
Abstract
Background
Whole-Exome Sequencing (WES) is a valuable tool for the molecular diagnosis of patients with a suspected genetic condition. In complex and heterogeneous diseases, the interpretation of WES variants is more challenging given the absence of diagnostic handles and other reported cases with overlapping clinical presentations.
Objective
To describe candidate variants emerging from trio-WES and possibly associated with the clinical phenotype in clinically heterogeneous conditions.
Methods
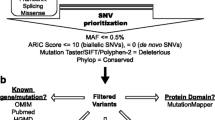
We performed WES in ten patients from eight families, selected because of the lack of a clear clinical diagnosis or suspicion, the presence of multiple clinical signs, and the negative results of traditional genetic tests.
Results
Although we identified ten candidate variants, reaching the diagnosis of these cases is challenging, given the complexity and the rarity of these syndromes and because affected genes are already associated with known genetic diseases only partially recapitulating patients’ phenotypes. However, the identification of these variants could shed light into the definition of new genotype–phenotype correlations. Here, we describe the clinical and molecular data of these cases with the aim of favoring the match with other similar cases and, hopefully, confirm our diagnostic hypotheses.
Conclusion
This study emphasizes the major limitations associated with WES data interpretation, but also highlights its clinical utility in unraveling novel genotype–phenotype correlations in complex and heterogeneous undefined clinical conditions with a suspected genetic etiology.


Similar content being viewed by others


Availability of data and materials
The data that support the findings of this study are available from the corresponding author, Prof. Monica Rosa Miozzo, upon reasonable request.
References
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249. https://doi.org/10.1038/nmeth0410-248
Bowling KM, Thompson ML, Amaral MD, Finnila CR, Hiatt SM, Engel KL, Cochran JN, Brothers KB, East KM, Gray DE et al (2017) Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med 9:43. https://doi.org/10.1186/s13073-017-0433-1
Chishti MS, Muhammad D, Haider M, Ahmad W (2006) A novel missense mutation in MSX1 underlies autosomal recessive oligodontia with associated dental anomalies in Pakistani families. J Hum Genet 51(10):872–878. https://doi.org/10.1007/s10038-006-0037-x
Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL (1998) Chlamydomonas Kinesin-II-dependent Intraflagellar Transport (IFT): IFT particles contain proteins required for ciliary assembly in caenorhabditis elegans sensory neurons. J Cell Biol 141(4):993–1008. https://doi.org/10.1083/jcb.141.4.993
Deciphering Developmental Disorders Study (2017) Prevalence and architecture of de novo mutations in developmental disorders. Nature 542(7642):433–438. https://doi.org/10.1038/nature21062
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M et al (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5):491–498. https://doi.org/10.1038/ng.806
Dobyns WB, Filauro A, Tomson BN, Chan AS, Ho AW, Ting NT, Oosterwijk JC, Ober C (2004) Inheritance of most X-linked traits is not dominant or recessive, just X-linked. Am J Med Genet 129(2):136–143. https://doi.org/10.1002/ajmg.a.30123
Dupont MA, Humbert C, Huber C, Siour Q, Guerrera IC, Jung V, Christensen A, Pouliet A, Garfa-Traoré M, Nitschké P et al (2019) Human IFT52 mutations uncover a novel role for the protein in microtubule dynamics and centrosome cohesion. Hum Mol Genet 28(16):2720–2737. https://doi.org/10.1093/hmg/ddz091
Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, Baxter RM, Zeng W, Mroske C, Parra MC et al (2015) Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med 17(7):578–586. https://doi.org/10.1038/gim.2014.154
