2021;269:119090. https://doi.org/10.1016/j.lfs.2021.119090.
Tian X, Xue Y, Xie G, Zhou Y, Xiao H, Ding F, Zhang M. (-)-Epicatechin ameliorates cigarette smoke-induced lung inflammation via inhibiting ROS/NLRP3 inflammasome pathway in rats with COPD. Toxicol Appl Pharmacol. 2021;429:115674. https://doi.org/10.1016/j.taap.2021.115674.
Cao Y, Zhou X, Yin Z, Yu X, Yang Q, Guo Q, Tian D, Xiong X, Xu G, Kuang X. The anti-inflammatory effect of BML-111 on COPD may be mediated by regulating NLRP3 inflammasome activation and ROS production. Prostaglandins Other Lipid Mediat. 2018;138:23–30. https://doi.org/10.1016/j.prostaglandins.2018.08.001.
Tong L, Shen S, Huang Q, Fu J, Wang T, Pan L, Zhang P, Chen G, Huang T, Li K, Liu Q, Xie S, Yang X, Moses RE, Li X, Li L. Proteasome-dependent degradation of Smad7 is critical for lung cancer metastasis. Cell Death Differ. 2020;27(6):1795–806. https://doi.org/10.1038/s41418-019-0459-6.
Luo X, Zang X, Yang L, Huang J, Liang F, Rodriguez-Canales J, Wistuba II, Gazdar A, Xie Y, Xiao G. Comprehensive computational pathological image analysis predicts lung cancer prognosis. J Thorac Oncol. 2017;12(3):501–9. https://doi.org/10.1016/j.jtho.2016.10.017.
Li B, Zhu L, Lu C, Wang C, Wang H, Jin H, Ma X, Cheng Z, Yu C, Wang S, Zuo Q, Zhou Y, Wang J, Yang C, Lv Y, Jiang L, Qin W. circNDUFB2 inhibits non-small cell lung cancer progression via destabilizing IGF2BPs and activating anti-tumor immunity. Nat Commun. 2021;12(1):295. https://doi.org/10.1038/s41467-020-20527-z.
Chen J, Liu A, Wang Z, Wang B, Chai X, Lu W, Cao T, Li R, Wu M, Lu Z, Pang W, Xiao L, Chen X, Zheng Y, Chen Q, Zeng J, Li J, Zhang X, Ren D, Huang Y. LINC00173.v1 promotes angiogenesis and progression of lung squamous cell carcinoma by sponging miR-511–5p to regulate VEGFA expression. Mol Cancer. 2020;19(1):98. https://doi.org/10.1186/s12943-020-01217-2.
Thai AA, Solomon BJ, Sequist LV, Gainor JF, Heist RS. Lung cancer. Lancet. 2021;398(10299):535–54. https://doi.org/10.1016/s0140-6736(21)00312-3.
Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553(7689):446–54. https://doi.org/10.1038/nature25183.
Duma N, Santana-Davila R, Molina JR. Non-small cell lung cancer: epidemiology, screening, diagnosis, and treatment. Mayo Clin Proc. 2019;94(8):1623–40. https://doi.org/10.1016/j.mayocp.2019.01.013.
Wu H-Y, Yang F-L, Li L-H, Rao YK, Ju T-C, Wong W-T, Hsieh C-Y, Pivkin MV, Hua K-F, Wu S-H. Ergosterol peroxide from marine fungus Phoma sp. induces ROS-dependent apoptosis and autophagy in human lung adenocarcinoma cells. Sci Rep. 2018;8(1):17956. https://doi.org/10.1038/s41598-018-36411-2.
Majumder D, Sarkar C, Debnath R, Tribedi P, Maiti D. Mechanistic insight into the synergism of IL-27 and IL-28B in regulation of benzo(a)pyrene-induced lung carcinogenesis associated ROS/NF-κB/NLRP3 crosstalk. Chem Biol Interact. 2022;354:109807. https://doi.org/10.1016/j.cbi.2022.109807.
Majumder D, Debnath R, Maiti D. IL-27 along with IL-28B ameliorates the pulmonary redox impairment, inflammation and immunosuppression in benzo(a)pyrene induced lung cancer bearing mice. Life Sci. 2020;260:118384. https://doi.org/10.1016/j.lfs.2020.118384.
Chen W, Liu Y, Wang H, Hnizdo E, Sun Y, Su L, Zhang X, Weng S, Bochmann F, Hearl FJ, Chen J, Wu T. Long-term exposure to silica dust and risk of total and cause-specific mortality in Chinese workers: a cohort study. PLoS Med. 2012;9(4):e1001206. https://doi.org/10.1371/journal.pmed.1001206.
Wu R, Högberg J, Adner M, Ramos-Ramírez P, Stenius U, Zheng H. Crystalline silica particles cause rapid NLRP3-dependent mitochondrial depolarization and DNA damage in airway epithelial cells. Part Fibre Toxicol. 2020;17(1):39. https://doi.org/10.1186/s12989-020-00370-2.
