
Abstract
Hundreds of millions of people are affected by hyperinsulinaemia, insulin resistance, obesity and the dysglycaemia that mark a common progression from metabolic health to type 2 diabetes. Although the relative contribution of these features and the order in which they appear may differ between individuals, the common clustering and seemingly progressive nature of type 2 diabetes aetiology has guided research and clinical practice in this area for decades. At the same time, lively debate around the causal relationships between these features has continued, as new data from human trials and highly controlled animal studies are presented. This ‘For debate’ article was prompted by the review in Diabetologia by Esser, Utzschneider and Kahn (https://doi.org/10.1007/s00125-020-05245-x), with the purpose of reviewing established and emerging data that provide insight into the relative contributions of hyperinsulinaemia and impaired glucose-stimulated insulin secretion in progressive stages between health, obesity and diabetes. It is concluded that these beta cell defects are not mutually exclusive and that they are both important, but at different stages.
Graphical abstract

Similar content being viewed by others


Introduction and scope of response
The review by Esser, Utzschneider and Kahn is a thorough treatment of a complicated subject [1]. Although the role of beta cells in type 2 diabetes was doubted in the past, truly elegant work by Kahn and his contemporaries put this to rest conclusively years ago. Today, we are left to debate when, how and why beta cells ‘fail’ along the progression to type 2 diabetes. Their review starts from the viewpoint that there are two opposing models of type 2 diabetes progression. In one model, which they refer to as ‘prevailing’, the authors state that ‘in the presence of insulin resistance, beta cell dysfunction that occurs early in the course of the disease process is the critical abnormality’. In another model, labelled as ‘alternative’, ‘primary beta cell overstimulation results in insulin hypersecretion that then leads to the development of obesity and insulin resistance, and ultimately to beta cell exhaustion’ [1]. These statements deserve careful parsing and reflection as to whether the models are mutually exclusive. I believe they are not. In this article I will comment on and debate the following questions, derived from these stated models:
-
1.
Both statements above imply that reduced glucose-stimulated insulin secretion is the primary (or only) relevant beta cell defect. What other beta cell differences are found in obesity, prediabetes, diabetes? Are these differences good or bad, or does it depend on the context?
-
2.
What is the evidence that hyperinsulinaemia can drive the earliest stages of disease susceptibility? Beyond correlations, determining causality requires specific types of loss-of-function and gain-of-function experiments. What have we learned from the specific manipulation of insulin?
-
3.
What is the role of insulin resistance in obesity, prediabetes, diabetes? Is insulin resistance the primary defect, preceding beta cell dysfunction: broadly defined
Before delving into questions of primacy, causality and specific defects, it is important to acknowledge that the aetiology of type 2 diabetes, including the order in which each pathological stage occurs, may not be the same for each individual or genetically distinct human population [2] and, as the authors point out, is understudied in young people [1]. I hope to convey the range of pathophysiological mechanisms that can contribute to dysglycaemia, rather than prescribe a single, firm pathophysiological pathway to disease. It is also important to acknowledge the work of others who have thoughtfully considered the relative importance of hyperinsulinaemia in the progression towards type 2 diabetes. Here I will try to avoid duplicating the arguments of Jessie Roth, Barbara Corkey, Walter Pories, Marc Prentki, Christopher Nolan, David Ludwig and many others [3,4,5,6,7,8].
What are the beta cell defects in obesity, insulin resistance and type 2 diabetes?
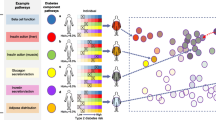
It is established that differences in beta cell function are fundamental to the pathogenesis of diabetes in populations with and without obesity and ectopic lipid accumulation [9]. Now it remains to be determined what the specific ‘dysfunctions’ are at the various stages of type 2 diabetes aetiology. First, let us define dysfunction, and let us acknowledge that physiological systems can both adapt and mal-adapt to stresses in ways that may appear similar. The title of the article by Esser et al frames a false dichotomy, as if hyperinsulinaemia is not itself, in large part, a beta cell dysfunction (see below). I therefore suggest using the broadest definition of beta cell dysfunction that includes inappropriate insulin secretion, excessive or insufficient, either in the fed or fasted state, in response to glucose or any other nutrient. We would expect different consequences of elevated insulin in distinct nutrient contexts. Second, let us consider the stages of the progression to diabetes, acknowledging the likelihood that these may differ in different individuals (Fig. 1). We have learned from deep phenotyping and human genetics that there are multiple sub-types of obesity, prediabetes and type 2 diabetes [2].

A multi-stage and individualised natural history of type 2 diabetes. Thick lines represent an approximate average of the latency of a key (patho)physiological variable associated with the progression to type 2 diabetes. Thinner lines represent a range of these phenotypic variables reflected in various sub-types of the disease. This is not an exhaustive set; for example, visceral adiposity is coincident with lipid accumulation in the liver, pancreas and other ectopic sites. Thin arrows denote highlighted effects of specific variables on each other. IFG, impaired fasting glucose; IGT, impaired glucose tolerance
The most prevalent feature of beta cell dysfunction found in obesity is reflected in an approximately twofold increase in insulin levels that can include changes in both the fasting and fed states, prior to insulin resistance, and well before relative glucose-stimulated insulin secretion is reduced [10,11,12,13]. For example, Hamley et al showed that ‘primary’ hyperinsulinaemia, resulting from a combination of insulin hypersecretion and reduced clearance, can be found in non-obese young adults, representing a precursor stage to impaired glucose tolerance and impaired fasting glucose [14]. Trico et al studied >1300 adolescents or adults, revealing a tertile of insulin hypersecretors who did not have clamp-measured insulin resistance but who exhibited increased incidence of impaired glucose tolerance and type 2 diabetes after 3 years of follow-up [15]. These studies, and others, illustrate that hyperinsulinaemia can be found prior to insulin resistance and/or obesity.
