
Abstract
Glutamate neurotoxicity is involved in various neurodegenerative disorders including brain ischemic stroke, trauma, and Alzheimer’s and Parkinson’s diseases. In addition to excitatory neuronal death, neuroinflammation accompanied by the activation of glial cells has been shown to be induced by these disorders. We previously reported the roles of nicotinic acetylcholine receptors (nAChRs) in the survival of central nervous system neurons during excitotoxic events and neuroinflammation. Nicotine and other nAChR agonists protected cortical neurons against glutamate neurotoxicity via α4- and α7-nAChRs in cultures of neurons obtained from the cerebral cortex of fetal rats. In addition, donepezil, a therapeutic acetylcholinesterase inhibitor currently being used for the treatment of Alzheimer’s disease, protected neuronal cells from glutamate neurotoxicity. Moreover, nicotine and donepezil induced the upregulation of nAChRs. Thus, we propose that nicotine as well as donepezil prevents glutamate neurotoxicity through Α4- and α7-nAChRs and the phosphatidylinositol 3-kinase (PI3K)/Akt pathway. In addition to the beneficial effect on neuronal cells, we have reported the responses of astrocytes to bradykinin, an inflammatory mediator, and the effect of nAChR stimulation on these responses using cultured cortical astrocytes. Bradykinin induced a transient increase of intracellular calcium concentration ([Ca2+]i) in cultured astrocytes. Both nicotine and donepezil reduced this bradykinin-induced [Ca2+]i increase. This reduction was inhibited not only by mecamylamine, an nAChR antagonist, but also by PI3K and Akt inhibitors. These results suggest that nAChR stimulation suppresses the inflammatory response induced by bradykinin via the PI3K-Akt pathway in astrocytes.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
4.1 Introduction
Nicotinic acetylcholine receptors (nAChRs) are distributed across various organs, such as the central nervous system (CNS), and are involved in changing cell functions and controlling cell survival via several intracellular signal transduction mechanisms. Many endogenous compounds such as glutamate are known to trigger neuronal death via apoptosis in degenerative diseases of the CNS (Bresnick 1989; Choi et al. 1987). To date, we have elucidated that excitatory neurotoxicity triggered by excitative glutamate can be inhibited by various endogenous factors. In research using cultured rat embryonic cortical neurons, nicotine was shown to exert neuroprotective effects against glutamate neurotoxicity via neuronal nAChRs (Akaike et al. 1994; Kaneko et al. 1997; Shimohama et al. 1998). These studies raised awareness of this issue among many researchers, and numerous groups subsequently reported that nAChR agonists inhibit acute glutamate-induced neuronal death.
Alzheimer’s disease (AD) is a common type of dementia. Its pathological characteristics include the formation of senile plaques primarily consisting of amyloid-β (Aβ) proteins, neurofibrillary tangles, and neurologic deficit; thus, AD has aspects of neurodegenerative disease (Whitehouse et al. 1982; Perry et al. 1995). Various disorders related to cholinergic nerves have also been found in the postmortem brain of AD patients, such as atrophy of the nucleus of origin of cholinergic nerves, reduction of acetylcholinergic nerves that project to the cortex from the Meynert nucleus, reduced activity of acetylcholine transferase activity in the cortex, and reduced expression of ACh and nAChRs (Bartus et al. 1982; Coyle et al. 1983). Since the cholinergic nervous system is involved in the functions of cognition and learning, attempts have been made to relieve symptoms by supplementing ACh, which led to the emergence of treatment drugs that act as acetylcholinesterase (AChE) inhibitors, such as donepezil. In addition to the neuronal injury, inflammation in the brain is involved in the pathogenesis of AD (Heppner et al. 2015; Heneka et al. 2015; Rogers 2008; Lee et al. 2010). In inflammatory conditions, it is reported that glial cells, such as astrocytes and microglia, are abnormally activated (Morris et al. 2013; Lee et al. 1995). This abnormal activation of glial cells contributes to the pathogenesis of various neurodegenerative disorders (Heppner et al. 2015; Heneka et al. 2015; Rogers 2008; Lee et al. 2010; Yan et al. 2014). Astrocytes are the most abundant cells in the CNS and play important roles in the maintenance of neuronal activity through the release of neurotrophic factors, the maintenance of ion gradients such as extracellular K+, and the construction of the blood–brain barrier (Giaume et al. 2010). However, a recent study revealed that, in various pathological conditions, astrocytes were abnormally activated and could be deleterious to adjacent neurons through the release of various inflammatory cytokines (Allan and Rothwell 2001). Therefore, inhibition of the abnormal activation of astrocytes is considered to be a therapeutic strategy for several CNS diseases involving inflammation.
