
Abstract
High-density lipoprotein (HDL) is considered to be an anti-atherogenic lipoprotein moiety. Generation of genetically modified (total body and tissue-specific knockout) mouse models has significantly contributed to our understanding of HDL function. Here we will review data from knockout mouse studies on the importance of HDL’s major alipoprotein apoA-I, the ABC transporters A1 and G1, lecithin:cholesterol acyltransferase, phospholipid transfer protein, and scavenger receptor BI for HDL’s metabolism and its protection against atherosclerosis in mice. The initial generation and maturation of HDL particles as well as the selective delivery of its cholesterol to the liver are essential parameters in the life cycle of HDL. Detrimental atherosclerosis effects observed in response to HDL deficiency in mice cannot be solely attributed to the low HDL levels per se, as the low HDL levels are in most models paralleled by changes in non-HDL-cholesterol levels. However, the cholesterol efflux function of HDL is of critical importance to overcome foam cell formation and the development of atherosclerotic lesions in mice. Although HDL is predominantly studied for its atheroprotective action, the mouse data also suggest an essential role for HDL as cholesterol donor for steroidogenic tissues, including the adrenals and ovaries. Furthermore, it appears that a relevant interaction exists between HDL-mediated cellular cholesterol efflux and the susceptibility to inflammation, which (1) provides strong support for the novel concept that inflammation and metabolism are intertwining biological processes and (2) identifies the efflux function of HDL as putative therapeutic target also in other inflammatory diseases than atherosclerosis.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Knockout mice
- High-density lipoprotein
- Reverse cholesterol transport
- Atherosclerosis
- Steroidogenesis
- Inflammation
- ABC transporters
- Scavenger receptor BI
- Apolipoprotein
- Lecithin-cholesterol acyltransferase
- Phospholipid transfer protein
1 Introduction
High levels of low-density lipoprotein (LDL) cholesterol are associated with an increased risk for cardiovascular disease. In contrast, plasma levels of cholesterol associated with high-density lipoprotein (HDL) are inversely correlated with the risk of cardiovascular disease (Gordon et al. 1977). Since standard treatment with statins to lower plasma levels of LDL cholesterol only reduces the risk of cardiovascular disease by ~30 % (Cholesterol Treatment Trialists’ (CTT) Collaboration et al. 2010; Cholesterol Treatment Trialists’ (CTT) Collaborators et al. 2012), raising plasma levels of HDL cholesterol has been considered a potential additional therapeutic strategy to overcome disease.
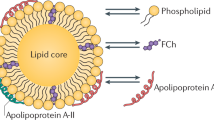
The term HDL refers to a class of alpha-migrating protein/lipid complexes that differ in size (5–12 nm), shape, and lipidation pattern. In order to be able to effectively modulate plasma HDL-cholesterol levels, it is of critical importance to understand the anti-atherogenic potential and metabolism of the different HDL particles. Due to the large biological/genetic variation and lifestyle of people, it is difficult to gain clear insight into the relation between the flux of cholesterol through the HDL pathway and the development of atherosclerosis—the primary underlying cause of cardiovascular disease—in the general human population. Small animal models, in particular mice, have been proven valuable tools to increase the understanding of the complexity of HDL metabolism and the consequences of interfering in specific pathways or atherosclerosis susceptibility. Here we will review data from several genetically modified (total body and tissue-specific knockout) mouse models to (1) show the importance of specific gene products in the life cycle of HDL and their contribution to HDL’s primary function, reverse cholesterol transport, i.e., the flux of cholesterol from peripheral cells back to the liver for subsequent excretion, and (2) highlight the potential of modulation of HDL metabolism as an approach to lower atherosclerotic disease burden.
2 Apolipoprotein A-I
Apolipoprotein A-I (apoA-I) is produced by the liver (Zannis et al. 1983) and intestine (Gordon et al. 1982) and represents the primary apolipoprotein constituent of HDL particles (Scanu et al. 1969). Several mutations in the apoA-I gene have been causally linked to HDL deficiency in humans (Tilly-Kiesi et al. 1995; Leren et al. 1997; Matsunaga et al. 1999; Hovingh et al. 2004; Dastani et al. 2006; Wada et al. 2009; Berge and Leren 2010; Lee et al. 2013).
The groups of Nobuyo Maeda (Williamson et al. 1992) and Jan Breslow (Plump et al. 1997) both inactivated the apoA-I gene in mice using dedicated homologous recombination strategies, leading to the absence of detectable amounts of apoA-I protein in plasma. In accordance with an important function for apoA-I in the formation and stability of HDL particles, genetic disruption of apoA-I in mice is associated with an 83 % decrease in the level of HDL cholesterol, contributing to a 68 % decrease in plasma total cholesterol levels (Williamson et al. 1992). The limited amount of HDL cholesterol present in apoA-I knockout mice is carried by particles that are enriched in triglycerides (Plump et al. 1997). However, HDL particle size and shape are similar to that of wild-type controls. ApoA-I-deficient HDL particles display increased levels of other apolipoprotein subspecies, including apolipoprotein A-II (apoA-II), apolipoprotein E (apoE), and apolipoprotein A-IV (apoA-IV) (Li et al. 1993; Plump et al. 1997; Moore et al. 2003). Homozygous apoA-I deficiency in mice not only lowers plasma levels of HDL cholesterol, but also decreases levels of cholesterol associated with apoB-containing very-low-density lipoproteins (VLDL) (Plump et al. 1997). The cholesterol ester to unesterified cholesterol ratio is lower in plasma of apoA-I-deficient mice, which can be attributed to a reduced endogenous cholesterol esterification rate (Parks et al. 1995). Furthermore, the cholesterol ester fatty acid composition of plasma is markedly different between the different genotypes, with apoA-I knockout mice displaying a higher degree of fatty acid saturation as compared to wild-type mice (Parks et al. 1995). ApoA-I, in addition to its structural role, thus also serves as an essential activator of the enzyme lecithin-cholesterol acyltransferase (LCAT) that esterifies cholesterol.
ApoA-I deficiency in mice does, in general, not affect tissue cholesterol levels (Plump et al. 1997). In the liver, decreased delivery of cholesterol esters to hepatocytes is compensated by a concomitant decrease in bile acid formation as judged from a decrease in hepatic mRNA expression levels of cholesterol 7alpha-hydroxylase (CYP7A1) in apoA-I knockout mice (Plump et al. 1997). In contrast, the adrenals are not able to cope with the diminished plasma cholesterol flux due to the lack of apoA-I. Cortical cells located in the zona glomerulosa and zona fasciculata from the adrenals of wild-type mice are filled with large lipid droplets containing cholesterol esters, while adrenals from apoA-I-deficient mice completely lack cholesterol esters and display a parallel decrease in microvillar channel width (Plump et al. 1996). Importantly, apoA-I-deficient mice show a diminished adrenal glucocorticoid output in response to steroidogenic triggers (Plump et al. 1996), suggesting that a lack of apoA-I-containing HDL particles impairs normal adrenal function in mice. Although the cholesterol ester content of the testis is similar between apoA-I knockout and wild-type mice, interstitial and theca cells within the ovaries of apoA-I knockout mice are similarly deprived of cholesterol esters (Plump et al. 1996). Currently no evidence is present for an altered female fertility, despite the clear ovarian cholesterol depletion phenotype. Combined, these findings indicate that apoA-I-containing HDL particles act as important cholesterol donors for several, but not all, steroidogenic tissues.
Human apoA-I mutation carriers may develop premature atherosclerosis (Hovingh et al. 2004; Dastani et al. 2006). The formation of atherosclerotic lesions in normolipidemic mice can be induced by feeding them a cholic acid-containing diet enriched in cholesterol and fat that increases levels of pro-atherogenic apoB-containing lipoproteins (Paigen et al. 1987; Ishida et al. 1991). Strikingly, deletion of apoA-I in wild-type mice does not alter the incidence or extent of atherogenic diet-induced lesion formation (Li et al. 1993). This may be explained by the fact that, under cholic acid-containing diet feeding conditions, levels of anti-atherogenic HDL as well as levels of pro-atherogenic VLDL/LDL are lower in apoA-I knockout mice as compared to wild-type controls (Li et al. 1993). To further determine the impact of apoA-I deficiency on atherosclerosis susceptibility, apoA-I knockout mice have been crossbred with genetically hyperlipidemic low-density lipoprotein receptor (LDLr) knockout mice that spontaneously develop atherosclerotic lesions on a chow diet, albeit at a slow rate (Ishibashi et al. 1994). Chow diet-fed apoA-I x LDLr double knockout (DKO) mice as compared to LDLr single knockout controls exhibit an increase in the plasma non-HDL-cholesterol to HDL-cholesterol ratio. This can be attributed to a marked increase in VLDL-cholesterol levels and a decrease in plasma HDL-cholesterol levels (Moore et al. 2003; Zabalawi et al. 2003). In accordance with the more atherogenic lipoprotein profile (higher VLDL cholesterol and lower HDL cholesterol), atherosclerotic lesion burden is increased in chow diet-fed apoA-I x LDLr DKO mice, as measured by en face analysis of the aorta, irrespective of the age of the mice (Moore et al. 2003). Feeding a Western-type high fat/high cholesterol diet induces a rapid progression of atherosclerotic lesion development in LDLr knockout mice (Ishibashi et al. 1994). In great contrast with the chow diet findings, the extent of atherosclerotic lesion formation was not significantly different between LDLr KO and apoA-I x LDLr DKO mice upon feeding an atherogenic diet containing 10 % saturated fat from palm oil and 0.1 % cholesterol for 16 weeks (Zabalawi et al. 2003). However, it should be noted that apoA-I x LDLr DKO mice show a much lower diet-induced increase in VLDL-cholesterol levels. Identical aortic cholesterol contents are thus found in the context of a ~70 % lower plasma total cholesterol level (Zabalawi et al. 2003). It therefore appears that ablation of apoA-I function and the concomitant decrease in plasma HDL-cholesterol levels do predispose for atherosclerotic lesion development in mice, but only after correcting for apoA-I genotype-associated changes in VLDL-cholesterol levels.
ApoA-I x LDLr DKO mice develop severe (fatal) skin lesions containing lipid-filled macrophages upon feeding the high cholesterol/high fat diet (Zabalawi et al. 2003). In addition, B-cells, T-cells, and dendritic cells within lymph nodes of apoA-I x LDLr DKO mice are enriched in cholesterol esters (Wilhelm et al. 2009). This is associated with an autoimmune phenotype characterized by relatively high autoantibody titers in plasma upon feeding an atherogenic high fat/high cholesterol diet (Wilhelm et al. 2009). ApoA-I-containing HDL particles thus also play an essential role in maintaining cholesterol homeostasis in the skin as well as in cells from the lymphoid system.
3 ATP-Binding Cassette Transporter A1
The cholesterol used to transform pre-beta1HDL into discoidal alpha-migrating HDL particles can be supplied through passive diffusion driven by a concentration gradient. However, it has become clear that energy-dependent flux of cholesterol and phospholipid across the membrane facilitated by members of the ATP-binding cassette (ABC) family of transporters contributes significantly to the lipidation of apoA-I.
Human carriers of a mutation in the full size ABC transporter ABCA1 gene suffer from familial HDL deficiency (Brooks-Wilson et al. 1999; Bodzioch et al. 1999; Hong et al. 2002). In accordance with a prominent role for ABCA1 in the control of HDL biogenesis, total body ABCA1-deficient mice are characterized by an almost complete lack of HDL cholesterol (>90 % decrease) in the context of similar levels of triglyceride-rich lipoproteins (Christiansen-Weber et al. 2000; Orsó et al. 2000; Calpe-Berdiel et al. 2005; Brunham et al. 2006). Mature alpha-migrating HDL particles are virtually absent in ABCA1 knockout mice. The remaining HDL cholesterol is carried by pre-beta HDL particles that are relatively enriched in triglycerides and exhibit an altered phospholipid species distribution (Orsó et al. 2000; Francone et al. 2003). ABCA1 knockout mice display a diminished LCAT activity (Francone et al. 2003), which highlights that lipidation of apoA-I via ABCA1 is required for LCAT activity and maturation of HDL.
Adrenocortical cells (Orsó et al. 2000) and testical Sertoli cells (Christiansen-Weber et al. 2000) of mice lacking ABCA1 show distinct accumulation of cholesterol esters. This can possibly be attributed to the observed increase in the activity of the cholesterol synthesis gene HMG-CoA reductase (Drobnik et al. 2001). However, ABCA1 may also act as a local regulator of steroidogenic tissue cholesterol homeostasis. In further support of a crucial role of ABCA1 in the control of normal steroidogenesis and development, pregnant ABCA1-deficient females exhibit lower plasma progesterone and estrogen levels (Christiansen-Weber et al. 2000). Furthermore, placentas of ABCA1 knockout mice often show malformations, which translates into a diminished birth and survival rate of ABCA1 knockout pups (Christiansen-Weber et al. 2000).
Livers and intestines of ABCA1 knockout mice are depleted of cholesterol esters (Orsó et al. 2000). Within the liver of wild-type mice, relatively high mRNA and protein expression levels of ABCA1 are found in hepatocytes and tissue macrophages (Kupffer cells) (Lawn et al. 2001; Hoekstra et al. 2003), while in the intestine ABCA1 expression seems to be mostly restricted to macrophages in the lamina propria of villi (Lawn et al. 2001). Dedicated gene targeting strategies have been employed to delineate the contribution of the different cell compartments to plasma HDL-cholesterol levels and total body cholesterol homeostasis. Conditional disruption of ABCA1 function specifically in parenchymal liver cells with the use of the Cre/Flox gene targeting system impairs the apoA-I-mediated efflux of cholesterol and phospholipid from hepatocytes without altering the efflux of cholesterol from macrophages to apoA-I (Timmins et al. 2005). Plasma HDL-cholesterol levels are ~80 % lower as a result of liver-specific ABCA1 deficiency (Timmins et al. 2005). Liver-specific deletion of ABCA1 is also associated with hypercatabolism of apoA-I by the kidneys resulting in an increased turnover of HDL particles and a marked decrease in steady-state plasma apoA-I levels (Timmins et al. 2005; Singaraja et al. 2006). Although no data on the effect of general ABCA1 deficiency in mice on apoA-I flux have been described, it can be assumed total body ABCA1 knockout mice similarly show an increased catabolism and clearance of apoA-I by the kidneys. ABCA1 deficiency in hepatocytes is associated with a rise in plasma levels and size of triglyceride-rich VLDL particles, which can be attributed to an increase in hepatic VLDL secretion and a parallel decrease in lipoprotein lipase (LPL)-mediated catabolism of triglycerides (Sahoo et al. 2004; Chung et al. 2010). ABCA1-mediated transport of cholesterol from hepatocytes to the plasma compartment thus not only controls HDL biogenesis, but also impacts on the synthesis and catabolism of non-HDL particles. Deletion of ABCA1 specifically in intestinal epithelial cells is associated with an accumulation of cholesterol esters in the intestine, a lower plasma apoA-I concentration, and a ~30 % decrease in plasma HDL-cholesterol levels (Brunham et al. 2006). In contrast to total body and hepatocyte-specific knockout mice, intestinal-specific ABCA1 knockout mice do not show major changes in non-HDL-cholesterol and plasma triglyceride levels (Brunham et al. 2006), which further highlights the role of hepatocyte ABCA1 in the control of plasma triglyceride levels. Importantly, mice genetically lacking ABCA1 in both the liver and intestine show a greater decrease in plasma HDL-cholesterol levels as compared to their single tissue knockout controls, almost reaching similarly low levels as those found in total body ABCA1 knockout mice (Brunham et al. 2006). Hepatocytes and the intestinal epithelium can thus be considered the primary cellular sources of the cholesterol that is used for lipidation of apoA-I and generation of HDL.
