
Abstract
Tooth agenesis constitutes the most common anomaly of dental development in humans. In the majority of familial cases of hypodontia alone or in association with other anomalies, the mode of inheritance is autosomal dominant. In the present study, we have identified two distantly related consanguineous Pakistani kindreds with an autosomal recessive form of oligodontia with associated dental anomalies. Locus in this case has been mapped on chromosome 4p16.1–p16.3. The maximum two-point LOD score of 2.85 (θ=0.0) was obtained at markers D4S2925 and D4S2285. A maximum multipoint LOD score exceeding 4 was obtained at the same markers. Recombination events observed in affected individuals localized the disease locus between markers D4S412 and D4S2935, spanning a 9.24-cM region on chromosome 4p16.1–p16.3. Sequence analysis of candidate gene MSX1 revealed a novel recessive missense mutation resulting in substitution of alanine to threonine amino acid (p. A219T), located in the MSX1 homeodomain, which is important for DNA binding and protein–protein interaction. The mutation, p. A219T, is the first recessive mutation identified in MSX1.
Similar content being viewed by others
Introduction
Congenital lack of one or a few permanent and/or deciduous teeth is among the well-recognized morphologic anomalies in humans. The teeth most often missing are second premolars, maxillary lateral incisors and third premolars (Aasheim and Ogaard 1993; Arte et al. 1996). Three different terms, hypodontia, oligodontia and anodontia, are generally used to describe the phenomenon of congenitally missing teeth. Arte and Pirinen (2004) have defined these terms by considering the number of teeth missing. The absence of one to six, more than six, and complete absence of teeth have been termed hypodontia, oligodontia, and anodontia, respectively.
A number of studies have showed the association of dental anomalies with congenitally missing teeth. Baccetti (1998) demonstrated the association of crown-size reduction with hypodontia. Ahmad et al. (1998) reported the association of hypodontia with dental anomalies such as malformation, enamel hypoplasia, failure of eruption of teeth, hypocalcification and dentinogenesis imperfecta. A condition called short root anomaly with short roots of some permanent teeth was described by Apajalahti et al. (1999). Association of hypodontia and oligodontia with taurodontism was reported by Arte et al. (2001). Some of the other abnormalities of dentition such as malposition of canines and ectopic eruption of first permanent molars are reported to have association with hypodontia (Svinhufvud et al. 1988; Bjerklin et al. 1992). Hypodontia and oligodontia have been reported in several syndromes. Complete agenesis of teeth has been observed in hypohydrotic ectodermal dysplasia (HED), which is a recessively inherited X-linked and autosomal condition. Mutations in genes EDA (ectodysplasin A, MIM 300451) (Passarge et al. 1966) and EDAR (ectodysplasin A receptor, MIM 604095) (Naeem et al. 2005) have been reported to be responsible for HED.
Tooth agenesis is caused by both environmental and genetic factors. Environmental factors include radiation therapy, chemotherapeutic agents and use of thalidomide during pregnancy (Axrup et al. 1966; Maguire et al. 1987; Nasman et al. 1997). In the majority of familial cases of hypodontia alone or its association with other anomalies, the mode of inheritance is autosomal dominant (Vastardis et al. 1996; van den Boogaard et al. 2000; Jumlongras et al. 2001; Lidral and Reising 2002; Jezewski et al. 2003; De Muynck et al. 2004). An autosomal recessive model of hypodontia with associated anomalies was reported in a large family by Ahmad et al. (1998). The locus in this case was mapped on chromosome 16q12.1. Several cases of recessively inherited lower incisor hypodontia (RIH) were reported by Pirinen et al. (2001), but no disease locus or gene was identified.
In the present study we have ascertained two distantly related consanguineous families from a remote region in Pakistan with autosomal recessive form of oligodontia associated with dental anomalies. A candidate gene approach and DNA sequencing led to the identification of a missense mutation (p. A219T) in MSX1 gene.