Many discussions of this topic do not distinguish between basal hyperinsulinaemia vs hyper-responsiveness to glucose stimulation. Interestingly, in mouse models with reduced insulin gene dosage, fasting insulin is reduced while glucose-stimulated insulin responsiveness remains relatively intact [16,17,18]. While the two types of hyperinsulinaemia can be separated experimentally, it remains unclear whether these (mal)adaptations are mechanistically distinct. Theoretically, fasting or fed hyperinsulinaemia can have multiple molecular mechanisms, including simple beta cell overstimulation driven by increased nutrient intake. Any modulation in the gain (e.g. a right-shift in the nutrient dose–response curve) would also change the efficiency of the insulin secretion in response to constant nutrient exposure. Similarly, basal hyperinsulinaemia can result from the inability to restrain constitutive insulin secretion in low glucose conditions, a cardinal feature of immature beta cells and insulinomas [3, 19]. Additional proposed molecular mechanisms of insulin hypersecretion include inappropriate redox signalling and/or mitochondrial proton leak, exposure to environmental toxins, disrupted circadian rhythm, and altered lipid metabolism related to local lipid accumulation in the pancreas [20,21,22,23,24]. It should also be noted that sustained insulin elevation occurs when the normal oscillatory pattern is disrupted, a process that can lead to liver insulin resistance and predisposition to type 2 diabetes [25]. The lower limit of insulin secretion is also related to the number of beta cells. We and others have shown that cell autonomous mechanisms link workload to beta cell proliferation and survival depending on the life stage [26, 27]. Finally, reduced insulin clearance plays a major role in hyperinsulinaemia [14, 28] and has been proposed as a significant driver of type 2 diabetes [29].
In those who progress to the early stages of impaired glucose tolerance, multiple beta cell dysfunctions can be identified. Cited by Esser, the seminal paper by Mitrakou et al describes ‘diminished early insulin release’ in individuals with impaired glucose tolerance, regardless of obesity; in fact, this paper shows that only the first phase (~0–60 min) was reduced, while all subsequent insulin measurements, and fasting insulin, were higher in the glucose-intolerant individuals (Fig. 2) [30]. We are in full agreement that a reduction in the first phase of glucose-stimulated insulin release must precede the development of dysglycaemia and eventual frank diabetes (Fig. 3). The question at hand concerns whether fasting or fed hyperinsulinaemia can have maladaptive consequences prior to the impairment of glucose tolerance.

Multiple insulin secretion abnormalities after the transition to impaired glucose tolerance. Note that while the acute (30 min) response to glucose is reduced and the peak is delayed in IGT, the insulin AUC is greater, with both elevated postprandial insulin and elevated fasting insulin relative to NGT. Approximated data traces are replotted from Mitrakou et al [30]. IGT, impaired glucose tolerance; NGT, normal glucose tolerance

Schematic of causes and cause-networks involved in dysglycaemia and type 2 diabetes. Integration of beta cell genetic resilience with other genetic factors and environmental stress in the initiation and progression of dysglycaemia. In this model, both hyperinsulinaemia and impaired GSIS are considered types of beta cell dysfunction. ER, endoplasmic reticulum; GSIS, glucose-stimulated insulin secretion
Physiological and pathophysiological consequences of hyperinsulinaemia
Can hyperinsulinaemia play a causal role in obesity and ectopic lipid deposition? We are delighted that Esser et al chose to highlight some of our pre-clinical studies using mice with genetically engineered specific reductions in circulating insulin, although some questions were raised. Several additional key papers from our laboratory, when considered, may address the concerns brought up by Esser et al (Table 1). Our initial work showing that reducing hyperinsulinaemia prevented diet-induced obesity, fatty liver and inflammation [16], rather than being built on the premise that hyperinsulinaemia is deleterious, took us by surprise and was originally designed to test the role of insulin in beta cell mass regulation. Subsequent studies extended these findings past the first year of life to the full murine lifespan [18], to the Lepob/ob background [31], and to correlations within genotypes [32]. We have also replicated the relationship between insulin gene dosage in body-weight tracking data from mice used to examine the role of hyperinsulinaemia in pancreatic cancer initiation [33], as well as mice missing only a single allele of Ins1 (supplemental data in [34]). This last study compares mice having the full complement of insulin alleles (Ins1+/+;Ins2+/+) with littermates lacking only a single Ins1 allele (Ins1+/−;Ins2+/+) (the smallest genetic insulin manipulation possible) and still finds small but significant differences in body weight in young mice [34]. Moreover, Dionne et al. found that the effects of reduced insulin gene and restriction of energy intake on adiposity were not additive [35]. Page et al used a model of inducible insulin reduction to demonstrate fat loss in mice that had already been made obese [36]. While some insulin gene configurations do not result in robust differences in circulating insulin [32], due to gene compensation [37], we have observed reduced adiposity in every model where we were able to reduce circulating insulin. Our results are consistent with other models in which insulin secretion has been suppressed and weight gain blunted [38].