Loss-of-function mutations in the ABCA1 gene could not be associated with increased coronary heart disease in the general population (Frikke-Schmidt et al. 2008a). However, several studies have suggested that premature atherosclerosis and cardiovascular disease are a common finding in human carriers of functional mutations in the ABCA1 gene (Huang et al. 2001; Hong et al. 2002; Frikke-Schmidt et al. 2008b). In contrast, total body ABCA1 deficiency does not alter the susceptibility to atherosclerosis in hyperlipidemic apoE knockout and LDL receptor (LDLr) knockout mice fed either regular chow or an atherogenic high fat/high cholesterol diet (Aiello et al. 2002). However, deletion of ABCA1 function in the context of genetic hyperlipidemia is not only associated with HDL deficiency but also with a significant decrease in the plasma concentration of cholesterol carried by pro-atherogenic VLDL/LDL (Aiello et al. 2002). As is also the case for apoA-I, normal ABCA1 function in mice thus only protects against atherosclerosis after correcting for ABCA1 genotype-associated confounding differences in atherogenic lipoprotein levels. The atheroprotective effect of ABCA1 may be attributed to its general impact on plasma HDL-cholesterol levels or specifically due to its potential role in the efflux of cholesterol from macrophages. Lipid-filled foam cells can be found in all macrophage-rich tissues of hyperlipidemic total body ABCA1 knockout mice (Aiello et al. 2002), which already hints to an important in vivo role for ABCA1 in macrophage cholesterol efflux. The capacity of ABCA1 knockout mice to facilitate reverse cholesterol transport, the transport from peripheral cells—i.e., macrophages—back to the liver for subsequent excretion, has been validated by measuring the recovery in the plasma compartment and feces of radiolabeled cholesterol from intraperitoneal administered macrophages. In line with the hypothesis that ABCA1-mediated cholesterol efflux is an important determinant of reverse cholesterol transport, a lower amount of radiolabeled cholesterol is excreted under the condition of both total body and macrophage-specific ABCA1 deficiency (Calpe-Berdiel et al. 2005; Wang et al. 2007).
Aiello et al. (2002) and Van Eck et al. (2002) utilized bone marrow transplantation to explore the specific effect of ABCA1-dependent macrophage cholesterol efflux on atherosclerosis susceptibility. In accordance with a negligible impact of macrophage ABCA1 on total HDL biogenesis, bone marrow ABCA1 deficiency does not alter plasma HDL-cholesterol levels (Haghpassand et al. 2001; Aiello et al. 2002; Van Eck et al. 2002). Atherosclerotic lesion development in apoE knockout mice is markedly enhanced—at multiple sites—in response to disruption of ABCA1 expression in bone marrow (Aiello et al. 2002). Transplantation of ABCA1 knockout bone marrow into lethally irradiated LDLr knockout mice similarly stimulates atherosclerotic lesion development (Van Eck et al. 2002). ABCA1-mediated efflux of cholesterol from leukocytes is thus an important protective mechanism to inhibit macrophage foam cell formation and atherosclerosis in mice. Remarkably, macrophage-specific ABCA1 deficiency does not affect the extent of atherosclerosis in LDLr knockout mice (Brunham et al. 2009). It can therefore be concluded that ABCA1’s function in macrophages does not confer protection against atherosclerosis, but that rather an efficient efflux of cholesterol from lymphocytes or other immune cells is essential to overcome disease.
Relative HDL deficiency in liver-specific ABCA1 knockout mice on a hyperlipidemic LDLr knockout background is not associated with any enhanced susceptibility for the initial development of macrophage-rich atherosclerotic lesions, while the development of more advanced (collagen-containing) plaques is inhibited (Bi et al. 2013). In accordance with a prominent role for macrophage ABCA1 in reverse cholesterol transport, no change is observed in the flux of cholesterol from macrophages to the feces in LDLr knockout mice lacking ABCA1 function only in hepatocytes (Bi et al. 2013). Strikingly, apoE knockout mice that contain hepatocyte-specific ABCA1 deficiency do show an increased susceptibility to atherosclerosis, despite lowered plasma VLDL/LDL-cholesterol levels (Brunham et al. 2009). A critical review of these data has suggested that the discrepancy in the atherosclerosis findings from liver-specific ABCA1 knockout mice may be attributed to differences in inflammatory status of the two hyperlipidemic mouse models (Van Eck and Van Berkel 2013), which makes proper interpretation of the findings difficult. However, due to the inconsistency of the aforementioned results, it seems clear that the decrease in plasma HDL-cholesterol levels due to deletion of ABCA1 function should not be considered the driving force for the increase in atherosclerosis in total body ABCA1 knockout mice.
A consistent decrease in plasma VLDL-cholesterol levels is observed in LDLr knockout mice that have been transplanted with ABCA1-deficient bone marrow upon feeding an atherogenic Western-type diet (Van Eck et al. 2002; Lammers et al. 2011, 2012). This finding suggests that modulation of the leukocyte cholesterol efflux rate can directly impact on the metabolism of VLDL particles in vivo. However, to date, the mechanism underlying this effect remains to be resolved. It is becoming more and more evident that immunology and metabolism are not just two separate disciplines, but that they intervene and impact each other at multiple levels (Mathis and Shoelson 2011). ABCA1-deficient macrophages display a higher sensitivity to the bacterial membrane component lipopolysaccharide (LPS) (Zhu et al. 2008; Yvan-Charvet et al. 2008) and myeloid cell-specific ABCA1 knockout mice are protected against bacterial infection (Zhu et al. 2012). Furthermore, ABCA1 deficiency in bone marrow is associated with an increase in blood leukocyte counts (Van Eck et al. 2002). Given the apparent link between ABCA1 and immune function in vivo, future research should be aimed at uncovering to what extent ABCA1 contributes to the interplay between inflammation and cholesterol metabolism.
4 ATP-Binding Cassette Transporter G1
ABCG1, formerly known as the ABC8/white gene, belongs to the family of ABC half transporters that need to dimerize to execute their function. ABCG1 expression is mostly restricted to macrophage-rich tissues, including liver, spleen, and lung (Klucken et al. 2000), where it acts as an intracellular sterol transporter (Tarling and Edwards 2011). ABCG1 mRNA expression can actually be used as a measure for the hepatic macrophage content in rodent livers, despite detectable levels of ABCG1 expression in hepatocytes (Hoekstra et al. 2003; Ye et al. 2008; Li et al. 2012). The capacity to efflux cholesterol to HDL is diminished in ABCG1-deficient macrophages, while the apoA-I-mediated cholesterol efflux rate is similar as compared to wild-type macrophages (Out et al. 2006). ABCG1 and ABCA1 thus interact with different HDL subspecies (mature HDL vs. pre-beta HDL) to facilitate their cholesterol efflux function. To date, no association between ABCG1 mutations and HDL-cholesterol levels has been described in humans, suggesting that ABCG1 does not significantly contribute to HDL biogenesis. In accordance, under basal chow diet feeding conditions, ABCG1 knockout mice do not exhibit a change in plasma total or HDL-cholesterol levels (Out et al. 2007; Wiersma et al. 2009).
Chow-fed ABCG1-deficient mice exhibit a severe pulmonary lipidosis phenotype, starting from the age of ~6 months, which is characterized by accumulation of phospholipids, cholesterol crystals, and inflammatory infiltrates in sub-pleural areas of the lung (Baldán et al. 2006a, 2008; Wojcik et al. 2008). The distinct lung phenotype in total body ABCG1 knockout mice can be specifically attributed to the lack of ABCG1 in bone marrow-derived cells, presumably macrophages. Lethally irradiated normolipidemic mice transplanted with ABCG1-deficient bone marrow also show inflammatory cell infiltrates in their lungs, while normalization of macrophage ABCG1 function prevents the occurrence of pulmonary lipidosis in total body ABCG1 knockout mice (Wojcik et al. 2008). Alveolar macrophages from ABCG1 knockout mice have a foamy appearance and display compensatory upregulation of the cholesterol efflux gene ABCA1 (Baldán et al. 2006a). In vitro cultured macrophages lacking ABCG1 are more susceptible to surfactant-induced cholesterol accumulation (Baldán et al. 2006a). ABCG1-mediated cholesterol efflux to HDL is thus essential for the protection against specifically surfactant-induced foam cell formation under normolipidemic conditions, since no accumulation of lipid has been noted in other macrophage-rich tissues in chow-fed ABCG1-deficient mice (Out et al. 2008a). The pulmonary lipidosis phenotype is exacerbated upon challenging the mice with a cholic acid-containing atherogenic diet (Out et al. 2007). Under atherogenic diet feeding conditions, ABCG1 knockout mice also show marked lipid accumulation in cells within and surrounding the germinal centers of the spleen (Out et al. 2007). This finding highlights that ABCG1 is not solely involved in maintaining cholesterol homeostasis locally within the lungs.
Variations in the human ABCG1 gene have been linked to an altered risk for atherosclerotic disease that cannot be explained by changes in lipoprotein levels (Furuyama et al. 2009). In accordance with a role for murine ABCG1 in the protection against atherosclerosis, a higher extent of lesion formation, in the context of similar plasma lipoprotein levels, is detected in ABCG1 knockout mice as compared to wild-type littermate controls upon feeding atherogenic diet (Out et al. 2007). The bone marrow transplantation technique has been applied to elucidate the specific contribution of macrophages in the atheroprotective effect of ABCG1. In line with the prominent role of macrophage ABCG1 in the prevention of pulmonary lipidosis, extensive lipid accumulation is observed also in lungs of hyperlipidemic mice transplanted with ABCG1 knockout bone marrow (Out et al. 2006; Baldán et al. 2006b). However, contrasting effects on atherosclerosis outcome have been noted upon bone marrow-specific ABCG1 deletion. Macrophage ABCG1 appears to protect against atherosclerosis in LDLr knockout mice as evident from the initial studies by Out et al. (2006). In marked contrast, similar studies by Baldán et al. in LDLr and apoE knockout mice (Baldán et al. 2006b) and Ranalletta et al. in LDLr-deficient mice (Ranalletta et al. 2006) have identified macrophage ABCG1 as being a pro-atherogenic factor. Importantly, following critical review of all the present data and additional bone marrow transplantation studies, Meurs et al. have been able to clarify the evident discrepancy in the findings regarding the role of macrophage ABCG1 in atherosclerosis. The effect of disruption of ABCG1 function in macrophages on atherogenesis appears to be highly dependent on the stage of lesion development (Meurs et al. 2012). Diminished ABCG1-mediated efflux of cholesterol to HDL stimulates the formation of foam cells and accelerates lesion development in initial plaques that primarily contain macrophages. During later stages of the disease, macrophage ABCG1 deficiency inhibits plaque progression as a result of activation of compensatory atheroprotective mechanisms, i.e., hypersecretion of apoE from macrophages (Ranalletta et al. 2006), and/or an increased apoptosis rate of lipid-laden macrophage foam cells (Baldán et al. 2006b).
5 Lecithin-Cholesterol Acyltransferase
Discoidal small HDL particles are able to carry a relatively limited load of cholesterol. In order for nascent HDL species to become fully enriched in cholesterol, the unesterified cholesterol acquired upon cellular efflux has to be esterified for subsequent storage in the core of more mature spherical HDL particles. The hepatocyte-derived enzyme lecithin-cholesterol acyltransferase (LCAT) is considered to be the sole mediator of cholesterol esterification in HDL in humans, since familial LCAT deficiency is characterized by a >95 % decrease in plasma HDL-cholesterol ester levels (Glomset et al. 1970). As such, homozygous carriers of loss-of-function mutations in LCAT present with HDL deficiency, while half of normal HDL-cholesterol values are observed in heterozygous carriers (Santamarina-Fojo et al. 2000; Hovingh et al. 2005). Two different strains of LCAT knockout mice have been generated through genetic deletion of respectively exon 1 or exons 2–5. In accordance with a major role for LCAT in the maturation of HDL, both types of LCAT-deficient mice show severe HDL-cholesterol deficiency (>90 % decreased; Ng et al. 1997; Sakai et al. 1997). This coincides with a significant decrease in plasma apoA-I levels (Ng et al. 1997), probably as a result of apoA-I hypercatabolism by the kidneys. The remaining plasma HDL cholesterol in LCAT knockout mice is contained in its unesterified form (Ng et al. 1997; Sakai et al. 1997).
Besides the clear HDL deficiency phenotype, LCAT knockout mice display significant changes in the metabolism of triglyceride-rich lipoproteins. Homozygous LCAT-deficient mice show hypertriglyceridemia and an increase in plasma VLDL-cholesterol levels on a chow diet (Sakai et al. 1997; Lambert et al. 2001; Ng et al. 2002). This phenotype is preserved when mice are fed a Western-type diet enriched in fat (Li et al. 2007). Furthermore, extensive triglyceride deposition can be observed within livers of LCAT knockout mice upon Western-type diet feeding (Li et al. 2007). Mechanistic studies in LCAT x LDLr double knockout mice have indicated that the hypertriglyceridemia can be attributed to an increase in secretion of triglyceride-rich VLDL particles and a decrease in the LPL-mediated lipolysis rate (Ng et al. 2004). As evidenced by lower fasting glucose and insulin levels, LCAT knockout mice exhibit an improved glucose tolerance and insulin sensitivity when crossbred onto the hyperlipidemic LDLr knockout background (Ng et al. 2004; Li et al. 2007). LCAT-mediated cholesterol ester formation thus not only contributes to the formation of mature HDL particles, but also (indirectly) impacts on fatty acid and glucose metabolism.
A genetic defect in LCAT does, in general, not affect the tissue cholesterol balance in mice (Ng et al. 1997). However, adrenals of LCAT knockout mice show gross morphological changes associated with neutral lipid depletion, i.e., they have a more red/brownish color instead of the normal white appearance after perfusion (Ng et al. 1997). Specifically adrenocortical cells within the zona fasciculata, but not zona glomerulosa, are deprived from neutral lipid stores (Hoekstra et al. 2013a). This results in an overall ~80 % decrease in adrenal cholesterol ester content (Ng et al. 1997). Probably as a compensatory response to overcome cholesterol insufficiency, marked increases in the expression of genes involved in the de novo synthesis and extracellular acquisition of cholesterol can be detected in LCAT knockout adrenals (Ng et al. 1997). Importantly, in support of the notion that HDL act as cholesterol donors for the synthesis of glucocorticoids by the adrenals, LCAT knockout mice display a 40–50 % reduction in the maximal glucocorticoid output (Hoekstra et al. 2013a). A minor decrease in tissue cholesterol content is also noted in ovaries, but not testis, in response to LCAT deficiency in mice (Tomimoto et al. 2001), which does not translate into apparent changes in female fertility.
