
Abstract
Accumulated evidence shows that activation of microglia is associated with a change in morphology, from ramified to globular, which also represents a transition to M1 microglia. M1 microglia contribute to the induction and development of various neuroinflammatory disorders, including stroke, spinal cord injury, multiple sclerosis, Parkinson’s disease, Alzheimer’s disease psychiatric disorders, neuropathic pain and epilepsy. Thus, inhibition of microglial activation would be crucial in treating neurological disorders. Recent studies suggest a number of attractive molecular targets for blocking microglial activation. Among them, the nicotinic ACh receptor (nAChR), which especially contains the α7 subunit, contributes to the regulation of microglial activity through the inhibition of the synthesis of proinflammatory molecules. In addition, the glutamate transporter GLAST expressed in microglia is upregulated by α7 nAChR stimulation, which is mediated through both inositol triphosphate-Ca2+/calmodulin-dependent protein kinase II and fibroblast growth factor-2 pathways. It is possible, then, that activation of microglial α7 nAChR could be neuroprotective through inhibition of the production of proinflammatory molecules and enhancement of glutamate clearance from the synapse. This chapter will give an overview of the role of the α7 nAChR in microglial functioning and its potential as a therapeutic target for neurological disorders.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Microglia
- α7 nicotinic ACh receptor
- Glutamate transporter
- GLAST
- Ca2+
- Calmodulin-dependent protein kinase II
- Fibroblast growth factor-2
5.1 Microglia
Neuroinflammation is involved in the induction of various neurodegenerative and neuropsychiatric disorders including stroke, spinal cord injury, multiple sclerosis, Parkinson’s disease, Alzheimer’s disease, depressive disorders, schizophrenia, neuropathic pain and epilepsy (Blank and Prinz 2013; Frank-Cannon et al. 2009; Yrjänheikki et al. 1998). Neuroinflammation is mainly mediated by CNS glial cells such as microglia and astrocytes. Microglia, originally derived from the reticuloendothelial system, have a pivotal role as the main effector cells of the immune system (Kettenmann and Verkhratsky 2008). Although present in all region of the CNS, microglia are not uniformly distributed, representing between 0.5 and 16.6% of all cells in human and mouse brain (Lawson et al. 1990; Mittelbronn et al. 2001). Microglia act as a type of macrophage in peripheral tissues. Microglia are highly ramified, with long processes and small cell bodies, under the normal physiological state (Kettenmann et al. 2011). The recent emergence of live cell imaging technology reveals that microglia have highly motile processes that continuously survey the surrounding environment (Nimmerjahn et al. 2005; Davalos et al. 2005). This state represents the “resting” phenotype, which is involved in maintaining homeostasis (Kettenmann et al. 2011). Therefore, changes in microglial activity and functionality are indicative of pathological conditions.
5.2 Neuroinflammatory and Neuroprotective Roles of Microglia
It is widely known that activation of microglia, in response to illness, infection and injury, lead to morphological changes, from the highly ramified configuration to a globular, amoeboid shape (Kitamura et al. 1978; Stence et al. 2001; Thomas 1992). Activated microglia demonstrate increased proliferation, migration to the site of injury, scavenging of exogenous substances, cellular debris and pathogens, and production of proinflammatory molecules, including cytokines, chemokines, prostaglandins, nitric oxide and reactive oxygen species (Suzuki et al. 2004; Hide et al. 2000; Koizumi et al. 2007; Stence et al. 2001; Nolte et al. 1996; Morioka et al. 2013; Garrido-Gil et al. 2013; Fernandes et al. 2014). Cells that exhibit this phenotype are identified as “M1 microglia” (Kigerl et al. 2009). In fact, it has been demonstrated that hyper- or chronic activation of microglia could lead to the initiation of neurodegenerative disorders (Moehle and West 2015; Henkel et al. 2009). In addition, treatment with the microglial inhibitor minocycline, a tetracycline antibiotic, reduces inflammation in animal models of neurodegeneration (Wu et al. 2002; Hou et al. 2016).
At the same time, microglia also contribute to tissue recovery. Microglia produce anti-inflammatory and neuroprotective molecules, such as brain-derived neurotrophic factor (BDNF), glial cell-derived neurotrophic factor (GDNF), transforming growth factor-β (TGF-β), tumor necrosis factor (TNF), interleukin-4 (IL-4) and interleukin-10 (IL-10) (Suzuki et al. 2004; Lai and Todd 2008; Polazzi and Monti 2010; Amantea et al. 2015). Microglia showing anti-inflammatory and neuroprotective properties are called “M2 microglia”. Stimulation of microglia with IL-4 and interleukin-13 (IL-13), which are secreted by Th2 lymphocytes (Freilich et al. 2013), induces the M2 phenotype. M2 microglia express markers such as heparin-binding lectin, cysteine-rich protein FIZZ-1 and arginase-1 (Freilich et al. 2013). Transient middle cerebral artery occlusion induces brain tissue infraction and cell apoptosis. The number of apoptotic cells in infarcted tissue in transgenic mice in which microglia were selectively ablated was significantly more than that of wild-type mice (Lalancette-Hébert et al. 2007). Microglial phenotype is altered depending on environmental conditions―thus, microglial functioning show apparently opposing properties, either pro-inflammatory or anti-inflammatory (Ponomarev et al. 2007). Previous findings also indicate that blockade of microglial activity alone may not be sufficient as a treatment for neuroinflammatory disorders, so further elucidating changes in microglial phenotypes and properties under specific pathological conditions is crucial. Although there are a number of studies on the role of M1 microglia in proinflammatory responses and their involvement in neurological disorders, their roles in neuroprotection and their function in neuroinflammatory and neurodegenerative diseases have yet to be fully elaborated. In this vein, more research is necessary on identifying the neuroprotective molecules released by microglia under pathological conditions.
