
Abstract
Dynamic behaviors of cells require optimized functions and regulations of protein complexes within a cell. In vitro reconstitution is a powerful method to dissect the molecular functions and structure of reconstituted protein complexes. However, owing to the difficulties associated with in vitro reconstitution of complicated supra-molecular complexes, such as the mitotic spindle and cortical force-generating machinery, key molecular features underlying their functions and regulations are not fully understood. Recently, several light-inducible dimerization tools have been developed to control localization of target proteins within a cell in a spatiotemporally controlled manner. In this chapter, I have summarized the recent progress in optogenetic tools and fluorescent proteins/probes and have determined strategies to achieve light-induced in cell reconstitution of protein complexes coupled with visualization of its downstream events. Moreover, I have discussed the advantages and potential of the optogenetic reconstitution system in diverse cell and developmental biology fields.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
To understand the basis of life, it is critical to visualize the dynamic behaviors of molecules within a cell. Since the discovery of green fluorescent protein (GFP) (Shimomura 1979), multiple fluorescent proteins or dyes have been developed (Rodriguez et al. 2017), and it has become possible to simultaneously visualize intracellular dynamics of multiple proteins in living cells. In addition, by combining recently developed genome editing technologies such as clustered regularly interspaced short palindromic repeats (CRISPR) (Ran et al. 2013), it is possible to monitor the dynamic behaviors of endogenous proteins even in animal cells, including human cultured cells.
In the last few decades, many genetic approaches, such as mutant screens and gene disruption, have been used in combination with live cell imaging to identify genes that code key proteins required for cellular functions (Hartwell 1978; Yanagida 2014; Goshima et al. 2007; Neumann et al. 2010). In addition, biochemical and proteomic approaches have defined functional protein complexes that underlie complicated cellular functions (Hutchins et al. 2010; Cheeseman et al. 2004; Obuse et al. 2004). Once key molecules or complexes are identified, biophysical and structural studies are performed to reveal the detailed molecular properties sufficient for their functions (Cheeseman et al. 2006; Dimitrova et al. 2016; Zhang et al. 2017; McKenney et al. 2014; Schlager et al. 2014). Furthermore, by combining mathematical simulations (Kimura and Onami 2005), nanodevices (Thery et al. 2005), and synthetic approaches (Good et al. 2013; Laan et al. 2012; Nguyen et al. 2014), novel molecular features that underlie complicated dynamic cellular events have been uncovered. However, it is still difficult to fully reconstitute macro-molecular complexes, such as the mitotic spindle, which consists of hundreds of proteins (Goshima et al. 2007; Sauer et al. 2005), and to manipulate their functions under physiological condition.
Recently, several light-induced tools have been developed to manipulate intracellular localization of target proteins with a spatiotemporal precision in living cells (Levskaya et al. 2009; Kennedy et al. 2010; Strickland et al. 2012; Guntas et al. 2015). In combination with other techniques, this optogenetic technology has great potential to reconstitute functional protein complexes, which are otherwise difficult to reconstitute in vitro, and to directly assess their functions within a cell. Here, I have presented an optogenetic reconstitution system to achieve light-induced in cell reconstitution of protein complexes coupled with visualization and manipulation of their cellular functions.
