
Abstract
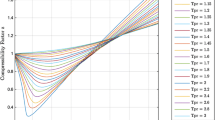
Several computer models of quartz were developed and tested. A simple model based on a potential energy function, derived in large part from quantum mechanical calculations on the molecule H6Si2O7, was found to reproduce the compressibility curve for quartz up to pressures of 8 GPa. The potential includes quadratic expressions for the SiO bond lengths, the OSiO angles and a parameter spanning the SiOSi angle together with an exponential OO repulsion term for non co-dimer O atoms. The variations in the cell edges and in the SiOSi angle, as a function of pressure, parallel observed trends when the bond lengths and angles calculated for the molecule are used as rgressor values. Poisson ratios calculated using the model match those observed. Two configurations for quartz related by the Dauphiné twin law are generated as minimum energy structures of the model with about equal frequencies as observed in nature. It is shown that the model, devised for quartz, can also be applied to the silica polymorph cristobalite, giving reasonable estimates of its compressibility curve, structural parameters and its negative Poisson ratio. When the observed bond lengths and angles are used as regressor values, the model generates the absolute coordinates of the atoms and the cell dimensions for quartz to within 0.005 Å and those of cristobalite to within 0.001 Å, on average, both at zero pressure. When applied to coesite, the model yields a zero pressure structure that is close to that observed but which is significantly softer than observed. The resulting SiO bond lengths are linearly correlated with f s (O), as observed for coesite, despite the use of a single bond length and a single SiOSi angle as regressor values in the calculation. When the structures are optimized assuming P1 space group symmetry and triclinic cell dimensions, the resulting frameworks of silicate tetrahedra exhibit the translational, rotational and reflection symmetries observed for quartz, cristobalite and coesite. The fact that the resulting frameworks exhibit observed space group symmetries is evidence that the symmetry adopted by the silica polymorphs can be explained by short ranged forces.
Similar content being viewed by others

References
Akimoto S-I (1972) The system MgO-FeO-SiO2 at high pressures and temperatures-Phase equilibria and elastic properties. Tectonophysics 13:161–187
Bassett WA, Barnett JD (1970) Isothermal compression of stishovite and coesite up to 85 kilobars at room temperature by X-ray diffraction. Phys Earth Planet Inter 3:54–60
Birch F (1938) The effect of pressure upon the elastic parameters of isotropic solids, according to Murnaghan's theory of finite strain. J Appl Phys 9:279–288
Birch F (1952) Elasticity and constitution of the earth's interior. J Geophys Res 57:227–286
Boisen MB Jr, Gibbs GV, Downs RT, D'Arco P (1990) The dependence of the SiO bond length on structural parameters in coesite, the silica polymorphs and the clathrasils. Am Mineral 75:748–754
Busing WR (1981) WMIN, A computer program to model molecules and crystals in terms of potential energy functions. Oak Ridge Natl Lab, Pub 5747
Catlow CRA, Price GD (1990) Computer modeling of solid-state inorganic materials. Nature 347:243–248
Chelikowsky JR, King HE Jr, Troullier N, Martins JL, Glinnemann J (1990) Structural properties of α-quartz near the amorphous transition. Phys Rev Let 65:3309–3312
D'Amour H, Denner W, Schulz H (1979) Structure determination of α-quartz up to 68 × 108 Pa. Acta Crystallogr B35:550–556
Dennis JE Jr, Schnabel RB (1983) Numerical Methods for Unconstrained Optimization and Nonlinear Equations. Prentice-Hall, Englewood Cliffs, pp 378
Downs RT (1992) Librational displacements of silicate tetrahedra in response to temperature and pressure. PhD Dissertation. Virginia Polytechnic Institute and State University, Blacksburg, Virginia, pp 76
Feynman RP, Leighton RB, Sands M (1963) The Feynman lectures on physics, Vol 1. Addison-Wesley, Reading Mass, pp 52–12
Frondel C (1982) The system of mineralogy, Vol. III, silica minerals. Wiley, New York, pp 329
Gibbs GV (1982) Molecules as models for bonding in silicates. Am Mineral 67:421–450
