
Abstract
Chronic GVHD (cGVHD) is the most relevant cause of late non-relapse morbidity and subsequent mortality (approximately 25%) following allo-HSCT (Grube et al. 2016).
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


1 Introduction
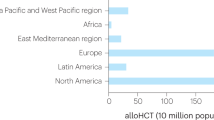
Chronic GVHD (cGVHD) is the most relevant cause of late non-relapse morbidity and subsequent mortality (approximately 25%) following allo-HSCT (Grube et al. 2016). Its incidence is approximately 50% among all patients following allo-HSCT and has increased during the last two decades due to increasing patient age and increasing use of unrelated and/or mismatched donors, RIC regimens, and PBSC (Arai et al. 2015). While the incidence of cGVHD is lower (20–40%) in children, its incidence rises to 60% as age increases (Baird et al. 2010).
The pathophysiology of cGVHD is different from aGVHD and mainly characterized by impaired immune tolerance mechanisms affecting innate and adaptive immunity. Both autoreactive and alloreactive donor-derived T and B cells play a role (Cooke et al. 2017). Other pathophysiological factors are indirect presentations of alloantigens through antigen-presenting donor cells and mechanisms of chronic inflammation with subsequent scar formation and fibrosis. One important aspect of GVHD pathophysiology is the variability of immune reconstitution, which is age-related and dependent on thymic function and hormones. This adds to the unpredictability of the effects of transplant procedures and complications in a very heterogenous cohort of children and adolescents with malignant and nonmalignant diseases.
Known risk factors for adult and pediatric cGVHD are unrelated and/or mismatched donor, PBSCs as donor source, older donor age, female donor into male recipient, and the use of total body irradiation (Baird et al. 2010). By far the strongest predictor is the history and severity of acute GVHD.
In addition to the harm it causes, cGVHD also has a protective effect, as patients with cGVHD have lower rates of recurrence of their underlying malignant disease (Grube et al. 2016). Overall survival of patients transplanted for malignant diseases developing mild cGVHD is therefore better compared to patients without cGVHD. Even OS of patients with moderate cGVHD is not different from patients without cGVHD, as the slightly increased mortality associated with cGVHD is counterbalanced by lower disease-associated mortality (Kuzmina et al. 2012).
In contrast, the long-term mortality rate of patients with severe cGVHD is as high as 50% taken into account that the severity is less relevant compared to certain risk factors for mortality consisting of low platelets at diagnosis of cGVHD, the direct progression of acute GVHD into cGVHD (progressive onset), and certain organ manifestations (lung, gastrointestinal and cholestatic liver involvement) (Grube et al. 2016). One important pediatric aspect involves the high proportion (up to 50%) of nonmalignant underlying diseases as HSCT indication. While malignant diseases benefit from the graft-versus-malignancy effect induced by GVHD, it only offers harm for the nonmalignant diseases. In daily clinical routine, this fact influences GVHD prophylaxis and treatment both in regard to intensity and duration of immunosuppressants (Lawitschka et al., data of a survey by the EBMT pediatric diseases WP, submitted). However, prospective pediatric data of immune reconstitution in GVHD patients evaluating the influence of underlying diseases are scarce.
2 Clinical Manifestations
cGVHD usually begins between 3 months and 2 years after HSCT, but earlier onset (at least 1 month after transplantation) is possible (Jagasia et al. 2015). Besides classical manifestations, cGVHD can imitate almost any autoimmune disease, such as myasthenia gravis and myositis. As cGVHD can affect a number of organs, and patients often do not report changes until functional impairment is recognized, regular examination of all organs potentially affected is essential. The following section describes the most common clinical organ manifestations of cGVHD. In general, pediatric manifestations are similar to adult cGVHD; when indicated, specific aspects are shortly described.
