
Abstract
A more complete understanding of biological processes at cellular and molecular levels requires the ability to study them in time and space. Such observation became increasingly easy with the use of genetically encoded fluorescent markers, such as GFP, and the latest developments in optical microscopy. This combination of tools permits the structure and function of biological tissues to be imaged in zebrafish non-invasively. For example, it has become possible to image cardiac structure when GFP is fused to cytoskeletal elements, such as alpha-catenin; similarly, neural activity can be followed when genetically encoded calcium sensors are used to eavesdrop on sets of neurons. Such intravital imaging has furthered our understanding of the relationships between cardiac structure and function, as well as between neuronal activity patterns and complex behaviors such as sleep. Imaging these processes in 3 dimensions – at whole tissue scale and at subcellular resolution – is challenging to perform at sufficient speeds to capture the dynamics under study. Here we review some emerging approaches that combine the speed of Light Sheet microscopy with sets of computational image processing and image analysis tools, offering clear paths to overcome these challenges. We will take advantage of the zebrafish as an excellent system for imaging, and offer examples drawn from recent efforts to perform 3D imaging at single-cell resolution of the beating heart, and of whole-brain neural activity.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others
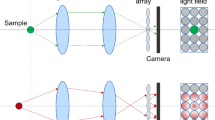

1 Intravital Imaging
A more complete understanding of biological processes at cellular and molecular levels requires the ability to study them in time and space. Such observation became increasingly easy with the use of genetically encoded fluorescent markers, such as GFP, and the latest developments in optical microscopy. This combination of tools permits the structure and function of biological tissues to be imaged in zebrafish non-invasively. For example, it has become possible to image cardiac structure when GFP is fused to cytoskeletal elements, such as alpha-catenin; similarly, neural activity can be followed when genetically encoded calcium sensors are used to eavesdrop on sets of neurons. Such intravital imaging has furthered our understanding of the relationships between cardiac structure and function, as well as between neuronal activity patterns and complex behaviors such as sleep. Imaging these processes in 3 dimensions – at whole tissue scale and at subcellular resolution – is challenging to perform at sufficient speeds to capture the dynamics under study. Here we review some emerging approaches that combine the speed of Light Sheet microscopy with sets of computational image processing and image analysis tools, offering clear paths to overcome these challenges. We will take advantage of the zebrafish as an excellent system for imaging, and offer examples drawn from recent efforts to perform 3D imaging at single-cell resolution of the beating heart, and of whole-brain neural activity.
2 Volumetric Cardiac Imaging in Embryonic Zebrafish
2.1 Zebrafish As a Model System for Cardiovascular Research
Congenital heart diseases (CHDs), not only represent the most prevalent birth defects in humans but also one of the leading causes of infant mortality and morbidity (Pierpont et al. 2007). Although several vertebrate model systems allow mechanistic investigations of cardiac development and of cardiac diseases, zebrafish (Danio rerio) offers a powerful model organism for cardiovascular development studies using imaging tools. Because of its small size, passive diffusion of oxygen can support the normal development of zebrafish embryos that are completely lacking a functional cardiovascular system or blood circulation; this permits analyses of embryos with severe cardiovascular defects that would be impossible in other systems (Stainier et al. 1996). Because zebrafish embryos develop externally and are optically transparent, they are ideal for live, in vivo imaging of cellular and physiologic processes involved in cardiac morphogenesis (Hove et al. 2003). The zebrafish heart is small enough in size (~250 μm × 200 μm × 150 μm) to be imaged in its entirety at sub-cellular resolution.
Zebrafish and mammalian hearts exhibit several well conserved structures including atria, ventricles, cardiac valves and a cardiac conduction system which coordinates the contractions of the atrial and ventricular chambers and maintains a normal heart rate (Beis et al. 2005; Chi et al. 2008; Sedmera et al. 2003; Stainier et al. 1993). These conserved features make zebrafish studies of cardiac development (e.g. valve development) and physiology (e.g. cardiac conduction studies) relevant to human cardiac development and pathologies.
The rapid development and large offspring numbers make zebrafish ideal for forward genetic screens, which have identified numerous cardiovascular mutant phenotypes. These mutants provide excellent model systems to understand human cardiac disease mechanisms, and the similarities of the mutant phenotypes to key features of some human cardiomyopathies, have resulted in the identification of novel candidate genes responsible for human cardiomyopathies. Zebrafish cardiac mutants have identified regulatory mechanisms that play crucial roles during cardiogenic specification and differentiation, migration of cardiac progenitor cells, heart tube morphogenesis, and cardiac function. For instance, the zebrafish weak atrium (wea) mutant has shed light on the importance of blood flow through the developing ventricle for the establishment of proper ventricle morphology (Auman et al. 2007).
Reverse genetic approaches allow functional tests of genes suspected to play important roles in cardiac function, development or pathology. For example, several potential genetic risk markers for atrio-ventricular (AV) valve and septum defects, identified from a study of 190 heart disease patients, result in malformation of the AV canal and other defects when expressed in the zebrafish (Vermot et al. 2009; Wienholds et al. 2003).