LCAT knockout mice on a normolipidemic background are virtually protected against cholic acid containing diet-induced formation of atherosclerotic lesions (Lambert et al. 2001). Chow diet-fed LCAT x apoE double knockout mice also display a decrease in atherosclerotic lesion formation, as compared with apoE single knockout controls (Lambert et al. 2001; Ng et al. 2002). Ablation of LCAT function is associated with marked HDL deficiency under all hyperlipidemic conditions. Strikingly, mixed results on the effect of LCAT deficiency on atherosclerosis susceptibility, however, have been noted in hyperlipidemic mice upon feeding diets enriched in fat. LCAT x LDLr double knockout mice display a lower atherosclerotic lesion burden upon feeding a cholic acid-containing diet (Lambert et al. 2001). In contrast, LCAT deficiency stimulates the formation of atherosclerotic lesions feeding in LDLr knockout mice upon feeding a Western-type diet devoid of cholic acid, as judged by the aortic cholesterol content (Furbee et al. 2002; Lee et al. 2004). Furthermore, LCAT x apoE double knockout mice also exhibit an increase in their aortic cholesterol content upon feeding the Western-type high fat diet (Furbee et al. 2002). Importantly, lower plasma levels of non-HDL cholesterol are found in the hyperlipidemic LCAT knockout mice under cholic acid-containing diet feeding conditions (Lambert et al. 2001), while non-HDL-cholesterol levels are actually increased in response to LCAT deficiency upon Western-type diet feeding (Furbee et al. 2002; Lee et al. 2004). It thus seems that the diverse effects of LCAT deficiency on atherosclerosis outcome in the different mouse models cannot be attributed to the HDL deficiency, but are rather a consequence of the genotype-associated changes in plasma levels of apoB-containing lipoproteins (Kunnen and Van Eck 2012).
6 Phospholipid Transfer Protein
Transfer of cholesterol esters generated by LCAT to the core of the HDL particles and the subsequent maturation of the HDL particles require the action of phospholipid transfer protein (PLTP), which supplies phospholipids allowing surface expansion of the shell of the particles. The phospholipids are liberated during the lipolysis of triglycerides in the core of apoB-containing lipoproteins by the action of lipoprotein lipase (Tall et al. 1985). In addition, PLTP facilitates the fusion of HDL3 particles to enlarged particles (Lusa et al. 1996). During this process lipid-poor apoA-I particles are released that can act as substrate for ABCA1-mediated cholesterol efflux from macrophages. In humans, common variants of PLTP have been identified that are associated with alterations in serum HDL cholesterol and the accumulation of small HDL particles (Albers et al. 2012). Furthermore, PLTP expression and activity is regulated during several diseases, including sepsis, multiple sclerosis, cancer, and cardiovascular disease (Albers et al. 2012), but it is largely unknown if the regulation of PLTP is a causative factor or simply a consequence of the processes underlying the diseases. The generation of PLTP knockout mice by the group of Alan Tall 15 years ago has greatly contributed to the general understanding of the role of PLTP in HDL metabolism. PLTP was inactivated in mice by replacing exon 2 containing the translation initiation codon, the signal peptide, and the first 16 amino acids of mature PLTP with a neomycin-resistant gene (Jiang et al. 1999). The plasma transfer activity of the major phospholipids, including phosphatidylcholine, phosphatidylethanolamine, phosphatidylinositol, and sphingomyelin into HDL, was completely blocked, while also the transfer of free cholesterol was impaired in PLTP knockout mice. Deletion of PLTP in mice led to markedly decreased levels of HDL phospholipids and cholesterol (65–70 %), illustrating the importance of transfer of surface phospholipids from apoB-containing lipoproteins by PLTP for maintaining HDL levels (Jiang et al. 1999). Furthermore, similarly as described for LCAT knockout mice the decrease in HDL cholesterol coincided with a significant decrease in plasma apoA-I levels. Hepatocytes isolated from PLTP knockout mice synthesized normal amounts of apoA-I, albeit with reduced amounts of phosphatidylcholine (Siggins et al. 2007), indicating that the reduced plasma apoA-I levels were not due to impaired production. Qin et al. showed by in vivo turnover studies using autologous HDL that the reduced apoA-I levels are likely the consequence of increased catabolism of HDL in the PLTP knockout mice (Qin et al. 2000).
On regular chow diet, PLTP-deficient mice absorb less cholesterol in the intestine (Liu et al. 2007), while hepatic phospholipids are increased and triglycerides are reduced in mice lacking PLTP (Siggins et al. 2007).
Feeding the mice a high saturated fat diet, containing 20 % hydrogenated coconut oil and 0.15 % cholesterol, led to the accumulation of surface components of apoB-containing lipoproteins as evidenced by a massive increase in VLDL and LDL phospholipids and cholesterol in the absence of changes in apoB (Jiang et al. 1999). Subsequent studies showed that the animals accumulated phospholipid and free cholesterol-rich lamellar particles containing apoA-IV and apoE (Qin et al. 2000). The accumulation of the lamellar particles specifically in coconut oil-fed PLTP knockout mice has been attributed to markedly reduced removal of these particles via scavenger receptor BI (SR-BI) by parenchymal liver cells (Kawano et al. 2002) and impaired secretion of biliary cholesterol and phospholipid (Yeang et al. 2010). Interestingly, these particles did not accumulate in the circulation when the mice were fed regular chow diet or a Western diet, containing 20 % milk fat and 0.15 % cholesterol (Kawano et al. 2002). In contrast, while feeding a Western diet, containing milk fat and 0.15 % cholesterol, PLTP deficiency was associated with an attenuated diet-induced hypercholesterolemia due to twofold lower concentrations of cholesterol transported by apoB-containing lipoproteins and HDL. Hepatic and intestinal lipid levels were not affected under these conditions, but cholesterol absorption in the intestine was reduced (Shelly et al. 2008).
Both on chow and while feeding the milk fat Western diet, PLTP knockout mice display reduced systemic inflammation as evidenced by lower levels of interleukin 6 and reduced expression of intercellular adhesion molecule 1 (ICAM1) and vascular adhesion molecule 1 (VCAM1) in aorta, indicating that mice lacking PLTP might have a reduced atherosclerosis susceptibility (Shelly et al. 2008).
To study the effect of PLTP deficiency on atherosclerotic lesion development, PLTP knockout mice were crossbred with human apoB transgenic mice, apoE knockout mice, and LDLr knockout mice (Jiang et al. 2001). Both in the apoB transgenic and the apoE knockout background, PLTP deficiency led, on top of the reduction in HDL lipids, to substantially lower VLDL lipid levels. Interestingly, this effect on VLDL lipids was not found in the LDLr knockout background, indicating the involvement of the LDLr in the lowering of VLDL lipids. In line, apoB production was reduced in the apoB transgenic and apoE knockout background, but not in the LDLr knockout background (Jiang et al. 2001). The content of the antioxidant vitamin E was significantly increased in all three backgrounds in the absence of PLTP, while autoantibodies to oxidized LDL were largely decreased (Jiang et al. 2002). HDL isolated from PLTP knockout mice crossbred to both the apoB transgenic and the LDLr knockout background displayed improved anti-inflammatory properties and reduced the ability of LDL to induce monocyte chemotaxis (Yan et al. 2004). After 6 months of feeding apoB transgenic mice Western diet and after feeding apoE knockout mice chow for 3 months, atherosclerotic lesion size was reduced fivefold in mice lacking PLTP. In contrast, in LDLr knockout mice PLTP deficiency led to a twofold reduction in atherosclerotic lesion size after 8 weeks Western diet, whereas no significant effects were observed after 12 weeks Western diet feeding (Jiang et al. 2001). Thus, while PLTP deficiency can reduce early atherosclerotic lesion development in mice lacking the LDLr, the pro-atherogenic effects of PLTP should be primarily attributed to its effects on the production of apoB-containing lipoproteins in mice fed regular chow or a Western diet. Notably, when apoE knockout mice lacking PLTP were challenged with a coconut oil-enriched, high fat diet for 7 weeks plasma levels of free cholesterol were 23 % higher due to the accumulation of the lamellar free cholesterol and phospholipid-rich particles. Under these conditions no effects on atherosclerotic lesion development were found (Yeang et al. 2010).
PLTP is widely distributed with expression in placenta > pancreas > lung > kidney > heart > liver > skeletal muscle > brain (Day et al. 1994). In addition, PLTP is found in endothelial cells (Day et al. 1994) and in smooth muscle cell and macrophage foam cells in atherosclerotic lesions (Desrumaux et al. 2003; Laffitte et al. 2003; O'Brien et al. 2003). Interestingly, Vikstedt and colleagues showed that the expression of PLTP is sixfold higher in Kupffer cells, macrophages of the liver as compared to hepatocytes (Vikstedt et al. 2007). Mice with a hepatocyte-specific deletion of PLTP were generated by injection of PLTP-flox/flippase animals with adenovirus-associated virus expressing Cre-recombinase under control of the thyroxine binding globulin promoter (Yazdanyar et al. 2013). PLTP activity was reduced by approximately 25 % in these animals with a hepatocyte-specific deletion of PLTP, leading to a 20 % reduction of HDL cholesterol. In addition, non-HDL cholesterol was 29 % lower due to an impaired production of apoB-containing lipoproteins (Yazdanyar et al. 2013). Conversely, liver-specific expression of PLTP in a PLTP knockout background promoted the secretion of apoB-containing lipoproteins by the liver (Yazdanyar and Jiang 2012). The contribution of macrophage PLTP to plasma PLTP activity was determined by bone marrow transplantation studies in LDLr knockout mice. Selective deletion of PLTP in bone marrow-derived cells led to a decrease in plasma PLTP activity on chow and Western diet (Vikstedt et al. 2007). Vikstedt et al. showed that after 9 weeks Western diet feeding atherosclerotic lesion size was 29 % smaller in LDLr knockout mice transplanted with PLTP-deficient bone marrow, which coincided with decreased serum VLDL-cholesterol and phospholipid levels, while HDL phospholipid and apoA-I were increased (Vikstedt et al. 2007). In contrast, Valenta et al. also found lower levels of cholesterol in apoB-containing lipoproteins upon disruption of PLTP in bone marrow-derived cells of LDLr knockout mice, but surprisingly atherosclerotic lesion development was increased (Valenta et al. 2006). No effect on atherosclerosis was observed upon transplantation of PLTP-deficient bone marrow into LDLr knockout mice overexpressing human apoA-I (Valenta et al. 2006). The studies by Valenta et al. suggest that locally in the arterial wall PLTP produced by macrophages can be anti-atherogenic. A possible explanation is that PLTP production by macrophages promotes ABCA1-mediated cholesterol efflux, as evidenced by decreased efflux to apoA-I from PLTP-deficient macrophages (Lee-Rueckert et al. 2006). In agreement, macrophages isolated from PLTP knockout mice were shown to accumulate more cholesterol upon incubation with native or acetylated LDL (Ogier et al. 2007). Importantly, the higher levels of lipid-poor apoA-I in LDLr knockout mice overexpressing human apoA-I can overcome the pro-atherogenic effects of deletion of PLTP in macrophages, probably by stimulating the cholesterol efflux capacity of the macrophages.
In summary, the effects of PLTP on atherosclerotic lesion development are determined by a balance between systemic effects influencing the levels of anti-atherogenic HDL and pro-atherogenic apoB-containing lipoproteins and the antioxidant vitamin E and local effects of PLTP produced by macrophages in the arterial wall influencing macrophage apoE production and ABCA1-mediated cholesterol efflux.
7 Scavenger Receptor BI
During the final step of the reverse cholesterol transport process, mature HDL particles deliver their cholesterol load to the liver for subsequent excretion via the bile. The transmembrane glycoprotein scavenger receptor BI (SR-BI) is considered to be the sole mediator of selective uptake of cholesterol esters from HDL by hepatocytes in mice (Out et al. 2004). Several genetic variations at the SR-BI locus—either translating in an altered SR-BI protein expression or functionality—have been associated with a change in plasma HDL-cholesterol levels (Hsu et al. 2003; West et al. 2009; Vergeer et al. 2011; Brunham et al. 2011; Chadwick and Sahoo 2012), which highlights the crucial role for SR-BI in human HDL metabolism. To uncover the role of SR-BI in lipoprotein metabolism, the group of Monty Krieger developed a mouse model carrying a targeted mutation in the SR-BI gene that leads to complete absence of functional SR-BI protein in all tissues (Rigotti et al. 1997). In addition, a strain of SR-BIatt mice has been generated by the group of Dennis Huszar in which the SR-BI promoter is mutated, resulting in a lowered protein expression of SR-BI in liver, testis, and adrenals (Varban et al. 1998). Furthermore, hypomorphic SR-BI knockout mice have been constructed that do not suffer from complete SR-BI deficiency, but show a restricted expression pattern of the protein in liver, kidney, aorta, and steroidogenic tissues (Huby et al. 2006).
Total body SR-BI knockout mice are characterized by a diet-independent hypercholesterolemia, which can be primarily attributed to a marked rise in plasma HDL-cholesterol levels (Rigotti et al. 1997; Van Eck et al. 2003, 2008). The fractional catabolic rate, plasma decay, and subsequent uptake of HDL-cholesterol esters by the liver are diminished in SR-BI knockout mice (Out et al. 2004). SR-BIatt mice similarly show a diminished fractional catabolic rate and hepatic selective uptake of HDL-associated cholesterol, also leading to a higher steady-state level of HDL cholesterol in the plasma of these mice (Varban et al. 1998; Ji et al. 1999). In accordance with a role for hepatic SR-BI in the regulation of plasma HDL-cholesterol levels, both total body and hepatocyte-specific restriction of SR-BI expression in hypomorphic SR-BI knockout mice is also associated with an increase in plasma HDL-cholesterol levels (Huby et al. 2006). Although SR-BI is thought to primarily mediate the uptake HDL-cholesterol esters, in particular free cholesterol levels are higher in plasma in the different SR-BI-deficient mice (Van Eck et al. 2003; Huby et al. 2006; El Bouhassani et al. 2011). The increase in plasma free cholesterol to cholesterol ester ratio underlies anemia and thrombocytopenia phenotypes in SR-BI knockout mice (Holm et al. 2002; Meurs et al. 2005; Dole et al. 2008; Korporaal et al. 2011) and can be attributed to a reduction in the LCAT activity (El Bouhassani et al. 2011). As judged by the unchanged plasma apoA-I levels in total body SR-BI knockout mice, only the size and cholesterol content but not the absolute number of HDL particles is increased in response to SR-BI deficiency (Rigotti et al. 1997). The enlarged HDL particles observed in plasma of SR-BI knockout mice are highly enriched in apoE. In contrast, apoA-II cannot be detected in the HDL fraction of SR-BI-deficient mice, which suggests a generally altered apolipoprotein composition (Rigotti et al. 1997). In line with the notion that SR-BI facilitates HDL-mediated reverse cholesterol transport, the recovery of radiolabeled cholesterol in feces upon injection of either peritoneal or J774 macrophages is lower in SR-BI knockout mice as compared to their wild-type littermate controls, despite an increased appearance of radioactivity in the plasma (HDL) compartment (Zhang et al. 2005a; Zhao et al. 2011).