The work of Templeman using mice with reduced insulin production also demonstrated a causal role for hyperinsulinaemia in age-associated insulin resistance [18]. Our work is consistent with another study that mildly blunted insulin secretion indirectly [39] and with experiments by Czech and co-workers showing that abrogating hyperinsulinaemia in a variety of models improved insulin sensitivity and reduced associated inflammation [40], a topic we have recently reviewed [41]. Moreover, chronic insulin infusion is sufficient to cause weight gain and glucose intolerance in lean rats [42]. The molecular mechanisms by which hyperinsulinaemia and sustained insulin receptor (INSR) activation (i.e. loss of insulin oscillations) could induce insulin resistance are known, including both INSR and post-receptor desensitisation [43]. Improved insulin sensitivity in models with reduced insulin production likely accounts for the maintenance of normoglycaemia. It is important to recognise that there is a narrow concentration range of insulin that can modulate lipid homeostasis without affecting glucose tolerance. This fits with the mechanistically independent effects of insulin on glucose uptake and on lipid storage, active at different insulin concentrations, with the later process’s enzymatic pathways sensitive to much smaller changes in insulin levels. For example, while 1 nmol/l insulin is sufficient for near maximal adipocyte differentiation [44], that dose (already in the high physiological range) does not stimulate glucose uptake in vitro [45]. In vivo human studies consistently show that while insulin’s effects on glucose disposal are relatively insensitive and linear across a range spanning 350–900 pmol/l, lipolysis is more highly sensitive to insulin, with a non-linear dose response [46]. In our animal models, we expect that once blood glucose is significantly affected by unphysiological shifts in circulating insulin, multiple counter-regulatory systems come into play.
Human clinical studies where insulin is directly modulated up or down confirm the causal relationship between hyperinsulinaemia and obesity/insulin resistance, as reviewed elsewhere [47, 48]. Despite improving glycaemic control and cardiovascular outcomes, and despite confounders present in these patient populations, clinical trials of insulins consistently demonstrate weight gain [49]. Gregory et al inferred the ability of chronic exogenous insulin to induce insulin resistance, independently of mild hyperglycaemia, by conducting parallel clamp studies on individuals with type 1 diabetes and MODY2 [50]. Meta-analysis confirms that suppression of insulin secretion alone is sufficient to treat obesity [51], consistent with our studies using mice with inducible partial insulin gene deletion [36, 52]. Esser also considered these studies and seemingly dismissed one study employing the somatostatin analogue octreotide because weight loss correlated with improved insulin sensitivity [53]. These findings are entirely consistent with the concept that hyperinsulinaemia is upstream of both obesity and insulin resistance, at least in some individuals. However, I do agree that further refinement of insulin inhibition therapies will be required before they can be recommended instead of lifestyle approaches to achieve the same goal.
Esser et al highlight several lines of clinical investigation that they suggest fail to support a causal role for insulin hypersecretion in the pathogenesis of dysglycaemia, including insights from interventional studies in animals and humans involving lifestyle, pharmacological inhibition of insulin secretion, insulin sensitisers, insulin secretagogues and bariatric surgery. While noting that many dietary weight-loss interventions also suppress insulin hypersecretion, the authors suggest an apparent paradox: ‘if hyperinsulinaemia was primary, one may have expected that the ‘fixed’ insulin hypersecretion, at least initially, would not have been able to adapt to the reduced energy intake or weight loss so that hypoglycaemia would have been reported, which was not the case’. As noted above, insulin sensitivity in peripheral tissues can rapidly adapt to changes in ambient insulin. In fact, muscle insulin receptor mRNA expression is significantly downregulated by hyperinsulinaemia within hours, and restored just as quickly upon removal of the desensitising stimulus [54]. The absence of hypoglycaemia, which is vigorously defended against by multiple redundant mechanisms, both central and peripheral, does in no way negate the more direct evidence for a causal pathological contribution from hyperinsulinaemia. Together, multiple lines of evidence, from exquisitely controlled animal models to clinical studies, support the assertion that hyperinsulinaemia is a contributor to obesity and insulin resistance (Fig. 3). Hyperinsulinaemia may eventually play a role in dysglycaemia through a combination of indirect and direct mechanisms. Hyperinsulinaemia need not be the only contributor to be critical and uniquely actionable.
What is the role of insulin resistance in type 2 diabetes?
Many investigators still believe that type 2 diabetes starts with primary insulin resistance. In the first model, the phrase ‘in the presence of insulin resistance’ points to a requirement for insulin resistance to ‘unmask’ underlying beta cell pathology. It is also commonly held that insulin resistance is pathological, even in its mildest forms, a concept that has been elegantly debated elsewhere [7, 55, 56]. While the idea that insulin resistance precedes and causes hyperinsulinaemia has long been held as dogma, attempting to define what comes first is critically dependent on technology and ease of tissue access. It is no surprise that early studies in adults identified alterations in glucose uptake in skeletal muscle prior to defects in glucose-stimulated insulin secretion. Skeletal muscle is easily accessible, while islets are buried within the pancreas and insulin secretion partially obscured by variation in insulin clearance [28]. Moreover, clamp studies can employ supraphysiological insulin levels and exaggerate the contribution of skeletal muscle glucose clearance [57]. Nevertheless, recent large studies challenge this order of events, identifying people with primary hyperinsulinaemia [14, 15]. Most population-wide studies are unable to distinguish primary hyperinsulinaemia from insulin resistance because insulin resistance is inferred from unexplained hyperinsulinaemia (e.g. HOMA-IR, etc.), rather than measured directly. The challenges of such a circular definition of insulin resistance for large-scale studies have recently been illuminated by mathematical modelling [58].