2.2 Cardiac Development: Symbiosis of Function and Form
Examining the relationship between function and form of the heart can offer important insights into cardiac morphogenesis as well as late-onset or acquired cardiac diseases (Hove et al. 2003; Bartman and Hove 2005; Beis et al. 2005). Since the anatomy of the heart is not fully formed until several days after its function has been initiated, it seems likely that the heart’s early mechanical function plays a role in its own morphogenesis. The beating of the heart generates several types of forces at different spatial and temporal scales. While both endocardial and myocardial cells undergo cyclical compression and expansion at the tissue scale, individual endothelial cells directly experience the shear stresses and oscillatory flows generated by moving blood (Vermot et al. 2009). Several studies have suggested that changes in myocardial function result in cardiac remodeling (Hove et al. 2003; Bartman and Hove 2005; Beis et al. 2005).
3D imaging of fixed heart tissue has prompted the proposal of models for how cardiac function affects cardiac development. For example, Auman et al. used confocal imaging of fixed zebrafish to propose (Auman et al. 2007) that cardiomyocyte contractility, in addition to previously suggested hemodynamics (Hove et al. 2003), plays a role in determining the structures of cardiomyocytes in different parts of the zebrafish heart. Because stopping the heart can lead to changes in cardiac cell morphology, imaging of fixed or immobile hearts is not an ideal means to accurately assess changes in cell shape and size. Similarly, 3D Ca2+ −mapping in heart tissue using light sheet microscopy has improved our understanding of cardiac conduction patterns, but in cells of an immobilized heart (Weber et al. 2017). The development of techniques that allow visualization of the cardiac chambers as well as the blood flow through the heart in 3 dimensions over the time-period of the heart development would best advance our understanding of how the proper functioning of the developing heart affects cardiac morphology and vice versa.
2.3 Cardiac Imaging Is a 4-Dimensional Challenge
Visualizing the beating heart by using in vivo dynamic imaging has become possible due to the availability of several stable transgenic zebrafish lines expressing fluorescent proteins in defined cardiac and blood cells. To study cardiac developmental mechanisms, in vivo dynamic imaging methods must provide high spatial and temporal resolution, and must allow imaging of an individual zebrafish embryos throughout significant fractions of their development. Only then will it become possible to address the relationship between blood flow, shear stress, wall motion and normal cardiac development.
Despite the flexibility offered by the small size and transparent tissues of zebrafish embryos, it is still a fundamental challenge to create four-dimensional (4D) images (i.e. a volumetric (3D) movie of the beating embryonic zebrafish heart through the full depth of the beating organ) at volume rates sufficient to resolve fast cardiac motion. In order to create a high spatial resolution 3D volume rendering, data from every voxel (up to several million voxels) must be acquired in a sequential fashion. The fluorescent labeling used to visualize the tissues imposes its own limitations. For example, the excitation needed to stimulate the dye can also bleach the dyes and cause other potential photo-toxic events. Image contrast results from the detected fluorescence from the sample (often called signal) against the autofluorescence of the tissue and detector signal due to electrical noise in the absence of fluorescence (often called noise). The signal to noise ratio (SNR) must be high enough to allow the observer to differentiate labeled tissue from the background. A large number of photons must be recorded per pixel to accurately image the sample, as fluorescence emission is a Poisson process and the probability that the actual intensity at a pixel is equal to the expected intensity is proportional to √N, where N is the number of photons generated by fluorescence excitation. Typically, these concerns favor longer detection times so that signal can be collected per pixel; however, this makes the acquisition of a single volumetric image an inordinately slow process (Vermot et al. 2008).
Creating a volumetric movie of the beating heart, requires the acquisition of many high spatial resolution volumetric images at a high temporal resolution. High temporal resolution is required because cardiac walls move at velocities up to 2 mm/s and blood cells flow at velocities up to 17 mm/s. To maintain subcellular resolution, each volumetric image must be acquired in a time-period so short that the cardiac motion does not blur the image. This predicts a required volume acquisition rate of more than 1000 volumes/s. Even if such high imaging speeds were possible, the extremely short detector integration time would require an extremely high fluorescence signal rate, which would, in turn, require a very bright fluorescence excitation light. As this would lead to photobleaching and phototoxicity, such an approach would likely compromise the imaging quality and, more importantly, perturb the developing organism (Taylor et al. 2012).
Confocal Slit Microscopy made it possible to reach imaging speeds of 120 frames/s through parallel acquisition of data from a line of voxels at a time (Liebling et al. 2005). In this technique the high Numerical Aperture (NA) detection objective is used to create a blade shaped illumination beam focused to a line (instead of a single point in a conventional confocal laser scanning microscope) and all the pixels in this illuminated line are imaged in parallel. Axial sectioning is achieved by using a pinhole slit. As with other confocal microscopy techniques, the imaging speed is mostly limited by significant photobleaching due to excitation of fluorescence outside the detection plane.
The imaging speed provided by Confocal Slit microscopy (up to 3 volumes/s) is still nowhere near the speeds required (~1000 volumes/s) to capture the entire beating embryonic zebrafish heart without blurring due to wall motion or blood flow. One approach to imaging the heart is to slow down the heart wall motion and the blood flow by using chemical agents; however, this can’t provide an accurate measure of the role of cardiac function on cardiac development at realistic heart rates. A better approach is to use computational techniques that utilize the quasi-periodic nature of the heart to reconstruct a volumetric representation of the beating heart from images acquired at speeds greater than 60 frames/s.