SR-BI knockout mice also display a significant increase in non-HDL-cholesterol levels (Rigotti et al. 1997; Van Eck et al. 2003, 2008). Hepatic VLDL secretion and LPL-mediated lipolysis rates are not affected by SR-BI deficiency (Van Eck et al. 2008). SR-BI has been shown to be directly involved in the selective uptake of cholesterol esters from LDL and VLDL particles by hepatocytes (Rhainds et al. 2003; Van Eck et al. 2008). It can therefore be anticipated that the increase in the level of cholesterol associated with apoB-containing lipoproteins in SR-BI knockout mice is directly resulting from a diminished clearance by the liver.
Two variations in the SR-BI gene have been linked to changes in fertility in humans (Yates et al. 2011). Without dietary addition of the lipid-lowering drug probucol, SR-BI knockout female mice are virtually sterile, with a ~90 % reduction in fertility and pup yield as compared to wild-type controls (Miettinen et al. 2001). The development of SR-BI knockout embryos is impaired in ex vivo cultures (Trigatti et al. 1999). Furthermore, developmental abnormalities are also observed in SR-BI knockout embryos in vivo (Santander et al. 2013). Although steroidogenic tissues, i.e., ovaries and adrenals, from SR-BI knockout mice exhibit an apparent decrease in their cholesterol ester stores (Rigotti et al. 1997; Trigatti et al. 1999), levels of the steroid hormone progesterone as well as the number of oocytes and cyclic activity are unaffected by murine SR-BI deficiency (Trigatti et al. 1999). The lack of pups generated from SR-BI-deficient breedings can thus rather be attributed to embryonic defects than to infertility of the mothers.
Human carriers of functional mutations in the SR-BI gene exhibit a decrease in their adrenal steroidogenesis rate (Vergeer et al. 2011). Similarly, depletion of adrenocortical cholesterol stores due to SR-BI deficiency translates into a significant change of the adrenal steroid function in mice. Basal glucocorticoid levels in plasma are similar between SR-BI knockout and wild-type mice (Hoekstra et al. 2008). However, total body SR-BI deficiency is associated with primary glucocorticoid insufficiency. Total body SR-BI knockout mice display increased plasma ACTH levels and are unable to increase their adrenal glucocorticoid output in response to established steroidogenic triggers (Cai et al. 2008; Hoekstra et al. 2008, 2009). It appears that SR-BI-mediated uptake of HDL-cholesterol esters by the adrenals is necessary for a proper steroid output, since the glucocorticoid function is also impaired in adrenocortical cell-specific SR-BI knockout mice (Hoekstra et al. 2013b). In further support, hepatocyte-specific SR-BI deficiency does not lower adrenal cholesterol levels or impair the adrenal corticosterone response to fasting (El Bouhassani et al. 2011).
No atherosclerotic lesion development is normally observed in normolipidemic wild-type mice upon feeding a regular chow diet or a high cholesterol high fat Western-type diet that does not contain cholic acid. No atherosclerotic plaques can also be detected in the aortic root of chow-fed SR-BI knockout mice (Zhao et al. 2011). In contrast, SR-BI knockout mice with increased HDL-cholesterol levels do readily develop macrophage-rich fatty streak lesions after 20 weeks of Western-type diet feeding (Van Eck et al. 2003). SR-BI knockout mice display an increase in non-HDL cholesterol under these feeding conditions (Van Eck et al. 2003). However, the measured levels of apoB-containing lipoproteins upon Western-type diet feeding are still considered to be too low to stimulate the formation of atherosclerotic lesions. This suggests that other SR-BI-related mechanisms may underlie the susceptibility for Western-type diet-induced atherogenesis in SR-BI knockout mice. Follow-up studies in liver-specific hypomorphic SR-BI knockout mice have indicated that the SR-BI-mediated atheroprotection originates from both hepatic and peripheral cell sources (Huby et al. 2006; El Bouhassani et al. 2011). A similarly increased susceptibility for atherosclerosis upon ablation of total body SR-BI function has been noted in response to feeding a cholic acid-containing atherogenic diet (Zhang et al. 2005b). In accordance with a role for SR-BI in the clearance of apoB-containing lipoproteins, high fat diet-fed SR-BIatt x LDLr knockout mice exhibit higher plasma LDL-cholesterol levels and an increased extent of atherosclerosis as compared to LDLr knockout controls (Huszar et al. 2000). In contrast, total body SR-BI deficiency in high fat diet-fed LDLr knockout mice is associated with lower VLDL/LDL levels, but a similarly enhanced formation of atherosclerotic lesions in the context of a higher plasma HDL-cholesterol concentration (Covey et al. 2003). Loss of total body SR-BI function in chow diet-fed hyperlipidemic apoE knockout mice is associated with severe cardiac dysfunction, i.e., a reduced contractility and ejection fraction, and occlusion of the coronary arteries, leading to premature death of these animals at the age of 6–8 weeks (Braun et al. 2002). Occlusive coronary atherosclerosis and an increased extent of atherosclerosis in the aortic root are also seen upon genetic deletion of SR-BI in atherogenic diet-fed hypomorphic APOER61h/h mice (Zhang et al. 2005b). An optimal total body SR-BI function thus protects against atherosclerosis and myocardial infarction, through (1) its role in hepatic uptake of lipoprotein-derived cholesterol and (2) via its action extrahepatic/peripheral cells.
In rodents, high expression of SR-BI is not only found in liver and steroidogenic tissues (Acton et al. 1996), but also in tissue macrophages such as liver Kupffer cells (Fluiter et al. 1998; Malerød et al. 2002; Hoekstra et al. 2003), which suggests that SR-BI can also contribute to macrophage cholesterol homeostasis locally within atherosclerotic lesions. Studies using isolated SR-BI-deficient peritoneal macrophages have suggested that SR-BI may facilitate cholesterol efflux to mature HDL (Van Eck et al. 2004). However, macrophage SR-BI does not contribute to reverse cholesterol in vivo, based upon radiolabeled cholesterol recovery studies (Wang et al. 2007). Bone marrow transplantation studies have been executed to delineate the specific role of macrophage SR-BI in the protection against atherosclerosis. Transplantation of SR-BI knockout bone marrow into LDLr knockout mice stimulates atherogenesis without altering plasma lipoprotein cholesterol levels (Covey et al. 2003; Van Eck et al. 2004). Inactivation of macrophage SR-BI also promotes the development of atherosclerotic plaques in the context of unchanged lipid levels in apoE knockout mice (Zhang et al. 2003). In addition, the cardiac hypertrophy and dysfunction and coronary atherosclerosis development are attenuated upon restoration of macrophage SR-BI function in apoE x SR-BI double knockout mice (Pei et al. 2013). Strikingly, in marked contrast to the findings from hyperlipidemic mice, disruption of macrophage SR-BI expression in normolipidemic C57BL/6 mice results in a lower susceptibility for atherosclerosis upon feeding a cholic acid-containing atherogenic diet (Van Eck et al. 2004). C57Bl/6 mice fed a cholic acid-containing atherogenic diet develop small initial atherosclerotic lesions, suggesting that SR-BI might be pro-atherogenic in this early stage of lesion development. In line, early atherosclerotic lesion development in LDLr knockout mice challenged with Western diet for only 4 weeks was reduced upon disruption of SR-BI in bone marrow-derived cells. These combined studies suggest that macrophage SR-BI plays a dual role in foam cell formation and atherogenesis, which may be dependent on the stage of atherosclerotic lesion development.
8 Insights from Intercrossing of the Different Knockout Mice
Since some of the aforementioned proteins execute both different and overlapping functions in the formation of HDL particles and reverse cholesterol transport, it is conceivable that they may act synergistically and/or be able to compensate for each other’s loss. To uncover interactions between specific gene products several double knockout mice have been generated via intercrossing. In line with a differential role in HDL formation and maturation, studies in mice lacking both ABCA1 and LCAT function have suggested that these two proteins act synergistically. Hepatic cholesterol accumulation is higher, while peripheral tissue cholesterol levels are lower in ABCA1 x LCAT double knockout mice as compared to the respective single knockout mice, despite an even higher HDL fractional catabolic rate (Hossain et al. 2009). In parallel, probucol-induced inhibition of ABCA1 function further lowers adrenal cholesterol levels in LCAT knockout mice (Tomimoto et al. 2001). The ABCA1-mediated formation of HDL particles rather than the SR-BI-mediated cholesterol uptake is the driving force in the maintenance of plasma HDL-cholesterol levels, as HDL-cholesterol levels in ABCA1 x SR-BI double knockout mice are still close to zero (Zhao et al. 2011). In contrast, ablation of either ABCA1 or SR-BI function is equally effective in lowering the rate of in vivo macrophage reverse cholesterol transport (Zhao et al. 2011). The mild pulmonary lipidosis phenotype of ABCA1 knockout mice is severely aggravated in response to deletion of SR-BI function, which suggests that macrophage SR-BI is able to partially compensate for the inability of ABCA1 to mediate cholesterol efflux (Zhao et al. 2011). In accordance, specific disruption of both ABCA1 and SR-BI in bone marrow-derived cells of LDLr knockout mice leads to an added increase in (tissue) macrophage foam cell formation and Western-type diet-induced atherosclerotic lesion development, in the context of lower plasma cholesterol levels, when compared to both single knockout bone marrow transplanted controls (Zhao et al. 2010). Importantly, ABCG1 seems to play a more essential role in the compensatory response to macrophage ABCA1 deficiency. Mass efflux of cholesterol from macrophages to both apoA-I and HDL is virtually absent as a result of combined deletion of ABCA1 and ABCG1 (Out et al. 2008a). Already on a chow diet ABCA1 x ABCG1 double knockout mice display marked accumulation of cholesterol not only within the lungs (reminiscent of the ABCG1 knockout phenotype), but also in primary and secondary lymphoid organs and macrophage-rich areas of the liver and intestine (Out et al. 2008a). Furthermore, the double deletion exacerbates high fat diet-induced lymphocytosis (Yvan-Charvet et al. 2008). Transplantation of bone marrow from ABCA1 x ABCG1 double knockout mice into LDLr knockout mice is associated with resistance to Western-type diet-induced hyperlipidemia (Out et al. 2008b). However, despite the rather low plasma cholesterol levels under Western-type diet feeding conditions, ABCA1 x ABCG1 double knockout bone marrow recipient mice show significant plaque development and exhibit distinct lipid deposition in macrophage-rich tissues (Out et al. 2008b). This highlights the necessity for these transporters to overcome macrophage foam cell formation and atherosclerosis. Subsequent bone marrow transplantations into mice with a heterozygous mutation in the LDLr gene (LDLr+/−) have validated the increased susceptibility to macrophage cholesterol accumulation and atherosclerosis in response to the combined deficiency in macrophage ABCA1 and ABCG1 upon challenge with a cholate-containing diet (Yvan-Charvet et al. 2007). Furthermore, these studies have suggested that the combined deficiency of these ABC transporters in bone marrow also impacts on the inflammatory function and susceptibility to apoptosis of macrophages as well as the cyclic activity of hematopoietic stem cells (Yvan-Charvet et al. 2008, 2010a).
9 Conclusions from the Gene Knockout Mouse Studies
Generation of the different knockout mice has significantly contributed to our understanding of HDL’s metabolism and function (see Fig. 1 for overview). It has become clear that the initial generation and maturation of HDL particles as well as the selective uptake of its cholesterol by the liver are important parameters in the life cycle of HDL. In line with the proposed atheroprotective function of HDL, murine HDL deficiency consistently predisposes to the development of atherosclerotic lesions. However, it should be noted that many of the detrimental atherosclerosis effects observed in response to HDL deficiency in mice cannot be solely attributed to the low HDL levels per se, as the low HDL levels are in most models paralleled by changes in non-HDL-cholesterol levels. Although in the human setting, low HDL-cholesterol levels predict increased cardiovascular risk at all plasma non-HDL-cholesterol levels, the mouse data firmly establish that HDL and non-HDL metabolism are heavily intertwined.

Major phenotypic changes in mouse models of disturbed HDL metabolism. An overview is given of the effects of deletion of apolipoprotein A-I (apoA-I), the ABC transporters A1 (ABCA1) and G1 (ABCG1), lecithin-cholesterol acyltransferase (LCAT), phospholipid transfer protein (PLTP), and scavenger receptor BI (SR-BI) for HDL’s metabolism, adrenal function, female fertility, and its protection against atherosclerosis in mice. = no effect, ↓ decreased, ↑ increased, C cholesterol, PL phospholipids, TG triglycerides, KO knockout
Based upon the bone marrow transplantation studies in hyperlipidemic mice, it appears that the cholesterol efflux function of HDL is of critical importance to overcome foam cell formation and the development of atherosclerotic lesions. We therefore consider it therapeutically highly relevant to increase the expression and/or activity of the cholesterol transporters in macrophages. In this regard, special focus should be given to macrophage ABCA1 as this transporter seems to be the driving force in the atheroprotection.
Although HDL is predominantly studied for its atheroprotective action, it can be appreciated that the mouse data suggest an essential role for HDL as cholesterol donor for steroidogenic tissues, including the adrenals and ovaries. Furthermore, it appears that a relevant interaction exists between HDL-mediated cellular cholesterol efflux and the susceptibility to inflammation, which (1) provides strong support for the novel concept that inflammation and metabolism are intertwining biological processes and (2) identifies the efflux function of HDL as putative therapeutic target also in other inflammatory diseases than atherosclerosis, including sepsis and diabetes.