It is now broadly agreed that beta cell ‘failure’ is the critical event in the progression to diabetes but it remains possible that pathological insulin resistance may be an initial causal factor in some individuals (Figs 1, 3). To address this question we must consider the consequences of insulin resistance on its own. The most specific way to study insulin resistance is to examine genetic reduction or specific pharmacological inhibition of insulin’s cognate receptor. Secondary signalling mediators, such as IRS1/2, phosphoinositide 3 (PI3)-kinases and Akt1/2/3, are less informative for this question because they integrate multiple signals beyond insulin, including the insulin-like growth factors, other tyrosine kinase receptors and possibility glucose itself [59]. While individuals with heterozygous INSR mutations exhibit insulin resistance on a par with that found in some people with type 2 diabetes and significant hyperinsulinaemia, they tend to exhibit only mild glucose intolerance; frank diabetes is either not observed or is delayed by decades [60, 61]. According to the T2D Knowledge Portal (t2d.hugeamp.org, accessed 1 January 2021), common variation near INSR is more closely associated with blood pressure, height, adjusted waist/hip ratio, serum urate, triacylglycerols and adjusted hip circumference than with type 2 diabetes. Insr heterozygous mice exhibit hyperinsulinaemia and normal glucose tolerance [62, 63]. Blockade of insulin receptor signalling using a specific monoclonal antibody results in transient hyperglycaemia at high doses but lowers fasting glucose after 3 weeks of treatment in mice. Pharmacological modulation of INSR in rodents, using the mixed agonist/antagonist S961, leads to hyperinsulinaemia and glucose intolerance but not diabetes [64]. S961 can increase beta cell proliferation in the normoglycaemic state but the molecular mechanisms remain unclear. In mice, high-fat-diet-induced beta cell proliferation precedes insulin resistance [65] and requires hyperinsulinaemia [16]. Thus, global insulin resistance, defined in the purest molecular terms, alone does not lead to diabetes under most conditions tested, in the absence of beta cell impairment [66]. Esser et al have done an excellent job of reviewing results from mouse models of tissue-specific insulin resistance and summarising that frank diabetes has not been found, except for the reported effects of Insr loss in beta cells [1]. In our hands, even beta cell-specific Insr loss does not lead to diabetes but, rather, mild hyperinsulinaemia [34]. Muscle Insr knockout alone is not sufficient to cause insulin resistance or glucose intolerance, although lipids are shunted to other tissues [67]. Adipose Insr knockout mice are protected from obesity and diabetes, and live longer than control mice [68].
Inferring the causal components along the progression from obesity to type 2 diabetes from the effects of medications is complicated. Metformin and thiazolidinediones are not useful tools for directly testing the role of general insulin sensitivity, since these drugs have multiple known modes of action, have insulin-sensitising actions that are dependent on tissue type, and affect beta cell function indirectly and directly [69]. Notably, metformin and pioglitazone reverse hyperinsulinaemia in isolated non-diabetic human islets [70, 71]. Sulfonylureas increase body weight, likely because the induced insulin secretion is not glucose dependent and can therefore be inappropriately timed. The effect of glucagon-like peptide-1 (GLP1) receptor agonists, on the other hand, is meal-dependent, ensuring that insulin release is appropriately timed, and they do not cause weight gain [72]. Insulin resistance is a ‘concept’ based on differences between what would be expected from a simple model and what is observed. Many hypotheses around insulin resistance are currently untestable in the absence of specific approaches to manipulate it. Experimentally and specifically manipulating ‘insulin resistance’ in humans is difficult and will necessarily affect different aspects of insulin signalling, perhaps in distinct ways between key tissues.
Clinical translation and unanswered questions
There are several open questions that should be discussed:
-
1.
When is the best time to intervene to prevent and reverse obesity and/or type 2 diabetes? There is still a paucity of longitudinal studies of large human cohorts starting in or before childhood, where most of the risk of type 2 diabetes is set [73]. We agree with Esser et al that it will be critical to conduct more studies in youth to investigate the early events that ultimately lead to type 2 diabetes. The authors review several studies on the progression to type 2 diabetes in youth, which largely confirm the loss of beta cell responsiveness to glucose as a prerequisite for disease.
-
2.
Is there a genetic basis for hyperinsulinaemia and its sequelae in humans? The genetics of common type 2 diabetes are complex. While it is clear that most of the hundreds of genes near genome-wide association study (GWAS) signals are enriched for islet expression, the specific effects of most of these SNPs on basal insulin, stimulated insulin, and beta cell mass, as well as the directionality of these effects, remains to be studied [74]. Mendelian randomisation has been used to infer that hyperinsulinaemia causes obesity [75] and insulin resistance [76], depending on the tool SNPs employed. We await the complete genetic dissection of signals within the insulin locus that are associated with anthropomorphic traits. It will also be important to understand the epigenetic control of insulin and its mediators.