We have elected to combine computational reconstruction approaches with the fast imaging provided by Selective Plane Illumination Microscopy (SPIM), as it offers the speed of Confocal Slit Microscopy, but is dramatically less bleaching. SPIM provides diffraction limited (sub-micron resolution) imaging at high frame rates (up to 200 frames/s) and sufficient Signal to Noise ratio, by parallelizing the detection of all the voxels in a single optical section. Optical sectioning is achieved by illuminating the sample with a thin sheet of light created by a separate objective lens along a path orthogonal to the optical axis of the detection objective, thus eliminating fluorescence excitation outside of the imaged optical section. The limitation of SPIM lies in the requirement that the sample must be optically accessible from two orthogonal directions.
2.4 Principles of Cardiac Gated Imaging in Zebrafish
To build a 3D representation of the beating heart using SPIM or other fast 2D imaging techniques, multiple images of 2D optical slices in a volume are acquired sequentially over a period spanning multiple heartbeat periods. The different strategies for collecting a 4D rendering (3D over time) can be understood if we consider the needs for capturing a sequence of 3-dimensional images representing the different phases of the cardiac cycle. Each 3D image is composed of 2D image slices; thus, a 4D movie of the heart is a 2D matrix containing one 2D image in each cell (Fig. 3.1). The two axes of this matrix are the phase (φ) of the beating heart and the axial position (z) of the image slice. The cells in this matrix could be filled with images that are acquired in almost any order, as long as computational algorithms can utilize the quasi-periodic nature of the heart’s motion to assign the 2D slices into the correct cell in the matrix. Once the matrix has been filled, the 2D cardiac images acquired during different heart beat periods are stitched into a single cardiac volume image at a single phase of the beating heart. The 4D rendering is derived from the 3D cardiac images for each phase point during the cardiac cycle, stitched. 4D cardiac imaging strategies can be classified into three groups: prospective cardiac gating, retrospective cardiac gating, and phase stamping.

Spatiotemporal Phase (φ) – slice position (z) representation of a 4D (3D + time) heart. If each cell in the 2-dimensional φ – z matrix consists one 2D image slice corresponding to that phase and slice position, then the matrix can be used to generate a volumetric movie of the beating heart. There are three ways to fill up this matrix with 2D image slices: prospective, retrospective and Macroscopic Phase Stamping (MaPS). In prospective gating images corresponding to entries along one column are acquired sequentially, creating a 3D image of heart at one phase point. In retrospective gating images corresponding to one row are acquired sequentially, creating a movie (2D + time) of the heart at one z plane. In MaPS, sequentially acquired images correspond to entries along diagonal lines of the matrix, thus filling the matrix with the least amount of photobleaching and hardware complexity
2.4.1 Prospective Gating
In prospective gating a reference signal is used to identify exactly when the heart will be in a desired phase in its cycle (Taylor et al. 2012). The reference signal could be frame data streamed from a bright-field imaging camera (Taylor et al. 2012), laser Doppler velocimetry data from a non-invasive needle probe (Jenkins et al. 2007), or scattered light modulated in intensity by the heartbeat collected from a region of tissue (such as esophagus or thorax) using a fiber optic cable (Brau et al. 2002; Sablong et al. 2014). Real-time analysis of this signal allows image acquisition of each 2D “z-slice”, required to build up a 3D image for a given phase (φ) point (time point in the heartbeat), to be triggered when the heart is in that desired position. Thus, if we represent the 4D beating heart as a φ-z matrix (with phases along the columns and z along the rows) where each entry is a 2D z-slice, then prospective gating collects images corresponding to entries along one column at a time, creating a 3D image of heart, one phase point at a time (Fig. 3.1).
Taylor et al. (2012) used prospective gating of SPIM images to generate volumetric movies of an embryonic zebrafish heart and observed peak cardiac wall velocities of 160 μm/s. The authors used brightfield images of the heart acquired using the SPIM detection objective as the reference signal for gating. By using one heartbeat period worth of frames as the representative frames, they determined the phase of the heartbeat for each brightfield frame, as it was acquired, by comparing it with these representative frames, to trigger the acquisition of a fluorescent frame at the desired phase point. The number of fluorescent frames acquired is exactly equal to the number of fluorescent frames used in the final 3D reconstruction which minimizes the photodamage. The phase prediction module can also be used to trigger an ablation laser for high precision targeting of moving cells within a normally-beating heart while observing the effects of ablation.
2.4.2 Retrospective Gating
In retrospective gating, movies are acquired for each 2D slice, over the course of multiple heart beat cycles and relative temporal shifts between adjacent 2D slices are calculated in order to temporally align movies from neighboring slices (Liebling et al. 2005). The 2D slice movies are first truncated to an integer number of heartbeats. Then pairwise correlation between the wavelet coefficients of frames of neighboring movies are used to determine the phase offset between neighboring 2D movies (Liebling et al. 2005). While, prospective gating collects images along the columns of the φ-z matrix, retrospective gating collects images along the rows (Fig. 3.1). Liebling et al. later improved upon their technique by using “temporal warping” of slice sequences by taking the variability of the cardiac rate between different slice sequences into account. The improved approach could reduce the quantization error introduced while assigning a discrete phase to any frame in one slice sequence based on the phase of the most similar frames in the neighboring slice sequence.
The simplicity of the imaging hardware in retrospective gating makes it a desirable technique for cardiac imaging. Retrospective gating has been combined with 2-photon SPIM to achieve high penetration depth while maintaining the high imaging speed required for cardiac imaging (Trivedi et al. 2015). This could allow imaging of older zebrafish embryos that have a more complex cardiac structure. Retrospective gating requires higher number of acquired frames (~2–5 min of continuous recording at 100 frames/s) compared to prospective gating; however, the total elapsed imaging time is generally shorter for retrospective approaches. With prospective gating, only one point in the phase-z matrix is acquired per heartbeat, which can extend elapsed imaging times up to 20 min, depending upon the number of desired z-slices.
