
Abstract
During the course of an immune response to a virus such as influenza, B cells undergo activation, clonal expansion, isotype switching, and somatic hypermutation (SHM). Members of an antigen-experienced B-cell clone can have different sequence features including SHM in the immunoglobulin heavy-chain V (IGHV) gene and can use the same IGVH gene in combination with different constant regions or isotypes (e.g., IgM, IgG, IgA). To study these features of expanded clones in an immune response by AIRR-seq, we provide a bulk RNA-based sequencing experimental procedure with unique molecular identifiers (UMIs) and the accompanying bioinformatics analytical workflow.
Nidhi Gupta and Susanna Marquez are shared first authors.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
This protocol enables users to generate indexed libraries with full-length transcripts that are ready for sequencing on Illumina platforms (Fig. 1). It allows for the analysis of both immunoglobulin heavy (IGH) and kappa/lambda light-chain (IGK /IGL ) gene rearrangements and has a sample input range from 10 ng to 1 μg of total RNA from peripheral blood mononuclear cells (PBMCs) or 1 to 100 ng of total RNA from purified B cells.

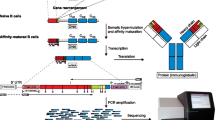
Overview of the SMARTer human BCR procedure. cDNA is synthesized from RNA isolated from PBMCs or B cells, followed by two rounds of PCR and finally purified and pooled to prepare the libraries for sequencing
The protocol leverages SMART technology (switching mechanism at 5′ end of RNA template) and employs a 5’ RACE-like approach to capture complete V(D)J variable regions of BCR /IG transcripts. It also incorporates unique molecular identifiers (UMIs). First-strand cDNA synthesis is oligo-dT primed and catalyzed by SMARTScribe™ reverse transcriptase (RT), which adds non-templated nucleotides at the 5′ end of each mRNA template. The SMART UMI Oligo anneals to these non-templated nucleotides, serves as a template for incorporation of a PCR handle into the first-strand cDNA, and uniquely tags each cDNA molecule with a UMI (UMIs allow for the generation of consensus sequences during data analysis, thereby minimizing PCR and sequencing errors). Following reverse transcription, two rounds of PCR are performed to amplify cDNAs. To capture the entire V(D)J region, primers in these PCRs anneal to sequence added by the SMART UMI Oligo at the 5′ end and the IG constant region(s) at the 3′ end. The second PCR takes the product from the first PCR as a template and uses semi-nested primers to amplify the entire IG variable region and a small portion of the constant region (Fig. 2).

A schematic of dT-primed first-strand cDNA synthesis followed by two rounds of successive PCR for amplification of cDNA sequences. After post-PCR purification, size selection, and quality analysis, the library is ready for sequencing
We also provide a computational workflow to analyze the sequencing data with the Immcantation framework (immcantation.org). The workflow covers preprocessing, isotype assignment, quality control and filtering, gene annotation, gene usage, population structure determination, and lineage reconstruction.
2 Materials
2.1 General Reagents
All components are available in SMARTer Human BCR IgG IgM /IgK/IgL Profiling Kit (Takara Bio, see Note 1).
-
1.
Control RNA (human spleen total RNA, 1 μg/μL).
-
2.
SMART UMI Oligo.
-
3.
dT Primer.
-
4.
5× first-strand buffer.
-
5.
100 U/μL SMARTScribe reverse transcriptase.
-
6.
Nuclease-free water.
-
7.
40 U/μL RNase inhibitor.
-
8.
BCR enhancer.
-
9.
10 mM Tris–HCl elution buffer (pH 8.5).
-
10.
hBCR PCR1 Universal Forward.
-
11.
hBCR PCR1 IgG reverse.
-
12.
hBCR PCR1 IgM reverse.
-
13.
hBCR PCR1 IgK reverse.
-
14.
hBCR PCR1 IgL reverse.
-
15.
1.25 U/μL PrimeSTAR GXL DNA polymerase.
-
16.
5× PrimeSTAR GXL buffer.
-
17.
2.5 mM dNTP mixture.
-
18.
hBCR PCR2 IgG reverse 1–4.
-
19.
hBCR PCR2 IgM reverse 1–4.
-
20.
hBCR PCR2 IgK reverse 1–4.
-
21.
hBCR PCR2 IgL reverse 1–4.
-
22.
hBCR PCR2 Forward 1–12.
2.2 Primers
2.2.1 Human BCR Indexing Primer Set HT for Illumina Sequences
Illumina indexes are incorporated into human BCR profiling libraries through both forward and reverse PCR primers. The corresponding Illumina indexes are listed below.
2.2.2 BCR PCR2 Forward Primer i7 HT Index
Primers are listed with the name, Illumina ID, and index sequence.
-
1.
hBCR PCR2 Universal Forward 1, D701, ATTACTCG.
-
2.
hBCR PCR2 Universal Forward 2, D702, TCCGGAGA.
-
3.
hBCR PCR2 Universal Forward 3, D703, CGCTCATT.
-
4.
hBCR PCR2 Universal Forward 4, D704, GAGATTCC.
-
5.
hBCR PCR2 Universal Forward 5, D705, ATTCAGAA.
-
6.
hBCR PCR2 Universal Forward 6, D706, GAATTCGT.
-
7.
hBCR PCR2 Universal Forward 7, D707, CTGAAGCT.
-
8.
hBCR PCR2 Universal Forward 8, D708, TAATGCGC.
-
9.
hBCR PCR2 Universal Forward 9, D709, CGGCTATG.
-
10.
hBCR PCR2 Universal Forward 10, D710, TCCGCGAA.
-
11.
hBCR PCR2 Universal Forward 11, D711, TCTCGCGC.
-
12.
hBCR PCR2 Universal Forward 12, D712, AGCGATAG.
2.2.3 BCR Indexing Reverse Primer Set HT for Illumina Index Sequences
Primers are listed with name, Illumina ID, and index sequences, as read on a MiSeq instrument.
-
1.
hBCR PCR2 IgG Reverse 1, D501, TATAGCCT.
-
2.
hBCR PCR2 IgM Reverse 1, D501, TATAGCCT.
-
3.
hBCR PCR2 IgK Reverse 1, D501, TATAGCCT.
-
4.
hBCR PCR2 IgL Reverse 1, D501, TATAGCCT.
-
5.
hBCR PCR2 IgG Reverse 2, D502, ATAGAGGC.
-
6.
hBCR PCR2 IgM Reverse 2, D502, ATAGAGGC.
-
7.
hBCR PCR2 IgK Reverse 2, D502, ATAGAGGC.
-
8.
hBCR PCR2 IgL Reverse 2, D502, ATAGAGGC.
-
9.
hBCR PCR2 IgG Reverse 3, D503, CCTATCCT.
-
10.
hBCR PCR2 IgM Reverse 3, D503, CCTATCCT.
-
11.
hBCR PCR2 IgK Reverse 3, D503, CCTATCCT..
-
12.
hBCR PCR2 IgL Reverse 3, D503, CCTATCCT.
-
13.
hBCR PCR2 IgG Reverse 4, D504, GGCTCTGA.
-
14.
hBCR PCR2 IgM Reverse 4, D504, GGCTCTGA.
-
15.
hBCR PCR2 IgK Reverse 4, D504, GGCTCTGA.