5.3 Nicotinic Acetylcholine Receptors and Microglia
Nicotinic acetylcholine receptors (nAChRs) are ligand-gated ion channels, consisting of hetero- or homo-pentameric subunits. These receptors have important roles in neurobiological processes such as memory, learning, locomotion, attention and anxiety (Dajas-Bailador and Wonnacott 2004; Dani and Bertrand 2007; Zoli et al. 2015). In the mammalian brain, 12 genes each encode a subunit, and nine different nAChR subunits have been identified (α2-α10 and β2-β4) (Dani and Bertrand 2007). The homomeric α7 nAChR is one of the most abundantly expressed and widely distributed subtype in the brain (Gotti and Clementi 2004; Sargent 1993). The α7 nAChR is expressed not only in neurons, but also in non-neuronal cells such as astrocytes, microglia, oligodendrocyte precursor cells and brain endothelial cells (Liu et al. 2015; Kihara et al. 2001; Suzuki et al. 2006; Hawkins et al. 2005; Rogers et al. 2001). Human microglia express the α3, α5, α7 and β4 subunits (Rock et al. 2008), whereas the α7 nAChR is the only functional nAChR subtype in rat cortical microglia (Morioka et al. 2014). The rat cortical microglia, then, makes an ideal system to study the neurobiological role of the α7 nAChR.
The α7 nAChR appears to have a critical role in neuroprotection. Peripheral macrophages express α7 nAChR, which regulate the systemic response to inflammation (Wang et al. 2003). Microglial α7 nAChR could be responsible for modulating the response to inflammation in the mouse brain. Prevention of lipopolysaccharide (LPS)-induced TNF release from murine microglia is mediated through activation of the α7 nAChR (Shytle et al. 2004). Activation of microglial α7 nAChR suppresses the production of a number of proinflammatory molecules (Suzuki et al. 2006; Giunta et al. 2004; De Simone et al. 2005; Rock et al. 2008; Zhang et al. 2017). Furthermore, stimulation of the α7 nAChR suppresses the production of reactive oxygen species (ROS) in microglia stimulated with fibrillar β-amyloid peptide (Moon et al. 2008), in addition, treatment of cultured microglia with galantamine, a nAChR allosteric ligand, induces phagocytosis of β-amyloid in an α7 nAChR-dependent manner (Takata et al. 2010), suggesting a potential role of the α7 nAChR in the pathophysiology of Alzheimer’s disease. Stimulation of the α7 nAChR also increases expression of anti-inflammatory and neuroprotective molecules such as TGF-β1, IL-4, IL-10 and heme oxygenase-1 (De Simone et al. 2005; Parada et al. 2013; Rock et al. 2008; Zhang et al. 2017). Treatment of a microglial cell line BV2 with an α7 nAChR agonist increases autophagy, an anti-inflammatory response (Shao et al. 2017). Furthermore, treatment with nicotine inhibits LPS-induced H+ currents through α7 nAChR (Noda and Kobayashi 2017). It is known that H+ channel-mediated currents are required for NAPDH oxidase-dependent ROS generation in brain microglia, a key step in the neuroinflammatory pathway (Wu et al. 2012). While it is currently unknown whether the stimulation of α7 nAChR induces the switching of microglial phenotype from M1 to M2, studies clearly indicate the importance of microglia-expressed α7 nAChR in reducing neuroinflammation, and suggest that microglial α7 nAChR could be utilized as a therapeutic target for the treatment of neuropathological disorders (Table 5.1).
5.4 Glutamate Transporters and Microglia
Glutamate is not only one of the major excitatory neurotransmitters mediating memory, learning and acute pain perception, but is excitotoxic at high concentrations in the synapse. Therefore, the regulation of synaptic glutamate concentration, to prevent the overstimulation of post-synaptic neurons, is important in preventing excitotoxicity, and is mainly conducted through Na+/K+-dependent glutamate transporters located in glial cells and neurons (Robinson and Dowd 1997). Thus far, five glutamate transporters have been cloned and pharmacologically characterized: excitatory amino acid transporter (EAAT) 1 (glutamate/aspartate transporter; GLAST) and EAAT2 (glutamate transporter 1; GLT-1), which are mainly expressed in glial cells, and EAAT3 (excitatory amino acid carrier 1; EAAC1), EAAT4 and EAAT5, which are mainly expressed in neurons (Arriza et al. 1997; Fairman et al. 1995; Kanai and Hediger 1992; Pines et al. 1992). In general, astrocytic GLAST and GLT-1 are important for maintaining low concentration of glutamate (Shibata et al. 1997), and astrocytic glutamate uptake at synapses account for about 90% of total clearance under physiological conditions (Tanaka et al. 1997).
Microglia express functional glutamate transporters, which are involved in regulating glutamate homeostasis in synapses (Morioka et al. 2008). It has been shown that activated microglia express GLAST and GLT-1 both in vivo and in vitro (Noda et al. 1999). Microglial glutamate uptake at synapses is about 10% of that of astrocytes under physiological conditions (Persson et al. 2005; Shaked et al. 2005). Under excitotoxic conditions induced by high concentrations of glutamate, however, activity and expression of microglial glutamate transporters are enhanced by excluding excess glutamate. For example, GLT-1 expression is increased in activated microglia following nerve injury (López-Redondo et al. 2000). Furthermore, stimulation of cultured microglia with LPS increases GLT-1 expression and glutamate transport capacity (Persson et al. 2005). A clinical study demonstrated that microglial glutamate transporters are involved in the control of neuronal damage in traumatic brain injury. Upregulation of GLAST expression in microglia is observed in brain white matter 1 week after ischemia (Beschorner et al. 2007). In addition, the expression of glutamate transporters (GLAST, GLT-1 and EAAC1) is observed in microglia/macrophages within the infract region at 7 and 28 days after ischemia (Arranz et al. 2010). Thus, these observations indicate that microglial glutamate transporters could be crucial in reducing glutamate-mediated excitotoxicity. Although astrocytes generally have a crucial role in clearing glutamate from the synapses, the activity of glutamate transport in astrocytes is in fact downregulated under pathological conditions (Fine et al. 1996; Xin et al. 2009). Therefore, microglial glutamate transporters, which are upregulated under pathological conditions, serve as a back-up to astrocytic glutamate uptake (López-Redondo et al. 2000; Xin et al. 2009). However, microglial glutamate transporter function and the functional relationship between α7 nAChR and glutamate transporters in microglia, have yet to be elaborated.