2 Light-Induced Heterodimerization Tools
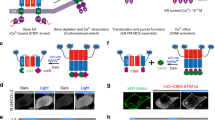
Since around 2009, several groups have developed light-induced protein–protein interaction tools using photoactivatable proteins/domains such as phytochrome B (Phy B) (Levskaya et al. 2009), cryptochrome 2 (CRY2) (Kennedy et al. 2010), and light, oxygen, and voltage (LOV) domains (Strickland et al. 2012; Guntas et al. 2015). In response to light, PhyB and CRY2 interact with their binding partners PIF and CIB1, respectively (Fig. 6.1). Thus, by tethering one module to appropriate sites, such as the plasma membrane, and by fusing the other module to the target protein as a tag, these tools work as a light-induced heterodimerization system (Fig. 6.1). In contrast, the LOV domain acts as a photoswitch that causes a conformational change in response to blue light and dissociates its C-terminal Jα helix from the core domain (Fig. 6.1). When a synthetic peptide is embedded in the Jα helix, this peptide is masked in the dark state but is exposed following light illumination. Thus, by designing the peptide and its binding partner, several light-induced dimerization tools have been developed, such as tunable light-controlled interacting protein tags (TULIP) (Strickland et al. 2012) and improved light-induced dimer (iLID) (Guntas et al. 2015). In iLID, SsrA peptide (seven residues) is embedded in the Jα helix and an SsrA-binding 13-kD protein, SspB, is used as the heterodimerization tag (Fig. 6.2). Although these photoactivatable proteins require cofactors, such as flavin mononucleotide, these cofactors exist in most mammalian cells or can be supplied externally in the culture medium (Zhang and Cui 2015). In addition to these photoactivatable proteins, photo-activated chemicals, such as photocaged dimerizer, have been recently developed to manipulate cell signaling with light (Fig. 6.1) (Ballister et al. 2014). Similar to GFP, these optogenetic tools are derived from plant and bacterial proteins and thus do not affect endogenous cellular functions in mammalian cells.

Toolkits to visualize and control molecules with light. (Left) Fluorogenic probes/dyes and fluorescent proteins are summarized. There are several commercially available SiR-based probes and HaloTag STELLA Fluor™. (Right) Light-induced heterodimerization systems. Photocaged dimerizer cTMP-Htag, LOV domain-based conformational change, CRY2–CIBN, PhyB–PIF, and BphP1–PpsR2 interactions are summarized. ∗ indicates light-sensing proteins containing cofactors

A design for light-induced membrane targeting and visualization of its downstream events. Blue-light-responding iLID is tethered at the plasma membrane using CAAX motif. In response to blue-light (450–488 nm) illumination, iLID causes conformational change and exposes its Jα helix, containing the SsrA peptide, which interacts with SspB protein, resulting in membrane recruitment of cytoplasmic SspB-fusion proteins. By co-expressing SspB–RFP fused protein of interest 1 (POI 1) and far-red fluorescent protein/probe tagged POI 2, it can be simultaneously visualized how membrane recruited POI 1 protein affects POI 2 protein following light illumination
These photoactivatable proteins and chemicals exhibit different characters and requirement for activation (Fig. 6.1; reviewed in (Zhang and Cui 2015)). Therefore, these characters must be considered for the experimental design. For example, to locally assemble protein complexes at the plasma membrane, optogenetic dimerizers with slow dissociation rate of target proteins may diffuse on the membrane following membrane targeting and fail to assemble the protein complexes at the specific site on the membrane. In contrast, to stably recruit signaling molecules on a specific organelle, such as kinetochore or centrosomes, optogenetic tools with slow dissociation rate would be more appropriate to generate robust cell signaling. Finally, simultaneous manipulation of two different target proteins is now possible by using different light-responsible tools such as blue- and near-infrared light-driven dimerizers (Kaberniuk et al. 2016).