-
16.
hBCR PCR2 IgL Reverse 4, D504, GGCTCTGA.
2.3 Equipment
-
1.
Pipettes: 10 μL, 20 μL, and 200 μL.
-
2.
Filter pipette tips: 2 μL, 20 μL, and 200 μL.
-
3.
Microcentrifuge tubes: 1.5 mL.
-
4.
Minicentrifuge 0.2 mL tubes or strips.
-
5.
NucleoSpin RNA Plus, mini kit for RNA purification with DNA removal column (Macherey-Nagel).
-
6.
Thermal cyclers, separate dedicated instruments for first-strand cDNA synthesis, and PCR amplification.
-
7.
Agilent 2100 Bioanalyzer – DNA 1000 kit; for validation, alternatively use the TapeStation.
-
8.
TapeStation, (Agilent), for validation, alternatively use the bioanalyzer.
-
9.
Qubit dsDNA HS Kit (Thermo Fisher Scientific).
-
10.
Nuclease-free thin wall PCR tubes, 96-well plates, or strips (USA Scientific).
-
11.
Nuclease-free low-adhesion 1.5 mL tubes.
-
12.
NucleoMag NGS clean-up and size select, Takara Bio 5 mL size for bead purification.
-
13.
100% ethanol, molecular biology grade.
-
14.
SMARTer-Seq™ Magnetic Separator, PCR Strip, Takara Bio 8-tube strips or Thermo Fisher Scientific 96-well plates.
-
15.
Low-speed benchtop centrifuge for 96-well plate.
2.4 Software
Immcantation suite Docker container. See step 3 (“obtain the software”) in Subheading 3.10.
3 Methods
3.1 Overview of Wet Bench Protocol
The major steps of the procedure are outlined in Fig. 3. This sequencing protocol has been optimized for 10 ng of total RNA per sequencing library, which corresponds to ~1000 cells. But the extraction of RNA is far more efficient and yields higher purity and higher-quality product at much higher numbers of input cells, on the scale of tens of thousands to millions. RNA extracted from PBMCs yields approximately 1 μg per mL or ~one million nucleated cells, of which only a small fraction (1–10%) are B cells.

Bulk RNA sequencing with unique molecular identifiers (UMIs). (a) This protocol begins with a single-cell suspension that can be isolated from whole blood, peripheral blood mononuclear cells, or by purification using either magnetic beads or flow cytometry. Total RNA is extracted from the cell population(s) of interest. (b) cDNA is reverse transcribed from RNA, and unique molecular identifiers (UMIs) are included in the SMART Oligo, tagging each parental cDNA molecule. (c) In PCR1, IgG, IgM, IG kappa, and IG lambda chains (including both the variable and part of the constant region) are separately amplified (only the IGH are shown). (d) In PCR2, Illumina indices are added to generate the sequencing libraries. (e) After size selection, QC, and normalization of library input, libraries are sequenced using the Illumina platform
3.2 RNA Extraction
The following is an illustration of RNA isolation from approximately 1 × 107 cultured cells using the NucleoSpin RNA Plus protocol.
-
1.
Cell collection: Transfer cells to an appropriate tube, and pellet by centrifugation for 5 min at 300 × g. Remove supernatant.
-
2.
Homogenize and lyse sample by adding 350 μL buffer LBP to the cell pellet. Mixing the cells with a lysis buffer is usually sufficient for complete lysis.
-
3.
Remove gDNA and filtrate lysate: Place the NucleoSpin® gDNA Removal Column (yellow ring) in a 2 mL collection tube, transfer the homogenized lysate to the NucleoSpin® gDNA Removal Column, and centrifuge for 30 s at 11,000 × g. Discard the column and continue with the flowthrough.
-
4.
Adjust RNA binding conditions: Add 100 μL binding solution BS to the flowthrough, and mix well by moderate vortexing or by pipetting up and down several times. After addition of binding solution BS, a stringy precipitate may become visible which will not affect the RNA isolation. Be sure to disaggregate any precipitate by mixing and load all of the precipitate on the column as described in the following step. Do not centrifuge the lysate after addition of binding solution before loading it onto the column in order to avoid pelleting the precipitate.
-
5.
Bind RNA: Transfer the whole lysate (~450 μL) to the NucleoSpin® RNA Plus Column (light blue ring) preassembled with a collection tube. Centrifuge for 15 s at 11,000 × g.
-
6.
Wash and dry silica membrane:
-
(a)
First wash: Add 200 μL buffer WB1 to the NucleoSpin® RNA Plus Column. Centrifuge for 15 s at 11,000 × g. Discard the flowthrough with the collection tube, and place the column into a new 2 mL collection tube.
-
(b)
Second wash: Add 600 μL buffer WB2 to the NucleoSpin® RNA Plus Column. Centrifuge for 15 s at 11,000 × g. Discard flowthrough, and place the column back into the collection tube.
-
(c)
Third wash: Add 250 μL buffer WB2 to the NucleoSpin® RNA Plus Column. Centrifuge for 2 min at 11,000 × g to dry the membrane completely. Place the column into a nuclease-free 1.5 mL collection tube.
-
(a)
-
7.
Elute RNA: Add 30 μL RNase-free H2O and centrifuge at 11,000 × g for 1 min. Add an additional 30 μL RNase-free H2O to the column, and centrifuge again at 11,000 × g for 1 min.
-
8.
After RNA extraction, if sample size is not limiting, we recommend evaluating total RNA quality using the Agilent RNA 6000 Pico Kit or an equivalent platform. Refer to the manufacturer for instructions (see Note 2).
3.3 First-Strand cDNA Synthesis
First-strand cDNA synthesis (from RNA) is primed by the dT Primer. Here we illustrate cDNA synthesis using the SMART UMI Oligo for template switching at the 5′ end of the transcript.
-
1.
Thaw the First-Strand Buffer at room temperature. Thaw BCR enhancer, SMART UMI Oligo, and dT Primer on ice. Gently vortex each reagent to mix and centrifuge briefly. Store all but the First-Strand Buffer on ice. Remove the SMARTScribe reverse transcriptase and RNase inhibitor from the freezer immediately before use, centrifuge briefly, and store on ice (see Note 3).
-
2.
Preheat the thermal cycler to 72 °C.
-
3.
On ice, prepare samples and controls in nuclease-free thin-wall PCR tubes, plates, or strips by adding the reagents in the order shown below.
Component
Volume (μL)
Sample or controla
1–9.5
Nuclease-free water
Up to 8.5
BCR enhancer
1
dT primer
2
Total volume
12.5
-
4.
Mix by gently vortexing and then centrifuge briefly.
-
5.
Incubate the tubes at 72 °C in the preheated, heated-lid thermal cycler for 3 min. During this incubation, prepare the RT Master Mix.
-
6.
At room temperature, prepare RT Master Mix by combining the following in the order shown. Wait to add the SMARTScribe reverse transcriptase to the master mix until just prior to use in step 10 of Subheading 3.3.
Component
Volume (μL)
First-Strand buffera
4
SMART UMI oligo
1
RNase inhibitor
0.5
SMARTScribe reverse transcriptase
2
Total volume
7.5
-
7.
Mix the RT Master Mix well by gently pipetting up and down, and then centrifuge briefly.
-
8.