Materials and methods
Family history
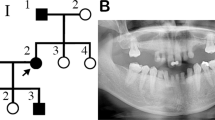
Two distantly related, consanguineous Pakistani families segregating an autosomal recessive form of oligodontia associated with dental anomalies were ascertained in which five males were affected. Prior to start of the study, approval was obtained from Quaid-i-Azam University Institutional Review Board (IRB). Informed consent was obtained from the individuals who agreed to participate in the study. The family members rarely marry outside the community, and consequently consanguineous unions are common. The pedigrees (Fig. 1) provided convincing evidence of autosomal recessive mode of inheritance, and consanguineous loops accounted for all the affected persons being homozygous for the mutant allele. All of the affected and normal individuals underwent examination at the Department of Oral and Maxillofacial Surgery, Karachi Medical Dental College and Abbasi Shaheed Hospital, Pakistan.

Pedigrees of two distantly related Pakistani families (a, b), with autosomal recessive oligodontia and associated dental anomalies. Filled symbols represent affected subjects. Open symbols represent unaffected individuals. The disease-associated haplotypes are shown beneath each symbol. Arrows on the haplotypes of the corresponding affected individuals indicate key recombination events
DNA extraction and genotyping
Blood samples were collected from five affected and eight unaffected members of the families. Genomic DNA was extracted from peripheral blood leucocytes in EDTA-containing tubes by the standard method (Grimberg et al. 1989).
Polymerase chain reaction (PCR) was carried out in 25-μl reaction volumes containing 40 ng of genomic DNA, 20 pmol of primers, 200 μM of each dNTP, 1U of Taq DNA polymerase (MBI Fermentas, UK), and 2.5 μl reaction buffer (KCl 50 mM, Tris–Cl pH 8.3, MgCl2 1.5 mM). The thermal cycling conditions used were 95°C for 5 min, followed by 40 cycles of 95°C for 1 min, 57–59°C for 1 min, 72°C for 1 min, and final extension at 72°C for 10 min. PCR was performed in a thermal cycler, Gene Amp PCR system 9700, obtained from Applied Biosystems (Foster City, CA, USA). PCR products were resolved on 8% non-denaturing polyacrylamide gel, and genotypes were assigned by visual inspection.
Linkage analysis
The pedigree and genotype data were checked with PEDCHECK (O’Connell and Weeks 1998). Two-point linkage analysis was carried out using the MLINK program of the FASTLINK computer package (Cottingham et al. 1993). Multipoint linkage analysis was performed using ALLEGRO (Gudbjartsson et al. 2002). The order of the fine mapping markers was determined following the National Center for Biotechnology Information (NCBI) Build 34 sequence-based physical map (http://www.ncbi.nlm.nih.gov), and the genetic map distances were derived from the Marshfield genetic map of the human genome (Broman et al. 1998). Haplotypes were constructed using SIMWALK2 (Weeks et al. 1995; Sobel and Lange 1996). An autosomal recessive mode of inheritance with complete penetrance and a disease allele frequency of 0.001 were used for the analysis. Equal allele frequencies were used for the fine mapping markers because it was not possible to estimate allele frequencies from the founders, since these markers were only genotyped in these families.
Mutation detection
To screen for the mutation in the MSX1 gene, both of its exons and splice junctions were PCR-amplified from genomic DNA and sequenced directly in an ABI prism 310 Automated DNA Sequencer, using the ABI Big Dye Terminator V 3.1 cycle sequencing kit (PE Applied Biosystem), following purification by rapid PCR purification system (Marligen Biosciences USA). To amplify 320-bp PCR fragments containing 250 bp of exon-2, the following primers were used: 5′ ACT TCT TGG GCT GAT CAT GC 3′ (intron-2, sense) and 5′ ATC TTC AGC TTC TCC AGC TC 3′ (exon-2, antisense).
Results
Clinical findings
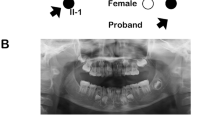
Dental examinations of the affected subjects in our families demonstrated a range of dental anomalies involving several maxillary and mandibular teeth. Panoramic radiographs (Fig. 2) showed the missing teeth and their abnormal structures. The affected male individual (V-2) in family a (Fig. 2a) had all four canines erupted and in place. The left central incisor was fully formed, while the lateral incisor on the same side appeared to be a retained deciduous tooth. The right central incisor had a deformed crown and root. Retained deciduous first maxillary premolars were also present. The rest of the maxilla had no alveolar bone. There appeared to be a midline fissure in the maxilla. In the lower jaw, the cuspids were present bilaterally, however there were three more teeth present in the anterior segment, which appeared to be lower permanent incisors, which were deformed in shape. Alveolar bone was missing bilaterally while the rest of the bone was healthy and robust looking.