Zheng H, Högberg J, Stenius U. ATM-activated autotaxin (ATX) propagates inflammation and DNA damage in lung epithelial cells: a new mode of action for silica-induced DNA damage? Carcinogenesis. 2017;38(12):1196–206. https://doi.org/10.1093/carcin/bgx100.
Peng L, Xing X, Li W, Qu L, Meng L, Lian S, Jiang B, Wu J, Shou C. PRL-3 promotes the motility, invasion, and metastasis of LoVo colon cancer cells through PRL-3-integrin beta1-ERK1/2 and-MMP2 signaling. Mol Cancer. 2009;8:110. https://doi.org/10.1186/1476-4598-8-110.
Ma W, Wang Z, Zhao Y, Wang Q, Zhang Y, Lei P, Lu W, Yan S, Zhou J, Li X, Yu W, Zhong Y, Chen L, Zheng T. Salidroside suppresses the proliferation and migration of human lung cancer cells through AMPK-dependent NLRP3 inflammasome regulation. Oxid Med Cell Longev. 2021;2021:6614574. https://doi.org/10.1155/2021/6614574.
Kaverina N, Borovjagin AV, Kadagidze Z, Baryshnikov A, Baryshnikova M, Malin D, Ghosh D, Shah N, Welch DR, Gabikian P, Karseladze A, Cobbs C, Ulasov IV. Astrocytes promote progression of breast cancer metastases to the brain via a KISS1-mediated autophagy. Autophagy. 2017;13(11):1905–23. https://doi.org/10.1080/15548627.2017.1360466.
Han B, Wang T, Xue Z, Wen T, Lu L, Meng J, Liu J, Wu S, Yu J, Xu H. Elemene nanoemulsion inhibits metastasis of breast cancer by ROS scavenging. Int J Nanomedicine. 2021;16:6035–48. https://doi.org/10.2147/ijn.S327094.
Wang T, Meng J, Wang C, Wen T, Jia M, Ge Y, Xie L, Hao S, Liu J, Xu H. Inhibition of murine breast cancer metastases by hydrophilic As4 S4 nanoparticles is associated with decreased ROS and HIF-1α downregulation. Front Oncol. 2019;9:333. https://doi.org/10.3389/fonc.2019.00333.
Wang J, Qi Q, Zhou W, Feng Z, Huang B, Chen A, Zhang D, Li W, Zhang Q, Jiang Z, Bjerkvig R, Prestegarden L, Thorsen F, Wang X, Li X, Wang J. Inhibition of glioma growth by flavokawain B is mediated through endoplasmic reticulum stress induced autophagy. Autophagy. 2018;14(11):2007–22. https://doi.org/10.1080/15548627.2018.1501133.
Teng J-F, Mei Q-B, Zhou X-G, Tang Y, Xiong R, Qiu W-Q, Pan R, Law BY-K, Wong VK-W, Yu C-L, Long H-A, Xiao X-L, Zhang F, Wu J-M, Qin D-L, Wu A-G. Polyphyllin VI induces caspase-1-mediated pyroptosis via the induction of ROS/NF-κB/NLRP3/GSDMD signal axis in non-small cell lung cancer. Cancers (Basel). 2020;12(1):193. https://doi.org/10.3390/cancers12010193.
Yin H, Liu Y-G, Li F, Wang L-Q, Zha J-H, Xia Y-C, Yu B-T, Wen D-H. Resibufogenin suppresses growth and metastasis through inducing caspase-1-dependent pyroptosis via ROS-mediated NF-κB suppression in non-small cell lung cancer. Anat Rec (Hoboken). 2021;304(2):302–12. https://doi.org/10.1002/ar.24415.
Liang X, Zhang D, Liu W, Yan Y, Zhou F, Wu W, Yan Z. Reactive oxygen species trigger NF-κB-mediated NLRP3 inflammasome activation induced by zinc oxide nanoparticles in A549 cells. Toxicol Ind Health. 2017;33(10):737–45. https://doi.org/10.1177/0748233717712409.
Li M, Du X, Yuan Z, Cheng M, Dong P, Bai Y. Lentinan triggers oxidative stress-mediated anti-inflammatory responses in lung cancer cells. Mol Cell Biochem. 2022;477(2):469–77. https://doi.org/10.1007/s11010-021-04293-0.
Liu J, Yao L, Zhang M, Jiang J, Yang M, Wang Y. Downregulation of LncRNA-XIST inhibited development of non-small cell lung cancer by activating miR-335/SOD2/ROS signal pathway mediated pyroptotic cell death. Aging. 2019;11(18):7830–46. https://doi.org/10.18632/aging.102291.