3 Visualization Tools Compatible with Optogenetic Manipulation
For simultaneous visualization of target protein and its downstream targets or events in response to light illumination, exciting fluorescent proteins or dyes without activating light-responsible proteins is required. For instance, when blue light is used to activate photoactivatable proteins, red or far-red light must be selected for visualization because shorter UV light also activates blue light-responsible elements (Fig. 6.2). To monitor the responses of the target protein and its downstream effectors following light illumination, at least two fluorescent proteins or dyes are required. Importantly, several far-red or near-infrared fluorescent proteins or cell permeable fluoregenic probes/dyes have been recently developed (Fig. 6.1), such as near-infrared fluorescent protein (iRFP) (Filonov et al. 2011), infrared fluorescent protein mutant (IFP2.0) (Yu et al. 2014), silicon–rhodamine-based fluorophore (SiR)-647 (Lukinavicius et al. 2014), SiR-700 (Lukinavicius et al. 2016), and HaloTag STELLA Fluor™ (http://www.promega.co.jp/halotag_imaging/). These fluoregenic probes/dyes are used in combination with SNAP-tag or HaloTag, or these dyes are directly conjugated with chemicals that bind to DNA, tubulin, or actin to visualize chromosomes (SiR–DNA) (Lukinavicius et al. 2015), tubulin (SiR–tubulin) (Lukinavicius et al. 2014), and actin cytoskeleton (SiR–actin) (https://spirochrome.com/). Thus, by combining these novel tools, simultaneous visualization of its downstream targets is now possible in parallel with light-induced manipulation of photoactivatable proteins. In contrast, when photocaged chemical dimerizer or near-infrared-activated proteins are used, GFP and other red fluorescent proteins or dyes can be used for multicolor imaging to visualize its downstream events and phenotypes.
4 Light-Induced Assembly/Reconstitution of Force-Generating Complexes During Mitosis
Recently, light-induced heterodimerization tools have been used to manipulate cell signaling and force-generating processes, such as organelle transport (van Bergeijk et al. 2015; Ballister et al. 2015) and cytokinesis (Wagner and Glotzer 2016). In these studies, protein complexes, including motor proteins such as dynein and myosin, are locally assembled following light illumination and their cellular functions are assessed within a cell. Another good target for optogenetic reconstitution is a cortical force-generating machinery (Kiyomitsu and Cheeseman 2012), which links the dynamic plus-end of astral microtubules emanating from the mitotic spindle with the plasma membrane and generates cortical pulling forces on astral microtubules to control spindle position and orientation during both symmetric and asymmetric cell division (Fig. 6.3a) (Kiyomitsu 2015; di Pietro et al. 2016). In most animal cells, this cortical machinery consists of evolutionally conserved protein complexes, including cytoplasmic dynein complex (Roberts et al. 2013), its binding partner dynactin complex, and cortically anchored nuclear mitotic apparatus protein (NuMA)–LGN–Gαi complex (Fig. 6.3b) (Kiyomitsu and Cheeseman 2012). Intrinsic or extrinsic polarity signals specify the cortical assembly site of the cortical machinery and drive the directional movement of the spindle (Fig. 6.3a, b) (Thery et al. 2005; Kiyomitsu and Cheeseman 2012). Although cortical microtubule interaction between dynein and dynamic microtubule end has been recently reconstituted in an in vitro system using purified dynein motor domain and micro-fabricated barriers (Laan et al. 2012), how functional force-generating machinery assembles in response to intrinsic and extrinsic signals (Fig. 6.3b, c) and generates large cortical spindle-pulling forces within a cell remains poorly understood.

Basic principles and essential steps to control spindle position and orientation. (a) A diagram summarizing the four key steps during spindle positioning. (b) A diagram showing how intrinsic and extrinsic signals are integrated at the cell cortex and generate cortical pulling forces to control spindle positioning. (c) Cortical interaction map of known cortical proteins. Line indicates physical interactions
Whereas dynein, dynactin and NuMA play other key roles in spindle assembly (Hueschen et al. 2017), LGN and Gαi specifically localize at the cell cortex and have no roles in functional spindle assembly (Kiyomitsu and Cheeseman 2012). Thus, depletion of LGN or Gαi by RNAi does not affect the integrity of the mitotic spindle and provides an appropriate condition to assemble cortical force-generating sub-complexes and directly assess their abilities in spindle positioning (Fig. 6.4a). Although the dynein heavy chain (~500 kD) and NuMA (~240 kD) are very large proteins, CRISPR-based genome editing enables to insert appropriate tags in their endogenous gene loci at either N- or C-terminal region (Natsume et al. 2016). In addition, by exogenously expressing truncation fragments, mutants, or siRNA-resistant proteins, it is also possible to assemble multiple different sub-complexes and analyze their functions in spindle positioning (Fig. 6.4b). Furthermore, HeLa cells or other cultured human cell lines divide symmetrically and likely do not express polarized factors such as Dishevelled (Fig. 6.3c) (Segalen et al. 2010), which is required for controlling spindle orientation during oriented/asymmetric cell division. Thus, by artificially expressing these polarized factors in symmetrically dividing cells and manipulating their localization using light, it is possible to dissect their roles in the assembly of cortical force-generating complex and spindle orientation.