As a therapeutic drug of AD, donepezil is an effective AChE inhibitor for slowing the progression of cognitive function disorders. In addition to AChE inhibitory activity, several other mechanisms have been noted for their possible relationships to the therapeutic effects of these drugs. AD is a neurodegenerative disease, and some of the therapeutic effects of donepezil may be attributable to its neuroprotective action; however, the assessment of its protective action and analysis of its mechanism of action have not been fully investigated, and much has yet to be clarified. In addition to the effect of donepezil on neurons, another group reported that donepezil inhibited the production of inflammatory cytokines induced by the treatment of Aβ oligomer in cultured microglia (Kim et al. 2014). However, the effect of donepezil on the function of astrocytes has not been elucidated.
This chapter summarizes the effects of nAChR stimulation using nicotine and nAChR ligands, including donepezil, on excitatory neuronal death in rat cultured neurons and neuroinflammation in astrocytes.
4.2 Neuroprotective Effect via Nicotine Receptors
On the basis of the above findings, we have focused on the neuroprotective action of donepezil and found that it exerts its protective effect against glutamate neurotoxicity, as determined through research seeking to reveal its mechanism of action. At the same time, we found that donepezil did not exhibit a protective effect when neurons were treated simultaneously with glutamate and that it expressed its protective action in a manner dependent on the duration and concentration of the pretreatment. This strongly suggests that donepezil exerts its protective effect by a mechanism of action other than AChE inhibition (Takada et al. 2003).
Thus, we continued our analysis with the objective of elucidating the mechanism of the neuroprotective effect of donepezil against glutamate neurotoxicity. It has been shown that nicotine exerted a protective effect against glutamate neurotoxicity via nAChR in the primary culture of cortical neurons by pretreatment (Akaike et al. 1994). Since donepezil exerted a neuroprotective effect in a manner dependent on the duration of the pretreatment, we examined the possible involvement of acetylcholine receptors in the protective action of donepezil. Upon treatment with glutamate after treatment for 24 h with the nAChR antagonist mecamylamine and donepezil, we found that mecamylamine treatment significantly prevented the protective effect of AChE inhibitors. Upon pretreatment with the muscarine acetylcholine receptor antagonist scopolamine and donepezil, there was no impact on the protective effect of donepezil. These results suggested that the protective effect of donepezil against glutamate neurotoxicity involves nAChRs.
Next, we examined nAChR subtypes which are involved in the protective action of donepezil. The brain-expressed primary subtypes among the currently known 12 nAChR subtypes are α4- and α7-nAChR. First, when we verified the expression of primary component subunits of α4- and α7-nAChR in the primary culture of rat embryonic cortical neurons used in our laboratory, we detected nAChR mRNA and proteins of α4 and α7 subunits, which are the subunits composing α7-nAChR. We also used dihydro-β-erythroidine (DHβE) and methyllycaconitine (MLA), which are α4- nAChR and α7-nAChR-specific antagonists, to examine the involvement of neuronal nAChR subtypes in the neuroprotective action of donepezil. We applied glutamate treatment after treatment with donepezil and DHβE or MLA for 24 h and found that the protective action of donepezil was significantly inhibited. The above results showed that the protective action of donepezil against glutamate neurotoxicity involved nAChRs (Takada et al. 2003).
4.3 Mechanisms of Neuroprotective Effects by Stimulating Nicotinic Receptors
Nicotine exerts a neuroprotective effect against neuronal apoptosis via α7-nAChR. The phosphatidylinositol 3-kinase (PI3K)/Akt signal pathway has been reported to be involved in this protective mechanism (Kihara et al. 2001; Shaw et al. 2002). In addition to the protective effect of nicotine against glutamate neurotoxicity reportedly being inhibited by various kinase inhibitors through the PI3K pathway, the phosphorylation of Akt by nicotine treatment and the increase in the expression of Bcl-2, an anti-apoptotic protein, have also been reported. Since donepezil exerts its protective effect through α7-nAChR, we considered the possibility of the involvement of the PI3K-Akt signal pathway in this protective action and investigated specific inhibitors of kinases that form the PI3K signal pathway. Janus-activated kinase 2 (JAK2), a non-receptor tyrosine kinase, and Fyn activate PI3K in conjunction with α7-nAChR, so we examined the involvements of AG490 and PP2, inhibitors of JAK2 and Fyn. After pretreating cerebral cortical cells with either AG 490 or PP2 along with donepezil 24 h ahead of time, the application of glutamate significantly inhibited the protective action of donepezil. Next, to clarify the involvement of PI3K, we examined the effect of the PI3K inhibitor LY294002 on donepezil and found the protective effect of donepezil to be significantly inhibited. After that, we examined whether the MAPK pathway might be involved by observing the effect of the MAPK inhibitor PD98059 on donepezil; we found that the protective effect of donepezil was unaffected. The results of using these different kinase inhibitors suggested that the protective effect of donepezil is expressed through the PI3K signal pathway.