10 Cholesterol Ester Transfer Protein Transgenic Mice
Despite the fact that a great deal regarding the function of specific gene products has been learned from the murine transgenic studies it should be acknowledged that one should take care when directly translating the data to the human situation. Humans, in contrast to mice, naturally express cholesterol ester transfer protein (CETP) that plays a major role in total body cholesterol homeostasis as it is able to exchange cholesterol esters and triglycerides between cholesterol-rich HDL and triglyceride-rich VLDL/LDL particles. In agreement with a major role for CETP in the modulation of plasma HDL-cholesterol levels, genetic CETP deficiency is associated with marked hyperalphalipoproteinemia, i.e., relatively high HDL-cholesterol levels, in humans (Rhyne et al. 2006; Calabresi et al. 2009). To overcome this apparent discrepancy between mice and humans in studies dealing with lipoprotein metabolism, the group of Alan Tall generated a mouse line (NFR-CETP) expressing the human CETP transgene under the control of its natural flanking regulatory elements (Jiang et al. 1992). Initial cloning studies (Drayna et al. 1987) indicated that CETP is primarily derived from liver, small intestine, adrenals, and spleen in humans. In accordance, relatively high CETP mRNA levels are found in liver, spleen, and small intestine of human NFR-CETP transgenic mice (Jiang et al. 1992). Of note, the Tall group also generated a transgenic mouse line expressing CETP under control of the methallothionein-I (MT) gene (Jiang et al. 1992). In MT-CETP transgenic mice, additional high expression of CETP can be detected in kidney, adipose tissue, and heart. Importantly, the expression of CETP in MT-CETP transgenic is not responsive to high cholesterol/high fat diet feeding, which contrasts the findings from NFR-CETP transgenic mice (Jiang et al. 1992) and species naturally expressing CETP, i.e., monkeys and rabbits (Quinet et al. 1990; Pape et al. 1991). Given the apparent limitations of MT-CETP transgenic mice, in our opinion the NFR-CETP mouse model should preferentially be used in studies aimed at translation to the human situation. Therefore, findings from NFR-CETP transgenic mice will only be reviewed in the next part.
Chow-fed CETP transgenic mice exhibit a ~15 % lower plasma total cholesterol level as compared to non-transgenic controls (Masucci-Magoulas et al. 1996; Harada et al. 2007). In contrast, plasma triglyceride levels are marginally increased in CETP carrying mice upon feeding chow diet (Zhou et al. 2006; Harada et al. 2007) due to a slightly diminished clearance of triglyceride-rich lipoproteins (Salerno et al. 2009). The decrease in total cholesterol in CETP transgenic mice can primarily be attributed to lower levels of cholesterol associated with HDL (Zhou et al. 2006). The fractional catabolic rate and uptake of HDL-cholesterol esters by the liver, adrenals, and adipose tissue are increased in CETP transgenic mice, which is associated with an increase in hepatic cholesterol stores (Harada et al. 2007). As a result, CETP transgenic mice exhibit a rise in the plasma total cholesterol over HDL-cholesterol ratio (Clee et al. 1997). The decrease in HDL-cholesterol levels cannot be attributed to a change in LCAT levels. Intriguingly, the presence of CETP actually enhances the saturation level of the LCAT-mediated cholesterol esterification reaction (Oliveira et al. 1997). This suggests that CETP generates particles that can serve as optimal substrates for LCAT.
Despite the marked increase in plasma HDL-cholesterol levels associated with complete CETP deficiency, variable effects on atherosclerosis and cardiovascular disease risk in response to genetic variations in the CETP gene have been noted in the human setting (Zhong et al. 1996; Agerholm-Larsen et al. 2000; Curb et al. 2004; Zheng et al. 2004; Rhyne et al. 2006). To provide clear insight into the contribution of CETP to atherosclerotic lesion development, NFR-CETP transgenic mice have been bred onto several different atherosclerosis-susceptible mouse backgrounds. CETP expression in heterozygous LDLr knockout mice on a cholic acid-containing atherogenic diet does not significantly alter plasma HDL- or non-HDL-cholesterol levels. As a result, CETP does not impact on the susceptibility to atherosclerosis in heterozygous LDLr knockout mice (Casquero et al. 2006). In contrast, CETP executes a clear pro-atherogenic effect in hyperlipidemic apoE knockout and homozygous LDLr knockout mice (Plump et al. 1999). CETP lowers plasma HDL-cholesterol levels in chow-fed apoE knockout mice without changing levels of cholesterol associated with non-HDL particles. In addition, triglyceride levels are increased in the plasma of CETP-expressing apoE knockout mice. CETP expression does not alter plasma HDL-cholesterol levels in LDLr knockout mice challenged with a Western-type high fat/high cholesterol diet. However, levels of cholesterol associated with pro-atherogenic apoB-containing lipoproteins are markedly increased in response to CETP expression in LDLr knockout mice. It thus appears that the increased atherosclerotic lesion development in apoE and LDLr knockout mice upon CETP expression can be directly related to an increased atherogenic index of the plasma compartment (higher non-HDL/HDL ratio) in both types of hyperlipidemic mice. In further support, a marked increase in plasma non-HDL-cholesterol levels and atherosclerotic lesion development—in the context of a marginal but significant decrease in HDL-cholesterol levels—was noted when we expressed CETP only in bone marrow-derived cells of LDLr knockout mice through bone marrow transplantation (Van Eck et al. 2007).
A clear drawback of using LDLr and apoE knockout mice for studies regarding lipoprotein metabolism is that they do not exhibit a normal clearance of apoE-containing VLDL/LDL particles. To determine the effect of CETP expression on lipoprotein metabolism and atherosclerosis development in a more relevant “humanized” setting, CETP transgenic mice have also been crossbred with hyperlipidemic apoE*3-Leiden mice. Transgenic apoE*3-Leiden mice express a mutation of the human apoE3 gene resulting in a slightly attenuated clearance of apoB-containing particles via the LDLr pathway (van den Maagdenberg et al. 1993). As a result, these mice (1) exhibit a lipoprotein cholesterol distribution profile that is highly similar to that found in humans (van den Maagdenberg et al. 1993; van Vlijmen et al. 1994), (2) are susceptible to atherosclerotic lesion development when fed a high fat/high cholesterol Western-type diet devoid of cholic acid (van Vlijmen et al. 1994), and (3) show a similar plasma lipid response as human subjects to several drug interventions (van der Hoogt et al. 2007; de Haan et al. 2008a; van der Hoorn et al. 2008). ApoE*3-Leiden x CETP double transgenic mice as compared to apoE*3-Leiden controls display a significant increase in plasma total cholesterol levels on both chow and under Western-type diet feeding conditions (Westerterp et al. 2006). Under these dietary conditions, VLDL-cholesterol levels are increased twofold, while HDL-cholesterol levels as well as plasma apoA-I levels are 25 % lower in apoE*3-Leiden x CETP transgenic mice (Westerterp et al. 2006). Importantly, apoE*3-Leiden x CETP transgenic mice exhibit increased atherosclerotic lesion development in the aortic root as compared to apoE*3-Leiden controls (Westerterp et al. 2006). Based upon these combined findings, CETP should clearly be considered a pro-atherogenic protein moiety.
In the last decade, therapeutic strategies have focused on inhibiting CETP activity through the use of a variety of CETP-binding drugs, i.e., torcetrapib, that form an inactive complex between CETP and HDL. Studies in apoE*3-Leiden x CETP transgenic mice have suggested that the HDL increase associated with torcetrapib treatment does not add to the beneficial impact of statin-induced lipid-lowering on atherosclerosis (de Haan et al. 2008b). Importantly, the ILLUMINATE and dal-OUTCOMES trials also failed to show the beneficial effect of CETP inhibition in statin-treated cardiovascular disease patients (Barter et al. 2007; Schwartz et al. 2012). Combination treatment with torcetrapib and statin increases the macrophage content in atherosclerotic lesions in apoE*3-Leiden x CETP transgenic mice as compared to statin treatment alone (de Haan et al. 2008b). A higher lesional macrophage content is generally associated with an increased susceptibility for plaque rupture and future cardiovascular events in the human situation (Redgrave et al. 2006; Marnane et al. 2014). An effect of CETP inhibition on plaque stability may therefore possibly have contributed to the negative outcome of the aforementioned torcetrapib and dalcetrapib trials.
Concluding Remark
The use of dedicated knockout and transgenic mouse models has significantly increased our knowledge on the different functions and metabolism of HDL. However, as evident from the recent failures of the CETP inhibition-based clinical trials, more research is clearly needed to better understand how we can use HDL as target in the treatment of patients at risk of cardiovascular disease and other immune-related disorders. The generation of “humanized” mouse models such as the apoE*3-Leiden x CETP transgenic mice will hopefully aid in the development of novel therapeutic approaches beneficially impacting on HDL functionality and atherosclerosis outcome also in the clinical setting.
Abbreviations
- ABCA1:
-
ATP-binding cassette transporter A1
- ABCG1:
-
ATP-binding cassette transporter G1
- apoA-I:
-
Apolipoprotein A-I
- apoA-II:
-
Apolipoprotein A-II
- apoA-IV:
-
Apolipoprotein A-IV
- apoB:
-
Apolipoprotein B
- apoE:
-
Apolipoprotein E
- CETP:
-
Cholesterol ester transfer protein
- HDL:
-
High-density lipoprotein
- LCAT:
-
Lecithin-cholesterol acyltransferase
- LDL:
-
Low-density lipoprotein
- LDLr:
-
Low-density lipoprotein receptor
- LPS:
-
Lipopolysaccharide
- PLTP:
-
Phospholipid transfer protein
- SR-BI:
-
Scavenger receptor BI
- VLDL:
-
Very-low-density lipoprotein
References
Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M (1996) Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271:518–520
Agerholm-Larsen B, Tybjaerg-Hansen A, Schnohr P, Steffensen R, Nordestgaard BG (2000) Common cholesteryl ester transfer protein mutations, decreased HDL cholesterol, and possible decreased risk of ischemic heart disease: the Copenhagen City heart study. Circulation 102:2197–2203
Aiello RJ, Brees D, Bourassa PA, Royer L, Lindsey S, Coskran T, Haghpassand M, Francone OL (2002) Increased atherosclerosis in hyperlipidemic mice with inactivation of ABCA1 in macrophages. Arterioscler Thromb Vasc Biol 22:630–637
Albers JJ, Vuletic S, Cheung MC (2012) Role of plasma phospholipid transfer protein in lipid and lipoprotein metabolism. Biochim Biophys Acta 1821:345–357
Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R, Cholesterol Treatment Trialists’ (CTT) Collaboration (2010) Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 376:1670–1681
Baldán A, Tarr P, Vales CS, Frank J, Shimotake TK, Hawgood S, Edwards PA (2006a) Deletion of the transmembrane transporter ABCG1 results in progressive pulmonary lipidosis. J Biol Chem 281:29401–29410
Baldán A, Pei L, Lee R, Tarr P, Tangirala RK, Weinstein MM, Frank J, Li AC, Tontonoz P, Edwards PA (2006b) Impaired development of atherosclerosis in hyperlipidemic Ldlr-/- and ApoE-/- mice transplanted with Abcg1-/- bone marrow. Arterioscler Thromb Vasc Biol 26:2301–2307
Baldán A, Gomes AV, Ping P, Edwards PA (2008) Loss of ABCG1 results in chronic pulmonary inflammation. J Immunol 180:3560–3568
Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B, Investigators ILLUMINATE (2007) Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 357:2109–2122
Berge KE, Leren TP (2010) Mutations in APOA-I and ABCA1 in Norwegians with low levels of HDL cholesterol. Clin Chim Acta 411:2019–2023
Bi X, Zhu X, Duong M, Boudyguina EY, Wilson MD, Gebre AK, Parks JS (2013) Liver ABCA1 deletion in LDLrKO mice does not impair macrophage reverse cholesterol transport or exacerbate atherogenesis. Arterioscler Thromb Vasc Biol 33:2288–2296
Bodzioch M, Orsó E, Klucken J, Langmann T, Böttcher A, Diederich W, Drobnik W, Barlage S, Büchler C, Porsch-Ozcürümez M, Kaminski WE, Hahmann HW, Oette K, Rothe G, Aslanidis C, Lackner KJ, Schmitz G (1999) The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet 22:347–351
Braun A, Trigatti BL, Post MJ, Sato K, Simons M, Edelberg JM, Rosenberg RD, Schrenzel M, Krieger M (2002) Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ Res 90:270–276
Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, Loubser O, Ouelette BF, Fichter K, Ashbourne-Excoffon KJ, Sensen CW, Scherer S, Mott S, Denis M, Martindale D, Frohlich J, Morgan K, Koop B, Pimstone S, Kastelein JJ, Genest J Jr, Hayden MR (1999) Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet 22:336–345
Brunham LR, Kruit JK, Iqbal J, Fievet C, Timmins JM, Pape TD, Coburn BA, Bissada N, Staels B, Groen AK, Hussain MM, Parks JS, Kuipers F, Hayden MR (2006) Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J Clin Invest 116:1052–1062
Brunham LR, Singaraja RR, Duong M, Timmins JM, Fievet C, Bissada N, Kang MH, Samra A, Fruchart JC, McManus B, Staels B, Parks JS, Hayden MR (2009) Tissue-specific roles of ABCA1 influence susceptibility to atherosclerosis. Arterioscler Thromb Vasc Biol 29:548–554