5.5 Nicotinic Acetylcholine Receptor and Glutamate Transporters
A number of studies have described significant interactions between nAChR and monoamine transporters, which comprise of noradrenaline, dopamine and serotonin transporters. For example, treatment with nicotine induced increased expression and functioning of these transporters in frontal cortical neurons and other cell types (Danielson et al. 2011; Itoh et al. 2010; Awtry and Werling 2003; Middleton et al. 2004). By contrast, few studies have demonstrated a positive functional interaction between the nAChR and glutamate transporters. Basal glutamate uptake in cultured glial cells derived from rat pups prenatally exposed to nicotine is higher than normal (Lim and Kim 2001). Furthermore, increased activity of astrocytic glutamate transporters (GLAST and GLT-1) is observed following neuronal nAChR stimulation, which increases synaptic levels of glutamate (Poitry-Yamate et al. 2002). Chronic treatment of Xenopus oocytes overexpressing EAAC1 with nicotine reduces EAAC1 activity (Yoon et al. 2014). Stimulation of cultured cerebellar astrocytes with nicotine modulates glutamate uptake, which is probably mediated through either a cAMP-independent or cAMP-dependent mechanism (Lim and Kim 2003).
5.6 Alpha7 Nicotinic Acetylcholine Receptors and Microglial Glutamate Transporters
Although nicotine modulates activity and expression of glutamate transporters in the CNS, the actual nAChR subtype involved and the intracellular signal cascade mediating the transporter’s response to nAChR stimulation are not clear. Furthermore, a potential role of α7 nAChR modulating microglial glutamate transporters has yet to be elaborated. A recent study showed that activation of the microglial α7 nAChR system is crucial in the regulation of glutamate transporters (Morioka et al. 2014, 2015). Cultured rat cortical microglia mainly express GLAST and not GLT-1, as shown by RT-PCR and pharmacological analysis using selective inhibitors for GLAST and GLT-1. Treatment with nicotine increases GLAST mRNA expression and glutamate transport activity and the effect of nicotine is blocked by pretreatment with a selective α7 nAChR antagonist, indicating that α7 nAChR mediates nicotine-induced GLAST expression. Understanding the role of the α7 subtype, this is the only nAChR subtype expressed in the cortical microglia.
The concentration of nicotine needed to induce GLAST expression is relatively high (300–1000 μM) compared to concentrations utilized in other in vitro assays. It is possible that, compared to α7 nAChR expressed in other cell types, cortical microglial α7 nAChR has unique properties. Microglial α7 nAChR demonstrates a different pattern of electrical current compared with that demonstrated by neurons, in which stimulation of cortical microglia with nicotine does not evoke current, although ATP treatment evokes current (Suzuki et al. 2006). Furthermore, the α7 nAChR has two isoforms with different pharmacological properties: a low and a high affinity nicotinic binding site (Severance et al. 2004). In fact, high concentrations of nicotine (>1000 μM) is used to stimulate microglia/macrophage α7 nAChR (Takata et al. 2010; Sun et al. 2013). Thus, in the case of microglial α7 nAChR, high concentrations of nicotine may be needed to activate microglia. Further investigation is necessary to elucidate precise pharmacological and functional properties of α7 nAChR.
Various intracellular signal molecules are involved following stimulation of α7 nAChR in vitro (Kihara et al. 2001; Arredondo et al. 2006; Maouche et al. 2013). In rat cortical microglia, stimulation of the α7 nAChR induced a rapid and transient increase in the concentration of cytosolic Ca2+ through the activation of phospholipase C (PLC) and the release of Ca2+ from inositol triphosphate (IP3)-sensitive intracellular stores, but not through the influx of extracellular Ca2+ (Suzuki et al. 2006). Increased cytosolic Ca2+ concentration through an IP3 receptor-dependent mechanism is one of the key events underlying nicotine-α7 nAChR-mediated GLAST expression, block of the IP3 receptor, but not removal of extracellular Ca2+, inhibits nicotine’s effect. Likewise, Mashimo et al. previously demonstrated that an IP3 receptor signaling cascade is crucial in the regulation of GLAST expression in Bergmann glial cells, which are a type of astrocyte found in the cerebellum (Mashimo et al. 2010).
A number of studies have indicated that several signaling molecules are activated following increased cytosolic Ca2+ concentration in microglia (Takata et al. 2010; Suzuki et al. 2006; Hide et al. 2000). The calmodulin-Ca2+/calmodulin-dependent protein kinase ΙΙ (CaMKII) pathway is activated following an α7 nAChR-mediated Ca2+ influx, eventually leading to microglia phagocytosis of amyloid β (Takata et al. 2010). CaMKII activation is crucial since inhibiting CaMKII blocks nicotine-induced GLAST expression and glutamate transport in cortical microglia. Others have confirmed that CaMKII activity has an important role in glutamate uptake in cortical astrocytes induced through other pharmacological stimuli (Smith and Navratilova 1999). By contrast, other signal molecules, including protein kinase A, protein kinase C, phosphatidylinositol 3-kinase, janus-activated kinase, Src tyrosine kinase and extracellular signal-regulated protein kinase, do not appear to have a major role in nicotine-mediated GLAST expression in microglia.
Increased cytosolic Ca2+ concentration is observed within 1–2 min following nicotine treatment (Suzuki et al. 2006). Thus, it is speculated that CaMKII is rapidly activated in parallel with increased intracellular Ca2+. However, upregulation of GLAST mRNA expression is observed only after 18 h of nicotine treatment. Therefore, this delay between increased cytosolic Ca2+ concentration and GLAST expression suggests the induction of intermediary molecules which could have a role in GLAST expression. In fact, the protein synthesis inhibitor cycloheximide blocks nicotine-induced GLAST mRNA expression, indicating the presence of a protein intermediary between increased Ca2+ concentration and GLAST expression.