Kuri-Cervantes L, Pampena MB, Meng W, Rosenfeld AM, Ittner CAG, Weisman AR, Agyekum RS, Mathew D, Baxter AE, Vella LA, Kuthuru O, Apostolidis SA, Bershaw L, Dougherty J, Greenplate AR, Pattekar A, Kim J, Han N, Gouma S, Weirick ME, Arevalo CP, Bolton MJ, Goodwin EC, Anderson EM, Hensley SE, Jones TK, Mangalmurti NS, Prak ETL, Wherry EJ, Meyer NJ, Betts MR. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci Immunol. 2020;5(49):eabd7114. https://doi.org/10.1126/sciimmunol.abd7114.
Kellner M, Noonepalle S, Lu Q, Srivastava A, Zemskov E, Black SM. ROS signaling in the pathogenesis of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). Adv Exp Med Biol. 2017;967:105–37. https://doi.org/10.1007/978-3-319-63245-2_8.
Ding C, Chen H, Liang B, Jiao M, Liang G, Zhang A. Biomimetic synthesis of the natural product salviadione and its hybrids: discovery of tissue-specific anti-inflammatory agents for acute lung injury. Chem Sci. 2019;10(17):4667–72. https://doi.org/10.1039/c9sc00086k.
Gao J, Feng Z, Wang X, Zeng M, Liu J, Han S, Xu J, Chen L, Cao K, Long J, Li Z, Shen W, Liu J. SIRT3/SOD2 maintains osteoblast differentiation and bone formation by regulating mitochondrial stress. Cell Death Differ. 2018;25(2):229–40. https://doi.org/10.1038/cdd.2017.144.
Zhang H, Chen S, Zeng M, Lin D, Wang Y, Wen X, Xu C, Yang L, Fan X, Gong Y, Zhang H, Kong X. Apelin-13 administration protects against LPS-induced acute lung injury by inhibiting NF-κB pathway and NLRP3 inflammasome activation. Cell Physiol Biochem. 2018;49(5):1918–32. https://doi.org/10.1159/000493653.
Zhao G, Yang L, Li L, Fan Z. NaHS alleviated cell apoptosis and mitochondrial dysfunction in remote lung tissue after renal ischemia and reperfusion via Nrf2 activation-mediated NLRP3 pathway inhibition. Biomed Res Int. 2021;2021:5598869. https://doi.org/10.1155/2021/5598869.
Chen Y, Huang Y, Xiong B, Luo H, Song X. Dexmedetomidine ameliorates renal ischemia reperfusion-mediated activation of the NLRP3 inflammasome in alveolar macrophages. Gene. 2020;758:144973. https://doi.org/10.1016/j.gene.2020.144973.
Puri G, Naura AS. Critical role of mitochondrial oxidative stress in acid aspiration induced ALI in mice. Toxicol Mech Methods. 2020;30(4):266–74. https://doi.org/10.1080/15376516.2019.1710888.
Puri G, Naura AS. Implication of mitochondrial ROS-NLRP3 inflammasome axis during two-hit mediated acute lung injury in mice. Free Radic Res. 2022;56(1):1–16. https://doi.org/10.1080/10715762.2021.2023740.
Kang J-Y, Xu M-M, Sun Y, Ding Z-X, Wei Y-Y, Zhang D-W, Wang Y-G, Shen J-L, Wu H-M, Fei G-H. Melatonin attenuates LPS-induced pyroptosis in acute lung injury by inhibiting NLRP3-GSDMD pathway via activating Nrf2/HO-1 signaling axis. Int Immunopharmacol. 2022;109:108782. https://doi.org/10.1016/j.intimp.2022.108782.
Xia Y, Cao Y, Sun Y, Hong X, Tang Y, Yu J, Hu H, Ma W, Qin K, Bao R. Calycosin alleviates sepsis-induced acute lung injury via the inhibition of mitochondrial ROS-mediated inflammasome activation. Front Pharmacol. 2021;12:690549. https://doi.org/10.3389/fphar.2021.690549.
Han X, Wu Y-C, Meng M, Sun Q-S, Gao S-M, Sun H. Linarin prevents LPS-induced acute lung injury by suppressing oxidative stress and inflammation via inhibition of TXNIP/NLRP3 and NF-κB pathways. Int J Mol Med. 2018;42(3):1460–72. https://doi.org/10.3892/ijmm.2018.3710.
Fang X-Z, Ge Y-L, Chen Z-Y, Shu H-Q, Yang Y-Y, Yu Y, Zhou X-J, Chen L, Cui S-N, Wang Y-X, Yao S-L, Shang Y. NecroX-5 alleviate lipopolysaccharide-induced acute respiratory distress syndrome by inhibiting TXNIP/NLRP3 and NF-κB. Int Immunopharmacol. 2020;81:106257. https://doi.org/10.1016/j.intimp.2020.106257.
Meng M. Digitoflavone (DG) attenuates LPS-induced acute lung injury through reducing oxidative stress and inflammatory response dependent on the suppression of TXNIP/NLRP3 and NF-κB. Biomed Pharmacother. 2017;94:712–25. https://doi.org/10.1016/j.biopha.2017.07.001.