Gibbs GV, Boisen MB Jr, Downs RT, Lasaga AC (1988) Mathematical modeling of the structures and bulk moduli of TX 2 quartz and cristobalite structure-types, T = C, Si, Ge and X = O, S. Mat Res Soc Symp Proc 121:155–166
Glinnemann J, King HE Jr, Schulz H, Hahn Th, LaPlaca SJ, Dacol F (1992) Crystal structures of the low-temperature quartz-type phases of SiO2 and GeO2 at elevated pressure. Z Kristallogr 198:177–212
Hazen RM, Finger LW (1982) Comparative Crystal Chemistry. Wiley, New York, pp 231
Hazen RM, Finger LW, Hemley RJ, Mao HK (1989) High-pressure crystal chemistry and amorphization of α-quartz. Solid State Commun 72:507–511
Jorgensen JD (1978) Compression mechanisms in α-quartz structures — SiO2 and GeO2. J Appl Phys 49:5473–5478
Keskar NR, Chelikowsky JR (1992) Negative Poisson ratios in crystalline SiO2 from first-principles calculations. Nature 358:222–224
Lager GA, Jorgensen JD, Rotella FJ (1982) Crystal structure and thermal expansion of α-quartz SiO2 at low temperatures. J Appl Phys 53:6751–6756
Lasaga AC, Gibbs GV (1987) Application of quantum mechanical potential surfaces to mineral physics calculations. Phys Chem Minerals 14:107–117
Lasaga AC, Gibbs GV (1988) Quantum mechanical potential surfaces and calculations on minerals and molecular clusters I STO-3G and 6–31G* results. Phys Chem Minerals 16:29–41
Lasage AC, Gibbs GV (1991) Quantum mechanical Hartree-Fock surfaces and calculations on minerals II. 6–31G* results. Phys Chem Minerals 17:485–491
Lazarev AN, Mirgorodsky AP (1991) Molecular force constants in dynamical model of α-quartz a calculation of the phonon spectrum, elastic and piezoelectric properties. Phys Chem Minerals 16:29–41
Levien L, Prewitt CT, Weidner DJ (1980) Structure and elastic properties of quartz at pressure. Am Mineral 65:920–930
Levien L, Prewitt CT (1981) High-pressure crystal structure and compressibility of coesite. Am Mineral 66:324–333
Liebau F, Böhm H (1982) On the co-existence of structurally different regions in the low-high-quartz and other displacive phase transformations. Acta Crystallogr A38:252–256
McSkimin HJ, Andreatch P Jr, Thurston RN (1965) Elastic moduli of quartz versus hydrostatic pressure at 25° and -195.8° C. J Appl Phys 36:1624–1632
Newton MD, Gibbs GV (1980) Ab initio calculated geometries and charge distributions for H4SiO4 and H6Si2O7 compared with experimental values for silicates and siloxanes. Phys Chem Minerals 6:221–246
O'Keeffe M, Newton MD, Gibbs GV (1980) Ab initio calculations of interactive force constants in H6Si2O7 and the bulk modulus of α quartz and α cristobalite. Phys Chem Minerals 6:305–312
Palmer DC, Downs RT (1991) High pressure behavior of cristobalite revealed by single crystal X-ray diffraction. EOS Transactions of the American Geophysical Union, 72(44):478
Sanders MJ, Leslie M, Catlow CRA (1984) Interatomic potentials for SiO2. J Chem Soc 19:1271–1273
Stixrude L, Bukowinski MST (1988) Simple covalent potential models of tetrahedral SiO2: Applications to α-quartz and coesite at pressure. Phys Chem Minerals 16:199–206
Tsuneyuki S, Tsukada M, Aoki H, Matsui Y (1988) First-principles interatomic potential of silica applied to molecular dynamics. Phys Rev Lett 61:869–872
Tsuneyuki S, Aoki H, Tsukada M, Matsui Y (1990) Molecular-dynamics study of the α to β structural phase transition of quartz. Phys Rev Lett 64:776–779
vanBeest BWH, Kramer GJ, Santen RA van (1990) Force fields for silicas and aluminophosphates based on ab initio calculations. Phys Rev Lett 64:1955–1958
Weidner DJ, Carleton HR (1977) Elasticity of coesite. Z Kristallogr 111:129–141
Yeganeh-Haeri A, Weidner DJ, Parise JB, Ko J, Vaughan MT, Liu X, Zhao Y, Wang Y, Pacalo R (1990) A New Polymorph of SiO2. EOS Trans. Am Geophys. Un 71:1671
Yeganeh-Haeri A, Weidner DJ, Parise JB (1992) Elasticity of α-cristobalite: A silicon dioxide with negative Poisson's ratio. Science 257:650–652
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Boisen, M.B., Gibbs, G.V. A modeling of the structure and compressibility of quartz with a molecular potential and its transferability to cristobalite and coesite. Phys Chem Minerals 20, 123–135 (1993). https://doi.org/10.1007/BF00207206
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF00207206