Stimulation of nAChRs contributes to the production of several molecules such as cytokines, chemokines, and neurotrophic factors (Hawkins et al. 2015; Maggio et al. 1998; Son and Winzer-Serhan 2009; Takarada et al. 2012). These substances in turn could enhance clearance of glutamate from the synapse by increasing GLAST expression. A number of studies have demonstrated that growth factors, including epidermal growth factor (EGF), fibroblast growth factor (FGF), insulin-like growth factor-1 (IGF-1) and TGF-β1, modulate GLAST expression in astrocytes (Figiel et al. 2003; Lee et al. 2009; Suzuki et al. 2001). In addition, treatment of cultured microglia with nicotine increases FGF-2 mRNA, but not EGF, IGF-1 and TGF-β1 mRNAs, via the stimulation of the α7 nAChR. FGF-2 protein is also increased after treatment with nicotine. Thus, these findings indicate that FGF-2 could be the crucial intermediary between α7 nAChR and GLAST upregulation.
In fact, treatment of cultured microglia with recombinant FGF-2 increases expression of GLAST and increases glutamate transport. In addition, pretreatment with a selective inhibitor of FGF receptor (FGFR) tyrosine kinase blocks the stimulatory effect of nicotine on GLAST expression and glutamate transport. The FGFR has four subtypes (FGFR1-FGFR4). Cultured cortical microglia express FGFR1 mRNA, but not FGFR2 mRNA, FGFR3 mRNA, and FGFR4 mRNA. In neurons, FGF2 exerts neuroprotection by activating FGFR1, thereby decreasing glutamate-induced damage of hippocampal neurons through the production of GDNF (Lenhard et al. 2002). Thus, the microglial α7 nAChR-FGF-2-FGFR1 pathway elicited by nicotine could be neuroprotective through the enhancement of glutamate clearance by GLAST upregulation. FGF-2 has a neuroprotective role in preclinical animal models of neuroinflammation and neurodegenerative disorders. FGF-2 secreted from injured neurons could lead to microglia transformation and neuroprotective activities such as migration and phagocytosis (Noda et al. 2014). Enhancing brain FGF-2 expression restores hippocampal functioning in a preclinical model of Alzheimer’s disease (Kiyota et al. 2011).
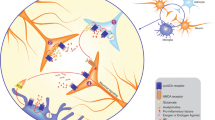
Although further investigation is needed to elaborate the relationship between transformed M2 microglia and neuroprotection, the findings so far indicate that the nicotine-α7 nAChR system modulates microglial GLAST function and regulates the clearance of synaptic glutamate (Fig. 5.1).

Schematic representation of nicotine-α7 nAChR mediating GLAST expression in microglia. Long-term treatment (more than 18 h) of microglia with nicotine (300–1000 μM) upregulates the expression of GLAST (mRNA and protein) through the stimulation of the α7 nAChR. The stimulation of α7 nAChR increases transient Ca2+ concentration through phospholipase C and inositol triphosphate (IP3)-dependent pathways, and subsequent activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII). The treatment of microglia with nicotine induces expression of fibroblast growth factor-2 (FGF-2) mRNA and protein. FGF-2 produced stimulates FGFR1 expressed in microglia in an autocrine and paracrine manner, and increases both GLAST expression and glutamate transport. Thus, clearance of synaptic glutamate is achieved via activation of a nicotine-α7 nAChR system, through regulation of GLAST expression and glutamate transport in microglia through IP3-Ca2+- CaMKII and FGF-2 pathways
5.7 Drug Development Targeting α7 nAChR for Neurological Disorders
Targeting the α7 nAChR is a potential strategy to treat neurological disorders which currently have no effective treatments. In fact, a selective α7 nAChR agonist reduces 6-hydroxydopamine-induced dopaminergic neuronal damage in a rat model of Parkinson’s disease (Suzuki et al. 2013; Bordia et al. 2015). Furthermore, the α7 nAChR is a potential target for the treatment of cognitive dysfunction associated with Alzheimer’s disease. Systemic treatment with selective α7 nAChR agonists, either PHA-543613 or galantamine, improves cognitive dysfunction in β-amyloid-treated mice (Sadigh-Eteghad et al. 2015). In addition, the α7 nAChR may be involved in regulating nociceptive transduction, as α7 nAChR agonists ameliorate experimental painful peripheral neuropathies (Di Cesare Mannelli et al. 2014; Freitas et al. 2013). Studies uncovering the relationship between α7 nAChR and microglial function in particular suggest the possibility that α7 nAChR expressed by microglia are a novel therapeutic target for the treatment of neurological disorders. For example, stimulation of the α7 nAChR enhances microglial β-amyloid clearance (Takata et al. 2010). Direct activation of microglial α7 nAChR is neuroprotective, through upregulation of heme oxtgenase-1, against oxygen and glucose deprivation in organotrophic hippocampal culture (Parada et al. 2013). Recent findings also demonstrate that stimulation of α7 nAChR enhances GLAST expression and glutamate transport in microglia, suggesting that enhancing glutamate reuptake at the synapse is crucial in maintaining normal functioning of the glutamatergic system. Furthermore, it is also possible that downregulation of the α7 nAChR itself is associated with the induction of neurological disorders. Thus, direct stimulation of α7 nAChR or gene therapy to enhance α7 nAChR expression, especially in microglia, could be useful for treatment of various neurological disorders.
5.8 Conclusions
Hyperactivation of microglia, especially transitioning to the M1 phenotype, contributes to the induction of neuropathology in the CNS, suggesting that targeting M1 microglia could be an appropriate treatment for neurological disorders. Although mechanisms regulating the switching of microglial phenotypes have yet to be fully elaborated, inducing the transitioning of microglia from the M1 phenotype to the M2 phenotype could be an alternate therapeutic approach. As described above, the α7 nAChR contributes to the regulation of a number of microglial functions, especially in the reduction of neuroinflammatory responses and the clearance of potentially excitotoxic levels of synaptic glutamate. Therefore, further understanding of the molecular and cellular mechanisms underlying α7 nAChR expressed in microglia could aid in the development of therapeutic strategies for neuroinflammatory and neurodegenerative diseases, which in general, are lacking in effective treatments.