Wu Y-C, Hsu S-P, Hu M-C, Lan Y-T, Yeh ETH, Yang F-M. PEP-sNASP peptide alleviates LPS-induced acute lung injury through the TLR4/TRAF6 axis. Front Med (Lausanne). 2022;9:832713. https://doi.org/10.3389/fmed.2022.832713.
Lu Y, Yu T, Liu J, Gu L. Vitexin attenuates lipopolysaccharide-induced acute lung injury by controlling the Nrf2 pathway. PLoS ONE. 2018;13(4):e0196405. https://doi.org/10.1371/journal.pone.0196405.
Dhar R, Rana MN, Zhang L, Li Y, Li N, Hu Z, Yan C, Wang X, Zheng X, Liu H, Cui H, Li Z, Tang H. Phosphodiesterase 4B is required for NLRP3 inflammasome activation by positive feedback with Nrf2 in the early phase of LPS- induced acute lung injury. Free Radic Biol Med. 2021;176:378–91. https://doi.org/10.1016/j.freeradbiomed.2021.10.007.
Liu Z, Qu M, Yu L, Song P, Chang Y. Artesunate inhibits renal ischemia-reperfusion-mediated remote lung inflammation through attenuating ROS-induced activation of NLRP3 inflammasome. Inflammation. 2018;41(4):1546–56. https://doi.org/10.1007/s10753-018-0801-z.
Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. 2018;14(2):243–51. https://doi.org/10.1080/15548627.2017.1402992.
Pan P, Chen J, Liu X, Fan J, Zhang D, Zhao W, Xie L, Su L. FUNDC1 regulates autophagy by inhibiting ROS-NLRP3 signaling to avoid apoptosis in the lung in a lipopolysaccharide-induced mouse model. Shock. 2021;56(5):773–81. https://doi.org/10.1097/shk.0000000000001835.
Wu D, Zhang H, Wu Q, Li F, Wang Y, Liu S, Wang J. Sestrin 2 protects against LPS-induced acute lung injury by inducing mitophagy in alveolar macrophages. Life Sci. 2021;267:118941. https://doi.org/10.1016/j.lfs.2020.118941.
Li J, Cao F, Yin H-L, Huang Z-J, Lin Z-T, Mao N, Sun B, Wang G. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):88. https://doi.org/10.1038/s41419-020-2298-2.
Xiao Z, Kong B, Fang J, Qin T, Dai C, Shuai W, Huang H. Ferrostatin-1 alleviates lipopolysaccharide-induced cardiac dysfunction. Bioengineered. 2021;12(2):9367–76. https://doi.org/10.1080/21655979.2021.2001913.
Fei W, Chen D, Tang H, Li C, Zheng W, Chen F, Song Q, Zhao Y, Zou Y, Zheng C. Targeted GSH-exhausting and hydroxyl radical self-producing manganese-silica nanomissiles for MRI guided ferroptotic cancer therapy. Nanoscale. 2020;12(32):16738–54. https://doi.org/10.1039/d0nr02396e.
Cao Z, Qin H, Huang Y, Zhao Y, Chen Z, Hu J, Gao Q. Crosstalk of pyroptosis, ferroptosis, and mitochondrial aldehyde dehydrogenase 2-related mechanisms in sepsis-induced lung injury in a mouse model. Bioengineered. 2022;13(3):4810–20. https://doi.org/10.1080/21655979.2022.2033381.
Chiu HW, Li LH, Hsieh CY, Rao YK, Chen FH, Chen A, Ka SM, Hua KF. Glucosamine inhibits IL-1β expression by preserving mitochondrial integrity and disrupting assembly of the NLRP3 inflammasome. Sci Rep. 2019;9(1):5603. https://doi.org/10.1038/s41598-019-42130-z.
Zeng Y, Qin Q, Li K, Li H, Song C, Li Y, Dai M, Lin F, Mao Z, Li Q, Long Y, Fan Y, Pan P. PKR suppress NLRP3-pyroptosis pathway in lipopolysaccharide-induced acute lung injury model of mice. Biochem Biophys Res Commun. 2019;519(1):8–14. https://doi.org/10.1016/j.bbrc.2019.08.054.
Kurundkar D, Kurundkar AR, Bone NB, Becker EJ, Liu W, Chacko B, Darley-Usmar V, Zmijewski JW, Thannickal VJ. SIRT3 diminishes inflammation and mitigates endotoxin-induced acute lung injury. JCI Insight. 2019;4(1):e120722. https://doi.org/10.1172/jci.insight.120722.
Jin H-Z, Yang X-J, Zhao K-L, Mei F-C, Zhou Y, You Y-D, Wang W-X. Apocynin alleviates lung injury by suppressing NLRP3 inflammasome activation and NF-κB signaling in acute pancreatitis. Int Immunopharmacol. 2019;75:105821. https://doi.org/10.1016/j.intimp.2019.105821.