Panoramic radiographs of affected subjects. a V-2 in family a, and b IV-1 in family b showing a number of dental anomalies involving several maxillary and mandibular teeth
Subject IV-1 from family b (Fig. 2b) had normal maxillary alveolus with right central incisor missing and a full set of dentition in the left maxillary arch. The maxillary left wisdom tooth was impacted. The teeth were smaller in size and appeared short in length. The chambers were not well demarcated and appeared fused with the canals. In the mandible both cuspids and first premolars were present and fully formed. The right mandibular second deciduous molar was retained with resorbed roots and the only other tooth present was the right lower third molar, which was unerrupted. Dental examination of the carriers (IV-3, IV-4 and V-4 in family a and III-2 and IV-4 in family b) showed no abnormality.
Assignment of hypodontia with associated anomalies to MSX1 locus
To identify the gene underlying the congenital hypodontia with the associated anomaly phenotypes, we followed a classical linkage analysis approach. We performed cosegregation and homozygosity analysis with microsatellite markers corresponding to candidate genes involved in the related phenotype. These included MSX1 (MIM 142983) at 4p16.1-p16.3 (D4S412, D4S2925, D4S2935, D4S2285) (Ivens et al. 1990), MSX2 (MIM 123101) at 5q34-q35 (D5S529, D5S2050, D5S625, D5S2108) (Jabs et al. 1993), epidermal growth factor (EGF, MIM 131530) at 4q25–27 (D4S1564, D4S193, D4S1611, D4S402) (Morton et al. 1986), epidermal growth factor receptor (EGFR, MIM 131550) at 7p12.1-p12.3 (D7S2561, D7S674, D7S506, D7S2475) (Haley et al. 1987), ectodermal dysplasia type 1 (ED1, MIM 305100) (MIM 300451) at Xq12-q13.1 (DXS7132, DXS1216, DXS8031, DXS453) (Srivastava et al. 1996), ectodermal dysplasia type 2 (ED2, MIM 129500) gene GJB6 (MIM 604418) at 13q12.11-q12.3 (D13S1316, D13S175, D13S633, D13S250, D13S787) (Kelley et al. 1999), ectodermal dysplasia type 3 (ED3, MIM 224900), ectodysplasin1 anhidrotic receptor (EDAR, MIM 604095) at 2q11-q13 (D2S1343, D2S2954, D2S340, D2S1889, D2S1893, D2S1891) (Monreal et al. 1999), and ectodermal dysplasia type 4 (ED4, MIM 225000) PVRL1 (MIM 600644) at 11q23-q24 (D11S1885, D11S4171, D11S4129, D11S924, D11S1299) (Lopez et al. 1995).
Evidence of linkage was obtained with markers D4S2925 and D4S2285 mapped in the MSX1 candidate region. For fine mapping, 12 additional markers (D4S2936, D4S412, D4S432, D4S3023, D4S431, D4S2366, D4S2935, D4S3007, D4S394, D4S2983, D4S1599 and D4S2949) were selected from the Marshfield genetic map (Broman et al. 1998). After genotyping all the family members with these markers, the data were analyzed using two-point and multipoint linkage analysis. Six of these markers were uninformative in this family and were not included in the analysis. The two-point analysis generated a maximum LOD score of 2.85 (θ=0.0) at markers D4S2925 and D4S2285 (Table 1). Multipoint analysis supported linkage to this region with maximum LOD score exceeding 4 at the same two markers.
Haplotypes were constructed to determine the critical recombination events. The disease haplotype (region of homozygosity) corresponds to the 9.24 cM region, spanning from D4S412 to D4S2935, according to the Marshfield genetic map (Broman et al. 1998). This region corresponds to a physical map distance of 3.26 Mb (International Human Genome Sequence Consortium 2001). The critical recombinations defining the co-segregating interval occurred in affected individuals. The centromeric boundary of this interval was defined by a recombination between markers D4S2285 and D4S2935 observed in affected individuals (V-2, V-3 and V-6) in family a. The telomeric boundary of this interval corresponds to a recombination event between markers D4S2925 and D4S412 in affected individual IV-1 in family b.