References
Amantea D, Micieli G, Tassorelli C, Cuartero MI, Ballesteros I, Certo M, Moro MA, Lizasoain I, Bagetta G (2015) Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front Neurosci 9:147. https://doi.org/10.3389/fnins.2015.00147
Arranz AM, Gottlieb M, Pérez-Cerdá F, Matute C (2010) Increased expression of glutamate transporters in subcortical white matter after transient focal cerebral ischemia. Neurobiol Dis 37(1):156–165. https://doi.org/10.1016/j.nbd.2009.09.019
Arredondo J, Chernyavsky AI, Jolkovsky DL, Pinkerton KE, Grando SA (2006) Receptor-mediated tobacco toxicity: cooperation of the Ras/Raf-1/MEK1/ERK and JAK-2/STAT-3 pathways downstream of alpha7 nicotinic receptor in oral keratinocytes. FASEB J 20(12):2093–2101. https://doi.org/10.1096/fj.06-6191com
Arriza JL, Eliasof S, Kavanaugh MP, Amara SG (1997) Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci U S A 94(8):4155–4160
Awtry TL, Werling LL (2003) Acute and chronic effects of nicotine on serotonin uptake in prefrontal cortex and hippocampus of rats. Synapse 50(3):206–211. https://doi.org/10.1002/syn.10259
Beschorner R, Simon P, Schauer N, Mittelbronn M, Schluesener HJ, Trautmann K, Dietz K, Meyermann R (2007) Reactive astrocytes and activated microglial cells express EAAT1, but not EAAT2, reflecting a neuroprotective potential following ischaemia. Histopathology 50(7):897–910. https://doi.org/10.1111/j.1365-2559.2007.02703.x
Blank T, Prinz M (2013) Microglia as modulators of cognition and neuropsychiatric disorders. Glia 61(1):62–70. https://doi.org/10.1002/glia.22372
Bordia T, McGregor M, Papke RL, Decker MW, McIntosh JM, Quik M (2015) The α7 nicotinic receptor agonist ABT-107 protects against nigrostriatal damage in rats with unilateral 6-hydroxydopamine lesions. Exp Neurol 263:277–284. https://doi.org/10.1016/j.expneurol.2014.09.015
Dajas-Bailador F, Wonnacott S (2004) Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci 25(6):317–324. https://doi.org/10.1016/j.tips.2004.04.006
Dani JA, Bertrand D (2007) Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol 47:699–729. https://doi.org/10.1146/annurev.pharmtox.47.120505.105214
Danielson K, Truman P, Kivell BM (2011) The effects of nicotine and cigarette smoke on the monoamine transporters. Synapse 65(9):866–879. https://doi.org/10.1002/syn.20914
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8(6):752–758. https://doi.org/10.1038/nn1472
De Simone R, Ajmone-Cat MA, Carnevale D, Minghetti L (2005) Activation of alpha7 nicotinic acetylcholine receptor by nicotine selectively up-regulates cyclooxygenase-2 and prostaglandin E2 in rat microglial cultures. J Neuroinflammation 2(1):4. https://doi.org/10.1186/1742-2094-2-4
Di Cesare Mannelli L, Pacini A, Matera C, Zanardelli M, Mello T, De Amici M, Dallanoce C, Ghelardini C (2014) Involvement of α7 nAChR subtype in rat oxaliplatin-induced neuropathy: effects of selective activation. Neuropharmacology 79:37–48. https://doi.org/10.1016/j.neuropharm.2013.10.034
Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG (1995) An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 375(6532):599–603. https://doi.org/10.1038/375599a0
Fernandes A, Miller-Fleming L, Pais TF (2014) Microglia and inflammation: conspiracy, controversy or control? Cell Mol Life Sci 71(20):3969–3985. https://doi.org/10.1007/s00018-014-1670-8
Figiel M, Maucher T, Rozyczka J, Bayatti N, Engele J (2003) Regulation of glial glutamate transporter expression by growth factors. Exp Neurol 183(1):124–135
Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, Gelbard HA (1996) Tumor necrosis factor alpha inhibits glutamate uptake by primary human astrocytes. Implications for pathogenesis of HIV-1 dementia. J Biol Chem 271(26):15303–15306
Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG (2009) Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener 4:47. https://doi.org/10.1186/1750-1326-4-47
Freilich RW, Woodbury ME, Ikezu T (2013) Integrated expression profiles of mRNA and miRNA in polarized primary murine microglia. PLoS One 8(11):e79416. https://doi.org/10.1371/journal.pone.0079416
Freitas K, Ghosh S, Ivy Carroll F, Lichtman AH, Imad Damaj M (2013) Effects of α7 positive allosteric modulators in murine inflammatory and chronic neuropathic pain models. Neuropharmacology 65:156–164. https://doi.org/10.1016/j.neuropharm.2012.08.022
Garrido-Gil P, Rodriguez-Pallares J, Dominguez-Meijide A, Guerra MJ, Labandeira-Garcia JL (2013) Brain angiotensin regulates iron homeostasis in dopaminergic neurons and microglial cells. Exp Neurol 250:384–396. https://doi.org/10.1016/j.expneurol.2013.10.013
Giunta B, Ehrhart J, Townsend K, Sun N, Vendrame M, Shytle D, Tan J, Fernandez F (2004) Galantamine and nicotine have a synergistic effect on inhibition of microglial activation induced by HIV-1 gp120. Brain Res Bull 64(2):165–170. https://doi.org/10.1016/j.brainresbull.2004.06.008
Gotti C, Clementi F (2004) Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol 74(6):363–396. https://doi.org/10.1016/j.pneurobio.2004.09.006
Hawkins BT, Egleton RD, Davis TP (2005) Modulation of cerebral microvascular permeability by endothelial nicotinic acetylcholine receptors. Am J Physiol Heart Circ Physiol 289(1):H212–H219. https://doi.org/10.1152/ajpheart.01210.2004
Hawkins JL, Denson JE, Miley DR, Durham PL (2015) Nicotine stimulates expression of proteins implicated in peripheral and central sensitization. Neuroscience 290C:115–125. https://doi.org/10.1016/j.neuroscience.2015.01.034