2.1 Skin
The skin is the most frequently involved organ with different morphology, depending on the different skin layers (epidermis, cutis, subcutis, and fasciae) involved. Some manifestations may overlap with acute GVHD like erythema, maculopapular rash, and pruritus. Cutaneous cGVHD may show many different non-sclerotic and sclerotic phenotypes often simulating well-known chronic inflammatory and autoimmune diseases (Strong Rodrigues et al. 2018).
Diagnostic features of NIH-defined cGVHD include poikiloderma, lichen planus-like, lichen sclerosus-like, morphea-like, and deep sclerotic eruptions, and no biopsy is needed to confirm the diagnosis. Distinctive for cGVHD, other or common skin manifestations like depigmentation and papulosquamous lesions or ichthyosis, keratosis pilaris, pigmental changes, loss of skin appendages, and sweat impairment are not sufficient for diagnosis and require histopathological confirmation if no diagnostic signs in the skin or other organs are present (Jagasia et al. 2015).
In pediatric patients, the incidence of viral reactivation and infection seems higher (although only proven for some viruses), and therefore infection has to be ruled out. Viral skin infections can worsen or activate cGVHD (Jacobsohn 2010). Premature graying of the hair is even in small children common, possibly together with seborrheic scalp changes. Of note, if sweat glands are destroyed, this may be of importance for phototherapy because of the inability to sweat with consequent hyperthermia.
2.2 Eyes
cGVHD of the eyes usually manifests as keratitis sicca. In addition to atrophy of the lacrimal gland with subsequent tear deficiency (sicca syndrome), the meibomian glands and eyelids are often affected by severe blepharitis which may initially present with tearing. Around the conjunctiva there are often not only fibrotic alterations but also chronic persistent inflammation with visible erythema of the conjunctiva. As dry eye symptoms are rarely communicated by children, light sensitivity is the predominant symptom, sometimes with excessive eye rubbing. Infections have to be ruled out. Referral to a pediatric experienced ophthalmologist is recommended.
2.3 Oral Mucosa
Oral manifestations may appear as erythema or lichenoid changes (the latter are regarded as diagnostic) of the oral mucosa as well as ulcera and mucoceles. Sicca symptoms may result from destruction of the salivary glands. Long-term cGVHD may lead to gingivitis, periodontitis, increased tooth decay, and tooth loss. In children excessive drinking during eating may be the first symptom of oral involvement. Not only mucosal problems but abnormal teeth development (e.g., hypodontia, root malformation, enamel hypoplasia) and caries are often seen as secondary symptoms in infants.
2.4 Liver
Liver involvement manifests as cholestasis and may resemble primary biliary cirrhosis, but hepatitic forms with high transaminases are also possible. Other factors, such as viral infections (hepatitis A, B, C, and E, CMV, EBV, ADV, and HHV6/7), drug toxicity, or total-parenteral nutrition-related cholestasis, should be excluded, but liver biopsy may be required to confirm the diagnosis, particularly in patients with no other symptoms of cGVHD and failure to respond to initial treatment of suspected GVHD (Stift et al. 2014).
2.5 Gastrointestinal Tract
GI manifestations can lead to dysphagia (esophagus), nausea and vomiting (stomach), or chronic diarrhea and malabsorption syndrome (intestines, pancreas). Occasionally cGVHD may also manifest as immune-mediated pancreatitis. Of note, except esophageal involvement, intestinal involvement is regarded as manifestation of acute GVHD, and patients are therefore classified as suffering from overlap syndrome in which concomitant symptoms of chronic and acute GVHD occur.
Infections like ADV or CMV gastroenteritis, secondary gluten or lactose intolerance, pancreatic insufficiency, and drug-related side effects (e.g., mycophenolate mofetil) have to be ruled out.
Malnutition and enteral fluid and protein loss in small children require regular laboratory monitoring.
2.6 Genitals
The symptoms of cGVHD are similar to those of genital lichen planus which may occur in males and females. Vaginal synechiae, ulceration, and fissures can subsequently occur. Genital manifestations are often associated with oral manifestations of cGVHD. As symptoms may not be reported spontaneously, females suffering from cGVHD require regular gynecological follow-up. In girls cGVHD may manifest with vulvovaginitis, in boys with balanitis or balanoposthitis. Of note, healing may occur with fibrosis possibly leading to synechia with the risk of hematocolpos during puberty in females and of phimosis in males.