Franklin by Genoox (2022) Franklin by Genoox. https://franklin.genoox.com Accessed 24 October 2022
Froyen G, Belet S, Martinez F, Santos-Rebouças CB, Declercq M, Verbeeck J, Donckers L, Berland S, Mayo S, Rosello M et al (2012) Copy-number gains of HUWE1 due to replication and recombination-based rearrangements. Am J Hum Genet 91(2):252–264. https://doi.org/10.1016/j.ajhg.2012.06.010
Global Genes (2019). Allies in Rare Disease—Global Genes. Glob Genes 2019. https://globalgenes.org/ Accessed 24 October 2022
Gubb SJA, Brcic L, Underwood JFG, Kendall KM, Caseras X, Kirov G, Davies W (2020) Medical and neurobehavioural phenotypes in male and female carriers of Xp22.31 duplications in the UK Biobank. Hum Mol Genet 29(17):2872–2881. https://doi.org/10.1093/hmg/ddaa174
Han JY, Lee IG (2020) Genetic tests by next-generation sequencing in children with developmental delay and/or intellectual disability. Korean J Pediatr 63(6):195–202. https://doi.org/10.3345/kjp.2019.00808
Hochstenbach R, van Binsbergen E, Engelen J, Nieuwint A, Polstra A, Poddighe P, Ruivenkamp C, Sikkema-Raddatz B, Smeets D, Poot M (2009) Array analysis and karyotyping: Workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands. Eur J Med Genet 52(4):161–169. https://doi.org/10.1016/j.ejmg.2009.03.015
Hong SY, Yang JJ, Li SY, Lee IC (2020) A wide spectrum of genetic disorders causing severe childhood epilepsy in Taiwan: a case series of ultrarare genetic cause and novel mutation analysis in a pilot study. J Pers Med 10(4):281. https://doi.org/10.3390/jpm10040281
Institute of Medicine (U.S.) Committee on Accelerating Rare Diseases Research and Orphan Product Development, Field MJ, Boat TF (2010) Rare diseases and orphan products: accelerating research and development. National Academy Press (US), doi: https://doi.org/10.17226/12953
Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, Massouras A (2019) VarSome: the human genomic variant search engine. Bioinformatics 35(11):1978–1980. https://doi.org/10.1093/bioinformatics/bty897
Kumps C, Dhaenens E, Vergult S, Leus J, Coster R, Jansen A, Devriendt K, Oostra A, Vanakker OM (2021) Phenotypic spectrum of the RBM10-mediated intellectual disability and congenital malformation syndrome beyond classic TARP syndrome features. Clin Genet 99(3):449–456. https://doi.org/10.1111/cge.13901
Kuperberg M, Lev D, Blumkin L, Zerem A, Ginsberg M, Linder I, Carmi N, Kivity S, Lerman-Sagie T, Leshinsky-Silver E (2016) Utility of whole exome sequencing for genetic diagnosis of previously undiagnosed pediatric neurology patients. J Child Neurol 31(14):1534–1539. https://doi.org/10.1177/0883073816664836
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W et al (2018) ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46(D1):D1062–D1067. https://doi.org/10.1093/nar/gkx1153
Le Tanno P, Breton J, Bidart M, Satre V, Harbuz R, Ray PF, Bosson C, Dieterich K, Jaillard S, Odent S et al (2017) PBX1 haploinsufficiency leads to syndromic congenital anomalies of the kidney and urinary tract (CAKUT) in humans. J Med Genet 54(7):502–510. https://doi.org/10.1136/jmedgenet-2016-104435
Li D, Ahrens-Nicklas RC, Baker J, Bhambhani V, Calhoun A, Cohen JS, Deardorff MA, Fernández-Jaén A, Kamien B, Jain M et al (2020) The variability of SMARCA4-related Coffin-Siris syndrome: do nonsense candidate variants add to milder phenotypes? Am J Med Genet A 182(9):2058–2067. https://doi.org/10.1002/ajmg.a.61732
Lindeboom RGH, Supek F, Lehner B (2016) The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat Genet 48(10):1112–1118. https://doi.org/10.1038/ng.3664
Lopes F, Barbosa M, Ameur A, Soares G, de Sá J, Dias AI, Oliveira G, Cabral P, Temudo T, Calado E et al (2016) Identification of novel genetic causes of Rett syndrome-like phenotypes. J Med Genet 53(3):190–199. https://doi.org/10.1136/jmedgenet-2015-103568