Immediately after the 3-min incubation at 72 °C (step 5 of Subheading 3.3), place the samples on ice for 2 min.
-
9.
Reduce the temperature of the thermal cycler to 42 °C.
-
10.
Add 7.5 μL of the RT Master Mix (step 6 of Subheading 3.3) to each reaction tube. Mix the contents of each tube by pipetting gently and centrifuge briefly.
-
11.
Place the tubes in a thermal cycler with a heated lid, preheated to 42 °C. Run the following program: 42 °C, 90 min; 70 °C, 10 s; 4 °C hold.
Stopping point: The tubes can be stored at 4 °C overnight.
3.4 First-Round Amplification
Semi-nested PCR amplifies the entire V(D)J region and a portion of the constant region of IG cDNA(s) and incorporates adapters and barcodes for Illumina sequencing platforms. Expression of different IG chains can vary significantly among B-cell populations. Thus, we recommend separately amplifying each chain of interest. Table 1 provides PCR cycling recommendations, but optimal parameters may vary for different sample types, input amounts, and thermal cyclers. We recommend trying a range of cycle numbers to determine the minimum number necessary to obtain the desired yield.
In the first round of PCR amplification, also referred to as PCR1, one performs separate IgG/IgM/IgK/IgL amplification. This PCR selectively amplifies full-length BCR V(D)J regions from first-strand cDNA. A portion of the first-strand cDNA is used for each amplification reaction. The hBCR PCR1 Universal Forward primer anneals to the 5′ end of transcripts via the SMART UMI Oligo sequence. The hBCR PCR1 IgG/IgM/IgK/IgL reverse primers anneal to sequences in the constant regions of IG heavy- and light-chain cDNAs.
-
1.
Thaw 5× PrimeSTAR GXL buffer, dNTP mix, primers, and nuclease-free water on ice. Gently vortex each reagent to mix and centrifuge briefly. Store on ice. Remove the PrimeSTAR GXL DNA polymerase from the freezer immediately before use, gently pipet to mix, centrifuge briefly, and store on ice.
-
2.
Prepare a PCR1 Master Mix for each IgG/IgM/IgK/IgL chain of interest, by combining the following in the order shown, on ice. Gently vortex to mix and centrifuge briefly (see Note 4).
Component
Volume (μL)
Nuclease-free water
29
5× PrimeSTAR GXL PCR buffer
10
dNTP mixture
4
hBCR PCR1 universal forward
1
hBCR PCR1 IgG reverse OR
1
hBCR PCR1 IgM reverse OR
hBCR PCR1 IgK reverse OR
hBCR PCR1 IgL reverse
PrimeSTAR GXL polymerase
1
Total volume
46
-
3.
Add 46 μL of the appropriate IgG/IgM/IgK/IgL PCR1 Master Mix to nuclease-free, thin-wall 0.2-mL PCR plate/tube(s).
-
4.
Add 4 μL of first-strand cDNA from Subheading 3.3 to the corresponding tube(s) containing PCR1 Master Mix. Gently vortex to mix, and centrifuge briefly.
-
5.
Place the plate/tube(s) in a preheated thermal cycler with a heated lid, and run the following program (lid temperature: 105 °C): 95°C 1 min; 98°C 10 s, 60°C 15 s, and 68°C 45 s (18 cycles); 4°C hold. *Consult Table 1 for PCR cycle number guidelines. Stopping point: The tubes may be stored at 4 °C overnight.
3.5 Second-Round PCR Amplification
In the second round of PCR amplification, termed PCR2, sequencing libraries are generated. PCR2 further amplifies the full-length IG V(D)J regions and adds Illumina indexes using a semi-nested approach. The hBCR PCR2 Universal Forward 1–12 primers add P7/i7 index sequences. The hBCR PCR2 IgG/IgM/IgK/IgL reverse 1–4 primers anneal to the constant region of the IG sequence and add P5/i5 index sequences (see Note 5).
-
1.
Thaw 5X PrimeSTAR GXL buffer, dNTP Mix, primers, and nuclease-free water on ice. Gently vortex each reagent to mix and centrifuge briefly. Store on ice. Remove the PrimeSTAR GXL DNA polymerase from the freezer immediately before use, gently pipet mix, centrifuge briefly, and store on ice.
-
2.
For each IgG/IgM/IgK/IgL chain of interest, prepare a PCR2 Master Mix by combining the following in the order shown, on ice. Gently vortex to mix and centrifuge briefly (see Note 6).
Component
Volume (μL)
Nuclease-free water
32
5X PrimeSTAR GXL PCR buffer
10
dNTP mixture
4
hBCR PCR2 IgG reverse 1–4 OR
1
hBCR PCR2 IgM reverse 1–4 OR
hBCR PCR2 IgK reverse 1–4 OR
hBCR PCR2 IgL reverse 1–4
PrimeSTAR GXL polymerase
1
Total volume
48
-
3.
For each reaction, add 48 μL of PCR2 Master Mix to nuclease-free, thin-wall, 0.2-mL PCR plate/tube(s).
-
4.
Add 1 μL of appropriate PCR1 product to each corresponding PCR 2 tube.
-
5.
Add 1 μL of the appropriate hBCR PCR2 Universal Forward 1–12 primer to each sample. Gently vortex to mix and centrifuge briefly.
-
6.
Place the plate/tube(s) in a preheated thermal cycler with a heated lid, and run the following program (lid temperature: 105 °C): 95°C 1 min; 98°C 10 s, 60°C 15 s, 68°C 45 s (X* cycles); 4°C Hold. *Consult Table 1 for PCR cycle number guidelines.
Stopping point: The tubes may be stored at 4 °C overnight.
3.6 Purification of Amplified Libraries
Here we illustrate amplified library purification using NucleoMag NGS clean-up and size select beads (see Note 7).
-
1.
Vortex NucleoMag beads until evenly mixed, and then add 25 μL of the NucleoMag beads to each sample.
-
2.
Mix thoroughly by gently pipetting the entire volume up and down at least ten times (see Note 8).
-
3.
Incubate at room temperature for 8 min to let the DNA bind to the beads.
-
4.
Briefly spin the samples to collect the liquid from the side of the tube or sample well. Place the samples on the magnetic separation device for ~5 min or longer until the liquid appears completely clear, and there are no beads left in the supernatant. The time required for the solution to clear will depend on the strength of the magnet (see Note 9).
-
5.
While the reaction tubes are sitting on the magnetic separation device, use a pipette to transfer the supernatant (which contains your library) to clean PCR tubes.
-
6.
Remove the tubes containing the beads from the magnetic separation device, and discard them.
-
7.
Add 10 μL of NucleoMag beads to each tube containing supernatant (see Note 10).
-
8.
Mix thoroughly by gently pipetting the entire volume up and down at least ten times.
-
9.
Incubate at room temperature for 8 min to let the DNA bind to the beads.
-
10.
Place the tubes on the magnetic separation device for ~10 min or until the solution is completely clear.
-
11.
While the tubes are sitting on the magnetic separation device, remove the supernatant with a pipette and discard it (the library is now bound to the beads).
-
12.
Keep the tubes on the magnetic separation device. Add 200 μL of freshly made 80% ethanol to each sample, without disturbing the beads, to wash away contaminants. Wait for 30 s, and use a pipette to carefully remove the supernatant containing contaminants. The library will remain bound to the beads during the washing process.