To further examine the role of donepezil in the PI3K signal transmission pathway, we observed the phosphorylation of Akt and the change in the amount of Bcl-2 expression in cerebral cortical cells treated with donepezil. Akt is known to be recruited by active PI3K in the vicinity of cell membranes, activated through phosphorylation, which in turn activates proteins and caspases of the Bcl-2 family of apoptotic control factors to regulate apoptosis. Using western blotting to observe Akt phosphorylation in cerebral cortical neurons treated for 1 h with donepezil, we found a marked increase of phosphorylated Akt in the neurons treated with donepezil. Likewise, in assessing change in the amount of Bcl-2 expression in cerebral cortical cells treated for 24 h with donepezil, an increase of Bcl-2 expression could be seen from the donepezil treatment. By integrating these results, the protective effect of donepezil against glutamate neurotoxicity is thought to be linked to the activation of PI3K by α7-nAChR via JAK2 and Fyn and, by virtue of activating p-Akt, to be expressed through initiation of an apoptosis control program involving an increase in Bcl-2 expression (Takada-Takatori et al. 2006).
4.4 Mechanism of the Nicotinic Acetylcholine Receptor Upregulation upon Long-Term Nicotine Stimulation
Unlike other receptors, the amount of protein increases in nAChR and its function is accelerated—that is, upregulation is provoked—as a result of long-term exposure to nicotine and other agonists. However, much remains unknown about the details of this mechanism. Thus, further research has been performed on the signals related to nAChR upregulation and increased sensitivity of the neuroprotective action in order to elucidate the mechanism that promotes the survival of neurons associated with nAChR and to understand the changes in cellular and receptor functions that are caused by long-term stimulation of nAChR, which has a neuroprotective effect. It is assumed that long-term nicotine stimulus is subjected to complex regulation by the desensitization of nAChR. Therefore, the study described below investigated this mechanism using donepezil, as an nAChR activator, which is currently indicated for long-term clinical use.
When we observed the amounts of nAChR expression in primary cortical neuron cultures as a result of long-term treatment with donepezil using western blot analysis, the expression level of α7-nAChR protein was increased. In addition, when we observed the immunohistochemical changes in the amount of nAChR expression as a result of long-term treatment with donepezil, we observed that the number of α7-nAChR-expressing cells increased in the cell membrane, which is important for the exertion of nAChR function. These results suggest that long-term stimulation of nAChR not only increases the amount of α7-nAChR that is expressed as a protein but also promotes a shift of that expression to the cell membrane, which is thought to be responsible for the function of α7-nAChR.
Next, to investigate the effect that increased expression of α7-nAChR has on the function of nAChR, we investigated the involvement of nicotine in increased intracellular concentrations of calcium ([Ca2+]i). Temporary increases in [Ca2+]i as a result of nicotine treatment increased even further as a result of nicotine stimulation in cells that were treated with donepezil for 4 days. This indicates that long-term treatment with donepezil enhanced the response to nicotine stimulation immediately after administration and induced upregulation (Kume et al. 2005).
Since it is clear that nAChR upregulation occurs as a result of long-term stimulation of nAChR receptors in cortical neuron cultures, we continued our investigation of the mechanism behind this effect. To investigate the involvement of nAChR stimulation and its downstream signal, we simultaneously carried out both long-term donepezil treatment and treatment with MLA, which is an α7-nAChR antagonist. The results indicated that the increases in the amount of nAChR proteins caused by long-term donepezil treatment were suppressed by simultaneous treatment with MLA. Therefore, we simultaneously carried out both long-term donepezil treatment and treatment with either LY294002, which is a PI3K inhibitor, or the MAPKK inhibitor PD98059 and found that simultaneous treatment with either LY294002 or PD98059 suppressed increases in the amount of nAChR proteins. These results indicate that the nAChR and the PI3K and MAPK signal pathways are involved in nAChR upregulation caused by long-term donepezil treatment. Next, to elucidate the mechanism of action of the promotion of nAChR function caused by long-term donepezil treatment, we investigated the involvement of the actions of nAChR antagonist, PI3K inhibitor, and MAPKK inhibitor on the increases in [Ca2+]i that are caused by nicotine. We carried out simultaneous long-term treatment with donepezil and treatment with the α7-nAChR antagonist MLA. This resulted in the suppression of further increases in [Ca2+]i as a result of nicotine. We then carried out simultaneous long-term treatment with donepezil and either the PI3K inhibitor LY294002 or the MAPKK inhibitor PD98059. This also resulted in the suppression of further increases in [Ca2+]i as a result of nicotine. These results suggest that nAChR as well as the PI3K and MAPK pathways are involved in the promotion of nAChR function in cell membranes, such as seen when [Ca2+]i increases as a result of long-term treatment with donepezil (Takada-Takatori et al. 2008a).