2.4.3 Macroscopic Phase Stamping
Both gating techniques have been used in cardiac imaging using modalities other than fluorescence microscopy (Thompson and McVeigh 2004; Larson et al. 2004; Grass et al. 2003; Treece et al. 2002). Despite being widely used, both these techniques suffer from limitations arising from the need to create 3D volumes from 2D slices in the absence of an imaging modality that can capture a 3D volume at a rate sufficient to minimize the motion of the heart to sub-pixel amounts. For the retrospective approach, the assumption of sample continuity in the z direction can break down in case of a peristaltic wave propagating through the cardiac tube along the imaging axis. Retrospective gating also lacks simultaneous and independent acquisition of the phase of the acquired images. The primary limitation for the prospective approach is the hardware complexity and the inability to trigger accurately off less distinct phase points due to the slower motion of cardiac tissue in the brightfield frames during the start of atrial and ventricular contraction, which leads to jitter in the final φ-z reconstruction.
We have developed Macroscopic Phase Stamping (MaPS) to provide a more robust reconstruction, by jointly refining low resolution brightfield images acquired synchronously with optically sectioned fluorescent images acquired with SPIM (Truong et al. 2014; Truong 2017). The brightfield images allow us to determine the macroscopic spatio-temporal phase of the beating heart in the fluorescent images in post-processing. In MaPS, the sample is repeatedly scanned along the axial direction, while the imaging plane is kept stationary during the synchronized fluorescent + brightfield image acquisition. Since both the z position and the phase of the beating heart advance from one image to the next, the phase-z matrix is filled diagonally (Fig. 3.1). The z position for the fluorescent frames is assigned based on the stage position.
MaPS combines the simplicity of the hardware needed for retrospective gating with the advantage of an independent phase channel in prospective gating, providing an improvement over both approaches. The hardware complexity is minimized because both cameras are synchronized only to each other and not to a cardiac event. The availability of the synchronized bright field images provides an independent channel to determine the periodicity of the heartbeat on which any type of cardiac gating relies. The brightfield images allow us to synchronize fluorescent images that are adjacent to each other in the z direction without using the fluorescent images themselves, resulting in more accurate reconstructions of the 3D structure of the heart.
The high resolution of the individual 2D slices afforded by SPIM added with the more accurate reconstruction process provides high spatial resolution 3D reconstruction volumes, in which individual cardiac cell boundaries are easily resolvable (Fig. 3.2a). The high spatio-temporal resolution has allowed us to segment individual cardiac cells and determine their displacement over the course of a heartbeat period (Fig. 3.2b–d). This will enable cardiac researchers to understand of how individual cell biomechanics relates to the overall biomechanics of the beating heart. Recent improvements in MaPS have reduced the error introduced due to phase assignments by using a finer quantization of the phase, reducing artefacts that might otherwise appear in the volumetric renderings when the heart wall moves at its fastest speeds.

(a) Macroscopic Projection of a 3D rendering of a 4D reconstructed cardiac movie at 84 hpf where the cardiac boundaries are visible through endogenous expression of citrine labeled alpha-catenin. The 4D cardiac movie was reconstructed through Macroscopic Phase Stamping of SPIM images. (b) Selectively segmented cell boundaries at different phases in the heart beat. (c) Selectively segmented cell boundaries along with their displacement during the heart beat (straight lines) from ventral (c) and lateral (d) views
Aperiodically beating mutant zebrafish hearts, which represent some of the best tools for understanding the causes and effects of human cardiac diseases such as arrhythmias, are also the most challenging to image with cardiac gating-based techniques described above. Imaging individual blood cells that flow aperiodically through a periodically beating heart is another goal that future developments in cardiac imaging tools would attempt to accomplish.
3 Large Scale In Vivo Brain Imaging with Two-Photon Light-Sheet Microscopy
One of the important characteristics of the brain is its ability to constantly change and adapt, driven in large part by its combined evoked and spontaneous activity, typically without any periodic pattern. Brain activity observation allows insight into these dynamic changes of the nervous system. Complex animal behaviors often involve activity that spans many brain regions. If we want to advance our understanding of complex behaviors such as learning or sleep, we necessarily have to look at multiple regions of the brain with single-cell resolution. This remains a challenge to overcome, as most current tools for functional brain imaging lack either resolution or field of view. For example, one of the best tools to study the activity of the roughly 100 billion neurons human brain, functional MRI using BOLD (blood-oxygen-level dependent) contrast, produces striking three-dimensional images; however, the signal presented in each voxel of thousands of neurons. At the other end of the spectrum are microscopy techniques, where the best light imaging instruments collect data from only a small fraction of the cortical neurons in mice. The best experiments collect data from a thousand out of the four million cortical neurons: merely 0.025% of the cells.
Zebrafish, with its compact brain, offers the potential of optical imaging that can observe neural activity at whole-brain scale with single-cell resolution. Functional imaging can be achieved using genetically-encoded fluorescent calcium indicators as an indicator of neuronal activity, as calcium is elevated in most neurons when they fire action potentials. Genetically encoded calcium indicators change their fluorescence intensity depending on calcium ion concentration in the neuron. Some dyes change fluorescence efficiency (intensity-based), while other sensors change spectral parameters (shifting emission spectra from green to red, for example). Fast imaging techniques, such as light-sheet microscopy or laser scanning microscopy, are need to capture these signals. The advantages and limitations of these imaging tools will be discussed below.