A design for light-induced reconstitution of the cortical force-generating sub-complexes. (a) In normal condition, symmetrically dividing cultured cells display symmetric cortical localization of cortical force-generating machinery that includes dynein, dynactin, and NuMA–LGN–Gαi complexes. The metaphase spindle is placed at the center of the cell. Following depletion of cortically localized LGN or Gαi, upstream factors of NuMA, light-induced cortical targeting of NuMA recruits dynein and assembles the cortical force-generating sub-complex. The abilities of cortical puling-force generation by light-induced sub-complexes can be assessed by analyzing the dynamics of the spindle. (b) By expressing and targeting different modules, it is possible to reconstitute different sub-complexes and understand the sufficiency and requirement of cortical pulling-force generation
5 Perspectives
Previous studies have identified key macro-molecular complexes that play critical roles in diverse cellular functions (Cheeseman et al. 2004; Obuse et al. 2004; Kiyomitsu and Cheeseman 2012; Goshima et al. 2008). Hence, it is definitely important to investigate their precise functions and structures in in vitro reconstitution systems. However, in many cases, it is difficult to fully reconstitute these macro-molecular complexes and their targets in vitro. Recent innovation of optogenetic tools has led to the development of in vivo reconstitution systems to understand the functional properties and structure of reconstituted sub-complexes within a cell. Light-induced targeting of proteins and their mutants is a powerful method to define both sufficiency and requirement of their molecular functions under more physiological conditions. Importantly, in cell reconstitution can be also used to manipulate cellular dynamics in a spatially and temporally controlled manner. For example, temporal reconstitution of force-generating sub-complex at specific cortical region during mitosis can induce spindle orientation or displacement and convert cell division mode from symmetric to asymmetric or vice versa in symmetrically or asymmetrically dividing cells. Such manipulation may lead to unexpected phenotypes and open new directions in the field of cell and developmental biology. Because light-based strategies have high penetrance, low toxicity, and high spatial and temporal precision, these characters and convenience are great advantages to manipulate target molecules in complicated in vivo situations, such as tissues in multicellular organisms.
References
Ballister ER, Aonbangkhen C, Mayo AM, Lampson MA, Chenoweth DM (2014) Localized light-induced protein dimerization in living cells using a photocaged dimerizer. Nat Commun 5:5475
Ballister ER, Ayloo S, Chenoweth DM, Lampson MA, Holzbaur ELF (2015) Optogenetic control of organelle transport using a photocaged chemical inducer of dimerization. Curr Biol 25:R407–R408
Cheeseman IM, Niessen S, Anderson S, Hyndman F, Yates JR 3rd, Oegema K, Desai A (2004) A conserved protein network controls assembly of the outer kinetochore and its ability to sustain tension. Genes Dev 18:2255–2268
Cheeseman IM, Chappie JS, Wilson-Kubalek EM, Desai A (2006) The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell 127:983–997
di Pietro F, Echard A, Morin X (2016) Regulation of mitotic spindle orientation: an integrated view. EMBO Rep 17:1106–1130