-
3.
Does assigning primary defects and causality matter clinically? For the last few decades, the prevailing view of type 2 diabetes pathogenesis has guided therapeutic approaches and drug development. Most drugs are designed to combat hyperglycaemia, counteract the deficiency in glucose-stimulated insulin secretion, and increase insulin sensitivity in the context of frank diabetes. A compelling case can be made that much of the important pathobiology has already occurred by the time glucose homeostasis is deranged and that some of these approaches may be double-edged swords. We now appreciate, through bariatric surgery and dietary intervention data, that type 2 diabetes is much more reversible than was once thought [77,78,79].
In conclusion, there is still much left to learn about the causal relationships between the different forms of hyperinsulinaemia, the different forms of insulin resistance, and the multiple (patho)physiological metabolic changes that occur along a path from health to obesity to diabetes. The field has come to consensus around the central role of dysfunctional beta cells in diabetes, and this author finds a majority of common ground with Drs Esser, Utzschneider, and Kahn. With this article, there is sincere hope to continue a conversation about causality that will ultimately improve treatment and the lives of people living with diabetes and those who are at risk.
Abbreviations
- INSR:
-
Insulin receptor
References
Esser N, Utzschneider KM, Kahn SE (2020) Early beta cell dysfunction vs insulin hypersecretion as the primary event in the pathogenesis of dysglycaemia. Diabetologia 63(10):2007–2021. https://doi.org/10.1007/s00125-020-05245-x
Wagner R, Heni M, Tabak AG et al (2021) Pathophysiology-based subphenotyping of individuals at elevated risk for type 2 diabetes. Nat Med 27:49–75. https://doi.org/10.1038/s41591-020-1116-9
Shanik MH, Xu Y, Skrha J, Dankner R, Zick Y, Roth J (2008) Insulin resistance and hyperinsulinemia: is hyperinsulinemia the cart or the horse? Diabetes Care 31(Suppl 2):S262–S268. https://doi.org/10.2337/dc08-s264
Corkey BE (2012) Banting lecture 2011: hyperinsulinemia: cause or consequence? Diabetes 61(1):4–13. https://doi.org/10.2337/db11-1483
Erion K, Corkey BE (2018) Beta-cell failure or beta-cell abuse? Front Endocrinol (Lausanne) 9:532. https://doi.org/10.3389/fendo.2018.00532
Pories WJ, Dohm GL (2012) Diabetes: have we got it all wrong? Hyperinsulinism as the culprit: surgery provides the evidence. Diabetes Care 35(12):2438–2442. https://doi.org/10.2337/dc12-0684
Nolan CJ, Prentki M (2019) Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: time for a conceptual framework shift. Diab Vasc Dis Res 16(2):118–127. https://doi.org/10.1177/1479164119827611
Ludwig DS, Ebbeling CB (2018) The carbohydrate-insulin model of obesity: beyond “calories in, calories out”. JAMA Intern Med 178(8):1098–1103. https://doi.org/10.1001/jamainternmed.2018.2933
Bell GI, Polonsky KS (2001) Diabetes mellitus and genetically programmed defects in beta-cell function. Nature 414(6865):788–791. https://doi.org/10.1038/414788a
Polonsky KS, Given BD, Van Cauter E (1988) Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. J Clin Invest 81(2):442–448. https://doi.org/10.1172/JCI113339
Ling JC, Mohamed MN, Jalaludin MY, Rampal S, Zaharan NL, Mohamed Z (2016) Determinants of high fasting insulin and insulin resistance among overweight/obese adolescents. Sci Rep 6:36270. https://doi.org/10.1038/srep36270
van Vliet S, Koh HE, Patterson BW et al (2020) Obesity is associated with increased basal and postprandial beta-cell insulin secretion even in the absence of insulin resistance. Diabetes 69(10):2112–2119. https://doi.org/10.2337/db20-0377
Le Stunff C, Bougneres P (1994) Early changes in postprandial insulin secretion, not in insulin sensitivity, characterize juvenile obesity. Diabetes 43(5):696–702. https://doi.org/10.2337/diab.43.5.696
Hamley S, Kloosterman D, Duthie T et al (2019) Mechanisms of hyperinsulinaemia in apparently healthy non-obese young adults: role of insulin secretion, clearance and action and associations with plasma amino acids. Diabetologia 62(12):2310–2324. https://doi.org/10.1007/s00125-019-04990-y
Trico D, Natali A, Arslanian S, Mari A, Ferrannini E (2018) Identification, pathophysiology, and clinical implications of primary insulin hypersecretion in nondiabetic adults and adolescents. JCI Insight 3(24):e124912. https://doi.org/10.1172/jci.insight.124912
Mehran AE, Templeman NM, Brigidi GS et al (2012) Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab 16(6):723–737. https://doi.org/10.1016/j.cmet.2012.10.019