-
13.
Repeat the ethanol wash (step 12 of this section) once more.
-
14.
Briefly spin the tubes (~2000 × g) to collect the remaining liquid at the bottom of each tube. Place the tubes on the magnetic separation device for 30 s, and then remove all remaining liquid with a pipette.
-
15.
Let the sample tubes rest open on the magnetic separation device at room temperature for ~2–2.5 min until the pellet appears dry and is no longer shiny. You may see a tiny crack in the pellet (see Note 11).
-
16.
Once the bead pellet has dried, remove the tubes from the magnetic separation device, and add 17 μL of elution buffer to cover the pellet. Mix thoroughly by pipetting up and down to ensure complete bead dispersion (see Note 12).
-
17.
Incubate at room temperature for at least 5 min to rehydrate.
-
18.
Briefly spin the samples to collect the liquid from the side of the tube or sample well. Place the samples back on the magnetic separation device for 2 min or longer until the solution is completely clear (see Note 13).
-
19.
Transfer clear supernatant containing purified BCR /IG library from each tube to a nuclease-free, low-adhesion tube. Label each tube with sample information and store at −20 °C.
Stopping point: The tubes may be stored at 4 °C overnight.
3.7 Library Validation
To assess the success of library preparation, purification, and size selection, we recommend quantifying the libraries with a Qubit dsDNA HS Kit and evaluating the libraries’ size distributions with an Agilent 2100 Bioanalyzer and the DNA 1000 Kit.
-
1.
Compare the results for your samples with Fig. 4 to verify whether each sample is suitable for further processing. High-quality libraries should yield no product for negative control reactions and a broad peak spanning 500 bp to 1200 bp, with a maximum between ~600 bp and ~900 bp for positive controls and samples containing PCR-amplified IG libraries. The position and shape of electropherogram peaks will vary depending on which chain sequences are included in the library, the nature of the originally included RNA sample, and the analysis method. In general, electropherogram peaks obtained with the Fragment Analyzer tend to be sharper than those obtained with the bioanalyzer.
-
2.
Following validation, libraries are ready for sequencing on Illumina platforms.

Validation of IG heavy- and IG light (kappa or lambda)-chain libraries from human spleen that were generated using the SMARTer Human BCR Profiling Kit. Purified and size-selected libraries were analyzed on an Agilent 2100 Bioanalyzer (Panels A–H). Panels A, C, E, and G show broad peaks between ~500 and 1200 bp and maximal peaks in the range of ~600–900 bp (typical results for a library generated from spleen RNA). RNA control (NRC) samples (Panels B, D, F, and H) show no library produced and a flat Bioanalyzer profile within the predicted amplicon range of 500–1200 bp
3.8 Pooling of Samples to Generate Libraries for Sequencing
Following library validation by Qubit and bioanalyzer, the desired library pools should be prepared for the sequencing run. Prior to pooling, libraries must be carefully quantified. By combining the quantification obtained with the Qubit with the average library size determined by the bioanalyzer, the concentration in ng/μL can be converted to nM. The following web tool is convenient for the conversion: http://www.molbiol.edu.ru/eng/scripts/01_07.html. Alternatively, libraries can be quantified by qPCR using the Library Quantification Kit from Takara Bio.
Most Illumina sequencing library preparation protocols require libraries with a final concentration of 4 nM, including the MiSeq instrument that we recommend for this protocol.
Prepare a pool of 4 nM as follows:
-
1.
Dilute each library to 4 nM in nuclease-free water. To avoid pipetting errors, use at least 2 μL of each original library for dilution.
-
2.
Pool the diluted libraries by combining an equal amount of each library in a low-bind 1.5-mL tube. Mix by vortexing at low speed or by pipetting up and down. Use at least 2 μL of each diluted library to avoid pipetting error.
-
3.
Use a 5 μL aliquot of the 4-nM-concentration-pooled libraries. Follow the library denaturation protocol according to the latest edition of your Illumina sequencing instrument’s user guide.
You should also plan to include a 10% PhiX control spike-in (PhiX Control v3, Illumina). The addition of the PhiX control is essential to increase the nucleotide diversity and achieve high-quality data generation (see Note 14).
Sequencing should be performed on an Illumina MiSeq sequencer using the 600-cycle MiSeq Reagent Kit v3 with paired-end, 2 × 300 base pair reads. When relying on Qubit quantification, we recommend diluting the pooled denatured libraries to a final concentration of 12.5 pM to achieve optimal cluster density. If using qPCR for quantification, one may need to use a lower final concentration.
The complexity of the human IG repertoire varies from person to person. We generally recommend a minimum of 200,000 reads for IG heavy-chain libraries (IgG and IgM) from an input of 10 ng PBMC RNA (or 1 ng B-cell RNA). and a minimum of 500,000 reads for IG light (IGK and IGL ) chains from an input of 10 ng PBMC RNA (or 1 ng B-cell RNA). For libraries generated from >10 ng PBMC RNA, higher sequencing depth is recommended. However, the optimal conditions may vary for different samples types, sample masses, sample complexities, and desired outcomes. We recommend trying a higher sequencing depth, then down sampling to determine the optimal sequencing depth.
As shown in Fig. 5, a human BCR profiling library contains a 12-nucleotide UMI that can be used to create consensus reads for sequences that share the same UMI , allowing correction for sequencing error correction.

SMARTer human BCR IgG IgM H/K/L profiling library structure. First 19 nt from Read2 can be trimmed off if UMI analysis is not performed
Upon completion of a sequencing run, data can be analyzed with Takara Bio Cogent NGS Immune Profiler Software or other software. In the following sections, we provide a workflow to analyze data with Immcantation, a suite that provides tools to perform preprocessing, population structure determination, and repertoire analysis. Immcantation is certified as compliant with AIRR Community software guidelines.
3.9 Data Analysis Overview
In this workflow, we show how to use Immcantation (immcantation.org) to analyze sequencing data generated following the experimental protocol described in Subheading 3.8. An overview is given in Fig. 6. In the “AIRR Community Guide to TR and IG Gene Annotation” and “AIRR Community Guide to Repertoire Analysis” chapters the goals of multiple common AIRR-seq analysis techniques are described in detail, which can be useful to interpret the analysis performed in this section.

Overview of data processing and analysis steps
3.10 Raw Data Processing
The command line tool pRESTO [1] provides utilities to execute all stages of sequencing data processing prior to germline gene assignment. The tools are modular and can be combined to build highly customizable workflows. pRESTO includes features for quality control, primer masking, annotation of reads with sequence-embedded barcodes, generation of unique molecular identifier (UMI) consensus sequences, assembly of paired-end reads, and identification of duplicate sequences.
-
1.
Remove PhiX
If spike-in PhiX was not removed by the sequencing facility, it is recommended [2] to filter out these reads.
-
2.
Understand the Read Layout
It is important to have a good understanding of the read layout and know what region each read covers, where the primers and barcodes are located, and how long they are. In this example, R1 starts in the constant region of the rearranged sequence and R2 upstream the V region. See Fig. 5 for details on the read layout.