Screening MSX1 for pathogenic mutation
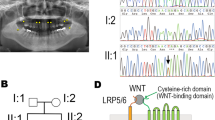
Because MSX1 was located in the candidate region identified in our families, the coding region and the splice junction sites of the same gene were sequenced in affected and normal individuals of these families. Sequence analysis of exon 2 of the gene from affected individuals V-2, V-3 and V-6 in family a, and IV-1 and IV-3 in family b revealed a G-to-A transition at nucleotide position 655 (Fig. 3) resulting in an alanine to threonine (p. A219T) amino acid substitution. This novel mutation was present in the heterozygous state in obligate carriers within these families (Fig. 3b). To ensure that the mutation does not represent a neutral polymorphism in the Pakistani population, a panel of 100 unrelated unaffected individuals (200 chromosomes) was screened for mutation using PCR followed by sequencing, and the mutation was not identified outside these families.

Automated DNA sequence analysis of MSX1 gene mutation. a DNA sequence analysis of wild-type allele. b DNA sequence analysis of a heterozygous carrier (IV-3) in family a. c DNA sequence analysis of an affected individual (V-2) in family a
Discussion
A tooth begins development as an epithelial bud, which undergoes complex morphogenesis and is regulated by interactions between the epithelial and mesenchymal tissue layers. These types of interactions are mediated by signal molecules such as bone morphogenetic proteins (BMP), fibroblast growth factors (FGF), sonic hedgehog (Shh) and tumor necrosis factor (TNF). These signals operate throughout development and regulate the expression of genes in the responding tissues. Mutations in many of these genes have been shown to cause dental defects in mice and humans (Thesleff 2000). In mice, although a null mutation in the MSX1 gene resulted in a complete failure of tooth development, mice heterozygous for MSX1 did not present with any tooth abnormalities (Satokata and Maas 1994; Houzelstein et al. 1997). However, mice carrying MSX1 null mutation die right after birth, and exhibit severe craniofacial abnormalities (Satokata and Maas 1994; Houzelstein et al. 1997). These phenotypes include cleft palate, an absence of alveolar processes, and an arrest of tooth development at the bud stage, thus predicting the phenotype observed in humans carrying MSX1 mutations.
Mutations in MSX1, a non-clustered homeobox protein, have been reported previously in families with autosomal dominant hypodontia (Vastardis et al. 1996; van den Boogaard et al. 2000; Jumlongras et al. 2001; Lidral and Reising 2002; Jezewski et al. 2003; De Muynck et al. 2004). The present study presents the first mutation identified in a Pakistani family with autosomal recessive hypodontia with associated dental anomalies. This missense mutation (p. A219T) is located in helix III of the MSX1 homeodomain. The conserved regions among the MSX proteins are the MSX homology regions I, II and III (MHR1 to -III), the extended homeodomain (EHD), and the homeodomain (Davidson 1995). The homeodomain consisting of 60 amino acids numbered 166–225 is subdivided into the N-terminal arm (NT arm) and helices I, II and III, which contribute to protein stability, DNA binding specificity, transcriptional repression and protein interactions (Shang et al. 1994; Isaac et al. 1995; Zhang et al. 1996, 1997). In the MSX1 homeodomain, there are several conserved residues that constitute a consensus sequence, which promotes tertiary structure and mediates DNA binding activity (Scott et al. 1989; Laughon 1991; Billeter 1993). Consensus residues in helices I–III furnish the hydrophobic core and preserve the amphipathic nature of the alpha helices, whereas other consensus residues in helix III and in the N-terminal arm contact DNA (Shang et al. 1994).
MSX1 has been shown to interact with distal-less homeobox (DLX) and TATA box binding protein (TBP) (Catron et al. 1995; Zhang et al. 1996). Both these types of interactions have been shown to be mediated by the homeodomain, specifically amino acids in the N-terminal region. The overlapping expression patterns of MSX1 and DLX genes and their involvement in epithelial–mesenchymal signaling cascades of murine odontogenesis suggest that MSX and DLX proteins form heterodimeric complexes in vivo which provide a mechanism for transcriptional regulation via functional antagonism (Zhang et al. 1997).