Templeman NM, Clee SM, Johnson JD (2015) Suppression of hyperinsulinaemia in growing female mice provides long-term protection against obesity. Diabetologia 58(10):2392–2402. https://doi.org/10.1007/s00125-015-3676-7
Templeman NM, Flibotte S, Chik JHL et al (2017) Reduced circulating insulin enhances insulin sensitivity in old mice and extends lifespan. Cell Rep 20(2):451–463. https://doi.org/10.1016/j.celrep.2017.06.048
Rezania A, Bruin JE, Arora P et al (2014) Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 32(11):1121–1133. https://doi.org/10.1038/nbt.3033
Staaf J, Ubhayasekera SJ, Sargsyan E et al (2016) Initial hyperinsulinemia and subsequent beta-cell dysfunction is associated with elevated palmitate levels. Pediatr Res 80(2):267–274. https://doi.org/10.1038/pr.2016.80
Taddeo EP, Alsabeeh N, Baghdasarian S et al (2020) Mitochondrial proton leak regulated by cyclophilin D elevates insulin secretion in islets at nonstimulatory glucose levels. Diabetes 69(2):131–145. https://doi.org/10.2337/db19-0379
Rakshit K, Qian J, Gaonkar KS, Dhawan S, Colwell CS, Matveyenko AV (2018) Postnatal ontogenesis of the islet circadian clock plays a contributory role in β-cell maturation process. Diabetes 67(5):911–922. https://doi.org/10.2337/db17-0850
Erion KA, Corkey BE (2017) Hyperinsulinemia: a cause of obesity? Curr Obes Rep 6(2):178–186. https://doi.org/10.1007/s13679-017-0261-z
Prentki M, Madiraju SR (2012) Glycerolipid/free fatty acid cycle and islet beta-cell function in health, obesity and diabetes. Mol Cell Endocrinol 353(1–2):88–100. https://doi.org/10.1016/j.mce.2011.11.004
O'Rahilly S, Turner RC, Matthews DR (1988) Impaired pulsatile secretion of insulin in relatives of patients with non-insulin-dependent diabetes. N Engl J Med 318(19):1225–1230. https://doi.org/10.1056/NEJM198805123181902
Szabat M, Page MM, Panzhinskiy E et al (2016) Reduced insulin production relieves endoplasmic reticulum stress and induces beta cell proliferation. Cell Metab 23(1):179–193. https://doi.org/10.1016/j.cmet.2015.10.016
Modi H, Johnson JD (2018) Folding mutations suppress early beta-cell proliferation. Elife 7:e43475. https://doi.org/10.7554/eLife.43475
Najjar SM, Perdomo G (2019) Hepatic insulin clearance: mechanism and physiology. Physiology (Bethesda) 34(3):198–215. https://doi.org/10.1152/physiol.00048.2018
Bergman RN, Piccinini F, Kabir M, Kolka CM, Ader M (2019) Hypothesis: role of reduced hepatic insulin clearance in the pathogenesis of type 2 diabetes. Diabetes 68(9):1709–1716. https://doi.org/10.2337/db19-0098
Mitrakou A, Kelley D, Mokan M et al (1992) Role of reduced suppression of glucose production and diminished early insulin release in impaired glucose tolerance. N Engl J Med 326(1):22–29. https://doi.org/10.1056/NEJM199201023260104
D'Souza AM, Johnson JD, Clee SM, Kieffer TJ (2016) Suppressing hyperinsulinemia prevents obesity but causes rapid onset of diabetes in leptin-deficient Lep(ob/ob) mice. Mol Metab 5(11):1103–1112. https://doi.org/10.1016/j.molmet.2016.09.007
Templeman NM, Mehran AE, Johnson JD (2016) Hyper-variability in circulating insulin, high fat feeding outcomes, and effects of reducing Ins2 dosage in male Ins1-null mice in a specific pathogen-free facility. PLoS One 11(4):e0153280. https://doi.org/10.1371/journal.pone.0153280
Zhang AMY, Magrill J, de Winter TJJ et al (2019) Endogenous hyperinsulinemia contributes to pancreatic cancer development. Cell Metab 30(3):403–404. https://doi.org/10.1016/j.cmet.2019.07.003
Skovsø S, Panzhinskiy E, Kolic J et al (2020) Beta-cell specific insulin resistance promotes glucose-stimulated insulin hypersecretion. bioRxiv: 2020.2010.2015.338160. https://doi.org/10.1101/2020.10.15.338160
Dionne DA, Skovso S, Templeman NM, Clee SM, Johnson JD (2016) Caloric restriction paradoxically increases adiposity in mice with genetically reduced insulin. Endocrinology 157(7):2724–2734. https://doi.org/10.1210/en.2016-1102
Page MM, Skovso S, Cen H et al (2018) Reducing insulin via conditional partial gene ablation in adults reverses diet-induced weight gain. FASEB J 32(3):1196–1206. https://doi.org/10.1096/fj.201700518R
Leroux L, Desbois P, Lamotte L et al (2001) Compensatory responses in mice carrying a null mutation for Ins1 or Ins2. Diabetes 50(Suppl 1):S150–S153. https://doi.org/10.2337/diabetes.50.2007.s150
Attane C, Peyot ML, Lussier R et al (2016) A beta cell ATGL-lipolysis/adipose tissue axis controls energy homeostasis and body weight via insulin secretion in mice. Diabetologia 59(12):2654–2663. https://doi.org/10.1007/s00125-016-4105-2
Carobbio S, Frigerio F, Rubi B et al (2009) Deletion of glutamate dehydrogenase in beta-cells abolishes part of the insulin secretory response not required for glucose homeostasis. J Biol Chem 284(2):921–929. https://doi.org/10.1074/jbc.M806295200