3.1 Brain Activity Monitoring in Behaving Zebrafish
The zebrafish larva offers an important compromise, as it is a vertebrate model relevant to higher animals and mammals, in which almost all of its 100,000 neurons of the central nervous system can be imaged. While such imaging is still challenging and requires the application of advanced optical light sheet microscopy (to be discussed later), it is the feasibility of in toto imaging that drives our choice to focus on the zebrafish nervous system here.
The benefits of zebrafish larva as model organism stem from the combination of its compact size and optical transparency, thus permitting high-resolution optical microscopy. In order to visualize neural activity, transgenic animals, positionally expressing a gene encoding a calcium-sensitive fluorophore (cf. GCaMP or its variants) have been used. These genetically encoded fluorescent calcium indicators change their color or their brightness when a cell’s calcium level increases, as it does following even single action potentials. GCaMP indicators, based on a fusion of a modified GFP fluorescent protein and the calmodulin calcium-binding domain, offer a large change in signal with changes in calcium levels. In addition, they offer the ability to use multiple colors, since GFP and other colored fluorescent proteins permit the creation of whole palette of calcium sensors, from blue to deep-red (Akerboom et al. 2013). GCaMP sensors are intensity-based, that is, they change their intensity as the calcium level changes. This is in contrast to other ratiometric calcium sensors, which change their fluorescent spectrum as the calcium level changes. Ratiometric indicators offer more accurate measurements of calcium concentration, but the need to measure the spectrum of the dye, or the ratio of its intensity at two emission (or excitation) wavelengths limits the number of simultaneous labels that can be deployed simultaneously. Several limitations should be considered, including temporal resolution of these sensors. While action potentials have duration of 10’s of milliseconds or less, calcium sensors give a signal in response to neuronal activity that lasts 100 ms/s. The speed of the signal can decline when sensor is concentrated in the cell soma, away from synapses where neural-related calcium transients are generated. In vertebrates that is not a general issue, the soma is electrically involved in the neural processing; however, in some invertebrate species, the soma can be physically and electrically distant from the synaptic activity. Currently, sensors of the GCaMP6 family are best option for imaging neural activity, when a single color is enough and two-photon excitation is required.
Modern bioengineering techniques allow labeling of neurons in specific brain region by expressing the reporter using a vector, an artificial DNA sequence, containing a tissue-specific promoter sequence, followed by protein coding sequence for one of the calcium sensors. A common zebrafish promoter used to drive gene in the nervous system is the promoter of the HuC gene, also known as elavl3. Often, the sensor fused with nuclear-localization sequence (as nls-GCaMP) (Kim et al. 2014), in order to increase the concentration of protein in nuclei, and hence increase signal-to-noise ratio. Concentration of the fluorescence signal in a discrete location such as the nuclei can be helpful for downstream image processing, as images of nuclear-localized fluorescent molecules are bright focal spots, easily segmented computationally. Concentrating the sensor molecules in nuclei decreases temporal resolution, as changes in calcium across the cell must propagate from its release site to the nucleus in order to affect sensor’s fluorescence, which can take up to a second. Faster dynamics are possible by localization of the sensor to other cellular compartments, such as synapses, by fusing it with synaptophysin (Li et al. 2011). This concentrates the sensor in compartment of interest (synapse) because synaptophysin is naturally localized there by the cell’s own machinery. However, such fusions also raise the possibility of perturbing the local environment of the synapse, since the synapse is a very crowded and tightly regulated compartment; furthermore, the protein molecule fused to the sensor might be perturbed and lose its original functionality.
Brain imaging using calcium indicators has been successfully applied to study zebrafish brain activity. For example, fluorescent calcium indicator dyes have been useful to investigate distribution of spontaneous activity-independent calcium transients in zebrafish spinal cord (Ashworth and Bolsover 2002). Intensity-based sensors, primarily members of GCaMP family, were used to observe neural activity while the animal is performing complex behaviors, such as prey capture (Muto et al. 2013), visual or auditory responses (Thompson and Scott 2016), optokinetic responses (Vladimirov et al. 2014), or spontaneous activity reorganization during development (Avitan et al. 2017).
An excellent demonstration of the power of in vivo brain imaging in zebrafish, was the description of the activity in the visual system (optic tectum) during prey capture (Muto et al. 2013) using wide field microscopy with visible light illumination. By observing the freely swimming fish surrounded by several paramecia, the authors were able to record calcium activity in tectal neurons and create maps of activity corresponding to the particular position of prey in the fish’s visual field. By simultaneous imaging of prey movement and calcium activity authors were able to map how prey’s trajectory maps onto optic tectum: the prey’s ventral-dorsal movement resulted in distinct tectal activity than prey movement in anterior-posterior direction. This work not only demonstrated the promise of brain imaging in zebrafish, but also revealed limitations of the particular imaging technique. First of all, the resolution of the imaging was low, averaging signals from tens of neurons. More importantly, this imaging approach used visible light to excite fluorescence, and hence potentially perturbed the animal’s visually-guided behaviors, as animal continuously illuminated with very bright blue light.