Liu Q, Ci X, Wen Z, Peng L. Diosmetin alleviates lipopolysaccharide-induced acute lung injury through activating the Nrf2 pathway and inhibiting the NLRP3 inflammasome. Biomol Ther (Seoul). 2018;26(2):157–66. https://doi.org/10.4062/biomolther.2016.234.
Jiang Y, Yang W, Gui S. Procyanidin B2 protects rats from paraquat-induced acute lung injury by inhibiting NLRP3 inflammasome activation. Immunobiology. 2018;223(10):555–61. https://doi.org/10.1016/j.imbio.2018.07.001.
Feng B, Zhao X, Zhao W, Jiang H, Ren Z, Chen Y, Yuan Y, Du Z. Ethyl 2-succinate-anthraquinone attenuates inflammatory response and oxidative stress via regulating NLRP3 signaling pathway. Front Pharmacol. 2021;12:719822. https://doi.org/10.3389/fphar.2021.719822.
Liu Q, Lv H, Wen Z, Ci X, Peng L. Isoliquiritigenin activates nuclear factor erythroid-2 related factor 2 to suppress the NOD-like receptor protein 3 inflammasome and inhibits the NF-κB pathway in macrophages and in acute lung injury. Front Immunol. 2017;8:1518. https://doi.org/10.3389/fimmu.2017.01518.
Yuan R, Li Y, Han S, Chen X, Chen J, He J, Gao H, Yang Y, Yang S, Yang Y. Fe-curcumin nanozyme-mediated reactive oxygen species scavenging and anti-inflammation for acute lung injury. ACS Cent Sci. 2022;8(1):10–21. https://doi.org/10.1021/acscentsci.1c00866.
Ren Y, Yang Z, Sun Z, Zhang W, Chen X, Nie S. Curcumin relieves paraquat-induced lung injury through inhibiting the thioredoxin interacting protein/NLR pyrin domain containing 3-mediated inflammatory pathway. Mol Med Rep. 2019;20(6):5032–40. https://doi.org/10.3892/mmr.2019.10612.
Ding H, Ci X, Cheng H, Yu Q, Li D. Chicoric acid alleviates lipopolysaccharide-induced acute lung injury in mice through anti-inflammatory and anti-oxidant activities. Int Immunopharmacol. 2019;66:169–76. https://doi.org/10.1016/j.intimp.2018.10.042.
Luo X-Q, Duan J-X, Yang H-H, Zhang C-Y, Sun C-C, Guan X-X, Xiong J-B, Zu C, Tao J-H, Zhou Y, Guan C-X. Epoxyeicosatrienoic acids inhibit the activation of NLRP3 inflammasome in murine macrophages. J Cell Physiol. 2020;235(12):9910–21. https://doi.org/10.1002/jcp.29806.
Katsumata K, Ishihara J, Mansurov A, Ishihara A, Raczy MM, Yuba E, Hubbell JA. Targeting inflammatory sites through collagen affinity enhances the therapeutic efficacy of anti-inflammatory antibodies. Sci Adv. 2019;5(11):eaay1971. https://doi.org/10.1126/sciadv.aay1971.
Désogère P, Tapias LF, Hariri LP, Rotile NJ, Rietz TA, Probst CK, Blasi F, Day H, Mino-Kenudson M, Weinreb P, Violette SM, Fuchs BC, Tager AM, Lanuti M, Caravan P. Type I collagen-targeted PET probe for pulmonary fibrosis detection and staging in preclinical models. Sci Transl Med. 2017;9(384):eaaf4696. https://doi.org/10.1126/scitranslmed.aaf4696.
Gu X, Wu G, Yao Y, Zeng J, Shi D, Lv T, Luo L, Song Y. Intratracheal administration of mitochondrial DNA directly provokes lung inflammation through the TLR9-p38 MAPK pathway. Free Radic Biol Med. 2015;83:149–58. https://doi.org/10.1016/j.freeradbiomed.2015.02.034.
Sakai T, Takagaki H, Yamagiwa N, Ui M, Hatta S, Imai J. Effects of the cytoplasm and mitochondrial specific hydroxyl radical scavengers TA293 and mitoTA293 in bleomycin-induced pulmonary fibrosis model mice. Antioxidants (Basel). 2021;10(9):1398. https://doi.org/10.3390/antiox10091398.
Migocka M, Maciaszczyk-Dziubinska E, Malas K, Posyniak E, Garbiec A. Corrigendum to: metal tolerance protein MTP6 affects mitochondrial iron and manganese homeostasis in cucumber. J Exp Bot. 2019;70(1):369. https://doi.org/10.1093/jxb/ery399.