Maddala R, Skiba NP, Lalane R, Sherman DL, Brophy PJ, Rao PV (2011) Periaxin is required for hexagonal geometry and membrane organization of mature lens fibers. Dev Biol 357(1):179–190. https://doi.org/10.1016/j.ydbio.2011.06.036
Manoukian S, Verderio P, Tabano S, Colapietro P, Pizzamiglio S, Grati FR, Calvello M, Peissel B, Burn J, Pensotti V et al (2013) X chromosome inactivation pattern in BRCA gene mutation carriers. Eur J Cancer 49(5):1136–1141. https://doi.org/10.1016/j.ejca.2012.10.013
Martinez-Granero F, Blanco-Kelly F, Sanchez-Jimeno C, Avila-Fernandez A, Arteche A, Bustamante-Aragones A, Rodilla C, Rodríguez-Pinilla E, Riveiro-Alvarez R et al (2021) Comparison of the diagnostic yield of aCGH and genome-wide sequencing across different neurodevelopmental disorders. NPJ Genom Med 6(1):25. https://doi.org/10.1038/s41525-021-00188-7
Meng L, Pammi M, Saronwala A, Magoulas P, Ghazi AR, Vetrini F, Zhang J, He W, Dharmadhikari AV, Qu C et al (2017) Use of exome sequencing for infants in intensive care units ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr 171(12):e173438. https://doi.org/10.1001/jamapediatrics.2017.3438
Miceli F, Soldovieri MV, Joshi N, Weckhuysen S, Cooper EC, Taglialatela M (2014) KCNQ3-Related Disorders. https://www.ncbi.nlm.nih.gov/books/NBK201978/
Mitsui SN, Yasue A, Masuda K, Naruto T, Minegishi Y, Oyadomari S, Noji S, Imoto I, Tanaka E (2016) Novel human mutation and CRISPR/Cas genome-edited mice reveal the importance of C-terminal domain of MSX1 in tooth and palate development. Sci Rep 6(1):38398. https://doi.org/10.1038/srep38398
Monroe GR, Frederix GW, Savelberg SMC, de Vries TI, Duran KJ, van der Smagt JJ, Terhal PA, van Hasselt PM, Kroes HY, Verhoeven-Duif NM et al (2016) Effectiveness of whole-exome sequencing and costs of the traditional diagnostic trajectory in children with intellectual disability. Genet Med 18(9):949–956. https://doi.org/10.1038/gim.2015.200
Moortgat S, Berland S, Aukrust I, Maystadt I, Baker L, Benoit V, Caro-Llopis A, Cooper NS, Debray FG, Faivre L et al (2018) HUWE1 variants cause dominant X-linked intellectual disability: a clinical study of 21 patients. Eur J Hum Genet 26(1):64–74. https://doi.org/10.1038/s41431-017-0038-6
Ng PC, Henikoff S (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31(13):3812–3814. https://doi.org/10.1093/nar/gkg509
Nyegaard M, Rendtorff ND, Nielsen MS, Corydon TJ, Demontis D, Starnawska A, Hedemand A, Buniello A, Niola F, Overgaard MT et al (2015) A novel locus harbouring a functional CD164 nonsense mutation identified in a large danish family with nonsyndromic hearing impairment. PLoS Genet. https://doi.org/10.1371/journal.pgen.1005386
OMIM (2022). Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD). https://omim.org/ Accessed 24 October 2022
Paderova J, Drabova J, Holubova A, Vlckova M, Havlovicova M, Gregorova A, Pourova R, Romankova V, Moslerova V, Geryk J et al (2018) Under the mask of Kabuki syndrome: Elucidation of genetic-and phenotypic heterogeneity in patients with Kabuki-like phenotype. Eur J Med Genet 61(6):315–321. https://doi.org/10.1016/j.ejmg.2018.01.005
Paradowska-Stolarz A (2015) MSX1 homeobox cleft lip and/or palate hypodontia MSX1 gene in the etiology orofacial deformities. Postepy Hig Med Dosw (online) 69:1499–1504
Piton A, Redin C, Mandel JL (2013) XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am J Hum Genet 93(2):368–383. https://doi.org/10.1016/j.ajhg.2013.06.013
Poplin R, Ruano-Rubio V, Depristo MA, Fennell TJ, Carneiro MO, van der Auwera GA, Kling DE, Gauthier LD, Levy-Moonshine A, Roazen D et al (2018) Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 201178, doi: https://doi.org/10.1101/201178