Dimitrova YN, Jenni S, Valverde R, Khin Y, Harrison SC (2016) Structure of the MIND complex defines a regulatory focus for yeast kinetochore assembly. Cell 167:1014–1027 e1012
Filonov GS, Piatkevich KD, Ting LM, Zhang J, Kim K, Verkhusha VV (2011) Bright and stable near-infrared fluorescent protein for in vivo imaging. Nat Biotechnol 29:757–761
Good MC, Vahey MD, Skandarajah A, Fletcher DA, Heald R (2013) Cytoplasmic volume modulates spindle size during embryogenesis. Science 342:856–860
Goshima G, Wollman R, Goodwin SS, Zhang N, Scholey JM, Vale RD, Stuurman N (2007) Genes required for mitotic spindle assembly in Drosophila S2 cells. Science 316:417–421
Goshima G, Mayer M, Zhang N, Stuurman N, Vale RD (2008) Augmin: a protein complex required for centrosome-independent microtubule generation within the spindle. J Cell Biol 181:421–429
Guntas G, Hallett RA, Zimmerman SP, Williams T, Yumerefendi H, Bear JE, Kuhlman B (2015) Engineering an improved light-induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proc Natl Acad Sci U S A 112:112–117
Hartwell LH (1978) Cell division from a genetic perspective. J Cell Biol 77:627–637
Hueschen CL, Kenny SJ, Xu K, Dumont S (2017) NuMA recruits dynein activity to microtubule minus-ends at mitosis. elife 6. https://doi.org/10.7554/eLife.29328
Hutchins JR, Toyoda Y, Hegemann B, Poser I, Heriche JK, Sykora MM, Augsburg M, Hudecz O, Buschhorn BA, Bulkescher J et al (2010) Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science 328:593–599
Kaberniuk AA, Shemetov AA, Verkhusha VV (2016) A bacterial phytochrome-based optogenetic system controllable with near-infrared light. Nat Methods 13:591–597
Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, Tucker CL (2010) Rapid blue-light-mediated induction of protein interactions in living cells. Nat Methods 7:973–975
Kimura A, Onami S (2005) Computer simulations and image processing reveal length-dependent pulling force as the primary mechanism for C. elegans male pronuclear migration. Dev Cell 8:765–775
Kiyomitsu T (2015) Mechanisms of daughter cell-size control during cell division. Trends Cell Biol 25:286–295
Kiyomitsu T, Cheeseman IM (2012) Chromosome- and spindle-pole-derived signals generate an intrinsic code for spindle position and orientation. Nat Cell Biol 14:311–317
Laan L, Pavin N, Husson J, Romet-Lemonne G, van Duijn M, Lopez MP, Vale RD, Julicher F, Reck-Peterson SL, Dogterom M (2012) Cortical dynein controls microtubule dynamics to generate pulling forces that position microtubule asters. Cell 148:502–514
Levskaya A, Weiner OD, Lim WA, Voigt CA (2009) Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 461:997–1001
Lukinavicius G, Reymond L, D’Este E, Masharina A, Gottfert F, Ta H, Guther A, Fournier M, Rizzo S, Waldmann H et al (2014) Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat Methods 11:731–733
Lukinavicius G, Blaukopf C, Pershagen E, Schena A, Reymond L, Derivery E, Gonzalez-Gaitan M, D’Este E, Hell SW, Gerlich DW et al (2015) SiR-hoechst is a far-red DNA stain for live-cell nanoscopy. Nat Commun 6:8497
Lukinavicius G, Reymond L, Umezawa K, Sallin O, D’Este E, Gottfert F, Ta H, Hell SW, Urano Y, Johnsson K (2016) Fluorogenic probes for multicolor imaging in living cells. J Am Chem Soc 138:9365–9368
McKenney RJ, Huynh W, Tanenbaum ME, Bhabha G, Vale RD (2014) Activation of cytoplasmic dynein motility by dynactin-cargo adapter complexes. Science 345:337–341
Natsume T, Kiyomitsu T, Saga Y, Kanemaki MT (2016) Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep 15:210–218