Brunham LR, Tietjen I, Bochem AE, Singaraja RR, Franchini PL, Radomski C, Mattice M, Legendre A, Hovingh GK, Kastelein JJ, Hayden MR (2011) Novel mutations in scavenger receptor BI associated with high HDL cholesterol in humans. Clin Genet 79:575–581
Cai L, Ji A, de Beer FC, Tannock LR, van der Westhuyzen DR (2008) SR-BI protects against endotoxemia in mice through its roles in glucocorticoid production and hepatic clearance. J Clin Invest 118:364–375
Calabresi L, Nilsson P, Pinotti E, Gomaraschi M, Favari E, Adorni MP, Bernini F, Sirtori CR, Calandra S, Franceschini G, Tarugi P (2009) A novel homozygous mutation in CETP gene as a cause of CETP deficiency in a Caucasian kindred. Atherosclerosis 205:506–511
Calpe-Berdiel L, Rotllan N, Palomer X, Ribas V, Blanco-Vaca F, Escolà-Gil JC (2005) Direct evidence in vivo of impaired macrophage-specific reverse cholesterol transport in ATP-binding cassette transporter A1-deficient mice. Biochim Biophys Acta 1738:6–9
Casquero AC, Berti JA, Salerno AG, Bighetti EJ, Cazita PM, Ketelhuth DF, Gidlund M, Oliveira HC (2006) Atherosclerosis is enhanced by testosterone deficiency and attenuated by CETP expression in transgenic mice. J Lipid Res 47:1526–1534
Chadwick AC, Sahoo D (2012) Functional characterization of newly-discovered mutations in human SR-BI. PLoS ONE 7:e45660
Christiansen-Weber TA, Voland JR, Wu Y, Ngo K, Roland BL, Nguyen S, Peterson PA, Fung-Leung WP (2000) Functional loss of ABCA1 in mice causes severe placental malformation, aberrant lipid distribution, and kidney glomerulonephritis as well as high-density lipoprotein cholesterol deficiency. Am J Pathol 157:1017–1029
Chung S, Timmins JM, Duong M, Degirolamo C, Rong S, Sawyer JK, Singaraja RR, Hayden MR, Maeda N, Rudel LL, Shelness GS, Parks JS (2010) Targeted deletion of hepatocyte ABCA1 leads to very low density lipoprotein triglyceride overproduction and low density lipoprotein hypercatabolism. J Biol Chem 285:12197–12209
Clee SM, Zhang H, Bissada N, Miao L, Ehrenborg E, Benlian P, Shen GX, Angel A, LeBoeuf RC, Hayden MR (1997) Relationship between lipoprotein lipase and high density lipoprotein cholesterol in mice: modulation by cholesteryl ester transfer protein and dietary status. J Lipid Res 38:2079–2089
Covey SD, Krieger M, Wang W, Penman M, Trigatti BL (2003) Scavenger receptor class B type I-mediated protection against atherosclerosis in LDL receptor-negative mice involves its expression in bone marrow-derived cells. Arterioscler Thromb Vasc Biol 23:1589–1594
Curb JD, Abbott RD, Rodriguez BL, Masaki K, Chen R, Sharp DS, Tall AR (2004) A prospective study of HDL-C and cholesteryl ester transfer protein gene mutations and the risk of coronary heart disease in the elderly. J Lipid Res 45:948–953
Dastani Z, Dangoisse C, Boucher B, Desbiens K, Krimbou L, Dufour R, Hegele RA, Pajukanta P, Engert JC, Genest J, Marcil M (2006) A novel nonsense apolipoprotein A-I mutation (apoA-I(E136X)) causes low HDL cholesterol in French Canadians. Atherosclerosis 185:127–136
Day JR, Albers JJ, Lofton-Day CE, Gilbert TL, Ching AF, Grant FJ, O'Hara PJ, Marcovina SM, Adolphson JL (1994) Complete cDNA encoding human phospholipid transfer protein from human endothelial cells. J Biol Chem 269:9388–9391
de Haan W, van der Hoogt CC, Westerterp M, Hoekstra M, Dallinga-Thie GM, Princen HM, Romijn JA, Jukema JW, Havekes LM, Rensen PC (2008a) Atorvastatin increases HDL cholesterol by reducing CETP expression in cholesterol-fed APOE*3-Leiden.CETP mice. Atherosclerosis 197:57–63
de Haan W, de Vries-van der Weij J, van der Hoorn JW, Gautier T, van der Hoogt CC, Westerterp M, Romijn JA, Jukema JW, Havekes LM, Princen HM, Rensen PC (2008b) Torcetrapib does not reduce atherosclerosis beyond atorvastatin and induces more proinflammatory lesions than atorvastatin. Circulation 117:2515–2522
Desrumaux CM, Mak PA, Boisvert WA, Masson D, Stupack D, Jauhiainen M, Ehnholm C, Curtiss LK (2003) Phospholipid transfer protein is present in human atherosclerotic lesions and is expressed by macrophages and foam cells. J Lipid Res 44:1453–1461
Dole VS, Matuskova J, Vasile E, Yesilaltay A, Bergmeier W, Bernimoulin M, Wagner DD, Krieger M (2008) Thrombocytopenia and platelet abnormalities in high-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol 28:1111–1116
Drayna D, Jarnagin AS, McLean J, Henzel W, Kohr W, Fielding C, Lawn R (1987) Cloning and sequencing of human cholesteryl ester transfer protein cDNA. Nature 327:632–634
Drobnik W, Lindenthal B, Lieser B, Ritter M, Christiansen Weber T, Liebisch G, Giesa U, Igel M, Borsukova H, Büchler C, Fung-Leung WP, Von Bergmann K, Schmitz G (2001) ATP-binding cassette transporter A1 (ABCA1) affects total body sterol metabolism. Gastroenterology 120:1203–1211
El Bouhassani M, Gilibert S, Moreau M, Saint-Charles F, Tréguier M, Poti F, Chapman MJ, Le Goff W, Lesnik P, Huby T (2011) Cholesteryl ester transfer protein expression partially attenuates the adverse effects of SR-BI receptor deficiency on cholesterol metabolism and atherosclerosis. J Biol Chem 286:17227–17238
Fluiter K, van der Westhuijzen DR, van Berkel TJ (1998) In vivo regulation of scavenger receptor BI and the selective uptake of high density lipoprotein cholesteryl esters in rat liver parenchymal and Kupffer cells. J Biol Chem 273:8434–8438
Francone OL, Subbaiah PV, van Tol A, Royer L, Haghpassand M (2003) Abnormal phospholipid composition impairs HDL biogenesis and maturation in mice lacking Abca1. Biochemistry 42:8569–8578
Frikke-Schmidt R, Nordestgaard BG, Stene MC, Sethi AA, Remaley AT, Schnohr P, Grande P, Tybjaerg-Hansen A (2008a) Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA 299:2524–2532
Frikke-Schmidt R, Nordestgaard BG, Jensen GB, Steffensen R, Tybjaerg-Hansen A (2008b) Genetic variation in ABCA1 predicts ischemic heart disease in the general population. Arterioscler Thromb Vasc Biol 28:180–186
Furbee JW Jr, Sawyer JK, Parks JS (2002) Lecithin:cholesterol acyltransferase deficiency increases atherosclerosis in the low density lipoprotein receptor and apolipoprotein E knockout mice. J Biol Chem 277:3511–3519
Furuyama S, Uehara Y, Zhang B, Baba Y, Abe S, Iwamoto T, Miura S, Saku K (2009) Genotypic effect of ABCG1 gene promoter −257T > G polymorphism on coronary artery disease severity in Japanese men. J Atheroscler Thromb 16:194–200
Glomset JA, Norum KR, King W (1970) Plasma lipoproteins in familial lecithin: cholesterol acyltransferase deficiency: lipid composition and reactivity in vitro. J Clin Invest 49:1827–1837
Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR (1977) High density lipoprotein as a protective factor against coronary heart disease. The Framingham study. Am J Med 62:707–714
Gordon JI, Smith DP, Andy R, Alpers DH, Schonfeld G, Strauss AW (1982) The primary translation product of rat intestinal apolipoprotein A-I mRNA is an unusual preproprotein. J Biol Chem 257:971–978
Haghpassand M, Bourassa PA, Francone OL, Aiello RJ (2001) Monocyte/macrophage expression of ABCA1 has minimal contribution to plasma HDL levels. J Clin Invest 108:1315–1320
Harada LM, Amigo L, Cazita PM, Salerno AG, Rigotti AA, Quintão EC, Oliveira HC (2007) CETP expression enhances liver HDL-cholesteryl ester uptake but does not alter VLDL and biliary lipid secretion. Atherosclerosis 191:313–318
Hoekstra M, Kruijt JK, Van Eck M, Van Berkel TJ (2003) Specific gene expression of ATP-binding cassette transporters and nuclear hormone receptors in rat liver parenchymal, endothelial, and Kupffer cells. J Biol Chem 278:25448–25453
Hoekstra M, Meurs I, Koenders M, Out R, Hildebrand RB, Kruijt JK, Van Eck M, Van Berkel TJ (2008) Absence of HDL cholesteryl ester uptake in mice via SR-BI impairs an adequate adrenal glucocorticoid-mediated stress response to fasting. J Lipid Res 49:738–745
Hoekstra M, Ye D, Hildebrand RB, Zhao Y, Lammers B, Stitzinger M, Kuiper J, Van Berkel TJ, Van Eck M (2009) Scavenger receptor class B type I-mediated uptake of serum cholesterol is essential for optimal adrenal glucocorticoid production. J Lipid Res 50:1039–1046
Hoekstra M, Korporaal SJ, van der Sluis RJ, Hirsch-Reinshagen V, Bochem AE, Wellington CL, Van Berkel TJ, Kuivenhoven JA, Van Eck M (2013a) LCAT deficiency in mice is associated with a diminished adrenal glucocorticoid function. J Lipid Res 54:358–364
Hoekstra M, van der Sluis RJ, Van Eck M, Van Berkel TJ (2013b) Adrenal-specific scavenger receptor BI deficiency induces glucocorticoid insufficiency and lowers plasma very-low-density and low-density lipoprotein levels in mice. Arterioscler Thromb Vasc Biol 33:e39–e46
Holm TM, Braun A, Trigatti BL, Brugnara C, Sakamoto M, Krieger M, Andrews NC (2002) Failure of red blood cell maturation in mice with defects in the high-density lipoprotein receptor SR-BI. Blood 99:1817–1824
Hong SH, Rhyne J, Zeller K, Miller M (2002) ABCA1(Alabama): a novel variant associated with HDL deficiency and premature coronary artery disease. Atherosclerosis 164:245–250
Hossain MA, Tsujita M, Akita N, Kobayashi F, Yokoyama S (2009) Cholesterol homeostasis in ABCA1/LCAT double-deficient mouse. Biochim Biophys Acta 1791:1197–1205
Hovingh GK, Brownlie A, Bisoendial RJ, Dube MP, Levels JH, Petersen W, Dullaart RP, Stroes ES, Zwinderman AH, de Groot E, Hayden MR, Kuivenhoven JA, Kastelein JJ (2004) A novel apoA-I mutation (L178P) leads to endothelial dysfunction, increased arterial wall thickness, and premature coronary artery disease. J Am Coll Cardiol 44:1429–1435
Hovingh GK, Hutten BA, Holleboom AG, Petersen W, Rol P, Stalenhoef A, Zwinderman AH, de Groot E, Kastelein JJ, Kuivenhoven JA (2005) Compromised LCAT function is associated with increased atherosclerosis. Circulation 112:879–884
Hsu LA, Ko YL, Wu S, Teng MS, Peng TY, Chen CF, Chen CF, Lee YS (2003) Association between a novel 11-base pair deletion mutation in the promoter region of the scavenger receptor class B type I gene and plasma HDL cholesterol levels in Taiwanese Chinese. Arterioscler Thromb Vasc Biol 23:1869–1874
Huang W, Moriyama K, Koga T, Hua H, Ageta M, Kawabata S, Mawatari K, Imamura T, Eto T, Kawamura M, Teramoto T, Sasaki J (2001) Novel mutations in ABCA1 gene in Japanese patients with Tangier disease and familial high density lipoprotein deficiency with coronary heart disease. Biochim Biophys Acta 1537:71–78
Huby T, Doucet C, Dachet C, Ouzilleau B, Ueda Y, Afzal V, Rubin E, Chapman MJ, Lesnik P (2006) Knockdown expression and hepatic deficiency reveal an atheroprotective role for SR-BI in liver and peripheral tissues. J Clin Invest 116:2767–2776
Huszar D, Varban ML, Rinninger F, Feeley R, Arai T, Fairchild-Huntress V, Donovan MJ, Tall AR (2000) Increased LDL cholesterol and atherosclerosis in LDL receptor-deficient mice with attenuated expression of scavenger receptor B1. Arterioscler Thromb Vasc Biol 20:1068–1073
Ishibashi S, Goldstein JL, Brown MS, Herz J, Burns DK (1994) Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J Clin Invest 93:1885–1893
Ishida BY, Blanche PJ, Nichols AV, Yashar M, Paigen B (1991) Effects of atherogenic diet consumption on lipoproteins in mouse strains C57BL/6 and C3H. J Lipid Res 32:559–568
Ji Y, Wang N, Ramakrishnan R, Sehayek E, Huszar D, Breslow JL, Tall AR (1999) Hepatic scavenger receptor BI promotes rapid clearance of high density lipoprotein free cholesterol and its transport into bile. J Biol Chem 274:33398–33402
Jiang XC, Agellon LB, Walsh A, Breslow JL, Tall A (1992) Dietary cholesterol increases transcription of the human cholesteryl ester transfer protein gene in transgenic mice. Dependence on natural flanking sequences. J Clin Invest 90:1290–1295
Jiang XC, Bruce C, Mar J, Lin M, Ji Y, Francone OL, Tall AR (1999) Targeted mutation of plasma phospholipid transfer protein gene markedly reduces high-density lipoprotein levels. J Clin Invest 103:907–914
Jiang XC, Qin S, Qiao C, Kawano K, Lin M, Skold A, Xiao X, Tall AR (2001) Apolipoprotein B secretion and atherosclerosis are decreased in mice with phospholipid-transfer protein deficiency. Nat Med 7:847–852
Jiang XC, Tall AR, Qin S, Lin M, Schneider M, Lalanne F, Deckert V, Desrumaux C, Athias A, Witztum JL, Lagrost L (2002) Phospholipid transfer protein deficiency protects circulating lipoproteins from oxidation due to the enhanced accumulation of vitamin E. J Biol Chem 277:31850–31856
Kawano K, Qin S, Vieu C, Collet X, Jiang XC (2002) Role of hepatic lipase and scavenger receptor BI in clearing phospholipid/free cholesterol-rich lipoproteins in PLTP-deficient mice. Biochim Biophys Acta 1583:133–140
Klucken J, Büchler C, Orsó E, Kaminski WE, Porsch-Ozcürümez M, Liebisch G, Kapinsky M, Diederich W, Drobnik W, Dean M, Allikmets R, Schmitz G (2000) ABCG1 (ABC8), the human homolog of the Drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc Natl Acad Sci USA 97:817–822
Korporaal SJ, Meurs I, Hauer AD, Hildebrand RB, Hoekstra M, Cate HT, Praticò D, Akkerman JW, Van Berkel TJ, Kuiper J, Van Eck M (2011) Deletion of the high-density lipoprotein receptor scavenger receptor BI in mice modulates thrombosis susceptibility and indirectly affects platelet function by elevation of plasma free cholesterol. Arterioscler Thromb Vasc Biol 31:34–42
Kunnen S, Van Eck M (2012) Lecithin:cholesterol acyltransferase: old friend or foe in atherosclerosis? J Lipid Res 53:1783–1799
Laffitte BA, Joseph SB, Chen M, Castrillo A, Repa J, Wilpitz D, Mangelsdorf D, Tontonoz P (2003) The phospholipid transfer protein gene is a liver X receptor target expressed by macrophages in atherosclerotic lesions. Mol Cell Biol 23:2182–2191