Primers from the vendor are not available. To identify isotypes, it is possible to use as primers the consensus sequences of the constant region available online from the protocols/Universal directory in the Immcantation repository (https://bitbucket.org/kleinstein/immcantation). These sequences have been created after analyzing the first 30 nucleotides of the human constant region sequences available from IMGT.
-
3.
Obtain the Software
Immcantation, with its dependencies, accessory scripts, and IgBLAST [4] and IMGT [3] reference germlines, is available as a Docker container on docker hub under immcantation/suite:x.x.x where x.x.x stands for a release number. This protocol is using the container release 4.3.0.
To start an interactive session inside the container and share local files in the current working directory with the /data folder in the container, use.
docker run -it -v $(pwd):/data:z --workdir /data immcantation/suite:4.3.0 bash
Once inside the container, you can use the commands versions report and builds report to know the versions of the software installed.
If you type pwd, you should get the result /data, as expected after starting the container with --workdir /data. If you type ls, you should see the files that you have in the local directory from which you launched the container. Being inside the container session, create the output directories presto and logs, and verify that the folder also becomes available locally in your computer:
mkdir presto mkdir logs
-
4.
Remove Low-Quality Sequences
To remove reads with a mean quality lower than 20, use the command.
FilterSeq.py quality -s data/S5_R1.fastq -q 20 --nproc 8 \ --outname CRR --outdir presto --log "logs/quality-crr.log" FilterSeq.py quality -s data/S5_R2.fastq -q 20 --nproc 8 \ --outname VRR --outdir presto --log "logs/quality-vrr.log"
Output data files for the constant region reads will use the prefix CRR, and data files for the V region reads, will use the prefix VRR.
-
5.
Identify Primers and UMI
The next step is to remove or mask primers and extract UMI barcodes from the sequence but keeping this information as annotations in the FASTQ file headers. We recommend to mask or remove primers so that sequencing errors in the primers do not affect downstream analyses. Here we remove barcodes and primers. We know that the kit used to generate the data has a 12-nucleotide-long UMI (−-start 12), followed by a linker sequence and a template switch (−-len 7). With this command, pRESTO will extract the first 12 bp and annotate the fastq file header with the field BARCODE.
MaskPrimers.py extract -s presto/VRR_quality-pass.fastq \ --start 12 --len 7 --barcode --bf BARCODE --mode cut \ --log "logs/primers-vrr.log" \ --outname VRR --outdir presto
An example output FASTQ header is as follows:
@M03355:144:000000000-CH2WP:1:1104:17528:20342 2:N:0:CGCTCATT+TATAGCCT|PRIMER=GTACGGG|BARCODE=TTGAAGTTATTC
-
6.
Annotate R1 with Internal C-Region
Use the following command to annotate the CRR FASTQ file with a constant region call. This step requires a reference FASTA file containing the reverse-complement of short sequences from the front of CH-1. The C-region sequences (-p) are available in the container. For each sequence, MaskPrimers.py align will look for good matches (maximum error of --maxerror 0.3) to the reference sequences in the first 100 nucleotides (--maxlen 100). The matching and preceding region will be cut out from the sequence. The matching sequence name will be added as an annotation into the FASTQ header, under the field C_CALL.
MaskPrimers.py align -s presto/CRR_quality-pass.fastq \ -p /usr/local/share/protocols/Universal/Human_IG_CRegion_RC.fasta \ --maxlen 100 --maxerror 0.3 \ --mode cut --skiprc --pf C_CALL \ --log "logs/cregion.log" --outname "CRR" --nproc 8
An example output FASTQ header is as follows:
@M03355:144:000000000-CH2WP:1:2116:18550:17244 1:N:0:CGCTCATT+TATAGCCT|SEQORIENT=F|C_CALL=IGHM
pRESTO tools save logs that can be converted into tabulated files with ParseLog.py. It is useful to use these files to generate diagnostic plots. To extract the information to make figures, inspect the C_CALLs made, and identify the starting position of the match, use the command below. It will create a tabulated file with the fields ID, PRIMER, ERROR, and PRSTART, which can be used to create such plots.
ParseLog.py -l "logs/cregion.log" -f ID PRIMER ERROR PRSTART --outdir logs
Once the log has been converted to a tabulated file, it can be easily loaded into R, to count the different isotypes that have been identified:
cregion_table <- read.delim("logs/cregion_table.tab") table(cregion_table$PRIMER)
Example output:
IGHA IGHD IGHE IGHG IGHM IGKC IGLC1 IGLC3 4 32 510 242981 220640 244015 250521 40647
These results match the expectations for this experimental protocol, because it uses a kit designed for IgM, IgG, IgK, and IgL. The isotype count can also be visualized (Fig. 7a):
# Create a color palette color_palette <- c( "IGHA"="882255", "IGHD"="#AA4499", "IGHE"="#88CCEE", "IGHG"="#CC6677", "IGHM"="#6699CC", "IGKC"="#44AA99", "IGLC1"="#888888", "IGLC3"="#DDCC77" ) isotypes <- sort(unique(cregion_table$PRIMER)) names(color_palette) <- isotypes # Plot isotype primer position cprimer_plot <- ggplot(cregion_table, aes(x=PRSTART, color=PRIMER)) + geom_freqpoly(size = 0.5,binwidth=1) + scale_color_manual(values = color_palette) + theme_minimal() + labs(x = "C-region alignment start", y = "Count", colour = "Primer") + theme(legend.key.height = unit(0.1, "lines"), legend.key.width = unit(0.5, "lines")) cprimer_plot
-
7.
Copy Annotations Between Reads
Propagation of annotations between mate pairs is accomplished with PairSeq.py, which also removes unpaired reads and sorts mate pairs in both files. In this example, the UMI barcode is part of read VRR, and C_CALL is part of read VCC. We need to transfer this information to be able to build consensus sequences for groups of reads sharing the same UMI and C_CALL.
PairSeq.py -1 presto/VRR_primers-pass.fastq \ -2 presto/CRR_primers-pass.fastq \ --1f BARCODE --2f C_CALL --coord illumina
-
8.
Generation of UMI Consensus Sequences
If UMIs are available, it is possible to correct sequencing errors maintaining true mutations introduced by SHM . Reads sharing a UMI barcode are reads that originated from the same RNA molecule. Ideally, if the primers used are different enough, and the UMIs have enough diversity, each UMI will represent one mRNA molecule, and each mRNA molecule will be represented by one UMI . BuildConsensus.py can then be used to generate a consensus sequence for a set of aligned reads sharing the same UMI . Finding more than one primer in a UMI group suggests sequences may not be aligned, as we expect reads originating from the same mRNA molecule should be amplified with the same primer. If the multiplex pool contains similar primers, they could be incorporated into the same UMI group during amplification, and the reads will have variations in the start positions. This situation can be mitigated by first aligning the reads.
-
(a)
Multiple Align UID Read Groups
If the reads are not aligned, a correction strategy is to use MUSCLE [5] and AlignSets.py to perform a multiple alignment of each UMI read group, before generating the consensus sequence in the next step.