Henkel JS, Beers DR, Zhao W, Appel SH (2009) Microglia in ALS: the good, the bad, and the resting. J NeuroImmune Pharmacol 4(4):389–398. https://doi.org/10.1007/s11481-009-9171-5
Hide I, Tanaka M, Inoue A, Nakajima K, Kohsaka S, Inoue K, Nakata Y (2000) Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J Neurochem 75(3):965–972
Hou Y, Xie G, Liu X, Li G, Jia C, Xu J, Wang B (2016) Minocycline protects against lipopolysaccharide-induced cognitive impairment in mice. Psychopharmacology (Berl) 233(5):905–916. https://doi.org/10.1007/s00213-015-4169-6
Itoh H, Toyohira Y, Ueno S, Saeki S, Zhang H, Furuno Y, Takahashi K, Tsutsui M, Hachisuka K, Yanagihara N (2010) Upregulation of norepinephrine transporter function by prolonged exposure to nicotine in cultured bovine adrenal medullary cells. Naunyn Schmiedeberg’s Arch Pharmacol 382(3):235–243. https://doi.org/10.1007/s00210-010-0540-7
Kanai Y, Hediger MA (1992) Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 360(6403):467–471. https://doi.org/10.1038/360467a0
Kettenmann H, Verkhratsky A (2008) Neuroglia: the 150 years after. Trends Neurosci 31(12):653–659. https://doi.org/10.1016/j.tins.2008.09.003
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91(2):461–553. https://doi.org/10.1152/physrev.00011.2010
Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG (2009) Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 29(43):13435–13444. https://doi.org/10.1523/JNEUROSCI.3257-09.2009
Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, Kume T, Akaike A (2001) alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J Biol Chem 276(17):13541–13546. https://doi.org/10.1074/jbc.M008035200
Kitamura T, Tsuchihashi Y, Fujita S (1978) Initial response of silver-impregnated “resting microglia” to stab wounding in rabbit hippocampus. Acta Neuropathol 44(1):31–39
Kiyota T, Ingraham KL, Jacobsen MT, Xiong H, Ikezu T (2011) FGF2 gene transfer restores hippocampal functions in mouse models of Alzheimer’s disease and has therapeutic implications for neurocognitive disorders. Proc Natl Acad Sci U S A 108(49):E1339–E1348. https://doi.org/10.1073/pnas.1102349108
Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, Joshi BV, Jacobson KA, Kohsaka S, Inoue K (2007) UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 446(7139):1091–1095. nature05704 [pii] 391038/nature05704
Lai AY, Todd KG (2008) Differential regulation of trophic and proinflammatory microglial effectors is dependent on severity of neuronal injury. Glia 56(3):259–270. https://doi.org/10.1002/glia.20610
Lalancette-Hébert M, Gowing G, Simard A, Weng YC, Kriz J (2007) Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci 27(10):2596–2605. https://doi.org/10.1523/JNEUROSCI.5360-06.2007
Lawson LJ, Perry VH, Dri P, Gordon S (1990) Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39(1):151–170
Lee ES, Sidoryk M, Jiang H, Yin Z, Aschner M (2009) Estrogen and tamoxifen reverse manganese-induced glutamate transporter impairment in astrocytes. J Neurochem 110(2):530–544. https://doi.org/10.1111/j.1471-4159.2009.06105.x
Lenhard T, Schober A, Suter-Crazzolara C, Unsicker K (2002) Fibroblast growth factor-2 requires glial-cell-line-derived neurotrophic factor for exerting its neuroprotective actions on glutamate-lesioned hippocampal neurons. Mol Cell Neurosci 20(2):181–197
Lim DK, Kim HS (2001) Changes in the glutamate release and uptake of cerebellar cells in perinatally nicotine-exposed rat pups. Neurochem Res 26(10):1119–1125
Lim DK, Kim HS (2003) Opposite modulation of glutamate uptake by nicotine in cultured astrocytes with/without cAMP treatment. Eur J Pharmacol 476(3):179–184
Liu Y, Zeng X, Hui Y, Zhu C, Wu J, Taylor DH, Ji J, Fan W, Huang Z, Hu J (2015) Activation of α7 nicotinic acetylcholine receptors protects astrocytes against oxidative stress-induced apoptosis: implications for Parkinson’s disease. Neuropharmacology 91:87–96. https://doi.org/10.1016/j.neuropharm.2014.11.028
López-Redondo F, Nakajima K, Honda S, Kohsaka S (2000) Glutamate transporter GLT-1 is highly expressed in activated microglia following facial nerve axotomy. Brain Res Mol Brain Res 76(2):429–435
Maggio R, Riva M, Vaglini F, Fornai F, Molteni R, Armogida M, Racagni G, Corsini GU (1998) Nicotine prevents experimental parkinsonism in rodents and induces striatal increase of neurotrophic factors. J Neurochem 71(6):2439–2446
Maouche K, Medjber K, Zahm JM, Delavoie F, Terryn C, Coraux C, Pons S, Cloëz-Tayarani I, Maskos U, Birembaut P, Tournier JM (2013) Contribution of α7 nicotinic receptor to airway epithelium dysfunction under nicotine exposure. Proc Natl Acad Sci U S A 110(10):4099–4104. https://doi.org/10.1073/pnas.1216939110
Mashimo M, Okubo Y, Yamazawa T, Yamasaki M, Watanabe M, Murayama T, Iino M (2010) Inositol 1,4,5-trisphosphate signaling maintains the activity of glutamate uptake in Bergmann glia. Eur J Neurosci 32(10):1668–1677. https://doi.org/10.1111/j.1460-9568.2010.07452.x
Middleton LS, Cass WA, Dwoskin LP (2004) Nicotinic receptor modulation of dopamine transporter function in rat striatum and medial prefrontal cortex. J Pharmacol Exp Ther 308(1):367–377. https://doi.org/10.1124/jpet.103.055335
Mittelbronn M, Dietz K, Schluesener HJ, Meyermann R (2001) Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol 101(3):249–255
Moehle MS, West AB (2015) M1 and M2 immune activation in Parkinson’s disease: foe and ally? Neuroscience 302:59–73. https://doi.org/10.1016/j.neuroscience.2014.11.018