2.7 Lung
Pulmonary manifestations occur as progressive, irreversible obstruction (bronchiolitis obliterans) and less frequently lymphocytic alveolitis resulting in interstitial fibrosis or bronchiolitis obliterans organizing pneumonia (BOOP) (see Chap. 52).
Since the onset of pulmonary symptoms may not be symptomatic and obstruction may be irreversible, regular evaluations of a serial pulmonary function test (PFT) with body plethysmography (from the age of 4–6 years on) and diffusion capacity (usually from 8–10 years of age on) are required in asymptomatic patients.
While interstitial fibrosis is well known after lung transplant (restrictive allograft syndrome), prospective data after allogeneic HSCT are lacking, but case reports indicate that restrictive immune-mediated lung disease after allo-HSCT may occur.
Patients require follow-up by a pediatric experienced pulmonologist. Of note, the possible overlap of (1) myopathy/hypotrophy of the respiratory muscles (glucocorticoid induced, ± central obesity, and/or physical inactivity), (2) restriction of the chest wall in the context of dermal sclerosis, and (3) unproportional chest growth after TBI and/or local irradiation may contribute with a restrictive ventilator dysfunction leading to a mixed picture.
Finally, a thorough diagnostic evaluation includes a lung CT scan and a BAL to rule out viral, bacterial, fungal, and mycobacterial infections.
Coexisting IgA deficiency and chronic sinusitis or sinubronchial syndrome should be considered in the diagnostic workup (Hildebrandt et al. 2011).
2.8 Joints and Fasciae
cGVHD-associated fasciitis (diagnostic for cGVHD) can result in restricted mobility of joints. This can also be caused by deep cutaneous sclerosis. Moreover, rheumatoid complaints may be associated with cGVHD. In children myositis, muscle weakness, cramping, edema, and pain are quite common. However, iatrogenic glucocorticoid-induced myopathy may overlap with fasciitis. Range-of-motion (ROM) examinations are recommended at baseline and at serial intervals with the P-ROM scale providing an easy-to-apply tool. (There is a pediatric adaption, ped P-ROM; see addendum).
3 Diagnosis
cGVHD is diagnosed on the basis of cGVHD symptoms of eight organs, laboratory values (for hepatic manifestations), and PFTs. Each organ is graded between 0 and 3. The overall severity of cGVHD is classified as mild, moderate, or severe based on this organ-specific grading (number of organs and severity). Overall severity is calculated on the basis of the number of organs affected and the severity of their involvement. Only in case that functional involvement is solely due to none GVHD causes the impairment is not scored (Jagasia et al. 2015). Biomarkers of cGVHD are currently explored but require validation before clinical use.
3.1 Organ Grading of cGVHD for Adults and Children (See Annex 1 and Addendum)
3.2 Grading of Overall Severity of cGVHD (Jagasia et al. 2015)
Overall severity | Mild | Moderate | Severe |
|---|---|---|---|
Number of involved organs | 1–2 | >3 | >3 |
Severity of involved organs | Mild (excluding lung) | Mild–moderate (lung only mild) | Severe (lung moderate or severe) |
If diagnostic symptoms of cGVHD are absent, histological confirmation of diagnosis may be required. This may be particularly the case in gastrointestinal, nonspecific cutaneous, hepatic, and pulmonary manifestations to rule out toxic or infectious causes or comorbidity. Clinicopathologic series indicate a significant risk for inappropriate diagnosis and subsequent treatment if diagnosis has been made solely by clinical manifestations (and lacking diagnostic symptoms) without histological confirmation.
4 Treatment
4.1 First-Line Therapy
First-line treatment (see Table 44.1) consists of steroids given alone or in combination with CNI and is based on randomized trials.