AlignSets.py muscle -s "presto/VRR_primers-pass_pair-pass.fastq" --exec /usr/local/bin/muscle --nproc 8 --log "logs/align-vrr.log" --outname "VRR" AlignSets.py muscle -s "presto/CRR_primers-pass_pair-pass.fastq" --exec /usr/local/bin/muscle --nproc 8 --log "logs/align-crr.log" --outname "CRR"
-
(b)
Build the Consensus Sequence
BuildConsensus.py will group sequences sharing the same barcode to build a consensus sequence. If a UMI group has a number of average mismatches larger than 0.1 (−-maxerror 0.1), it will be dismissed. Sequences with the same barcode have originated from the same original mRNA molecule, and they should also have the same isotype . --pf C_CALL and --prcons 0.6 are used to require that 60% of the UMI group have the same C_CALL.
BuildConsensus.py -s presto/CRR_align-pass.fastq \ --bf BARCODE --pf C_CALL --prcons 0.6 \ -n 1 -q 0 --maxerror 0.1 --maxgap 0.5 \ --nproc 8 --log "logs/consensus-crr.log" \ --outdir presto --outname "CRR"
Example output:
@TGTTGGTTGGGT|CONSCOUNT=5|PRCONS=IGHM|PRFREQ=0.8333333333333334
CONSCOUNT shows the number of sequences that contributed to build the consensus. In the example above, the consensus isotype (PRCONS) is IGHM, with a frequency of 0.83. In the starting UMI group, there were six sequences, and one of them was an IGKC. This sequence was not used to build the consensus.
The same process needs to be repeated for the other reads:
BuildConsensus.py -s presto/VRR_align-pass.fastq \ --bf BARCODE --pf C_CALL --prcons 0.6 \ -n 1 -q 0 --maxerror 0.1 --maxgap 0.5 \ --nproc 8 --log "logs/consensus-vrr.log" \ --outdir presto --outname "VRR"
-
(a)
-
9.
Synchronize Reads
This step puts pairs of reads in the same order.
PairSeq.py -1 "presto/VRR_consensus-pass.fastq" -2 \ “presto/CRR_consensus-pass.fastq" \ --coord presto
-
10.
Assemble Pairs
Consensus sequences are paired in two steps, starting with joining overlapping mate pairs. For read pairs failing this step, the tool proceeds to perform a reference guided alignment, using ungapped V-segment reference sequences to properly space nonoverlapping reads.
AssemblePairs.py sequential -1 "presto/VRR_consensus-pass_pair-pass.fastq" \ -2 "presto/CRR_consensus-pass_pair-pass.fastq" \ -r /usr/local/share/igblast/fasta/imgt_human_ig_v.fasta \ --coord presto --rc tail --1f CONSCOUNT --2f PRCONS CONSCOUNT \ --minlen 8 --maxerror 0.3 --alpha 1e-5 --scanrev \ --minident 0.5 --evalue 1e-5 --maxhits 100 --aligner blastn \ --nproc 8 --log "logs/assemble.log" \ --outname "S5"
Example output: @ACTAGGGTTCAT|CONSCOUNT = 4,4|PRCONS=IGHM .
PRCONS is the consensus C_CALL from the CRR file.
-
11.
Mask Low-Quality positions
Positions with a low consensus quality can be masked with Ns.
FilterSeq.py maskqual -s presto/S5_assemble-pass.fastq -q 30 --nproc 8 \ --outname "S5-MQ" --log "logs/maskqual.log"
-
12.
Track the Number of Sequences that Contributed to the Consensus
It is important to know the number of unique sequences that contributed to build the consensus, as this information will be used in a later step.
ParseHeaders.py collapse -s presto/S5-MQ_maskqual-pass.fastq -f CONSCOUNT --act min \ --outname "S5-final" mv "presto/S5-final_reheader.fastq" "presto/S5-final_total.fastq"
-
13.
Collapse Duplicates
The goal is to remove duplicated sequences to retain in the repertoire one representative sequence per cell. The argument “-n 0 –inner” will determine how to handle N and gap characters. In this example, we allow 0 ambiguous characters, ignoring any continuous N or gap characters that occur at any end of the sequence. “—uf” specifies fields that should be used to define groups of unique sequences. “--cf CONSCOUNT” requests to copy the field CONSCOUNT and then perform the action “--act sum,” to obtain a final unique sequence with CONSCOUNT equal to the sum of the CONSCOUNTS of the collapsed sequences.
CollapseSeq.py -s "presto/S5-final_total.fastq" -n 0 \ --uf PRCONS --cf CONSCOUNT --act sum --inner \ --keepmiss --outname "S5-final"
-
14.
Subset to Sequences Seen at Least Twice
We recommend filtering the data to focus the analysis on sequences with at least two contributing reads. Sequences with CONSCOUNT of 1 are generated with only one sequence contributing to the UMI group, and this suggests the existence of sequencing error.
SplitSeq.py group -s presto/S5-final_collapse-unique.fastq -f CONSCOUNT \ --num 2
-
15.
Explore the Logs
All pRESTO tools provide the option to generate detailed logs that can be used to generate diagnostic plots. The log files can be converted to tabulated text files with ParseLog.py. The tabulated text files can be loaded into R or python to generate plots.
-
(a)
Obtain Tabulated Data
The output files are parsed to generate tables of data for the repertoire.
ParseHeaders.py table -s "presto/S5-final_total.fastq" \ -f ID PRCONS CONSCOUNT --outname "final-total" \ --outdir logs ParseHeaders.py table -s "presto/S5-final_collapse-unique.fastq" \ -f ID PRCONS CONSCOUNT DUPCOUNT --outname "final-unique" \ --outdir logs ParseHeaders.py table -s "presto/S5-final_collapse-unique_atleast-2.fastq" \ -f ID PRCONS CONSCOUNT DUPCOUNT --outname "final-unique-atleast2" \ --outdir logs
To see a summary of the final isotype assignments:
log <- read.delim("logs/final-unique-atleast2_headers.tab") table(log$PRCONS) IGHE IGHG IGHM IGKC IGLC1 IGLC3 1 48485 50955 37578 46879 7609
-
(b)
Process the Log Files Generated at Each Step
Log files are also parsed into tabulated files.
ParseLog.py -l "logs/primers-vrr.log" -f ID BARCODE ERROR \ --outdir logs ParseLog.py -l "logs/consensus-vrr.log" "logs/consensus-crr.log" \ -f BARCODE SEQCOUNT CONSCOUNT PRIMER PRCONS PRCOUNT PRFREQ ERROR \ --outdir logs ParseLog.py -l "logs/assemble.log" \ -f ID REFID LENGTH OVERLAP GAP ERROR PVALUE EVALUE1 EVALUE2 IDENTITY FIELDS1 FIELDS2 \ --outdir logs ParseLog.py -l "logs/maskqual.log" -f ID MASKED \ --outdir logs
Fig. 7 
Evaluation of B-cell isotype and clonal evolution. (a) Count and position of isotype primers. (b) Gene usage by isotype . (c) Reconstructed lineage tree
-
(a)

3.11 Gene Annotation
Raw sequences which have passed general quality control filters should and then be annotated with gene information: for IGH sequences V, D, and J genes and for IGK /IGL only V and J genes. The IgBLAST executable and the reference database are available in the Immcantation container.
-
1.
Convert FASTQ to FASTA
IgBLAST takes as input FASTA file. The FASTQ files obtained at the end of the raw data processing section need to be converted to FASTA format. Simultaneously, rename PRCONS to C_CALL.
ParseHeaders.py rename -s presto/S5-final_collapse-unique_atleast-2.fastq --fasta -f PRCONS -k C_CALL
-
2.