Moon JH, Kim SY, Lee HG, Kim SU, Lee YB (2008) Activation of nicotinic acetylcholine receptor prevents the production of reactive oxygen species in fibrillar beta amyloid peptide (1-42)-stimulated microglia. Exp Mol Med 40(1):11–18. https://doi.org/10.3858/emm.2008.40.1.11
Morioka N, Abdin MJ, Kitayama T, Morita K, Nakata Y, Dohi T (2008) P2X(7) receptor stimulation in primary cultures of rat spinal microglia induces downregulation of the activity for glutamate transport. Glia 56(5):528–538. https://doi.org/10.1002/glia.20634
Morioka N, Tokuhara M, Harano S, Nakamura Y, Hisaoka-Nakashima K, Nakata Y (2013) The activation of P2Y6 receptor in cultured spinal microglia induces the production of CCL2 through the MAP kinases-NF-κB pathway. Neuropharmacology 75C:116–125. https://doi.org/10.1016/j.neuropharm.2013.07.017
Morioka N, Tokuhara M, Nakamura Y, Idenoshita Y, Harano S, Zhang FF, Hisaoka-Nakashima K, Nakata Y (2014) Primary cultures of rat cortical microglia treated with nicotine increases in the expression of excitatory amino acid transporter 1 (GLAST) via the activation of the α7 nicotinic acetylcholine receptor. Neuroscience 258:374–384. https://doi.org/10.1016/j.neuroscience.2013.11.044
Morioka N, Harano S, Tokuhara M, Idenoshita Y, Zhang FF, Hisaoka-Nakashima K, Nakata Y (2015) Stimulation of α7 nicotinic acetylcholine receptor regulates glutamate transporter GLAST via basic fibroblast growth factor production in cultured cortical microglia. Brain Res 1625:111–120. https://doi.org/10.1016/j.brainres.2015.08.029
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308(5726):1314–1318. https://doi.org/10.1126/science.1110647
Noda M, Kobayashi AI (2017) Nicotine inhibits activation of microglial proton currents via interactions with α7 acetylcholine receptors. J Physiol Sci 67(1):235–245. https://doi.org/10.1007/s12576-016-0460-5
Noda M, Nakanishi H, Akaike N (1999) Glutamate release from microglia via glutamate transporter is enhanced by amyloid-beta peptide. Neuroscience 92(4):1465–1474
Noda M, Takii K, Parajuli B, Kawanokuchi J, Sonobe Y, Takeuchi H, Mizuno T, Suzumura A (2014) FGF-2 released from degenerating neurons exerts microglial-induced neuroprotection via FGFR3-ERK signaling pathway. J Neuroinflammation 11:76. https://doi.org/10.1186/1742-2094-11-76
Nolte C, Möller T, Walter T, Kettenmann H (1996) Complement 5a controls motility of murine microglial cells in vitro via activation of an inhibitory G-protein and the rearrangement of the actin cytoskeleton. Neuroscience 73(4):1091–1107
Parada E, Egea J, Buendia I, Negredo P, Cunha AC, Cardoso S, Soares MP, López MG (2013) The microglial α7-acetylcholine nicotinic receptor is a key element in promoting neuroprotection by inducing heme oxygenase-1 via nuclear factor erythroid-2-related factor 2. Antioxid Redox Signal 19(11):1135–1148. https://doi.org/10.1089/ars.2012.4671
Persson M, Brantefjord M, Hansson E, Rönnbäck L (2005) Lipopolysaccharide increases microglial GLT-1 expression and glutamate uptake capacity in vitro by a mechanism dependent on TNF-alpha. Glia 51(2):111–120. https://doi.org/10.1002/glia.20191
Pines G, Danbolt NC, Bjørås M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI (1992) Cloning and expression of a rat brain L-glutamate transporter. Nature 360(6403):464–467. https://doi.org/10.1038/360464a0
Poitry-Yamate CL, Vutskits L, Rauen T (2002) Neuronal-induced and glutamate-dependent activation of glial glutamate transporter function. J Neurochem 82(4):987–997
Polazzi E, Monti B (2010) Microglia and neuroprotection: from in vitro studies to therapeutic applications. Prog Neurobiol 92(3):293–315. https://doi.org/10.1016/j.pneurobio.2010.06.009
Ponomarev ED, Maresz K, Tan Y, Dittel BN (2007) CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci 27(40):10714–10721. https://doi.org/10.1523/JNEUROSCI.1922-07.2007
Robinson MB, Dowd LA (1997) Heterogeneity and functional properties of subtypes of sodium-dependent glutamate transporters in the mammalian central nervous system. Adv Pharmacol 37:69–115
Rock RB, Gekker G, Aravalli RN, Hu S, Sheng WS, Peterson PK (2008) Potentiation of HIV-1 expression in microglial cells by nicotine: involvement of transforming growth factor-beta 1. J NeuroImmune Pharmacol 3(3):143–149. https://doi.org/10.1007/s11481-007-9098-7
Rogers SW, Gregori NZ, Carlson N, Gahring LC, Noble M (2001) Neuronal nicotinic acetylcholine receptor expression by O2A/oligodendrocyte progenitor cells. Glia 33(4):306–313
Sadigh-Eteghad S, Talebi M, Mahmoudi J, Babri S, Shanehbandi D (2015) Selective activation of α7 nicotinic acetylcholine receptor by PHA-543613 improves Aβ25-35-mediated cognitive deficits in mice. Neuroscience 298:81–93. https://doi.org/10.1016/j.neuroscience.2015.04.017
Sargent PB (1993) The diversity of neuronal nicotinic acetylcholine receptors. Annu Rev Neurosci 16:403–443. https://doi.org/10.1146/annurev.ne.16.030193.002155
Severance EG, Zhang H, Cruz Y, Pakhlevaniants S, Hadley SH, Amin J, Wecker L, Reed C, Cuevas J (2004) The alpha7 nicotinic acetylcholine receptor subunit exists in two isoforms that contribute to functional ligand-gated ion channels. Mol Pharmacol 66(3):420–429. https://doi.org/10.1124/mol.104.000059
Shaked I, Tchoresh D, Gersner R, Meiri G, Mordechai S, Xiao X, Hart RP, Schwartz M (2005) Protective autoimmunity: interferon-gamma enables microglia to remove glutamate without evoking inflammatory mediators. J Neurochem 92(5):997–1009. https://doi.org/10.1111/j.1471-4159.2004.02954.x