The substitution p. A219T occurs in helix III (amino acids 207–225) of the MSX1 homeodomain, which is important for DNA binding and DLX interaction. Therefore, it is more likely that p. A219T mutation may result in abnormal structure of protein along with reduced or no DNA binding activity. It is also possible that the p. A219T may interfere with dimerization of MSX1 and DLX proteins.
Hu et al. (1998) proposed that tooth agenesis in affected individuals, due to heterozygous mutations in MSX1, is a result of haplo insufficiency and not dominant negative mutations. The heterozygous missense mutations in humans selectively affect certain teeth including the second premolars and third molars (Vastardis et al. 1996). In mice, MSX1 expresses throughout the tooth mesenchyme (Jowett et al. 1993), and complete loss of MSX1 function results in failure of tooth development (Chen et al. 1996). However, mice heterozygous for MSX1 showed no abnormality in tooth development (Satokata and Maas 1994). Because dental examination of carriers and affected family members in our study showed abnormality only in the affected individuals, we propose that phenotype in our family is due to loss of function mutation.
References
Aasheim B, Ogaard B (1993) Hypodontia in 9-year-old Norwegians related to need of orthodontic treatment. Scand J Dent Res 101:257–260
Ahmad W, Brancolini V, ul Faiyaz MF, Lam H, ul Haque S, Haider M, Maimon A, Aita VM, Owen J, Brown D, Zegarelli DJ, Ahmad M, Ott J, Christiano AM (1998) A locus for autosomal recessive hypodontia with associated dental anomalies maps to chromosome 16q12.1. Am J Hum Genet 62:987–991
Apajalahti S, Arte S, Pirinen S (1999) Short root anomaly in families and its association with other dental anomalies. Eur J Oral Sci 107:97–101
Arte S Pirinen S (2004) Hypodontia. Orphanet encyclopedia (http://www.orpha.net/data/patho/GB/uk-hypodontia.pdf)
Arte S, Nieminen P, Pirinen S, Thesleff I, Peltonen L (1996) Gene defect in hypodontia: exclusion of EGF, EGFR, and FGF-3 as candidate genes. J Dent Res 75:1346–1352
Arte S, Nieminen P, Apajalahti S, Haavikko K, Thesleff I, Pirinen S (2001) Characteristics of incisor-premolar hypodontia in families. J Dent Res 80:1445–5140
Axrup K, D Avignon M, Hellgren K, Herikson CO, Juhlin IM, Larsson KS (1966) Children with thalidomide embryopathy: odontological observations and aspects. Acta Odontol Scand 24:3–21
Baccetti T (1998) A clinical and statistical study of etiologic aspects related to associated tooth anomalies in number, size, and position. Minerva Stomatol 47:655–663
Billeter M, Qian YQ, Otting G, Muller M, Gehring W, Wuthrich K (1993) Determination of the nuclear magnetic resonance solution structure of an antennapedia homeodomain–DNA complex. J Mol Biol 234:1084–1093
Bjerklin K, Kurol J, Valentin J (1992) Ectopic eruption of maxillary first permanent molars and association with other tooth and developmental disturbances. Eur J Orthod 14:369–375
Broman KW, Murray JC, Sheffield VC, White RL, Weber JL (1998) Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am J Hum Genet 63:861–869
Catron KM, Zhang H, Marshall SC, Inostroza JA, Wilson JM, Abate C (1995) Transcriptional repression by MSX-1 does not require homeodomain DNA-binding sites. Mol Cell Biol 15:861–871
Chen Y, Bei M, Woo I, Satokata I, Maas R (1996) MSX1 controls inductive signaling in mammalian tooth morphogenesis. Development 122:3035–3044
Cottingham R, Indury RM, Schaffer AA (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53:252–263
Davidson D (1995) The function and evolution of MSX genes: pointers and paradoxes. Trends Genet 11:405–411
De Muynck S, Schollen E, Matthijs G, Verdonck A, Devriendt K, Carels C (2004) A novel MSX1 mutation in hypodontia. Am J Med Genet A 128:401–403