Run IgBLAST
The wrapper tool AssignGenes.py, from Change-O [6], uses IgBLAST, and a reference database created with germlines from IMGT, to make V(D)J allele calls.
mkdir changeo AssignGenes.py igblast -s presto/S5-final_collapse-unique_atleast-2_reheader.fasta \ --organism human --loci ig \ -b /usr/local/share/igblast --format blast --nproc 8 \ --outdir changeo --outname "S5"
-
3.
Data Standardization
IgBLAST’s results need to be converted into an AIRR-formatted file (https://immcantation.readthedocs.io/en/stable/datastandards.html) suitable for downstream analysis.
MakeDb.py igblast -s presto/S5-final_collapse-unique_atleast-2_reheader.fasta \ -i changeo/S5_igblast.fmt7 \ --extended --failed --format airr \ -r /usr/local/share/germlines/imgt/human/vdj/ --outname S5
Some sequences don’t pass this MakeDb step with these settings. This could be because a junction could not be identified, there are Ns in the junction, there is a stop codon, or the reads are partial, among other possible reasons.
3.12 Quality Control After Gene Assignment
Once sequences have been annotated with allele calls, and the aligned rearranged sequence is available, further collapsing of duplicates and removal of low-quality sequences is possible. Here we demonstrate how to perform some common additional QC steps using R and Immcantation tools (alakazam [6]). The goal is to keep sequences with at least 200 informative positions, with coherent gene and locus calls, and with a limited number of ambiguous nucleotides. It is also common to focus the analysis in productive sequences. Here we will keep productive sequences and will remove sequences with junction length that is not a multiple of three. Finally, chimeric reads will be identified and removed.
-
1.
Identify Short Sequences.
library(airr) library(alakazam) library(stringi) library(dplyr) airr <- read_rearrangement("changeo/S5_db-pass.tsv") # Min length 200 nt long_seq <- stri_count(airr[['sequence_alignment']],regex="[^-.N]") >= 200
-
2.
Identify Reads with Coherent Gene, Primer, and Isotype Calls
The goal is to remove sequences with incoherent gene and isotype calls. For example, a sequence that has a V gene assigned, but an IG light-chain-constant region will be removed.
# Keep reads with coherent gene, primer and isotype calls same_locus <- getLocus(airr[['v_call']]) == airr[['locus']] & getLocus(airr[[c_call]]) == airr[['locus']]
-
3.
Identify Reads with an Acceptable Number of Ambiguous Nucleotides.
# Max 10% N num_n <- stri_count(airr[['sequence_alignment']],fixed="N") len <- stri_count(airr[['sequence_alignment']],regex="[^-.]") low_n <- num_n/len <= 0.10
-
4.
Identify Productive Sequences.
prod <- airr[['productive']]
-
5.
Identify Sequences with Junction Length Multiple of Three.
m3 <- airr[['junction_length']] %% 3 == 0
-
6.
Filter and Save.
filter_pass <- long_seq & same_locus & low_n & prod & m3 write_rearrangement(airr[filter_pass,], file="changeo/S5_filter-pass.tsv")
-
7.
Reconstruct Germline Sequences
Identify the V(D)J germline sequences from which each of the sequences is derived. These germlines will be used to analyze mutation patterns in a sliding window to identify chimeric sequences.
CreateGermlines.py -d changeo/S5_filter-pass.tsv \ -r /usr/local/share/germlines/imgt/human/vdj \ -g dmask --format airr
-
8.
Identify and Remove Chimeric Sequences
Chimeric sequences can be identified by analyzing their mutation frequencies. The function slideWindowDb, from shazam [6], identifies which sequences in the repertoire contain excessive mutations in a given length of consecutive nucleotides (a “window”) when compared to their respective germline sequence.
library(airr) library(shazam) airr <- read_rearrangement("changeo/S5_filter-pass_germ-pass.tsv") is_chimeric <- slideWindowDb( airr, sequenceColumn = "sequence_alignment", germlineColumn = "germline_alignment_d_mask", mutThresh=6, windowSize=10 ) table(is_chimeric) airr <- airr[!is_chimeric,]
-
9.
Collapse Duplicates
Once the sequences in the repertoire are aligned following the IMGT scheme, further collapsing of duplicate sequences can be done with the function collapseDuplicates.
library(dplyr) num_fields <- c("consensus_count", "duplicate_count") # Data comes one sample, so no need to add # sample identifier groups collapse_groups <- c("v_gene", "j_gene", "junction_length", "c_call", "productive") airr <- airr %>% mutate(v_gene=getGene(v_call), j_gene=getGene(j_call)) %>% group_by(.dots=collapse_groups) %>% do(collapseDuplicates(., id = "sequence_id", seq = "sequence_alignment", text_fields = NULL, num_fields = num_fields, seq_fields = NULL, add_count = TRUE, ignore = c("N", "-", ".", "?"), sep = ",", dry = FALSE, verbose = FALSE )) %>% ungroup() %>% select(-v_gene, -j_gene)
3.13 Identify Clonally Related Sequences
The goal is to partition sequences into clonal lineages. Each clonal lineage is a group of sequences derived from the same original cell. There are several methods to identify clonal lineages (see Subheading 3.9.2: Identification of B-Cell Clones in the chapter “AIRR Community Guide to Repertoire Analysis”). Here, we first group by V gene, J gene, and junction length. Then we compare the junctions and apply a threshold to separate sequences into clonal lineages.
-
1.
Calculate the Distance to the Nearest Distribution
Hierarchical clustering requires a measure of distance between pairs of sequences and a choice of linkage to define the distance between groups of sequences. The result is a hierarchy, and a threshold is needed to cut the tree into clonal groups.
# Subset to heavy chain sequences airr_heavy <- airr %>% filter(locus == "IGH") # Group by V gene, J gene and junction length, and calculate the distance # to the nearest sequence in the group airr_heavy <- distToNearest(airr_heavy, sequenceColumn="junction", vCallColumn="v_call", jCallColumn="j_call", model="ham", first=FALSE, normalize="len", nproc=params$nproc) write_rearrangement(airr_heavy, file="changeo/IB7_heavy_collapse-pass.tsv")
-
2.
Find a Threshold
It is possible to determine a threshold by analyzing the distribution of the distances. The distribution is usually bimodal. The first mode represents sequences that have a close relative. The second mode is representative of sequences without clonal relatives. The goal is to select a threshold that separates the two modes.
threshold <- findThreshold(airr_heavy[['dist_nearest']], method="density") plot(threshold, binwidth=0.02, silent=FALSE) clone_threshold <- round(threshold@threshold,) clone_threshold
-
3.
Identify Clonally Related Sequences
Once a threshold is selected, it is applied to identify groups of related sequences:
DefineClones.py -d changeo/S5_heavy_collapse-pass.tsv --model ham \ --dist 0.09 --mode gene --act set --nproc 8 \ --outname S5 --outdir changeo --format airr --log "logs/clone.log"
-
4.
Reconstruct Clonal Germline
The next step is to identify the V(D)J germline sequences from which each of the observed sequences is derived. These germlines are used as the reference to analyze mutations.