Shao BZ, Ke P, Xu ZQ, Wei W, Cheng MH, Han BZ, Chen XW, Su DF, Liu C (2017) Autophagy plays an important role in anti-inflammatory mechanisms stimulated by Alpha7 nicotinic acetylcholine receptor. Front Immunol 8:553. https://doi.org/10.3389/fimmu.2017.00553
Shibata T, Yamada K, Watanabe M, Ikenaka K, Wada K, Tanaka K, Inoue Y (1997) Glutamate transporter GLAST is expressed in the radial glia-astrocyte lineage of developing mouse spinal cord. J Neurosci 17(23):9212–9219
Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, Ehrhart J, Silver AA, Sanberg PR, Tan J (2004) Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem 89(2):337–343. https://doi.org/10.1046/j.1471-4159.2004.02347.x
Smith TL, Navratilova E (1999) Increased calcium/calmodulin protein kinase activity in astrocytes chronically exposed to ethanol: influences on glutamate transport. Neurosci Lett 269(3):145–148
Son JH, Winzer-Serhan UH (2009) Chronic neonatal nicotine exposure increases mRNA expression of neurotrophic factors in the postnatal rat hippocampus. Brain Res 1278:1–14. https://doi.org/10.1016/j.brainres.2009.04.046
Stence N, Waite M, Dailey ME (2001) Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia 33(3):256–266
Sun Y, Li Q, Gui H, Xu DP, Yang YL, Su DF, Liu X (2013) MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Res 23(11):1270–1283. https://doi.org/10.1038/cr.2013.116
Suzuki K, Ikegaya Y, Matsuura S, Kanai Y, Endou H, Matsuki N (2001) Transient upregulation of the glial glutamate transporter GLAST in response to fibroblast growth factor, insulin-like growth factor and epidermal growth factor in cultured astrocytes. J Cell Sci 114(Pt 20):3717–3725
Suzuki T, Hide I, Ido K, Kohsaka S, Inoue K, Nakata Y (2004) Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J Neurosci 24(1):1–7. 24/1/1 [pii] 10.1523/JNEUROSCI.3792-03.2004
Suzuki T, Hide I, Matsubara A, Hama C, Harada K, Miyano K, Andrä M, Matsubayashi H, Sakai N, Kohsaka S, Inoue K, Nakata Y (2006) Microglial alpha7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. J Neurosci Res 83(8):1461–1470. https://doi.org/10.1002/jnr.20850
Suzuki S, Kawamata J, Matsushita T, Matsumura A, Hisahara S, Takata K, Kitamura Y, Kem W, Shimohama S (2013) 3-[(2,4-Dimethoxy)benzylidene]-anabaseine dihydrochloride protects against 6-hydroxydopamine-induced parkinsonian neurodegeneration through α7 nicotinic acetylcholine receptor stimulation in rats. J Neurosci Res 91(3):462–471. https://doi.org/10.1002/jnr.23160
Takarada T, Nakamichi N, Kawagoe H, Ogura M, Fukumori R, Nakazato R, Fujikawa K, Kou M, Yoneda Y (2012) Possible neuroprotective property of nicotinic acetylcholine receptors in association with predominant upregulation of glial cell line-derived neurotrophic factor in astrocytes. J Neurosci Res 90(11):2074–2085. https://doi.org/10.1002/jnr.23101
Takata K, Kitamura Y, Saeki M, Terada M, Kagitani S, Kitamura R, Fujikawa Y, Maelicke A, Tomimoto H, Taniguchi T, Shimohama S (2010) Galantamine-induced amyloid-{beta} clearance mediated via stimulation of microglial nicotinic acetylcholine receptors. J Biol Chem 285(51):40180–40191. https://doi.org/10.1074/jbc.M110.142356
Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276(5319):1699–1702
Thomas WE (1992) Brain macrophages: evaluation of microglia and their functions. Brain Res Brain Res Rev 17(1):61–74
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421(6921):384–388. https://doi.org/10.1038/nature01339
Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S (2002) Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci 22(5):1763–1771
Wu LJ, Wu G, Akhavan Sharif MR, Baker A, Jia Y, Fahey FH, Luo HR, Feener EP, Clapham DE (2012) The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat Neurosci 15(4):565–573. https://doi.org/10.1038/nn.3059
Xin WJ, Weng HR, Dougherty PM (2009) Plasticity in expression of the glutamate transporters GLT-1 and GLAST in spinal dorsal horn glial cells following partial sciatic nerve ligation. Mol Pain 5:15. https://doi.org/10.1186/1744-8069-5-15
Yoon HJ, Lim YJ, Zuo Z, Hur W, Do SH (2014) Nicotine decreases the activity of glutamate transporter type 3. Toxicol Lett 225(1):147–152. https://doi.org/10.1016/j.toxlet.2013.12.002
Yrjänheikki J, Keinänen R, Pellikka M, Hökfelt T, Koistinaho J (1998) Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A 95(26):15769–15774
Zhang Q, Lu Y, Bian H, Guo L, Zhu H (2017) Activation of the α7 nicotinic receptor promotes lipopolysaccharide-induced conversion of M1 microglia to M2. Am J Transl Res 9(3):971–985
Zoli M, Pistillo F, Gotti C (2015) Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 96(Pt B):302–311. https://doi.org/10.1016/j.neuropharm.2014.11.003
Acknowledgement
This work was supported in part by grants from Smoking Research Foundation. We also thank Dr. Aldric T. Hama for his careful editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
This chapter is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
Copyright information
© 2018 The Author(s)
About this chapter

Cite this chapter
Morioka, N., Hisaoka-Nakashima, K., Nakata, Y. (2018). Regulation by Nicotinic Acetylcholine Receptors of Microglial Glutamate Transporters: Role of Microglia in Neuroprotection. In: Akaike, A., Shimohama, S., Misu, Y. (eds) Nicotinic Acetylcholine Receptor Signaling in Neuroprotection. Springer, Singapore. https://doi.org/10.1007/978-981-10-8488-1_5
Download citation
DOI: https://doi.org/10.1007/978-981-10-8488-1_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-8487-4
Online ISBN: 978-981-10-8488-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)