Grimberg J, Nawoschik S, Bellusico L, McKee R, Turck A, Eisenberg A (1989) A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acid Res 17:61–68
Gudbjartsson DF, Jonasson K, Frigge ML, Kong A (2002) Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25:12–13
Haley J, Whittle N, Bennett P, Kinchington D, Ullrich A, Waterfield M (1987) The human EGF receptor gene: structure of the 110 kb locus and identification of sequences regulating its transcription. Oncogene Res 1:375–396
Houzelstein D, Cohen A, Buckingham ME, Robert B (1997) Insertional mutation of the mouse MSX1 homeobox gene by an nlacZ reporter gene. Mech Dev 65:123–133
Hu G, Vastardis H, Bendall AJ, Wang Z, Logan M, Zhang H, Nelson C, Stein S, Greenfield N, Seidman CE, Seidman JG, Abate-Shen C (1998) Haploinsufficiency of MSX1: a mechanism for selective tooth agenesis. Mol Cell Biol 18:6044–6051
Isaac VE, Sciavolino P, Abate C (1995) Multiple amino acids determine the DNA binding specificity of the MSX-1 homeodomain. Biochemistry 34:7127–7134
Ivens A, Flavin N, Williamson R, Dixon M, Bates G, Buckingham M, Robert B (1990) The human homeobox gene HOX7 maps to chromosome 4p16.1 and may be implicated in Wolf-Hirschhorn syndrome. Hum Genet 84:473–476
Jabs EW, Muller U, Li X, Ma L, Luo W, Haworth I, S Klisak I, Sparkes R, Warman ML, Mulliken JB, Snead ML, Maxson R (1993) A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell 75:443–450
Jezewski PA, Vieira AR, Nishimura C, Ludwig B, Johnson M, O’Brien SE, Daak-Hirsch S, Schultz RE, Weber A, Nepomucena B, Romitti PA, Christensen K, Orioli IM, Castilla EE, Machida J, Natsume N, Murray JC (2003) Complete sequencing shows a role for MSX1 in non-syndromic cleft lip and palate. J Med Genet 40:399–407
Jowett AK, Vainio S, Ferguson MW, Sharpe PT, Thesleff I (1993) Epithelial–mesenchymal interactions are required for MSX1 and MSX2 gene expression in the developing murine molar tooth. Development 117:461–470
Jumlongras D, Bei M, Stimson JM, Wang WF, DePalma SR, Seidman CE, Felbor U, Maas R, Seidman JG (2001) A nonsense mutation in MSX1 causes Witkop syndrome. Am J Hum Genet 69:67–74
Kelley PM, Abe S, Askew JW, Smith SD, Usami S, Kimberling WJ (1999) Human connexin 30 (GJB6), a candidate gene for nonsyndromic hearing loss: molecular cloning, tissue-specific expression, and assignment to chromosome 13q12. Genomics 62:172–176
Laughon A (1991) DNA binding specificity of homeodomain. Biochemistry 30:11357–11367
Lidral AC, Reising BC (2002) The role of MSX1 in human tooth agenesis. J Dent Res 81:274–278
Lopez M, Eberle F, Mattei MG, Gabert J, Birg F, Bardin F, Maroc C, Dubreuil P (1995) Complementary DNA characterization and chromosomal localization of a human gene related to the poliovirus receptor-encoding gene. Gene 155:261–265
Maguire A, Craft AW, Evans RG, Amineddine H, Kernahan J, Macleod RI, Murray JJ, Welbury RR (1987) The long-term effects of treatment on the dental condition of children surviving malignant disease. Cancer 60:2570–2575
Monreal AW, Ferguson BM, Headon DJ, Street SL, Overbeek PA, Zonana J (1999) Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat Genet 22:366–369
Morton CC, Byers MG, Nakai H, Bell GI, Shows TB (1986) Human genes for insulin-like growth factors I and II and epidermal growth factor are located on 12q22-q24.1, 11p15, and 4q25-q27, respectively. Cytogenet Cell Genet 41:245–249
Naeem M, Muhammad D, Ahmad W (2005) Novel mutations in the EDAR gene in two Pakistani consanguineous families with autosomal recessive hypohidrotic ectodermal dysplasia. Br J Dermatol 153:46–50
Nasman M, Forsberg CM, Dahllof G (1997) Long-term dental development in children after treatment for malignant disease. Eur J Orthod 19:151–159