Pariano M, Pieroni S, Luca AD, Iannitti RG, Borghi M, Puccetti M, Giovagnoli S, Renga G, D’Onofrio F, Bellet MM, Stincardini C, Della-Fazia MA, Servillo G, Veerdonk FLVD, Costantini C, Romani L. Anakinra activates superoxide dismutase 2 to mitigate inflammasome activity. Int J Mol Sci. 2021;22(12):6531. https://doi.org/10.3390/ijms22126531.
Sun L, Zhu M, Feng W, Lin Y, Yin J, Jin J, Wang Y. Exosomal miRNA let-7 from menstrual blood-derived endometrial stem cells alleviates pulmonary fibrosis through regulating mitochondrial DNA damage. Oxid Med Cell Longev. 2019;2019:4506303. https://doi.org/10.1155/2019/4506303.
Sgalla G, Iovene B, Calvello M, Ori M, Varone F, Richeldi L. Idiopathic pulmonary fibrosis: pathogenesis and management. Respir Res. 2018;19(1):32. https://doi.org/10.1186/s12931-018-0730-2.
Jäger B, Seeliger B, Terwolbeck O, Warnecke G, Welte T, Müller M, Bode C, Prasse A. The NLRP3-inflammasome-Caspase-1 pathway is upregulated in idiopathic pulmonary fibrosis and acute exacerbations and is inducible by apoptotic A549 cells. Front Immunol. 2021;12:642855. https://doi.org/10.3389/fimmu.2021.642855.
Meng Y, Pan M, Zheng B, Chen Y, Li W, Yang Q, Zheng Z, Sun N, Zhang Y, Li X. Autophagy attenuates angiotensin II-induced pulmonary fibrosis by inhibiting redox imbalance-mediated NOD-like receptor family pyrin domain containing 3 inflammasome activation. Antioxid Redox Signal. 2019;30(4):520–41. https://doi.org/10.1089/ars.2017.7261.
Xu M, Wang X, Xu L, Zhang H, Li C, Liu Q, Chen Y, Chung KF, Adcock IM, Li F. Chronic lung inflammation and pulmonary fibrosis after multiple intranasal instillation of PM2.5 in mice. Environ Toxicol. 2021;36(7):1434–46. https://doi.org/10.1002/tox.23140.
Fan H-C, Chen C-Y, Hsu Y-C, Chou R-H, Teng C-LJ, Chiu C-H, Hsu CY, Muo C-H, Chang M-Y, Chang K-H. Increased risk of incident nasopharyngeal carcinoma with exposure to air pollution. PLoS ONE. 2018;13(9):e0204568. https://doi.org/10.1371/journal.pone.0204568.
Zheng R, Tao L, Jian H, Chang Y, Cheng Y, Feng Y, Zhang H. NLRP3 inflammasome activation and lung fibrosis caused by airborne fine particulate matter. Ecotoxicol Environ Saf. 2018;163:612–9. https://doi.org/10.1016/j.ecoenv.2018.07.076.
Liu X, Wang J, Dou P, Zhang X, Ran X, Liu L, Dou D. The ameliorative effects of arctiin and arctigenin on the oxidative injury of lung induced by ailica via TLR-4/NLRP3/TGF-β signaling pathway. Oxid Med Cell Longev. 2021;2021:5598980. https://doi.org/10.1155/2021/5598980.
Hindman B, Ma Q. Carbon nanotubes and crystalline silica stimulate robust ROS production, inflammasome activation, and IL-1β secretion in macrophages to induce myofibroblast transformation. Arch Toxicol. 2019;93(4):887–907. https://doi.org/10.1007/s00204-019-02411-y.
Zhou L, Li P, Zhang M, Han B, Chu C, Su X, Li B, Kang H, Ning J, Zhang B, Ma S, Su D, Pang Y, Niu Y, Zhang R. Carbon black nanoparticles induce pulmonary fibrosis through NLRP3 inflammasome pathway modulated by miR-96 targeted FOXO3a. Chemosphere. 2020;241:125075. https://doi.org/10.1016/j.chemosphere.2019.125075.
Er JZ, Koean RAG, Ding JL. Loss of T-bet confers survival advantage to influenza-bacterial superinfection. EMBO J. 2019;38(1):e99176. https://doi.org/10.15252/embj.201899176.
Cuypers L, Thijssen M, Shakibzadeh A, Sabahi F, Ravanshad M, Pourkarim MR. Next-generation sequencing for the clinical management of hepatitis C virus infections: does one test fits all purposes? Crit Rev Clin Lab Sci. 2019;56(6):420–34. https://doi.org/10.1080/10408363.2019.1637394.
Wang R, Zhu Y, Lin X, Ren C, Zhao J, Wang F, Gao X, Xiao R, Zhao L, Chen H, Jin M, Ma W, Zhou H. Influenza M2 protein regulates MAVS-mediated signaling pathway through interacting with MAVS and increasing ROS production. Autophagy. 2019;15(7):1163–81. https://doi.org/10.1080/15548627.2019.1580089.