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, Walkiewicz M, Bi W, Xiao R, Ding Y et al (2017) Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med 376(1):21–31. https://doi.org/10.1056/NEJMoa1516767
Qian Y, Zhou Y, Wu B, Chen H, Xu S, Wang Y, Zhang P, Li G, Xu Q, Zhou W et al (2021) Novel Variants of the SMARCA4 gene associated with autistic features rather than typical coffin-siris syndrome in eight Chinese pediatric patients. J Autism Dev Disord 52(11):5033–5041. https://doi.org/10.1007/s10803-021-05365-2
Rabbani B, Tekin M, Mahdieh N (2014) The promise of whole-exome sequencing in medical genetics. J Hum Genet 59(1):5–15. https://doi.org/10.1038/jhg.2013.114
Ramos P, Karnezis AN, Craig DW, Sekulic A, Russell ML, Hendricks WPD, Corneveaux JJ, Barrett MT, Shumansky K, Yang Y et al (2014) Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet 46(5):427–429. https://doi.org/10.1038/ng.2928
Redin C, Gérard B, Lauer J, Herenger Y, Muller J, Quartier A, Masurel-Paulet A, Willems M, Lesca G, El-Chehadeh S et al (2014) Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J Med Genet 51(11):724–736. https://doi.org/10.1136/jmedgenet-2014-102554
Reis LM, Semina EV (2019) Genetic landscape of isolated pediatric cataracts: extreme heterogeneity and variable inheritance patterns within genes. Hum Genet 138(8–9):847–863. https://doi.org/10.1007/s00439-018-1932-x
Reis LM, Tyler RC, Muheisen S, Raggio V, Salviati L, Han DP, Costakos D, Yonath H, Hall S, Power P, Semina EV (2013) Whole exome sequencing in dominant cataract identifies a new causative factor, CRYBA2, and a variety of novel alleles in known genes. Hum Genet 132(7):761–770. https://doi.org/10.1007/s00439-013-1289-0
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Richter T, Nestler-Parr S, Babela R, Khan ZM, Tesoro T, Molsen E, Hughes DA (2015) Rare disease terminology and definitions-a systematic global review: report of the ISPOR rare disease special interest group. Value in Health 18(6):906–914. https://doi.org/10.1016/j.jval.2015.05.008
Salsano E, Tabano S, Sirchia SM, Colapietro P, Castellotti B, Gellera C, Rimoldi M, Pensato V, Mariotti C, Pareyson D et al (2012) Preferential expression of mutant ABCD1 allele is common in adrenoleukodystrophy female carriers but unrelated to clinical symptoms. Orphanet J Rare Dis 7(1):10. https://doi.org/10.1186/1750-1172-7-10
Sands TT, Miceli F, Lesca G, Beck AE, Sadleir LG, Arrington DK, Schönewolf-Greulich B, Moutton S, Lauritano A, Nappi P et al (2019) Autism and developmental disability caused by KCNQ3 gain-of-function variants. Ann Neurol 86(2):181–192. https://doi.org/10.1002/ana.25522
Satokata I, Maas R (1994) Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet 6(4):348–356. https://doi.org/10.1038/ng0494-348
Sawyer SL, Hartley T, Dyment DA, Beaulieu CL, Schwartzentruber J, Smith A, Bedford HM, Bernard G, Bernier FP, Brais B et al (2016) Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet 89(3):275–284. https://doi.org/10.1111/cge.12654
Schnabel CA, Selleri L, Cleary ML (2003) Pbx1 is essential for adrenal development and urogenital differentiation. Genesis 37(3):123–130. https://doi.org/10.1002/gene.10235
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11(4):361–362. https://doi.org/10.1038/nmeth.2890
Stark Z, Tan TY, Chong B, Brett GR, Yap P, Walsh M, Yeung A, Peters H, Mordaunt D, Cowie S et al (2016) A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med 18(11):1090–1096. https://doi.org/10.1038/gim.2016.1
Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NST, Abeysinghe S, Krawczak M, Cooper DN (2003) Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 21(6):577–581. https://doi.org/10.1002/humu.10212
Tammimies K, Marshall CR, Walker S, Kaur G, Thiruvahindrapuram B, Lionel AC, Yuen RKC, Uddin M, Roberts W, Weksberg R et al (2015) Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA 314(9):895. https://doi.org/10.1001/jama.2015.10078