Pedersen DJ, Guilherme A, Danai LV et al (2015) A major role of insulin in promoting obesity-associated adipose tissue inflammation. Mol Metab 4(7):507–518. https://doi.org/10.1016/j.molmet.2015.04.003
Zhang AMY, Wellberg EA, Kopp JL, Johnson JD (2021) Hyperinsulinemia in obesity, inflammation, and cancer. Diabetes Metab J 45(3):285–311. https://doi.org/10.4093/dmj.2020.0250
Hamza SM, Sung MM, Gao F et al (2017) Chronic insulin infusion induces reversible glucose intolerance in lean rats yet ameliorates glucose intolerance in obese rats. Biochim Biophys Acta Gen Subj 1861(2):313–322. https://doi.org/10.1016/j.bbagen.2016.11.029
Marshall S, Olefsky JM (1980) Effects of insulin incubation on insulin binding, glucose transport, and insulin degradation by isolated rat adipocytes. Evidence for hormone-induced desensitization at the receptor and postreceptor level. J Clin Invest 66(4):763–772. https://doi.org/10.1172/JCI109914
Botezelli JD, Overby P, Lindo L et al (2020) Adipose depot-specific upregulation of Ucp1 or mitochondrial oxidative complex proteins are early consequences of genetic insulin reduction in mice. Am J Physiol Endocrinol Metab 319(3):E529–E539. https://doi.org/10.1152/ajpendo.00128.2020
Sarabia V, Ramlal T, Klip A (1990) Glucose uptake in human and animal muscle cells in culture. Biochem Cell Biol 68(2):536–542. https://doi.org/10.1139/o90-076
Jensen MD, Nielsen S (2007) Insulin dose response analysis of free fatty acid kinetics. Metabolism 56(1):68–76. https://doi.org/10.1016/j.metabol.2006.08.022
Kolb H, Kempf K, Rohling M, Martin S (2020) Insulin: too much of a good thing is bad. BMC Med 18(1):224. https://doi.org/10.1186/s12916-020-01688-6
Ludwig DS, Ebbeling CB, Bikman BT, Johnson JD (2020) Testing the carbohydrate-insulin model in mice: the importance of distinguishing primary hyperinsulinemia from insulin resistance and metabolic dysfunction. Mol Metab 35:100960–100960. https://doi.org/10.1016/j.molmet.2020.02.003
Hodish I (2018) Insulin therapy, weight gain and prognosis. Diabetes Obes Metab 20(9):2085–2092. https://doi.org/10.1111/dom.13367
Gregory JM, Smith TJ, Slaughter JC et al (2019) Iatrogenic hyperinsulinemia, not hyperglycemia, drives insulin resistance in type 1 diabetes as revealed by comparison with GCK-MODY (MODY2). Diabetes 68(8):1565–1576. https://doi.org/10.2337/db19-0324
Huang Z, Wang W, Huang L, Guo L, Chen C (2020) Suppression of insulin secretion in the treatment of obesity: a systematic review and meta-analysis. Obesity (Silver Spring) 28(11):2098–2106. https://doi.org/10.1002/oby.22955
Page MM, Johnson JD (2018) Mild suppression of hyperinsulinemia to treat obesity and insulin resistance. Trends Endocrinol Metab 29(6):389–399. https://doi.org/10.1016/j.tem.2018.03.018
Velasquez-Mieyer PA, Cowan PA, Arheart KL et al (2003) Suppression of insulin secretion is associated with weight loss and altered macronutrient intake and preference in a subset of obese adults. Int J Obes Relat Metab Disord 27(2):219–226. https://doi.org/10.1038/sj.ijo.802227
Cen H, Botezelli JD, Johnson JD (2021) Modulation of Insr and insulin receptor signaling by hyperinsulinemia in vitro and in vivo. Biorxiv. https://doi.org/10.1101/556571
Nolan CJ, Ruderman NB, Kahn SE, Pedersen O, Prentki M (2015) Insulin resistance as a physiological defense against metabolic stress: implications for the management of subsets of type 2 diabetes. Diabetes 64(3):673–686. https://doi.org/10.2337/db14-0694
Nolan CJ, Ruderman NB, Prentki M (2013) Intensive insulin for type 2 diabetes: the risk of causing harm. Lancet Diabetes Endocrinol 1(1):9–10. https://doi.org/10.1016/S2213-8587(13)70027-5
Kowalski GM, Bruce CR (2014) The regulation of glucose metabolism: implications and considerations for the assessment of glucose homeostasis in rodents. Am J Physiol Endocrinol Metab 307(10):E859–E871. https://doi.org/10.1152/ajpendo.00165.2014
Chawla S, Pund A, Vibishan B, Kulkarni S, Diwekar-Joshi M, Watve M (2018) Inferring causal pathways among three or more variables from steady-state correlations in a homeostatic system. PLoS One 13(10):e0204755. https://doi.org/10.1371/journal.pone.0204755
Stamateris RE, Sharma RB, Kong Y et al (2016) Glucose induces mouse beta-cell proliferation via IRS2, MTOR, and cyclin D2 but not the insulin receptor. Diabetes 65(4):981–995. https://doi.org/10.2337/db15-0529
Taylor SI (1992) Lilly lecture: molecular mechanisms of insulin resistance. Lessons from patients with mutations in the insulin-receptor gene. Diabetes 41(11):1473–1490. https://doi.org/10.2337/diab.41.11.1473
Semple RK, Savage DB, Cochran EK, Gorden P, O'Rahilly S (2011) Genetic syndromes of severe insulin resistance. Endocr Rev 32(4):498–514. https://doi.org/10.1210/er.2010-0020