Park H-S, Liu G, Liu Q, Zhou Y. Swine influenza virus induces RIPK1/DRP1-mediated interleukin-1 beta production. Viruses. 2018;10(8):419. https://doi.org/10.3390/v10080419.
Liu H, You L, Wu J, Zhao M, Guo R, Zhang H, Su R, Mao Q, Deng D, Hao Y. Berberine suppresses influenza virus-triggered NLRP3 inflammasome activation in macrophages by inducing mitophagy and decreasing mitochondrial ROS. J Leukoc Biol. 2020;108(1):253–66. https://doi.org/10.1002/jlb.3ma0320-358rr.
Liu C-C, Miao Y, Chen R-L, Zhang Y-Q, Wu H, Yang S-M, Shang L-Q. STIM1 mediates IAV-induced inflammation of lung epithelial cells by regulating NLRP3 and inflammasome activation via targeting miR-223. Life Sci. 2021;266:118845. https://doi.org/10.1016/j.lfs.2020.118845.
Dauber B, Wolff T. Activation of the antiviral kinase PKR and viral countermeasures. Viruses. 2009;1(3):523–44. https://doi.org/10.3390/v1030523.
Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BR. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol Cell Biol. 2000;20(4):1278–90. https://doi.org/10.1128/mcb.20.4.1278-1290.2000.
Bonnet MC, Weil R, Dam E, Hovanessian AG, Meurs EF. PKR stimulates NF-kappaB irrespective of its kinase function by interacting with the IkappaB kinase complex. Mol Cell Biol. 2000;20(13):4532–42. https://doi.org/10.1128/mcb.20.13.4532-4542.2000.
Darweesh M, Kamel W, Gavrilin MA, Akusjärvi G, Svensson C. Adenovirus VA RNAI blocks ASC oligomerization and inhibits NLRP3 inflammasome activation. Front Immunol. 2019;10:2791. https://doi.org/10.3389/fimmu.2019.02791.
Chen W, Ma Y, Zhang H, Guo Y, Guan M, Wang Y. Reduning plus ribavirin display synergistic activity against severe pneumonia induced by H1N1 influenza A virus in mice. J Tradit Chin Med. 2020;40(5):803–11. https://doi.org/10.19852/j.cnki.jtcm.2020.05.010.
Paulin LM, Gassett AJ, Alexis NE, Kirwa K, Kanner RE, Peters S, Krishnan JA, Paine R, Dransfield M, Woodruff PG, Cooper CB, Barr RG, Comellas AP, Pirozzi CS, Han M, Hoffman EA, Martinez FJ, Woo H, Peng RD, Fawzy A, Putcha N. Association of long-term ambient ozone exposure with respiratory morbidity in smokers. JAMA Intern Med. 2020;180(1):106–15. https://doi.org/10.1001/jamainternmed.2019.5498.
Xu M, Wang L, Wang M, Wang H, Zhang H, Chen Y, Wang X, Gong J, Zhang JJ, Adcock IM, Chung KF, Li F. Mitochondrial ROS and NLRP3 inflammasome in acute ozone-induced murine model of airway inflammation and bronchial hyperresponsiveness. Free Radic Res. 2019;53(7):780–90. https://doi.org/10.1080/10715762.2019.1630735.
He K, Hettinga A, Kale SL, Hu S, Xie MM, Dent AL, Ray A, Poholek AC. Blimp-1 is essential for allergen-induced asthma and Th2 cell development in the lung. J Exp Med. 2020;217(7):e20190742. https://doi.org/10.1084/jem.20190742.
Hu X, Shen Y, Zhao Y, Wang J, Zhang X, Tu W, Kaufman W, Feng J, Gao P. Epithelial aryl hydrocarbon receptor protects from mucus production by inhibiting ROS-triggered NLRP3 inflammasome in asthma. Front Immunol. 2021;12:767508. https://doi.org/10.3389/fimmu.2021.767508.
Theofani E, Semitekolou M, Samitas K, Mais A, Galani IE, Triantafyllia V, Lama J, Morianos I, Stavropoulos A, Jeong S-J, Andreakos E, Razani B, Rovina N, Xanthou G. TFEB signaling attenuates NLRP3-driven inflammatory responses in severe asthma. Allergy. 2022;77(7):2131–46. https://doi.org/10.1111/all.15221.
Xin R, Pan Y-L, Wang Y, Wang S-Y, Wang R, Xia B, Qin R-N, Fu Y, Wu Y-H. Nickel-refining fumes induce NLRP3 activation dependent on mitochondrial damage and ROS production in Beas-2B cells. Arch Biochem Biophys. 2019;676:108148. https://doi.org/10.1016/j.abb.2019.108148.
Xu L-T, Wang T, Fang K-L, Zhao Y, Wang X-N, Ren D-M, Shen T. The ethanol extract of flower buds of Tussilago farfara L. attenuates cigarette smoke-induced lung inflammation through regulating NLRP3 inflammasome, Nrf2, and NF-κB. J Ethnopharmacol. 2022;283:114694. https://doi.org/10.1016/j.jep.2021.114694.