The optic tectum or its equivalent, the superior colliculus in the mammalian midbrain, is present in all vertebrates and responsible for the animal’s orientation toward a subject and attention. In larval zebrafish, the optic tectum is only about 100 um thick and is positioned at the dorsal surface of the brain, making it an accessible system for studying neural function. This allowed Avitan et al (2017) to follow changes in functional mapping during development in larval zebrafish. Observing spontaneous activity in animals of age 4–7 days post fertilization (dpf), the authors discovered changes in single-cell as well as group activity patterns during development. Immobilization of the animal and use of two-photon laser scanning microscopy (LSM) allowed imaging with single-cell resolution, thus making it possible to robustly identify cellular communities containing as small as a few cells.
Advancements in imaging techniques, behavioral protocols, and genetic tools, as well as image analysis algorithms, allow whole-brain imaging of behaving zebrafish with single-cell resolution. These achievements prove how useful zebrafish and brain imaging can be together. More importantly it supports the use of zebrafish model and imaging for detailed studies of more complex behaviors, such as learning and sleep. Sleep behavior in particular requires careful approach, since it occurs over long period of time; hence we need a tool that would allow longitudinal (up to 24 h) imaging of brain activity in behaving zebrafish. Combined with already established sleep behavioral models, such as narcolepsy or insomnia models, such toolbox would be immensely useful for more detailed examination of underlying neural substrate of these disorders, as well as natural sleep.
3.2 Principles and Successes of Light-Sheet Microscopy for Zebrafish Brain Imaging
Light-sheet microscopy, or selective-plane illumination microscopy (SPIM), solves previous limitations by providing tool to collect large-scale imaging data with cellular resolution (Huisken 2004). Photo-toxicity is lower because only imaged volume is illuminated, thus allowing imaging either with higher signal-to-noise, or greater speed. SPIM imaging has reached a 1 volume per second imaging rate, sufficient for whole-brain GCaMP activity mapping. Light-sheet mode also provides opportunity to use two-photon illumination, using light invisible to the fish. This removes artifacts of high-power visible light illumination, such as visual system stimulation, and allows better use of optogenetic tools.
Even though a number of commercial SPIM systems are available, many labs choose to build their own light-sheet microscopes, often following designs available in numerous research publications. A project dedicated to creation of an open-source light-sheet microscope, OpenSPIM, provides detailed designs and step-by-step instructions for the assembly of a relatively cheap home-built microscope with a small footprint. For more flexible solutions it is important that the software for running microscopes is open source and free. The popular microscopy program, Micro-Manager provides a package that supports a myriad of hardware components, allowing robust control of almost any part of a microscope.
First experiments with whole-brain SPIM imaging in zebrafish larvae were performed on paralyzed animals (Ahrens et al. 2012) during adaptation of motor behavior. This work was further developed (Vladimirov et al. 2014) to show how ablation of particular neurons changes fictive whole-animal behavior. One limitation of these experiments is that study of such “fictive” behavior in a paralyzed animal, when motor response is not observed but rather recorded by measuring motor neurons’ activity, is achieved by inducing irreversible paralysis. This preparation limits experiment time, as animals can survive such treatment for only a few hours.
Sample preparation is important for experiments in living animals. Imaging of zebrafish brain using SPIM can be achieved with immobilization, but without total paralysis (Keller et al. 2008). Often soft agarose gel is used to restrict animal head under detection objective to perform long-term observation of neural activity with single-cell resolution. Such restraining can be deleterious from several points of view. For longer imaging in young animals, agarose embedding impedes proper development (Kaufmann et al. 2012). Such immobilization can also possibly change behavior by stimulating efferent neurons, neurons of the lateral line, and other sensory circuits. New methods are being developed in order to overcome this limitation and image freely swimming larval zebrafish (Kim et al. 2017), but only using high power visible light as fluorescence excitation source. When sufficiently advanced, these would provide much needed tool to observe neural activity while animal performs unrestricted behavior. Such experiments would allow observation of movement and prey capture behavior in real time, as well as long-term imaging, necessary for studying development or learning. Applied to imaging compact and accessible zebrafish brain, this would allow observation of brain activity in behaving vertebrate at unprecedented scale, unavailable to rodent model systems.
High temporal resolution is important for fast-pacing action potentials, as discussed above. In light-sheet microscopy pixels in different axial planes are acquired at different times, limiting temporal resolution. Light-field microscopy (LFM) offers a potential solution to this problem. LFM (Levoy et al. 2009) uses a microlens array interposed between the image plane and the camera to re-image light of sources from different axial positions to different sets of pixels on the camera. Computational deconvolution is used to reconstruct 3-dimensional image of the sample. This comes at a price of lower spatial resolution, but it allows the capturing of events at millisecond time scales across large volumes, extending to as large as nearly the entire zebrafish brain. LFM is a promising tool as it would allow imaging of very fast events across the brain, such as propagation of action potentials along axons.