Taschner M, Kotsis F, Braeuer P, Wolfgang Kuehn E, Lorentzen E (2014) Crystal structures of IFT70/52 and IFT52/46 provide insight into intraflagellar transport B core complex assembly. J Cell Biol 207(2):269–282. https://doi.org/10.1083/jcb.201408002
Valencia CA, Husami A, Holle J, Johnson JA, Qian Y, Mathur A, Wei C, Indugula SR, Zou F, Meng H et al (2015) Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center’s experience. Front Pediatr 3:67. https://doi.org/10.3389/fped.2015.00067
Vissers LELM, van Nimwegen KJM, Schieving JH, Kamsteeg EJ, Kleefstra T, Yntema HG, Pfundt R, van der Wilt GJ, Krabbenborg L, Brunner HG et al (2017) A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet Med 19(9):1055–1063. https://doi.org/10.1038/gim.2017.1
Wang K, Li M, Hakonarson H (2010) ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. https://doi.org/10.1093/nar/gkq603
Wright CF, FitzPatrick DR, Firth HV (2018) Paediatric genomics: diagnosing rare disease in children. Nat Rev Genet 19(5):253–268. https://doi.org/10.1038/nrg.2017.116
Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z et al (2013) Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 369(16):1502–1511. https://doi.org/10.1056/NEJMoa1306555
Yoder M, Hildebrand JD (2007) Shroom4 (Kiaa1202) is an actin-associated protein implicated in cytoskeletal organization. Cell Motil Cytoskelet 64(1):49–63. https://doi.org/10.1002/cm.20167
Yuan L, Yi J, Lin Q, Xu H, Deng X, Xiong W, Xiao J, Jiang C, Yuan X, Chen Y, Deng H (2016) Identification of a PRX variant in a Chinese family with congenital cataract by exome sequencing. QJM 109(11):731–735. https://doi.org/10.1093/qjmed/hcw058
Zhong Q, Gao W, Du F, Wang X (2005) Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 121(7):1085–1095. https://doi.org/10.1016/j.cell.2005.06.009
Acknowledgements
We would like to thank the probands and their families for adhering to the study and giving consent to publish their data, as well as the clinical investigators and research staff (in particular Erica Rosina, Benedetta Beltrami, Federica Gaudioso and Diana Simpatico) for their participation in the “CARE: Challenging Approaches to undiagnosed Rare diseases with Exome sequencing” project. The project was granted by Scientific Direction, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico of Milan.
Author information
Authors and Affiliations
Contributions
GM, OR, AM, CP: WES data analysis and interpretation, and manuscript drafting. JC: bioinformatic analysis. CS, PC: WES and Sanger experiments. EM, GM, FG, BR, EP, GS, RV, MFB, DM: clinical evaluation and patient follow-up. MRM, LF: project supervision and contribution to result interpretation.
Corresponding author
Ethics declarations
Conflict of interest
All authors (Giada Moresco, Ornella Rondinone, Alessia Mauri, Jole Costanza, Carlo Santaniello, Patrizia Colapietro, Emanuele Micaglio, Giovanni Marfia, Federico Grilli, Berardo Rinaldi, Elisabetta Prada, Giulietta Scuvera, Roberta Villa, Maria Francesca Bedeschi, Monica Rosa Miozzo, Donatella Milani and Laura Fontana) declare that they have no conflict of interest.
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico of Milan (Protocol code: PED-CARE-2018).
Patient consent for publication
Informed consent for publication of clinical data was obtained from all patients and their family, including pictures of affected individuals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Moresco, G., Rondinone, O., Mauri, A. et al. Pitfalls of whole exome sequencing in undefined clinical conditions with a suspected genetic etiology. Genes Genom 45, 637–655 (2023). https://doi.org/10.1007/s13258-022-01341-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-022-01341-x