4.5 Mechanism of Increased Sensitivity in the Neuronal Protective Effect of Nicotine That Accompanies Receptor Upregulation Caused by Long-Term Stimulation of Nicotine Receptors
Next, to investigate whether the increased sensitivity to the neuronal protective effect results from nAChR causing upregulation, we conducted long-term treatment with donepezil and investigated its effect on donepezil against glutamate neurotoxicity during a state of increased nAChR function. The concentrations of donepezil that are sufficiently low that they do not lead to expression of the normal protective action (1 nM) in the treatment of neurons that have been subjected to long-term treatment with donepezil resulted in the marked suppression of glutamate neurotoxicity. This indicates that the protective effect that is induced by long-term treatment with donepezil is dependent on both the treatment duration and the concentration. Therefore, to elucidate the mechanism of neuroprotection that occurs under the conditions of long-term treatment, we investigated the effect of simultaneous long-term treatment with donepezil, nAChR antagonist, and either PI3K or MAPK pathway inhibitor on increased sensitivity to the neuroprotective action of donepezil. First, when we examined simultaneous long-term treatment with donepezil and treatment with the α7-nAChR antagonist MLA, we observed that the neuroprotective action caused by low concentrations of donepezil administered after long-term treatment with donepezil was suppressed by the simultaneous treatment with MLA. Next, when we examined simultaneous long-term treatment with donepezil and treatment with either the PI3K inhibitor LY294002 or the MAPKK inhibitor PD98059, we found that both had the same suppressive effect. Finally, to elucidate the mechanism by which the survival signal is enhanced by the phosphorylation of Akt in the downstream PI3K pathway, we observed the phosphorylation of Akt in cortical neurons with long-term donepezil treatment and pretreatment using western blotting. The results indicated that, although the amount of phosphorylated Akt in neurons with long-term donepezil treatment and pretreatment increased markedly, when we carried out treatment with nAChR inhibitor and PI3K inhibitor simultaneously with donepezil pretreatment, increases in the amount of phosphorylated Akt were suppressed. These results indicate that when receptors are in a state of upregulation as a result of long-term nAChR stimulation, the promotive function of nAChR in cell membranes increases sensitivity to the neuroprotective effect of donepezil, which in turn indicates that the nAChR and either the PI3K or the MAPK signal pathways are involved in that action. When nAChR is in an upregulated state, the survival signal via the nAChR, PI3K-Akt, and MAPK pathways is more efficiently transmitted, and thus, we assume that this is why even lower concentrations of donepezil exerted the protective effect (Takada-Takatori et al. 2008b).
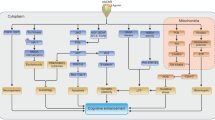
The mechanism of the neuroprotective effect of donepezil via nAChR that we assume is at work as a result of the above results is shown in Fig. 4.1. We hypothesize the following: As a result of the stimulation of α4- and α7-nAChR, donepezil exerts its neuroprotective effect via the PI3K-Akt signal pathway. Long-term nAChR stimulation induces the upregulation of nAChR in cell membranes, and the resulting promotion of nAChR function causes increased sensitivity to the neuroprotective effect (Takada-Takatori et al. 2009).