O’Connell JR, Weeks DE (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63:259–266
Passarge E, Nuzum CT, Schubert WK (1966) Anhidrotic ectodermal dysplasia as autosomal recessive trait in an inbred kindred. Humangenetik 3:181–185
Pirinen S, Kentala A, Nieminen P, Varilo T, Thesleff I, Arte S (2001) Recessively inherited lower incisor hypodontia. J Med Genet 38:551–556
Satokata I, Maas R (1994) MSX1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet 6:348–356
Scott MP, Tamkun JW, Hartzell GW 3rd (1989) The structure and function of the homeodomain. Biochim Biophys Acta 989:25–48
Shang Z, Isaac VE, Li H, Patel L, Catron KM, Curran T, Montelione GT, Abate C (1994) Design of a “minimAl” homeodomain: the N-terminal arm modulates DNA binding affinity and stabilizes homeodomain structure. Proc Natl Acad Sci USA 91:8373–8377
Sobel E, Lange K (1996) Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 58:1323–1337
Srivastava AK, Montonen O, Saarialho-Kere U, Chen E, Baybayan P, Pispa J, Limon J, Schlessinger D, Kere J (1996) Fine mapping of the EDA gene: a translocation breakpoint is associated with a CpG island that is transcribed. Am J Hum Genet 58:126–132
Svinhufvud E, Myllarniemi S, Norio R (1988) Dominant inheritance of tooth malpositions and their association to hypodontia. Clin Genet 34:373–381
Thesleff I (2000) Genetic basis of tooth development and dental defects. Acta Odontol Scand 58:191–194
van den Boogaard MJ, Dorland M, Beemer FA, van Amstel HK (2000) MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet 24:342–343. Erratum in: Nat Genet 2000 25:125
Vastardis H, Karimbux N, Guthua SW, Seidman JG, Seidman CE (1996) A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet 13:417–421
Weeks DE, Sobel E, O’Connell JR, Lange K (1995) Computer programs for multilocus haplotyping of general pedigrees. Am J Hum Genet 56:1506–1507
Zhang H, Catron KM, Abate-Shen C (1996) A role for the MSX-1 homeodomain in transcriptional regulation: residues in the N-terminal arm mediate TATA binding protein interaction and transcriptional repression. Proc Natl Acad Sci USA 93:1764–1769
Zhang H, Hu G, Wang H, Sciavolino P, Iler N, Shen M (1997) Heterodimerization of MSX and DLX homeoproteins results in functional antagonism. Mol Cell Biol 17:2920–2932
Acknowledgements
We wish to thank the family members for their cooperation. This work was made possible through a grant from Higher Education Commission (HEC), Islamabad, Pakistan. Muhammad Salman Chishti was supported by indigenous PhD fellowships from Higher Education Commission, Islamabad, Pakistan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chishti, M.S., Muhammad, D., Haider, M. et al. A novel missense mutation in MSX1 underlies autosomal recessive oligodontia with associated dental anomalies in Pakistani families. J Hum Genet 51, 872–878 (2006). https://doi.org/10.1007/s10038-006-0037-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-006-0037-x
Keywords
This article is cited by
-
Pitfalls of whole exome sequencing in undefined clinical conditions with a suspected genetic etiology
Genes & Genomics (2023)
-
Novel MSX1 variants identified in families with nonsyndromic oligodontia
International Journal of Oral Science (2021)
-
Novel human mutation and CRISPR/Cas genome-edited mice reveal the importance of C-terminal domain of MSX1 in tooth and palate development
Scientific Reports (2016)
-
MSX1 mutations and associated disease phenotypes: genotype-phenotype relations
European Journal of Human Genetics (2016)
-
Genetic background of nonsyndromic oligodontia: a systematic review and meta-analysis
Journal of Orofacial Orthopedics / Fortschritte der Kieferorthopädie (2013)