As mild cGVHD does not impair organ function, the use of topical IS (topical steroids, topical CNI, or phototherapy) should be considered. If this is impossible, PRD treatment at an initial dose of 0.5–1 mg/kg body weight/day is recommended. Topical IS can be used in addition to systemic IS, to improve efficacy, or to reduce systemic IS, but lack systemic efficacy.
For moderate or severe cGVHD, systemic treatment with PRD or methylPRD at an initial dose of 1 mg/kg body weight/day should be used. In individual cases lower doses of 0.5–1 mg/kg may be used (Jacobsohn 2010). The combination of steroids with a CNI (CSA or TAC) is particularly worth considering for severe cGVHD. Rituximab has been explored in first-line treatment of cGVHD in combination with steroids and CNI demonstrating an increased response rate on the expense of an increased risk for late infectious complications and delayed B-cell recovery. Currently, ECP and ibrutinib are evaluated in first-line treatment of cGVHD within randomized clinical trials.
As cGVHD often takes time to respond to IS treatment, response should not be assessed until at least 8 weeks have elapsed or until 3–6 months have elapsed in the presence of deep cutaneous sclerosis. Long-term IS treatment lasting at least 3–6 months is often required. Dose reduction of IS agents should be performed stepwise. Depending on the patient population, first-line therapy achieves complete remission of cGVHD in approximately 20% (adults) to 50% (children) of cases. If symptoms progress during the first 4 weeks of first-line therapy or there is no improvement in symptoms within 8–12 weeks, second-line therapy should be initiated.
4.2 Topical Therapy and Supportive Care
In principle, there is no difference between cGVHD treatment for children and adults. However, long-term steroid therapy in children causes major side effects in terms of growth, bone density, osteonecrosis, and organ development, making agents that reduce steroid use, entailing the use of topical drugs, particularly important. Age-based ancillary supportive care is essential in the management of pediatric cGVHD with the chance of sparing systemic therapy, often supported by highly compliant parents and/or family members as caregivers (Carpenter et al. 2015). In small children, the risk of systemic effects of topical steroid and CNI treatment must be considered. cGVHD is by itself remarkably immunosuppressive intensified by its treatment (especially high-dose corticosteroids) leading to a high risk for infections: (a) for viral reactivation like CMV, ADV, and EBV and (b) for fungal infection like candida and aspergillosis. Functional asplenia with occurrence of Howell-Jolly bodies and a higher incidence of pneumococcal sepsis has to be considered also. Breakdown of skin and mucosal barriers adds to this risk.
Revaccinations (see Chap. 29) with inactivated vaccines are strongly recommended after consolidation of cGVHD (Hilgendorf et al. 2011). Live vaccines should be avoided in this patient population. Ursodeoxycholic acid reduced liver GVHD and improved survival (Ruutu et al. 2014). Supplemental IVIG replacement is recommended in cGVHD patients with IgG <400 mg/dL or recurrent infections which is of special importance in children but does also apply to adults. In case of long-term substitution or the history of anaphylactic reactions, we prefer to substitute subcutaneously.
4.3 Second-Line Therapy
While first-line therapy is based on randomized trials, second-line therapy mostly is based on phase II trials, and retrospective analyses are available (see Table 44.2). In addition, because the data on disease severity and patient populations are very heterogeneous (in terms of age, conditioning, and stem cell source), the published response rates cannot be fully extrapolated to the majority of patients currently treated for cGVHD. Moreover, many substances have been used almost exclusively in combination with steroids.
In general, no more than three IS agents should be combined, as combinations of more drugs often does not lead to improved efficacy but results in a significantly increased risk of side effects and infections. Because of the substantial toxicity of long-term steroid treatment, strategies for dose reduction are very important. Since no predictors of response for a single agent in individual patients are yet available, the choice of agent depends mainly on side effect profiles and patients’ medical history. The response rates for specific agents range between 20% and 70% (photopheresis).