The mechanism of the neuroprotective effects of donepezil via nicotinic acetylcholine receptors
4.6 Effect of the Stimulation of Nicotinic Acetylcholine Receptor in Astrocytes on Inflammatory Response in the Brain
Not only direct nAChR receptor stimulation on neuronal cells but also an indirect effect through glial cells is considered to mediate the neuroprotective effect. Therefore, we investigated the effect of nAChR stimulation on the inflammatory responses of astrocytes, which play a crucial role in brain inflammation. As an inflammatory mediator in the brain, bradykinin is produced in the early stage of inflammation and induces the expression of several inflammatory genes (Lin et al. 2012; Hsieh et al. 2007; Schwaninger et al. 1999). In particular, bradykinin induces a variety of responses, such as a transient increase of intracellular calcium concentration ([Ca2+]i) (Akita and Okada 2011), the expression of matrix metalloprotease-9 (Lin et al. 2012) and cyclooxygenase-2 (COX-2) (Hsieh et al. 2007), and the release of interleukin-6 (IL-6) (Schwaninger et al. 1999) and glutamate (Liu et al. 2009) in astrocytes. In addition, it was previously reported that the cleavage of high-molecular-weight kininogen, the precursor of bradykinin, was increased in the cerebrospinal fluid of AD patients (Iores-Marçal et al. 2006; Bergamaschini et al. 1998) and that bradykinin receptor antagonists ameliorated the cognitive deficits in AD model mice (Bicca et al. 2015; Prediger et al. 2008; Lacoste et al. 2013). Accordingly, it can be speculated that inflammation induced by bradykinin in astrocytes is involved in the pathogenesis of AD. On the basis of these findings, to elucidate the effect of nAChR stimulation using nicotine and donepezil on the function in astrocytes, we investigated the responses of astrocytes to bradykinin and the effects of donepezil on these responses using cultured cortical astrocytes.
We first examined the effect of bradykinin on [Ca2+]i in cultured cortical astrocytes. Bradykinin induced a transient increase in [Ca2+]i in a concentration-dependent manner. Next, we verified the gene expression of B1 and B2 receptors in cultured cortical astrocytes. Both B1 and B2 receptors were expressed in our cultured astrocytes. We investigated the involvement of B1 and B2 receptors in the increase in [Ca2+]i induced by bradykinin using subtype-specific antagonists. Des-Arg9-[Leu8]-bradykinin, an antagonist of B1 receptors, did not affect the increase in [Ca2+]i induced by bradykinin, but HOE140, an antagonist of B2 receptors, almost completely inhibited the Ca2+ response induced by bradykinin. We further determined whether the [Ca2+]i increase induced by bradykinin was due to Ca2+ influx from the extracellular space or Ca2+ release from the intracellular Ca2+ store. Bradykinin-induced [Ca2+]i increase did not change upon excluding extracellular Ca2+. In contrast, depletion of Ca2+ stored in the endoplasmic reticulum (ER) by treating cells with thapsigargin, a blocker of Ca2+-ATPase on the ER, significantly reduced the Ca2+ response. These results suggest that bradykinin-induced [Ca2+]i response is attributable not to Ca2+ influx from the extracellular space but to Ca2+ release from the ER. Next, we examined the effect of nicotine and donepezil on the increase in [Ca2+]i induced by bradykinin. Simultaneous treatment of nicotine and donepezil did not affect the increase of [Ca2+]i. However, 24 h pretreatment of these drugs significantly reduced the Ca2+ response in a concentration-dependent manner, while these drugs did not affect the cell morphology and cell proliferation in astrocytes. These results suggest that nicotinic receptor stimulation suppressed the increase of [Ca2+]i induced by bradykinin and that pretreatment is required for this inhibitory effect.
We attempted to elucidate the mechanism of the inhibitory effect of nicotinic receptor stimulation on the increase of [Ca2+]i induced by bradykinin. We previously reported that donepezil exerted a neuroprotective effect on glutamate neurotoxicity via nAChRs in cultured cortical neurons (Takada-Takatori et al. 2006). Thus, we herein investigated the involvement of nAChRs in this effect of donepezil using nAChR antagonists. When cortical astrocytes were treated with a nAChR antagonist, mecamylamine, for 24 h before Ca2+ imaging, the inhibitory effect of donepezil was significantly antagonized. Previous studies reported that the most abundant nAChR subtypes in the brain are the α7- and α4- nAChRs (Paterson and Nordberg 2000) and that both of the receptor subtypes were also expressed in astrocytes (Oikawa et al. 2005). Thus, we examined the effects of MLA, an α7- nAChR antagonist, and DHβE, an α4- nAChR antagonist, on the inhibition by donepezil of the [Ca2+]i increase induced by bradykinin. When cortical astrocytes were pretreated with either MLA or DHβE for 24 h, the inhibitory effect of donepezil did not change. However, the effect of donepezil was significantly suppressed by treating cortical astrocytes with both MLA and DHβE for 24 h prior to bradykinin. These results suggested that both α7- and α4- nAChRs were involved in the inhibitory effect of donepezil on the increase of [Ca2+]i induced by bradykinin.