Neumann B, Walter T, Heriche JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U et al (2010) Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464:721–727
Nguyen PA, Groen AC, Loose M, Ishihara K, Wuhr M, Field CM, Mitchison TJ (2014) Spatial organization of cytokinesis signaling reconstituted in a cell-free system. Science 346:244–247
Obuse C, Iwasaki O, Kiyomitsu T, Goshima G, Toyoda Y, Yanagida M (2004) A conserved Mis12 centromere complex is linked to heterochromatic HP1 and outer kinetochore protein Zwint-1. Nat Cell Biol 6:1135–1141
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308
Roberts AJ, Kon T, Knight PJ, Sutoh K, Burgess SA (2013) Functions and mechanics of dynein motor proteins. Nat Rev Mol Cell Biol 14:713–726
Rodriguez EA, Campbell RE, Lin JY, Lin MZ, Miyawaki A, Palmer AE, Shu X, Zhang J, Tsien RY (2017) The growing and glowing toolbox of fluorescent and photoactive proteins. Trends Biochem Sci 42:111–129
Sauer G, Korner R, Hanisch A, Ries A, Nigg EA, Sillje HH (2005) Proteome analysis of the human mitotic spindle. Mol Cell Proteomics 4:35–43
Schlager MA, Hoang HT, Urnavicius L, Bullock SL, Carter AP (2014) In vitro reconstitution of a highly processive recombinant human dynein complex. EMBO J 33:1855–1868
Segalen M, Johnston CA, Martin CA, Dumortier JG, Prehoda KE, David NB, Doe CQ, Bellaiche Y (2010) The Fz-Dsh planar cell polarity pathway induces oriented cell division via Mud/NuMA in Drosophila and zebrafish. Dev Cell 19:740–752
Shimomura O (1979) Structure of the chromophore of AEQUOREA green fluorescent protein. FEBS Lett 104:220–222
Strickland D, Lin Y, Wagner E, Hope CM, Zayner J, Antoniou C, Sosnick TR, Weiss EL, Glotzer M (2012) TULIPs: tunable, light-controlled interacting protein tags for cell biology. Nat Methods 9:379–384
Thery M, Racine V, Pepin A, Piel M, Chen Y, Sibarita JB, Bornens M (2005) The extracellular matrix guides the orientation of the cell division axis. Nat Cell Biol 7:947–953
van Bergeijk P, Adrian M, Hoogenraad CC, Kapitein LC (2015) Optogenetic control of organelle transport and positioning. Nature 518:111–114
Wagner E, Glotzer M (2016) Local RhoA activation induces cytokinetic furrows independent of spindle position and cell cycle stage. J Cell Biol 213:641–649
Yanagida M (2014) The role of model organisms in the history of mitosis research. Cold Spring Harb Perspect Biol 6:a015768
Yu D, Gustafson WC, Han C, Lafaye C, Noirclerc-Savoye M, Ge WP, Thayer DA, Huang H, Kornberg TB, Royant A et al (2014) An improved monomeric infrared fluorescent protein for neuronal and tumour brain imaging. Nat Commun 5:3626
Zhang K, Cui B (2015) Optogenetic control of intracellular signaling pathways. Trends Biotechnol 33:92–100
Zhang K, Foster HE, Rondelet A, Lacey SE, Bahi-Buisson N, Bird AW, Carter AP (2017) Cryo-EM reveals how human cytoplasmic dynein is auto-inhibited and activated. Cell 169:1303–1314 e1318
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this paper

Cite this paper
Kiyomitsu, T. (2020). Optogenetic Reconstitution: Light-Induced Assembly of Protein Complexes and Simultaneous Visualization of Their Intracellular Functions. In: Toyama, Y., Miyawaki, A., Nakamura, M., Jinzaki, M. (eds) Make Life Visible. Springer, Singapore. https://doi.org/10.1007/978-981-13-7908-6_6
Download citation
DOI: https://doi.org/10.1007/978-981-13-7908-6_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-7907-9
Online ISBN: 978-981-13-7908-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)