Joshi RL, Lamothe B, Cordonnier N et al (1996) Targeted disruption of the insulin receptor gene in the mouse results in neonatal lethality. EMBO J 15(7):1542–1547. https://doi.org/10.1002/j.1460-2075.1996.tb00498.x
Accili D, Drago J, Lee EJ et al (1996) Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat Genet 12(1):106–109. https://doi.org/10.1038/ng0196-106
Vikram A, Jena G (2010) S961, an insulin receptor antagonist causes hyperinsulinemia, insulin-resistance and depletion of energy stores in rats. Biochem Biophys Res Commun 398(2):260–265. https://doi.org/10.1016/j.bbrc.2010.06.070
Mosser RE, Maulis MF, Moullé VS et al (2015) High-fat diet-induced β-cell proliferation occurs prior to insulin resistance in C57Bl/6J male mice. Am J Physiol Endocrinol Metab 308(7):E573–E582. https://doi.org/10.1152/ajpendo.00460.2014
Skovso S (2014) Modeling type 2 diabetes in rats using high fat diet and streptozotocin. J Diabetes Investig 5(4):349–358. https://doi.org/10.1111/jdi.12235
Bruning JC, Michael MD, Winnay JN et al (1998) A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 2(5):559–569. https://doi.org/10.1016/s1097-2765(00)80155-0
Bluher M, Kahn BB, Kahn CR (2003) Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 299(5606):572–574. https://doi.org/10.1126/science.1078223
Evans-Molina C, Robbins RD, Kono T et al (2009) Peroxisome proliferator-activated receptor gamma activation restores islet function in diabetic mice through reduction of endoplasmic reticulum stress and maintenance of euchromatin structure. Mol Cell Biol 29(8):2053–2067. https://doi.org/10.1128/MCB.01179-08
Cen J, Sargsyan E, Forslund A, Bergsten P (2018) Mechanisms of beneficial effects of metformin on fatty acid-treated human islets. J Mol Endocrinol 61(3):91–99. https://doi.org/10.1530/JME-17-0304
Zhang F, Sjoholm A, Zhang Q (2006) Pioglitazone acutely influences glucose-sensitive insulin secretion in normal and diabetic human islets. Biochem Biophys Res Commun 351(3):750–755. https://doi.org/10.1016/j.bbrc.2006.10.103
Drucker DJ (2018) Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab 27(4):740–756. https://doi.org/10.1016/j.cmet.2018.03.001
Hoffman DJ, Powell TL, Barrett ES, Hardy DB (2020) Developmental origins of metabolic disease. Physiol Rev. https://doi.org/10.1152/physrev.00002.2020
Krentz NAJ, Gloyn AL (2020) Insights into pancreatic islet cell dysfunction from type 2 diabetes mellitus genetics. Nat Rev Endocrinol 16(4):202–212. https://doi.org/10.1038/s41574-020-0325-0
Astley CM, Todd JN, Salem RM et al (2018) Genetic evidence that carbohydrate-stimulated insulin secretion leads to obesity. Clin Chem 64(1):192–200. https://doi.org/10.1373/clinchem.2017.280727
Zhao JV, Luo S, Schooling CM (2019) Sex-specific Mendelian randomization study of genetically predicted insulin and cardiovascular events in the UK Biobank. Commun Biol 2:332. https://doi.org/10.1038/s42003-019-0579-z
Xin Y, Davies A, Briggs A et al (2020) Type 2 diabetes remission: 2 year within-trial and lifetime-horizon cost-effectiveness of the Diabetes Remission Clinical Trial (DiRECT)/Counterweight-Plus weight management programme. Diabetologia 63(10):2112–2122. https://doi.org/10.1007/s00125-020-05224-2
Hanipah ZN, Schauer PR (2020) Bariatric surgery as a long-term treatment for type 2 diabetes/metabolic syndrome. Annu Rev Med 71:1–15. https://doi.org/10.1146/annurev-med-053117-123246
Hallberg SJ, Gershuni VM, Hazbun TL, Athinarayanan SJ (2019) Reversing type 2 diabetes: a narrative review of the evidence. Nutrients 11(4):766. https://doi.org/10.3390/nu11040766
Acknowledgements
I thank B. Corkey (Boston University, USA), M. Prenki (University of Montreal, Canada) and C. Doucette (University of Manitoba, Canada) for critical reading of this manuscript. I thank S. Kahn (University of Washington) for the informative (and informal) e-mail communication on this topic.
Authors’ relationships and activities
The author declares that there are no relationships or activities that might bias, or be perceived to bias, this work.
Funding
Work in the author’s laboratory on the effects of hyperinsulinaemia on metabolism and pancreatic cancer is supported by operating grants from CIHR (P-168857 and P-168854) and a Diabetes Investigator Grant from Diabetes Canada.
Author information
Authors and Affiliations
Contributions
JDJ is the sole author of this article and is the final guarantor of the work.
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Johnson, J.D. On the causal relationships between hyperinsulinaemia, insulin resistance, obesity and dysglycaemia in type 2 diabetes. Diabetologia 64, 2138–2146 (2021). https://doi.org/10.1007/s00125-021-05505-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-021-05505-4