Previous studies demonstrated that JAK2 was activated in the downstream signaling of nAChRs (Razani-Boroujerdi et al. 2007; Marrero and Bencherif 2009). Therefore, we used AG490, a JAK2 inhibitor, to investigate the involvement of JAK2 in the inhibitory effect of donepezil. Treatment with AG490 significantly suppressed the inhibitory effect of donepezil on the increase in [Ca2+]i induced by bradykinin.
We previously reported that the stimulation of nAChRs activated PI3K and that the phosphorylation and activation of Akt downstream of PI3K were induced (Kihara et al. 2001). Thus, we examined the involvement of the PI3K-Akt pathway in the inhibitory effect of donepezil on the Ca2+ response induced by bradykinin. LY294002, a PI3K inhibitor, and Akt inhibitor significantly suppressed the effect of donepezil. In addition, we investigated the effect of donepezil on the phosphorylation state of Akt in cultured cortical astrocytes. The phosphorylation level of Akt was significantly elevated by treating cells with donepezil for 6 h.
Taking the obtained findings together, nAChR stimulation using donepezil induced the activation of the PI3K-Akt pathway in both cultured astrocytes and cultured neurons. However, there are some differences between astrocytes and neurons with respect to the signaling pathway mediated by nAChRs. For example, we previously showed that Akt phosphorylation was induced 1 h after the treatment of donepezil in cultured neurons, although donepezil induced Akt phosphorylation 6 h after the treatment in cultured astrocytes. Moreover, treatment of MLA or DHβE alone did not inhibit the effect of donepezil, and the combination of MLA and DHβE significantly reduced this effect. Thus, further studies are needed to elucidate the differences of the time course of Akt phosphorylation and the involvement of nAChR subtypes between neurons and astrocytes. Here we demonstrated that the Akt inhibitor suppressed the effect of donepezil on the increase in [Ca2+]i induced by bradykinin. Other researchers reported that, as the downstream signaling of the PI3K-Akt pathway, the activated Akt induced the phosphorylation of inositol trisphosphate (IP3) receptors and lowered the function of IP3 receptors (Khan et al. 2006; Szado et al. 2008). Taken together, these findings suggest that the phosphorylation and hypofunction of IP3 receptors by Akt could be involved in the effect of donepezil.
A previous study showed that bradykinin induced the production of intracellular reactive oxygen species (ROS) after the [Ca2+]i increase in cultured astrocytes (Akita and Okada 2011). To elucidate the effect of donepezil on ROS production, we examined ROS production induced by bradykinin using a fluorescent ROS indicator, H2DCF-DA. Bradykinin induced ROS production in a time-dependent manner. Donepezil significantly inhibited the increase of intracellular ROS level induced by bradykinin. Previous studies reported that an increase in intracellular ROS level activated transcription factors, such as nuclear factor-kappa B (NF-κB) and activator protein 1, and this activation led to inflammatory responses in astrocytes (Yang et al. 2012; Park et al. 2004). In fact, bradykinin induced the expression of IL-6 and COX-2 downstream of ROS production and activation of NF-κB in cultured astrocytes (Hsieh et al. 2007; Schwaninger et al. 1999). We here demonstrated that donepezil inhibited ROS production induced by bradykinin. Taking these results and reports into account, we suggest that nAChR stimulation using donepezil decreases the inflammatory response induced by bradykinin by regulating the increase of intracellular ROS level.
4.7 Conclusion and Future Prospects
It was previously reported that nAChR is involved in AD and many neurodegenerative diseases. We investigated the involvement of nAChRs in the neuroprotective action that is induced through the use of AD drugs and found that PI3K-Akt and other intracellular information transmission systems play an important role in the neuroprotective effect and nAChR upregulation. In addition, we found that nAChR stimulation exerted an inhibitory effect on the [Ca2+]i increase induced by bradykinin via the PI3K-Akt pathway and also inhibited the increase of the ROS level in cultured cortical astrocytes. The cholinergic hypothesis was first proposed in the 1970s, but interesting new discoveries relating to the mechanism of action of AD drugs based on this hypothesis continue to be made. Research into the molecular mechanism that targets the signals that are related to nAChR, which is still only slightly understood, should make major contributions to the development of new drugs for neurodegenerative disorders including AD.