Certain drugs such as imatinib and retinoids are recommended only for manifestations associated with sclerosis (bronchiolitis obliterans [imatinib], sclerodermoid cutaneous alterations [retinoids, imatinib]), because of their specific mechanisms of action.
Response is assessed as for first-line therapy. Administration of drugs that have been shown to be ineffective should be stopped. As a rule, drugs shown to be ineffective should be tapered off stepwise with no more than one drug to be changed at a time in order to be able to evaluate their efficacy.
References
Arai S, Arora M, Wang T, et al. Increasing incidence of chronic graft-versus-host disease in allogeneic transplantation: a report from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant. 2015;21:266–74.
Baird K, Cooke K, Schultz KR. Chronic graft-versus-host disease (GVHD) in children. Pediatr Clin N Am. 2010;57:297–322.
Carpenter PA, Kitko CL, Elad S, et al. National Institutes of Health Consensus Development Project on criteria for clinical trials in chronic graft-versus-host disease: V. The 2014 Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant. 2015;21:1167–87.
Cooke KR, Luznik L, Sarantopoulos S, et al. The biology of chronic graft-versus-host disease: A Task Force Report from the National Institutes of Health Consensus Development Project on criteria for clinical trials in chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2017;23:211–34.
Grube M, Holler E, Weber D, et al. Risk factors and outcome of chronic graft-versus-host disease after allogeneic stem cell transplantation-results from a single-center observational study. Biol Blood Marrow Transplant. 2016;22:1781–91.
Hildebrandt GC, Fazekas T, Lawitschka A, et al. Diagnosis and treatment of pulmonary chronic GVHD: report from the consensus conference on clinical practice in chronic GVHD. Bone Marrow Transplant. 2011;46:1283–95.
Hilgendorf I, Freund M, Jilg W, et al. Vaccination of allogeneic haematopoietic stem cell transplant recipients: report from the international consensus conference on clinical practice in chronic GVHD. Vaccine. 2011;29:2825–33.
Jacobsohn DA. Optimal management of chronic graft-versus-host disease in children. Br J Haematol. 2010;150:278–92.
Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. Biol Blood Marrow Transplant. 2015;21:389.e1–401.e1.
Kuzmina Z, Eder S, Bohm A, et al. Significantly worse survival of patients with NIH-defined chronic graft-versus-host disease and thrombocytopenia or progressive onset type: results of a prospective study. Leukemia. 2012;26:746–56.
Ruutu T, Juvonen E, Remberger M, et al. Improved survival with ursodeoxycholic acid prophylaxis in allogeneic stem cell transplantation: long-term follow-up of a randomized study. Biol Blood Marrow Transplant. 2014;20:135–8.
Stift J, Baba HA, Huber E, et al. Consensus on the histopathological evaluation of liver biopsies from patients following allogeneic hematopoietic cell transplantation. Virchows Arch. 2014;464:175–90.
Strong Rodrigues K, Oliveira-Ribeiro C, de Abreu Fiuza Gomes S, Knobler R. Cutaneous graft-versus-host disease: diagnosis and treatment. Am J Clin Dermatol. 2018;19:33–50.
Wolff D, Bertz H, Greinix H, et al. The treatment of chronic graft-versus-host disease: consensus recommendations of experts from Germany, Austria, and Switzerland. Dtsch Arztebl Int. 2011;108:732–40.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Appendices
Appendix 1



Appendix 2



Appendix 3

Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2019 EBMT and the Author(s)
About this chapter

Cite this chapter
Wolff, D., Lawitschka, A. (2019). Chronic Graft-Versus-Host Disease. In: Carreras, E., Dufour, C., Mohty, M., Kröger, N. (eds) The EBMT Handbook. Springer, Cham. https://doi.org/10.1007/978-3-030-02278-5_44
Download citation
DOI: https://doi.org/10.1007/978-3-030-02278-5_44
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-02277-8
Online ISBN: 978-3-030-02278-5
eBook Packages: MedicineMedicine (R0)