Qing C, Ziyun L, Xuefei Y, Xinyi Z, Xindong X, Jianhua F. Protective effects of 18β-glycyrrhetinic acid on neonatal rats with hyperoxia exposure. Inflammation. 2022;45(3):1224–38. https://doi.org/10.1007/s10753-021-01616-7.
Li X, Gong Y, Li D, Xiang L, Ou Y, Jiang L, Shu P, Liu X, Guo F, Qin D, Mo Z, Qin Q, Wang X, Wang Y. Low-dose radiation therapy promotes radiation pneumonitis by activating NLRP3 inflammasome. Int J Radiat Oncol Biol Phys. 2020;107(4):804–14. https://doi.org/10.1016/j.ijrobp.2020.02.643.
Zheng P, Kang J, Xing E, Zheng B, Wang X, Zhou H. Lung inflation with hydrogen during the cold ischemia phase alleviates lung ischemia-reperfusion injury by inhibiting pyroptosis in rats. Front Physiol. 2021;12:699344. https://doi.org/10.3389/fphys.2021.699344.
Xu K-Y, Wu C-Y, Tong S, Xiong P, Wang S-H. The selective Nlrp3 inflammasome inhibitor Mcc950 attenuates lung ischemia-reperfusion injury. Biochem Biophys Res Commun. 2018;503(4):3031–7. https://doi.org/10.1016/j.bbrc.2018.08.089.
Song J-Q, Jiang L-Y, Fu C-P, Wu X, Liu Z-L, Xie L, Wu X-D, Hao S-Y, Li S-Q. Heterozygous SOD2 deletion deteriorated chronic intermittent hypoxia-induced lung inflammation and vascular remodeling through mtROS-NLRP3 signaling pathway. Acta Pharmacol Sin. 2020;41(9):1197–207. https://doi.org/10.1038/s41401-019-0349-y.
Lee C-W, Chi M-C, Hsu L-F, Yang C-M, Hsu T-H, Chuang C-C, Lin W-N, Chu P-M, Lee I-T. Carbon monoxide releasing molecule-2 protects against particulate matter-induced lung inflammation by inhibiting TLR2 and 4/ROS/NLRP3 inflammasome activation. Mol Immunol. 2019;112:163–74. https://doi.org/10.1016/j.molimm.2019.05.005.
Cao X, Tian S, Fu M, Li Y, Sun Y, Liu J, Liu Y. Resveratrol protects human bronchial epithelial cells against nickel-induced toxicity via suppressing p38 MAPK, NF-κB signaling, and NLRP3 inflammasome activation. Environ Toxicol. 2020;35(5):609–18. https://doi.org/10.1002/tox.22896.
Zhang W, Wang W, Xu M, Xie H, Pu Z. GPR43 regulation of mitochondrial damage to alleviate inflammatory reaction in sepsis. Aging. 2021;13(18):22588–610. https://doi.org/10.18632/aging.203572.
Luo X, Chang X, Zhou H, Lin H, Fan H. Glaesserella parasuis induces inflammatory response in 3D4/21 cells through activation of NLRP3 inflammasome signaling pathway via ROS. Vet Microbiol. 2021;256:109057. https://doi.org/10.1016/j.vetmic.2021.109057.
Huang Y, Hua M, Cui X. Fungal β-glucan activates the NLRP3 inflammasome in human bronchial epithelial cells through ROS production. Inflammation. 2018;41(1):164–73. https://doi.org/10.1007/s10753-017-0674-6.
Dai J, Chen Y, Jiang F. Allicin reduces inflammation by regulating ROS/NLRP3 and autophagy in the context of A fumigatus infection in mice. Gene. 2020;762:145042. https://doi.org/10.1016/j.gene.2020.145042.
Wang J, Sahoo M, Lantier L, Warawa J, Cordero H, Deobald K, Re F. Caspase-11-dependent pyroptosis of lung epithelial cells protects from melioidosis while caspase-1 mediates macrophage pyroptosis and production of IL-18. PLoS Pathog. 2018;14(5):e1007105. https://doi.org/10.1371/journal.ppat.1007105.
Funding
This work was partially supported by the National Natural Science Foundation of China (Grant No. 82260820) and the Natural Science Foundation of Jilin Province, Jilin, China (Grant Nos. YDZJ202201ZYTS155, 20190701034GH, and 20200201034JC).
Author information
Authors and Affiliations
Contributions
YC conceived of the review and wrote the manuscript; YZ and NL organized the literature and discussion; and ZJ and XL critically revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, Y., Zhang, Y., Li, N. et al. Role of mitochondrial stress and the NLRP3 inflammasome in lung diseases. Inflamm. Res. 72, 829–846 (2023). https://doi.org/10.1007/s00011-023-01712-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-023-01712-4