References
Akaike A, Tamura Y, Yokota T et al (1994) Nicotine-induced protection of cultured cortical neurons against N-methyl-D-aspartate receptor-mediated glutamate cytotoxicity. Brain Res 644:181–187
Akita T, Okada Y (2011) Regulation of bradykinin-induced activation of volume-sensitive outwardly rectifying anion channels by Ca2+ nanodomains in mouse astrocytes. J Physiol 589:3909–3927
Allan SM, Rothwell NJ (2001) Cytokines and acute neurodegeneration. Nat Rev Neurosci 2:734–744
Bartus RT, Dean RL 3rd, Beer B et al (1982) The cholinergic hypothesis of geriatric memory dysfunction. Science 217:408–414
Bergamaschini L, Parnetti L, Pareyson D et al (1998) Activation of the contact system in cerebrospinal fluid of patients with Alzheimer disease. Alzheimer Dis Assoc Disord 12:102–108
Bicca MA, Costa R, Loch-Neckel G et al (2015) B2 receptor blockage prevents Aβ-induced cognitive impairment by neuroinflammation inhibition. Behav Brain Res 278:482–491
Bresnick GH (1989) Excitotoxins: a possible new mechanism for the pathogenesis of ischemic retinal damage. Arch Ophthalmol 107:339–341
Choi DW, Maulucci-Gedde M, Kriegstein AJ (1987) Glutamate neurotoxicity in cortical cell culture. J Neurosci 7:357–368
Coyle JT, Price DL, DeLong MR et al (1983) Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science 219:1184–1190
Giaume C, Koulakoff A, Roux L et al (2010) Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat Rev Neurosci 11:87–99
Heneka MT, Carson MJ, El Khoury J et al (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14:388–405
Heppner FL, Ransohoff RM, Becher B (2015) Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16:358–372
Hsieh HL, Wang HH, Wu CY et al (2007) BK-induced COX-2 expression via PKC-δ-dependent activation of p42/p44 MAPK and NF-κB in astrocytes. Cell Signal 19:330–340
Iores-Marçal LM, Viel TA, Buck HS et al (2006) Bradykinin release and inactivation in brain of rats submitted to an experimental model of Alzheimer’s disease. Peptides 27:3363–3369
Kaneko S, Maeda T, Kume T et al (1997) Nicotine protects cultured cortical neurons against glutamate-induced cytotoxicity via α7 neuronal receptors and neuronal CNS receptors. Brain Res 765:135–140
Khan MT, Wagner L 2nd, Yule DI et al (2006) Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J Biol Chem 281:3731–3737
Kihara T, Shimohama S, Sawada H et al (2001) α7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A β-amyloid-induced neurotoxicity. J Biol Chem 276:13541–13546
Kim HG, Moon M, Choi JG et al (2014) Donepezil inhibits the amyloid-β oligomer-induced microglial activation in vitro and in vivo. Neurotoxicology 40:23–32
Kume T, Sugimoto M, Takada Y et al (2005) Up-regulation of nicotinic acetylcholine receptors by central-type acetylcholinesterase inhibitors in rat cortical neurons. Eur J Pharmacol 527:77–85
Lacoste B, Tong XK, Lahjouji K et al (2013) Cognitive and cerebrovascular improvements following kinin B1 receptor blockade in Alzheimer’s disease mice. J Neuroinflammation 4:10–57
Lee SC, Dickson DW, Brosnan CF (1995) Interleukin-1, nitric oxide and reactive astrocytes. Brain Behav Immun 9:345–354
Lee YJ, Han SB, Nam SY, Oh KW et al (2010) Inflammation and Alzheimer’s disease. Arch Pharm Res 33:1539–1556
Lin CC, Hsieh HL, Shih RH et al (2012) NADPH oxidase 2-derived reactive oxygen species signal contributes to bradykinin-induced matrix metalloproteinase-9 expression and cell migration in brain astrocytes. Cell Commun Signal 10:35
Liu HT, Akita T, Shimizu T et al (2009) Bradykinin-induced astrocyte-neuron signaling: glutamate release is mediated by ROS-activated volume-sensitive outwardly rectifying anion channels. J Physiol 587:2197–2209
Marrero MB, Bencherif M (2009) Convergence of α7 nicotinic acetylcholine receptor-activated pathways for anti-apoptosis and anti-inflammation: central role for JAK2 activation of STAT3 and NF-κB. Brain Res 1256:1–7
Morris GP, Clark IA, Zinn R et al (2013) Microglia: a new frontier for synaptic plasticity, learning and memory, and neurodegenerative disease research. Neurobiol Learn Mem 105:40–53
Oikawa H, Nakamichi N, Kambe Y et al (2005) An increase in intracellular free calcium ions by nicotinic acetylcholine receptors in a single cultured rat cortical astrocyte. J Neurosci Res 79:535–544