In our work, we were able to observe neural activity during sleep and wake in zebrafish (Fig. 3.3) for more than 24 h for the first time (Lee et al. 2017). Application of two-photon light-sheet microscopy and opto- and chemo-genetic activation of sleep-promoting NPVF circuit, allowed us to demonstrate how sleep behavior manifest itself on brain activity level in natural and induced sleep (Fig. 3.3c). Light-sheet imaging though allows extension of this simple analysis in order to quantify functional inter-neuronal connections, correlation of activity between neurons, and other metrics that are usually used to understand functionality of the brain. Extension of this work (presented at the Society for Neuroscience Annual Meeting, 2017), shows that functional correlation between regions differs in sleep and wake. In particular we observed that activity of cerebellum lobes becomes more correlated during sleep, suggesting a pathway to study role of sleep in memory consolidation in zebrafish. Another exciting application allowed by development of this toolbox is the investigation of effects of melatonin and other hypnotic chemicals on brain activity during sleep and wake. This should permit comparative analyses of chemically-induced sleep with natural healthy sleep.

(a) Single-plane imaging of brain in 6dpf zebrafish Tg(HuC:H2B-GCaMP6s). Neurons are labeled with nuclei-concentrated calcium indicator GCaMP6s. (b) Neurons are localized using PCA/ICA toolbox (Mukamel et al. 2009). (c) Day/night oscillation in fluorescent signals across whole brain of the animal. Imaging does not perturb circadian clock and allows observation of spontaneous and light-induced activity
4 Conclusion
The development of imaging tools, discussed here, allows unprecedented access to biological processes in living zebrafish. Light-sheet and light-field imaging provide information about anatomy and function of rapidly beating heart with subcellular resolution and permit whole-brain mapping of activity with single-neuron resolution. Future work will benefit from combination of large-scale, high-resolution imaging with genomics and other genome-scale methods. Many of these tools are already available, but a collaborative environment where biologists who can maintain optimal conditions for sample survival, physicists and engineers who can create the imaging instruments, and mathematicians who can design analytical tools can work together in order to foresee the possible challenges and find alternative solutions, will be required to carry out interesting and meaningful experiments.
References
Ahrens MB, Li JM, Orger MB et al (2012) Brain-wide neuronal dynamics during motor adaptation in zebrafish. Nature 485:471–477. https://doi.org/10.1038/nature11057
Akerboom J, Carreras Calderón N, Tian L et al (2013) Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front Mol Neurosci 6(2). https://doi.org/10.3389/fnmol.2013.00002
Ashworth R, Bolsover SR (2002) Spontaneous activity-independent intracellular calcium signals in the developing spinal cord of the zebrafish embryo. Brain Res Dev Brain Res 139:131–137
Auman HJ, Coleman H, Riley HE et al (2007) Functional modulation of cardiac form through regionally confined cell shape changes. PLoS Biol 5:e53. https://doi.org/10.1371/journal.pbio.0050053
Avitan L, Pujic Z, Mölter J et al (2017) Spontaneous activity in the zebrafish tectum reorganizes over development and is influenced by visual experience. Curr Biol CB 27:2407–2419.e4. https://doi.org/10.1016/j.cub.2017.06.056
Bartman T, Hove J (2005) Mechanics and function in heart morphogenesis. Dev Dyn 233:373–381. https://doi.org/10.1002/dvdy.20367
Beis D, Bartman T, Jin S-W et al (2005) Genetic and cellular analyses of zebrafish atrioventricular cushion and valve development. Dev Camb Engl 132:4193–4204. https://doi.org/10.1242/dev.01970
Brau ACS, Wheeler CT, Hedlund LW, Johnson GA (2002) Fiber-optic stethoscope: a cardiac monitoring and gating system for magnetic resonance microscopy. Magn Reson Med 47:314–321
Chi NC, Shaw RM, Jungblut B, Huisken J, Ferrer T, Arnaout R, Scott I, Beis D, Xiao T, Baier H, Jan LY, Tristani-Firouzi M, Stainier DYR, Hogan BLM (2008) Genetic and physiologic dissection of the vertebrate cardiac conduction system. PLoS Biol 6(5):e109
Grass M, Manzke R, Nielsen T et al (2003) Helical cardiac cone beam reconstruction using retrospective ECG gating. Phys Med Biol 48:3069–3084
Hove JR, Köster RW, Forouhar AS et al (2003) Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature 421:172–177. https://doi.org/10.1038/nature01282
Huisken J (2004) Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science 305:1007–1009. https://doi.org/10.1126/science.1100035
Jenkins MW, Chughtai OQ, Basavanhally AN et al (2007) In vivo gated 4D imaging of the embryonic heart using optical coherence tomography. J Biomed Opt 12:030505. https://doi.org/10.1117/1.2747208
Kaufmann A, Mickoleit M, Weber M, Huisken J (2012) Multilayer mounting enables long-term imaging of zebrafish development in a light sheet microscope. Dev Camb Engl 139:3242–3247. https://doi.org/10.1242/dev.082586
Keller PJ, Schmidt AD, Wittbrodt J, Stelzer EHK (2008) Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science 322:1065–1069. https://doi.org/10.1126/science.1162493