Lambert G, Sakai N, Vaisman BL, Neufeld EB, Marteyn B, Chan CC, Paigen B, Lupia E, Thomas A, Striker LJ, Blanchette-Mackie J, Csako G, Brady JN, Costello R, Striker GE, Remaley AT, Brewer HB Jr, Santamarina-Fojo S (2001) Analysis of glomerulosclerosis and atherosclerosis in lecithin cholesterol acyltransferase-deficient mice. J Biol Chem 276:15090–15098
Lammers B, Zhao Y, Hoekstra M, Hildebrand RB, Ye D, Meurs I, Van Berkel TJ, Van Eck M (2011) Augmented atherogenesis in LDL receptor deficient mice lacking both macrophage ABCA1 and ApoE. PLoS ONE 6:e26095
Lammers B, Zhao Y, Foks AC, Hildebrand RB, Kuiper J, Van Berkel TJ, Van Eck M (2012) Leukocyte ABCA1 remains atheroprotective in splenectomized LDL receptor knockout mice. PLoS ONE 7:e48080
Lawn RM, Wade DP, Couse TL, Wilcox JN (2001) Localization of human ATP-binding cassette transporter 1 (ABC1) in normal and atherosclerotic tissues. Arterioscler Thromb Vasc Biol 21:378–385
Lee RG, Kelley KL, Sawyer JK, Farese RV Jr, Parks JS, Rudel LL (2004) Plasma cholesteryl esters provided by lecithin: cholesterol acyltransferase and acyl-coenzyme a:cholesterol acyltransferase 2 have opposite atherosclerotic potential. Circ Res 95:998–1004
Lee EY, Klementowicz PT, Hegele RA, Asztalos BF, Schaefer EJ (2013) HDL deficiency due to a new insertion mutation (ApoA-INashua) and review of the literature. J Clin Lipidol 7:169–173
Lee-Rueckert M, Vikstedt R, Metso J, Ehnholm C, Kovanen PT, Jauhiainen M (2006) Absence of endogenous phospholipid transfer protein impairs ABCA1-dependent efflux of cholesterol from macrophage foam cells. J Lipid Res 47:1725–1732
Leren TP, Bakken KS, Daum U, Ose L, Berg K, Assmann G, von Eckardstein A (1997) Heterozygosity for apolipoprotein A-I(R160L)Oslo is associated with low levels of high density lipoprotein cholesterol and HDL-subclass LpA-I/A-II but normal levels of HDL-subclass LpA-I. J Lipid Res 38:121–131
Li H, Reddick RL, Maeda N (1993) Lack of apoA-I is not associated with increased susceptibility to atherosclerosis in mice. Arterioscler Thromb 13:1814–1821
Li L, Naples M, Song H, Yuan R, Ye F, Shafi S, Adeli K, Ng DS (2007) LCAT-null mice develop improved hepatic insulin sensitivity through altered regulation of transcription factors and suppressors of cytokine signaling. Am J Physiol Endocrinol Metab 293:E587–E594
Li Z, Wang Y, van der Sluis RJ, van der Hoorn JW, Princen HM, Van Eck M, Van Berkel TJ, Rensen PC, Hoekstra M (2012) Niacin reduces plasma CETP levels by diminishing liver macrophage content in CETP transgenic mice. Biochem Pharmacol 84:821–829
Liu R, Iqbal J, Yeang C, Wang DQ, Hussain MM, Jiang XC (2007) Phospholipid transfer protein-deficient mice absorb less cholesterol. Arterioscler Thromb Vasc Biol 27:2014–2021
Lusa S, Jauhiainen M, Metso J, Somerharju P, Ehnholm C (1996) The mechanism of human plasma phospholipid transfer protein-induced enlargement of high-density lipoprotein particles: evidence for particle fusion. Biochem J 313:275–282
Malerød L, Juvet K, Gjøen T, Berg T (2002) The expression of scavenger receptor class B, type I (SR-BI) and caveolin-1 in parenchymal and nonparenchymal liver cells. Cell Tissue Res 307:173–180
Marnane M, Prendeville S, McDonnell C, Noone I, Barry M, Crowe M, Mulligan N, Kelly PJ (2014) Plaque inflammation and unstable morphology are associated with early stroke recurrence in symptomatic carotid stenosis. Stroke 45:801–806
Masucci-Magoulas L, Plump A, Jiang XC, Walsh A, Breslow JL, Tall AR (1996) Profound induction of hepatic cholesteryl ester transfer protein transgene expression in apolipoprotein E and low density lipoprotein receptor gene knockout mice. A novel mechanism signals changes in plasma cholesterol levels. J Clin Invest 97:154–161
Mathis D, Shoelson SE (2011) Immunometabolism: an emerging frontier. Nat Rev Immunol 11:81
Matsunaga A, Sasaki J, Han H, Huang W, Kugi M, Koga T, Ichiki S, Shinkawa T, Arakawa K (1999) Compound heterozygosity for an apolipoprotein A1 gene promoter mutation and a structural nonsense mutation with apolipoprotein A1 deficiency. Arterioscler Thromb Vasc Biol 19:348–355
Meurs I, Hoekstra M, van Wanrooij EJ, Hildebrand RB, Kuiper J, Kuipers F, Hardeman MR, Van Berkel TJ, Van Eck M (2005) HDL cholesterol levels are an important factor for determining the lifespan of erythrocytes. Exp Hematol 33:1309–1319
Meurs I, Lammers B, Zhao Y, Out R, Hildebrand RB, Hoekstra M, Van Berkel TJ, Van Eck M (2012) The effect of ABCG1 deficiency on atherosclerotic lesion development in LDL receptor knockout mice depends on the stage of atherogenesis. Atherosclerosis 221:41–47
Miettinen HE, Rayburn H, Krieger M (2001) Abnormal lipoprotein metabolism and reversible female infertility in HDL receptor (SR-BI)-deficient mice. J Clin Invest 108:1717–1722
Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, Barnes EH, Voysey M, Gray A, Collins R, Baigent C, Cholesterol Treatment Trialists’ (CTT) Collaborators (2012) The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet 380:581–590
Moore RE, Kawashiri MA, Kitajima K, Secreto A, Millar JS, Pratico D, Rader DJ (2003) Apolipoprotein A-I deficiency results in markedly increased atherosclerosis in mice lacking the LDL receptor. Arterioscler Thromb Vasc Biol 23:1914–1920
Ng DS, Francone OL, Forte TM, Zhang J, Haghpassand M, Rubin EM (1997) Disruption of the murine lecithin: cholesterol acyltransferase gene causes impairment of adrenal lipid delivery and up-regulation of scavenger receptor class B type I. J Biol Chem 272:15777–15781
Ng DS, Maguire GF, Wylie J, Ravandi A, Xuan W, Ahmed Z, Eskandarian M, Kuksis A, Connelly PW (2002) Oxidative stress is markedly elevated in lecithin: cholesterol acyltransferase-deficient mice and is paradoxically reversed in the apolipoprotein E knockout background in association with a reduction in atherosclerosis. J Biol Chem 277:11715–11720
Ng DS, Xie C, Maguire GF, Zhu X, Ugwu F, Lam E, Connelly PW (2004) Hypertriglyceridemia in lecithin-cholesterol acyltransferase-deficient mice is associated with hepatic overproduction of triglycerides, increased lipogenesis, and improved glucose tolerance. J Biol Chem 279:7636–7642
O'Brien KD, Vuletic S, McDonald TO, Wolfbauer G, Lewis K, Tu AY, Marcovina S, Wight TN, Chait A, Albers JJ (2003) Cell-associated and extracellular phospholipid transfer protein in human coronary atherosclerosis. Circulation 108:270–274
Ogier N, Klein A, Deckert V, Athias A, Bessède G, Le Guern N, Lagrost L, Desrumaux C (2007) Cholesterol accumulation is increased in macrophages of phospholipid transfer protein-deficient mice: normalization by dietary alpha-tocopherol supplementation. Arterioscler Thromb Vasc Biol 27:2407–2412
Oliveira HC, Ma L, Milne R, Marcovina SM, Inazu A, Mabuchi H, Tall AR (1997) Cholesteryl ester transfer protein activity enhances plasma cholesteryl ester formation. Studies in CETP transgenic mice and human genetic CETP deficiency. Arterioscler Thromb Vasc Biol 17:1045–1052
Orsó E, Broccardo C, Kaminski WE, Böttcher A, Liebisch G, Drobnik W, Götz A, Chambenoit O, Diederich W, Langmann T, Spruss T, Luciani MF, Rothe G, Lackner KJ, Chimini G, Schmitz G (2000) Transport of lipids from golgi to plasma membrane is defective in tangier disease patients and Abc1-deficient mice. Nat Genet 24:192–196
Out R, Hoekstra M, Spijkers JA, Kruijt JK, van Eck M, Bos IS, Twisk J, Van Berkel TJ (2004) Scavenger receptor class B type I is solely responsible for the selective uptake of cholesteryl esters from HDL by the liver and the adrenals in mice. J Lipid Res 45:2088–2095
Out R, Hoekstra M, Hildebrand RB, Kruit JK, Meurs I, Li Z, Kuipers F, Van Berkel TJ, Van Eck M (2006) Macrophage ABCG1 deletion disrupts lipid homeostasis in alveolar macrophages and moderately influences atherosclerotic lesion development in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol 26:2295–2300
Out R, Hoekstra M, Meurs I, de Vos P, Kuiper J, Van Eck M, Van Berkel TJ (2007) Total body ABCG1 expression protects against early atherosclerotic lesion development in mice. Arterioscler Thromb Vasc Biol 27:594–599
Out R, Jessup W, Le Goff W, Hoekstra M, Gelissen IC, Zhao Y, Kritharides L, Chimini G, Kuiper J, Chapman MJ, Huby T, Van Berkel TJ, Van Eck M (2008a) Coexistence of foam cells and hypocholesterolemia in mice lacking the ABC transporters A1 and G1. Circ Res 102:113–120
Out R, Hoekstra M, Habets K, Meurs I, de Waard V, Hildebrand RB, Wang Y, Chimini G, Kuiper J, Van Berkel TJ, Van Eck M (2008b) Combined deletion of macrophage ABCA1 and ABCG1 leads to massive lipid accumulation in tissue macrophages and distinct atherosclerosis at relatively low plasma cholesterol levels. Arterioscler Thromb Vasc Biol 28:258–264
Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA (1987) Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis 68:231–240
Pape ME, Rehberg EF, Marotti KR, Melchior GW (1991) Molecular cloning, sequence, and expression of cynomolgus monkey cholesteryl ester transfer protein. Inverse correlation between hepatic cholesteryl ester transfer protein mRNA levels and plasma high density lipoprotein levels. Arterioscler Thromb 11:1759–1771
Parks JS, Li H, Gebre AK, Smith TL, Maeda N (1995) Effect of apolipoprotein A-I deficiency on lecithin: cholesterol acyltransferase activation in mouse plasma. J Lipid Res 36:349–355
Pei Y, Chen X, Aboutouk D, Fuller MT, Dadoo O, Yu P, White EJ, Igdoura SA, Trigatti BL (2013) SR-BI in bone marrow derived cells protects mice from diet induced coronary artery atherosclerosis and myocardial infarction. PLoS ONE 8:e72492
Plump AS, Erickson SK, Weng W, Partin JS, Breslow JL, Williams DL (1996) Apolipoprotein A-I is required for cholesteryl ester accumulation in steroidogenic cells and for normal adrenal steroid production. J Clin Invest 97:2660–2671
Plump AS, Azrolan N, Odaka H, Wu L, Jiang X, Tall A, Eisenberg S, Breslow JL (1997) ApoA-I knockout mice: characterization of HDL metabolism in homozygotes and identification of a post-RNA mechanism of apoA-I up-regulation in heterozygotes. J Lipid Res 38:1033–1047
Plump AS, Masucci-Magoulas L, Bruce C, Bisgaier CL, Breslow JL, Tall AR (1999) Increased atherosclerosis in ApoE and LDL receptor gene knock-out mice as a result of human cholesteryl ester transfer protein transgene expression. Arterioscler Thromb Vasc Biol 19:1105–1110
Qin S, Kawano K, Bruce C, Lin M, Bisgaier C, Tall AR, Jiang X (2000) Phospholipid transfer protein gene knock-out mice have low high density lipoprotein levels, due to hypercatabolism, and accumulate apoA-IV-rich lamellar lipoproteins. J Lipid Res 41:269–276
Quinet EM, Agellon LB, Kroon PA, Marcel YL, Lee YC, Whitlock ME, Tall AR (1990) Atherogenic diet increases cholesteryl ester transfer protein messenger RNA levels in rabbit liver. J Clin Invest 85:357–363
Ranalletta M, Wang N, Han S, Yvan-Charvet L, Welch C, Tall AR (2006) Decreased atherosclerosis in low-density lipoprotein receptor knockout mice transplanted with Abcg1-/- bone marrow. Arterioscler Thromb Vasc Biol 26:2308–2315
Redgrave JN, Lovett JK, Gallagher PJ, Rothwell PM (2006) Histological assessment of 526 symptomatic carotid plaques in relation to the nature and timing of ischemic symptoms: the Oxford plaque study. Circulation 113:2320–2328
Rhainds D, Brodeur M, Lapointe J, Charpentier D, Falstrault L, Brissette L (2003) The role of human and mouse hepatic scavenger receptor class B type I (SR-BI) in the selective uptake of low-density lipoprotein-cholesteryl esters. Biochemistry 42:7527–7538
Rhyne J, Ryan MJ, White C, Chimonas T, Miller M (2006) The two novel CETP mutations Gln87X and Gln165X in a compound heterozygous state are associated with marked hyperalphalipoproteinemia and absence of significant coronary artery disease. J Mol Med (Berl) 84:647–650
Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M (1997) A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci USA 94:12610–12615
Sahoo D, Trischuk TC, Chan T, Drover VA, Ho S, Chimini G, Agellon LB, Agnihotri R, Francis GA, Lehner R (2004) ABCA1-dependent lipid efflux to apolipoprotein A-I mediates HDL particle formation and decreases VLDL secretion from murine hepatocytes. J Lipid Res 45:1122–1131
Sakai N, Vaisman BL, Koch CA, Hoyt RF Jr, Meyn SM, Talley GD, Paiz JA, Brewer HB Jr, Santamarina-Fojo S (1997) Targeted disruption of the mouse lecithin: cholesterol acyltransferase (LCAT) gene. Generation of a new animal model for human LCAT deficiency. J Biol Chem 272:7506–7510
Salerno AG, Patrício PR, Berti JA, Oliveira HC (2009) Cholesteryl ester transfer protein (CETP) increases postprandial triglyceridaemia and delays triacylglycerol plasma clearance in transgenic mice. Biochem J 419:629–634
Santamarina-Fojo S, Lambert G, Hoeg JM, Brewer HB Jr (2000) Lecithin-cholesterol acyltransferase: role in lipoprotein metabolism, reverse cholesterol transport and atherosclerosis. Curr Opin Lipidol 11:267–275
Santander NG, Contreras-Duarte S, Awad MF, Lizama C, Passalacqua I, Rigotti A, Busso D (2013) Developmental abnormalities in mouse embryos lacking the HDL receptor SR-BI. Hum Mol Genet 22:1086–1096
Scanu A, Toth J, Edelstein C, Koga S, Stiller E (1969) Fractionation of human serum high density lipoprotein in urea solutions. Evidence for polypeptide heterogeneity. Biochemistry 8:3309–3316
Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Mundl H, Nicholls SJ, Shah PK, Tardif JC, Wright RS, dal-OUTCOMES Investigators (2012) Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med 367:2089–2099