CreateGermlines.py -d changeo/S5_clone-pass.tsv \ -r /usr/local/share/germlines/imgt/human/vdj \ -g dmask --format airr --cloned --outname S5-airr
3.14 Gene Usage by Isotype
When isotype information is available, it is possible to investigate biases in gene usage at the isotype level.
library(alakazam) library(airr) library(dplyr) airr <- read_rearrangement("changeo/S5-airr_germ-pass.tsv") # Gene usage by Isotype for one sample with only heavy chain data v_usage_isotype <- countGenes(airr, "v_call",group="c_call", fill=T) most_used_v <- v_usage_isotype %>% filter(c_call != "IGHE") %>% group_by(c_call) %>% slice_max(.,seq_freq,n=1) # Plot the most used V gene(s) library(scales) gene_usage_plot <- ggplot(v_usage_isotype %>% filter(c_call != "IGHE" & gene %in% most_used_v[['gene']]), aes(x=gene,y=seq_freq, color=c_call)) + scale_color_manual(values=color_palette) + scale_y_continuous(labels=percent) + geom_point(size=2) + theme_minimal() + xlab("Gene") + ylab("Frequency") + guides(color=guide_legend(title="Isotype")) gene_usage_plot
The gene usage by isotype can be visualized in Fig. 7b.
3.15 Clonal Lineage Tree Analysis
Dowser [7] provides tools for building and visualizing IG lineage trees using multiple methods and implements statistical tests for discrete trait analysis of B-cell migration, differentiation, and isotype switching.
-
1.
Format
First, data must be formatted into a data table of AIRR clone objects. The formatClones function will change non-nucleotide characters to N characters, collapse sequences that are either identical or differ only by ambiguous characters, and remove uninformative sequence sites in which all sequences have N characters.
library(dowser) clones <- formatClones(airr, traits=c("c_call"), num_fields=c("duplicate_count"), columns=c("d_call"), minseq=10)
-
2.
Build the Trees
There are several lineage reconstruction methods implemented in dowser. Maximum parsimony trees (topologies that minimize the number of mutations needed along the tree) can be built with the getTrees function.
# build maximum parsimony trees clones <- getTrees(clones)
-
3.
Visualize
The function plotTrees makes plotting lineages easy. Branch lengths by default represent the number of mutations per site between nodes. It is also possible to show numerical or categorical information associated with the tree tips, such as the duplicate count or the isotype .
# Plot the trees. Save them in a list of plots. # Use tip metadata: c_call and duplicate_count tree_plots <- plotTrees(clones, tips="c_call", tipsize="duplicate_count", tip_palette=c(color_palette, "Germline"="#000000"))
Example output is given in Fig. 7c.
4 Notes
-
1.
Other reagents may be substituted but require additional optimization to ensure adequate performance of the protocol. Store BCR enhancer at −20 °C. Once thawed, the buffer can be stored at 4 °C. Store nuclease-free water at −20 °C. Once thawed, the water can be stored at 4 °C. Store elution buffer at −20 °C. Once thawed, the buffer can be stored at room temperature.
-
2.
The success of the experiment depends on the quality of the input RNA. The RNA should be of high integrity (RIN > 7) to enable oligo(dT) priming. Prior to cDNA synthesis, ensure that the RNA is in nuclease-free water, is intact, and is free of contaminants. Input RNA should also be free from poly(A) carrier RNA that will interfere with oligo(dT)-primed cDNA synthesis.
-
3.
The First-Strand Buffer may form precipitates. Thaw this buffer at room temperature, and vortex before using to ensure all components are completely in solution.
-
4.
Each reverse PCR primer is used in a separate PCR Master Mix. Alternatively, plan to add 1 μL of each primer individually instead of including them in the PCR1 Master Mix, particularly if the number of samples is low.
-
5.
Different combinations of hBCR PCR2 Universal Forward 1–12 and hBCR PCR2 IgG/IgM/IgK/IgL reverse 1–4 indices must be used for each sample if samples are to be pooled and loaded on a single flow cell.
-
6.
Each PCR primer is used in a separate PCR Master Mix. Alternatively, plan to add 1 μL of each primer individually instead of including them in the PCR2 Master Mix, particularly if the number of samples is low.
-
7.
Aliquot NucleoMag beads into 1.5-mL tubes upon receipt in the laboratory. Before each use, bring bead aliquots to room temperature for at least 30 min, and mix well to disperse. Prepare fresh 80% ethanol for each experiment. You will need 400 μL per sample. You will need a magnetic separation device for 0.2-mL tubes, strip tubes, or a 96-well plate.
-
8.
The beads are viscous; pipette the entire volume and push it out slowly. Do not vortex. Vortexing will generate bubbles, making subsequent handling of the beads difficult.
-
9.
Ensure that the solution is completely clear, as any bead carryover will decrease the efficiency of size selection. There is no disadvantage to separating the samples for longer than 5 min.
-
10.
Ensure that the beads are fully resuspended before use. If the beads appear to have settled at the bottom of the tube, vortex to ensure that they are completely mixed.
-
11.
Be sure to dry the pellet only until it is just dry. The pellet will look matte with no shine.
-
12.
Be sure that the beads are completely resuspended. The beads can sometimes stick to the sides of the tube.
-
13.
Gently pipette any remaining beads that are in suspension toward the magnet where the rest of the beads have already pelleted. Continue the incubation until there are no beads left in the supernatant.
-
14.
Follow Illumina guidelines on how to denature, dilute, and combine a PhiX control library with your own pool of libraries. Make sure to use a fresh and reliable stock of the PhiX control library.
References
Vander Heiden JA, Yaari G, Uduman M, Stern JNH, O’Connor KC, Hafler DA et al (2014) pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics 30:1930–1932. https://doi.org/10.1093/bioinformatics/btu138
Mukherjee S, Huntemann M, Ivanova N, Kyrpides NC, Pati A (2015) Large-scale contamination of microbial isolate genomes by Illumina PhiX control. Stand Genomic Sci 10:18. https://doi.org/10.1186/1944-3277-10-18
Giudicelli V, Chaume D, Lefranc M-P (2005) IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res 33:D256–D261. https://doi.org/10.1093/nar/gki010
Ye J, Ma N, Madden TL, Ostell JM (2013) IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 41:W34–W40. https://doi.org/10.1093/nar/gkt382
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, Kleinstein SH (2015) Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31:3356–3358. https://doi.org/10.1093/bioinformatics/btv359
Hoehn KB, Pybus OG, Kleinstein SH (2020) Phylogenetic analysis of migration, differentiation, and class switching in B cells. Immunology. https://doi.org/10.1101/2020.05.30.124446
Acknowledgments
The authors would like to thank Eline Luning Prak for assistance with manuscript content and formatting of text and Chaim Schramm, Johannes Truck, and Wenwen Xiang for constructive criticism of the manuscript. Conflict of interest: NG and AF are employees at Takara Bio, Inc., San Jose, CA, USA, that produces the kit described in this protocol.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Gupta, N. et al. (2022). Bulk Sequencing from mRNA with UMI for Evaluation of B-Cell Isotype and Clonal Evolution: A Method by the AIRR Community. In: Langerak, A.W. (eds) Immunogenetics. Methods in Molecular Biology, vol 2453. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2115-8_19
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2115-8_19
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2114-1
Online ISBN: 978-1-0716-2115-8
eBook Packages: Springer Protocols