Park J, Choi K, Jeong E et al (2004) Reactive oxygen species mediate chloroquine-induced expression of chemokines by human astroglial cells. Glia 47:9–20
Paterson D, Nordberg A (2000) Neuronal nicotinic receptors in the human brain. Prog Neurobiol 61:75–111
Perry EK, Morris CM, Court JA et al (1995) Alteration in nicotine binding sites in Parkinson’s disease, Lewy body dementia and Alzheimer’s disease: possible index of early neuropathology. Neuroscience 64:385–395
Prediger RD, Medeiros R, Pandolfo P et al (2008) Genetic deletion or antagonism of kinin B(1) and B(2) receptors improves cognitive deficits in a mouse model of Alzheimer’s disease. Neuroscience 151:631–643
Razani-Boroujerdi S, Boyd RT, Dávila-García MI et al (2007) T cells express α7-nicotinic acetylcholine receptor subunits that require a functional TCR and leukocyte-specific protein tyrosine kinase for nicotine-induced Ca2+ response. J Immunol 179:2889–2998
Rogers J (2008) The inflammatory response in Alzheimer’s disease. J Periodontol 79:1535–1543
Schwaninger M, Sallmann S, Petersen N et al (1999) Bradykinin induces interleukin-6 expression in astrocytes through activation of nuclear factor- κB. J Neurochem 73:1461–1466
Shaw S, Bencherif M, Marrero MB et al (2002) Janus kinase 2, an early target of α7 nicotinic acetylcholine receptor-mediated neuroprotection against Aβ-(1-42) amyloid. J Biol Chem 277:44920–44924
Shimohama S, Greenwald DL, Shafron DH et al (1998) Nicotinic α7 receptors protect against glutamate neurotoxicity and neuronal ischemic damage. Brain Res 779:359–363
Szado T, Vanderheyden V, Parys JB et al (2008) Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci U S A 105:2427–2432
Takada Y, Yonezawa A, Kume T et al (2003) Nicotinic acetylcholine receptor-mediated neuroprotection by donepezil against glutamate neurotoxicity in rat cortical neurons. J Pharmacol Exp Ther 306:772–777
Takada-Takatori Y, Kume T, Sugimoto M et al (2006) Acetylcholinesterase inhibitors used in treatment of Alzheimer’s disease prevent glutamate neurotoxicity via nicotinic acetylcholine receptors and phosphatidylinositol 3-kinase cascade. Neuropharmacology 51:474–486
Takada-Takatori Y, Kume T, Ohgi Y et al (2008a) Mechanisms of α7-nicotinic receptor up-regulation and sensitization to donepezil-induced by chronic donepezil treatment. Eur J Pharmacol 590:150–156
Takada-Takatori Y, Kume T, Ohgi Y et al (2008b) Mechanism of neuroprotection by donepezil pretreatment in rat cortical neurons chronically treated with donepezil. J Neurosci Res. 2008 86:3575–3583
Takada-Takatori Y, Kume T, Izumi Y (2009) Roles of nicotinic receptors in acetylcholinesterase inhibitor-induced neuroprotection and nicotinic receptor up-regulation. Biol Pharm Bull 32:318–324
Whitehouse PJ, Price DL, Struble RG et al (1982) Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science 215:1237–1239
Yan J, Fu Q, Cheng L et al (2014) Inflammatory response in Parkinson’s disease (review). Mol Med Rep 10:2223–2233
Yang CM, Lin CC, Lee IT et al (2012) Japanese encephalitis virus induces matrix metalloproteinase-9 expression via a ROS/c-Src/PDGFR/PI3K-Akt/MAPKs-dependent AP-1 pathway in rat brain astrocytes. J Neuroinflammation 18:9–12
Acknowledgment
This study was supported in part by JSPS KAKENHI Grant Numbers JP24590111 and JP24390139 to Toshiaki Kume, and JP25860069 and JP17K08323 to Yuki Takada-Takatori. It was also supported in part by a grant from the Smoking Research Foundation and the Naito Foundation of Japan.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
This chapter is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
Copyright information
© 2018 The Author(s)
About this chapter

Cite this chapter
Kume, T., Takada-Takatori, Y. (2018). Nicotinic Acetylcholine Receptor Signaling: Roles in Neuroprotection. In: Akaike, A., Shimohama, S., Misu, Y. (eds) Nicotinic Acetylcholine Receptor Signaling in Neuroprotection. Springer, Singapore. https://doi.org/10.1007/978-981-10-8488-1_4
Download citation
DOI: https://doi.org/10.1007/978-981-10-8488-1_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-8487-4
Online ISBN: 978-981-10-8488-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)