Shelly L, Royer L, Sand T, Jensen H, Luo Y (2008) Phospholipid transfer protein deficiency ameliorates diet-induced hypercholesterolemia and inflammation in mice. J Lipid Res 49:773–781
Siggins S, Bykov I, Hermansson M, Somerharju P, Lindros K, Miettinen TA, Jauhiainen M, Olkkonen VM, Ehnholm C (2007) Altered hepatic lipid status and apolipoprotein A-I metabolism in mice lacking phospholipid transfer protein. Atherosclerosis 190:114–123
Singaraja RR, Stahmer B, Brundert M, Merkel M, Heeren J, Bissada N, Kang M, Timmins JM, Ramakrishnan R, Parks JS, Hayden MR, Rinninger F (2006) Hepatic ATP-binding cassette transporter A1 is a key molecule in high-density lipoprotein cholesteryl ester metabolism in mice. Arterioscler Thromb Vasc Biol 26:1821–1827
Tall AR, Krumholz S, Olivecrona T, Deckelbaum RJ (1985) Plasma phospholipid transfer protein enhances transfer and exchange of phospholipids between very low density lipoproteins and high density lipoproteins during lipolysis. J Lipid Res 26:842–851
Tarling EJ, Edwards PA (2011) ATP binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. Proc Natl Acad Sci USA 108:19719–19724
Tilly-Kiesi M, Zhang Q, Ehnholm S, Kahri J, Lahdenperä S, Ehnholm C, Taskinen MR (1995) ApoA-IHelsinki (Lys107–> 0) associated with reduced HDL cholesterol and LpA-I:A-II deficiency. Arterioscler Thromb Vasc Biol 15:1294–1306
Timmins JM, Lee JY, Boudyguina E, Kluckman KD, Brunham LR, Mulya A, Gebre AK, Coutinho JM, Colvin PL, Smith TL, Hayden MR, Maeda N, Parks JS (2005) Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J Clin Invest 115:1333–1342
Tomimoto S, Tsujita M, Okazaki M, Usui S, Tada T, Fukutomi T, Ito S, Itoh M, Yokoyama S (2001) Effect of probucol in lecithin-cholesterol acyltransferase-deficient mice: inhibition of 2 independent cellular cholesterol-releasing pathways in vivo. Arterioscler Thromb Vasc Biol 21:394–400
Trigatti B, Rayburn H, Viñals M, Braun A, Miettinen H, Penman M, Hertz M, Schrenzel M, Amigo L, Rigotti A, Krieger M (1999) Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc Natl Acad Sci USA 96:9322–9327
Valenta DT, Ogier N, Bradshaw G, Black AS, Bonnet DJ, Lagrost L, Curtiss LK, Desrumaux CM (2006) Atheroprotective potential of macrophage-derived phospholipid transfer protein in low-density lipoprotein receptor-deficient mice is overcome by apolipoprotein AI overexpression. Arterioscler Thromb Vasc Biol 26:1572–1578
van den Maagdenberg AM, Hofker MH, Krimpenfort PJ, de Bruijn I, van Vlijmen B, van der Boom H, Havekes LM, Frants RR (1993) Transgenic mice carrying the apolipoprotein E3-Leiden gene exhibit hyperlipoproteinemia. J Biol Chem 268:10540–10545
van der Hoogt CC, de Haan W, Westerterp M, Hoekstra M, Dallinga-Thie GM, Romijn JA, Princen HM, Jukema JW, Havekes LM, Rensen PC (2007) Fenofibrate increases HDL-cholesterol by reducing cholesteryl ester transfer protein expression. J Lipid Res 48:1763–1771
van der Hoorn JW, de Haan W, Berbée JF, Havekes LM, Jukema JW, Rensen PC, Princen HM (2008) Niacin increases HDL by reducing hepatic expression and plasma levels of cholesteryl ester transfer protein in APOE*3Leiden.CETP mice. Arterioscler Thromb Vasc Biol 28:2016–2022
Van Eck M, Van Berkel TJ (2013) ATP-binding cassette transporter A1 in lipoprotein metabolism and atherosclerosis: a new piece of the complex puzzle. Arterioscler Thromb Vasc Biol 33:2281–2283
Van Eck M, Bos IS, Kaminski WE, Orsó E, Rothe G, Twisk J, Böttcher A, Van Amersfoort ES, Christiansen-Weber TA, Fung-Leung WP, Van Berkel TJ, Schmitz G (2002) Leukocyte ABCA1 controls susceptibility to atherosclerosis and macrophage recruitment into tissues. Proc Natl Acad Sci USA 99:6298–6303
Van Eck M, Twisk J, Hoekstra M, Van Rij BT, Van der Lans CA, Bos IS, Kruijt JK, Kuipers F, Van Berkel TJ (2003) Differential effects of scavenger receptor BI deficiency on lipid metabolism in cells of the arterial wall and in the liver. J Biol Chem 278:23699–23705
Van Eck M, Bos IS, Hildebrand RB, Van Rij BT, Van Berkel TJ (2004) Dual role for scavenger receptor class B, type I on bone marrow-derived cells in atherosclerotic lesion development. Am J Pathol 165:785–794
Van Eck M, Ye D, Hildebrand RB, Kar Kruijt J, de Haan W, Hoekstra M, Rensen PC, Ehnholm C, Jauhiainen M, Van Berkel TJ (2007) Important role for bone marrow-derived cholesteryl ester transfer protein in lipoprotein cholesterol redistribution and atherosclerotic lesion development in LDL receptor knockout mice. Circ Res 100:678–685
Van Eck M, Hoekstra M, Out R, Bos IS, Kruijt JK, Hildebrand RB, Van Berkel TJ (2008) Scavenger receptor BI facilitates the metabolism of VLDL lipoproteins in vivo. J Lipid Res 49:136–146
van Vlijmen BJ, van den Maagdenberg AM, Gijbels MJ, van der Boom H, HogenEsch H, Frants RR, Hofker MH, Havekes LM (1994) Diet-induced hyperlipoproteinemia and atherosclerosis in apolipoprotein E3-Leiden transgenic mice. J Clin Invest 93:1403–1410
Varban ML, Rinninger F, Wang N, Fairchild-Huntress V, Dunmore JH, Fang Q, Gosselin ML, Dixon KL, Deeds JD, Acton SL, Tall AR, Huszar D (1998) Targeted mutation reveals a central role for SR-BI in hepatic selective uptake of high density lipoprotein cholesterol. Proc Natl Acad Sci USA 95:4619–4624
Vergeer M, Korporaal SJ, Franssen R, Meurs I, Out R, Hovingh GK, Hoekstra M, Sierts JA, Dallinga-Thie GM, Motazacker MM, Holleboom AG, Van Berkel TJ, Kastelein JJ, Van Eck M, Kuivenhoven JA (2011) Genetic variant of the scavenger receptor BI in humans. N Engl J Med 364:136–1345
Vikstedt R, Ye D, Metso J, Hildebrand RB, Van Berkel TJ, Ehnholm C, Jauhiainen M, Van Eck M (2007) Macrophage phospholipid transfer protein contributes significantly to total plasma phospholipid transfer activity and its deficiency leads to diminished atherosclerotic lesion development. Arterioscler Thromb Vasc Biol 27:578–586
Wada M, Iso T, Asztalos BF, Takama N, Nakajima T, Seta Y, Kaneko K, Taniguchi Y, Kobayashi H, Nakajima K, Schaefer EJ, Kurabayashi M (2009) Marked high density lipoprotein deficiency due to apolipoprotein A-I Tomioka (codon 138 deletion). Atherosclerosis 207:157–161
Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH, Tall AR, Rader DJ (2007) Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest 117:2216–2224
West M, Greason E, Kolmakova A, Jahangiri A, Asztalos B, Pollin TI, Rodriguez A (2009) Scavenger receptor class B type I protein as an independent predictor of high-density lipoprotein cholesterol levels in subjects with hyperalphalipoproteinemia. J Clin Endocrinol Metab 94:1451–1457
Westerterp M, van der Hoogt CC, de Haan W, Offerman EH, Dallinga-Thie GM, Jukema JW, Havekes LM, Rensen PC (2006) Cholesteryl ester transfer protein decreases high-density lipoprotein and severely aggravates atherosclerosis in APOE*3-Leiden mice. Arterioscler Thromb Vasc Biol 26:2552–2559
Wiersma H, Nijstad N, de Boer JF, Out R, Hogewerf W, Van Berkel TJ, Kuipers F, Tietge UJ (2009) Lack of Abcg1 results in decreased plasma HDL cholesterol levels and increased biliary cholesterol secretion in mice fed a high cholesterol diet. Atherosclerosis 206:141–147
Wilhelm AJ, Zabalawi M, Grayson JM, Weant AE, Major AS, Owen J, Bharadwaj M, Walzem R, Chan L, Oka K, Thomas MJ, Sorci-Thomas MG (2009) Apolipoprotein A-I and its role in lymphocyte cholesterol homeostasis and autoimmunity. Arterioscler Thromb Vasc Biol 29:843–849
Williamson R, Lee D, Hagaman J, Maeda N (1992) Marked reduction of high density lipoprotein cholesterol in mice genetically modified to lack apolipoprotein A-I. Proc Natl Acad Sci USA 89:7134–7138
Wojcik AJ, Skaflen MD, Srinivasan S, Hedrick CC (2008) A critical role for ABCG1 in macrophage inflammation and lung homeostasis. J Immunol 180:4273–4282
Yan D, Navab M, Bruce C, Fogelman AM, Jiang XC (2004) PLTP deficiency improves the anti-inflammatory properties of HDL and reduces the ability of LDL to induce monocyte chemotactic activity. J Lipid Res 45:1852–1858
Yates M, Kolmakova A, Zhao Y, Rodriguez A (2011) Clinical impact of scavenger receptor class B type I gene polymorphisms on human female fertility. Hum Reprod 26:1910–1916
Yazdanyar A, Jiang XC (2012) Liver phospholipid transfer protein (PLTP) expression with a PLTP-null background promotes very low-density lipoprotein production in mice. Hepatology 56:576–584
Yazdanyar A, Quan W, Jin W, Jiang XC (2013) Liver-specific phospholipid transfer protein deficiency reduces high-density lipoprotein and non-high-density lipoprotein production in mice. Arterioscler Thromb Vasc Biol 33:2058–2064
Ye D, Hoekstra M, Out R, Meurs I, Kruijt JK, Hildebrand RB, Van Berkel TJ, Van Eck M (2008) Hepatic cell-specific ATP-binding cassette (ABC) transporter profiling identifies putative novel candidates for lipid homeostasis in mice. Atherosclerosis 196:650–658
Yeang C, Qin S, Chen K, Wang DQ, Jiang XC (2010) Diet-induced lipid accumulation in phospholipid transfer protein-deficient mice: its atherogenicity and potential mechanism. J Lipid Res 51:2993–3002
Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, Welch C, Tall AR (2007) Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest 117:3900–3908
Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR (2008) Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation 118:1837–1847
Yvan-Charvet L, Pagler TA, Seimon TA, Thorp E, Welch CL, Witztum JL, Tabas I, Tall AR (2010a) ABCA1 and ABCG1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ Res 106:1861–1869
Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, Tall AR (2010b) ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 328:1689–1693
Zabalawi M, Bhat S, Loughlin T, Thomas MJ, Alexander E, Cline M, Bullock B, Willingham M, Sorci-Thomas MG (2003) Induction of fatal inflammation in LDL receptor and ApoA-I double-knockout mice fed dietary fat and cholesterol. Am J Pathol 163:1201–1213
Zannis VI, Karathanasis SK, Keutmann HT, Goldberger G, Breslow JL (1983) Intracellular and extracellular processing of human apolipoprotein A-I: secreted apolipoprotein A-I isoprotein 2 is a propeptide. Proc Natl Acad Sci USA 80:2574–2578
Zhang W, Yancey PG, Su YR, Babaev VR, Zhang Y, Fazio S, Linton MF (2003) Inactivation of macrophage scavenger receptor class B type I promotes atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation 108:2258–2263
Zhang Y, Da Silva JR, Reilly M, Billheimer JT, Rothblat GH, Rader DJ (2005a) Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J Clin Invest 115:2870–2874
Zhang S, Picard MH, Vasile E, Zhu Y, Raffai RL, Weisgraber KH, Krieger M (2005b) Diet-induced occlusive coronary atherosclerosis, myocardial infarction, cardiac dysfunction, and premature death in scavenger receptor class B type I-deficient, hypomorphic apolipoprotein ER61 mice. Circulation 111:3457–3464
Zhao Y, Pennings M, Hildebrand RB, Ye D, Calpe-Berdiel L, Out R, Kjerrulf M, Hurt-Camejo E, Groen AK, Hoekstra M, Jessup W, Chimini G, Van Berkel TJ, Van Eck M (2010) Enhanced foam cell formation, atherosclerotic lesion development, and inflammation by combined deletion of ABCA1 and SR-BI in Bone marrow-derived cells in LDL receptor knockout mice on western-type diet. Circ Res 107:e20–e31
Zhao Y, Pennings M, Vrins CL, Calpe-Berdiel L, Hoekstra M, Kruijt JK, Ottenhoff R, Hildebrand RB, van der Sluis R, Jessup W, Le Goff W, Chapman MJ, Huby T, Groen AK, Van Berkel TJ, Van Eck M (2011) Hypocholesterolemia, foam cell accumulation, but no atherosclerosis in mice lacking ABC-transporter A1 and scavenger receptor BI. Atherosclerosis 218:314–322
Zheng KQ, Zhang SZ, Zhang L, Huang DJ, Liao LC, Hou YP (2004) A novel missense mutation (L296 Q) in cholesteryl ester transfer protein gene related to coronary heart disease. Acta Biochim Biophys Sin (Shanghai) 36:33–36
Zhong S, Sharp DS, Grove JS, Bruce C, Yano K, Curb JD, Tall AR (1996) Increased coronary heart disease in Japanese-American men with mutation in the cholesteryl ester transfer protein gene despite increased HDL levels. J Clin Invest 97(12):2917–2923
Zhou H, Li Z, Silver DL, Jiang XC (2006) Cholesteryl ester transfer protein (CETP) expression enhances HDL cholesteryl ester liver delivery, which is independent of scavenger receptor BI, LDL receptor related protein and possibly LDL receptor. Biochim Biophys Acta 1761:1482–1488
Zhu X, Lee JY, Timmins JM, Brown JM, Boudyguina E, Mulya A, Gebre AK, Willingham MC, Hiltbold EM, Mishra N, Maeda N, Parks JS (2008) Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem 283:22930–22941
Zhu X, Westcott MM, Bi X, Liu M, Gowdy KM, Seo J, Cao Q, Gebre AK, Fessler MB, Hiltbold EM, Parks JS (2012) Myeloid cell-specific ABCA1 deletion protects mice from bacterial infection. Circ Res 111:1398–1409
Acknowledgments
This work was supported by the European COST Action BM0904 “HDL–From Biological Understanding to Clinical Exploitation,” the Netherlands Heart Foundation (Established Investigator Grant 2007 T056 to M. Van Eck and Senior Postdoc Grants 2008 T70 and 2012 T80 to M. Hoekstra), and VICI Grant 91813603 from the Netherlands Organization for Scientific Research.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this chapter
Cite this chapter
Hoekstra, M., Van Eck, M. (2015). Mouse Models of Disturbed HDL Metabolism. In: von Eckardstein, A., Kardassis, D. (eds) High Density Lipoproteins. Handbook of Experimental Pharmacology, vol 224. Springer, Cham. https://doi.org/10.1007/978-3-319-09665-0_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-09665-0_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-09664-3
Online ISBN: 978-3-319-09665-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)