Kim CK, Miri A, Leung LC et al (2014) Prolonged, brain-wide expression of nuclear-localized GCaMP3 for functional circuit mapping. Front Neural Circuits 8:138. https://doi.org/10.3389/fncir.2014.00138
Kim DH, Kim J, Marques JC et al (2017) Pan-neuronal calcium imaging with cellular resolution in freely swimming zebrafish. Nat Methods 14:1107–1114. https://doi.org/10.1038/nmeth.4429
Larson AC, White RD, Laub G et al (2004) Self-gated cardiac cine MRI. Magn Reson Med 51:93–102. https://doi.org/10.1002/mrm.10664
Lee DA, Andreev A, Truong TV et al (2017) Genetic and neuronal regulation of sleep by neuropeptide VF. eLife 6. https://doi.org/10.7554/eLife.25727
Levoy M, Zhang Z, McDowall I (2009) Recording and controlling the 4D light field in a microscope using microlens arrays. J Microsc 235:144–162. https://doi.org/10.1111/j.1365-2818.2009.03195.x
Li H, Foss SM, Dobryy YL et al (2011) Concurrent imaging of synaptic vesicle recycling and calcium dynamics. Front Mol Neurosci 4:34. https://doi.org/10.3389/fnmol.2011.00034
Liebling M, Forouhar AS, Gharib M et al (2005) Four-dimensional cardiac imaging in living embryos via postacquisition synchronization of nongated slice sequences. J Biomed Opt 10:054001. https://doi.org/10.1117/1.2061567
Mukamel EA, Nimmerjahn A, Schnitzer MJ (2009) Automated analysis of cellular signals from large-scale calcium imaging data. Neuron 63:747–760. https://doi.org/10.1016/j.neuron.2009.08.009
Muto A, Ohkura M, Abe G et al (2013) Real-time visualization of neuronal activity during perception. Curr Biol CB 23:307–311. https://doi.org/10.1016/j.cub.2012.12.040
Pierpont ME, Basson CT, Benson DW et al (2007) Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation 115:3015–3038. https://doi.org/10.1161/CIRCULATIONAHA.106.183056
Sablong R, Rengle A, Ramgolam A et al (2014) An optical fiber-based gating device for prospective mouse cardiac MRI. IEEE Trans Biomed Eng 61:162–170. https://doi.org/10.1109/TBME.2013.2278712
Sedmera D, Reckova M, deAlmeida A, Sedmerova M, Biermann M, Volejnik J, Sarre A, Raddatz E, McCarthy RA, Gourdie RG, Thompson RP (2003) Functional and morphological evidence for a ventricular conduction system in zebrafish and hearts. Am J Phys Heart Circ Phys 284(4):H1152–H1160
Stainier DY, Lee RK, Fishman MC (1993) Cardiovascular development in the zebrafish. I. Myocardial fate map and heart tube formation. Dev Camb Engl 119:31–40
Stainier DY, Fouquet B, Chen JN et al (1996) Mutations affecting the formation and function of the cardiovascular system in the zebrafish embryo. Dev Camb Engl 123:285–292
Taylor JM, Girkin JM, Love GD (2012) High-resolution 3D optical microscopy inside the beating zebrafish heart using prospective optical gating. Biomed Opt Express 3:3043–3053. https://doi.org/10.1364/BOE.3.003043
Thompson RB, McVeigh ER (2004) Flow-gated phase-contrast MRI using radial acquisitions. Magn Reson Med 52:598–604. https://doi.org/10.1002/mrm.20187
Thompson AW, Scott EK (2016) Characterisation of sensitivity and orientation tuning for visually responsive ensembles in the zebrafish tectum. Sci Rep 6:34887. https://doi.org/10.1038/srep34887
Treece GM, Prager RW, Gee AH, Berman L (2002) Correction of probe pressure artifacts in freehand 3D ultrasound. Med Image Anal 6:199–214
Trivedi V, Truong TV, Trinh LA, Holland DB, Liebling M, Fraser SE (2015) Dynamic structure and protein expression of the live embryonic heart captured by 2-photon light sheet microscopy and retrospective registration. Biomed Opt Express 6(6):2056
Truong TV (2017) Optimized live volumetric imaging with light sheet microscopy and related strategies. Microsc Microanal 23:2320–2321. https://doi.org/10.1017/S1431927617012260
Truong TV, Trivedi V, Trinh L et al (2014) Live 4D imaging of the embryonic vertebrate heart with two-photon light sheet microscopy and simultaneous optical phase stamping. Biophys J 106:435a–436a. https://doi.org/10.1016/j.bpj.2013.11.2453
Vermot J, Fraser SE, Liebling M (2008) Fast fluorescence microscopy for imaging the dynamics of embryonic development. HFSP J 2:143–155. https://doi.org/10.2976/1.2907579
Vermot J, Forouhar AS, Liebling M et al (2009) Reversing blood flows act through klf2a to ensure normal valvulogenesis in the developing heart. PLoS Biol 7:e1000246. https://doi.org/10.1371/journal.pbio.1000246
Vladimirov N, Mu Y, Kawashima T et al (2014) Light-sheet functional imaging in fictively behaving zebrafish. Nat Methods 11:883–884. https://doi.org/10.1038/nmeth.3040
Weber M, Scherf N, Meyer AM et al (2017) Cell-accurate optical mapping across the entire developing heart. eLife 6. https://doi.org/10.7554/eLife.28307
Wienholds E, van Eeden F, Kosters M et al (2003) Efficient target-selected mutagenesis in zebrafish. Genome Res 13:2700–2707. https://doi.org/10.1101/gr.1725103
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this paper

Cite this paper
Andreev, A., Fraser, S.E., Madaan, S. (2020). Eavesdropping on Biological Processes with Multi-dimensional Molecular Imaging. In: Toyama, Y., Miyawaki, A., Nakamura, M., Jinzaki, M. (eds) Make Life Visible. Springer, Singapore. https://doi.org/10.1007/978-981-13-7908-6_3
Download citation
DOI: https://doi.org/10.1007/978-981-13-7908-6_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-7907-9
Online ISBN: 978-981-13-7908-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)