
Abstract
Accumulation of amyloid β protein (Aβ) in the brain causes cognitive impairment in Alzheimer’s disease (AD). The nature of the relationship between smoking and AD or dementia has been controversial. However, a recent meta-analysis revealed that smoking is a risk factor for AD. With regard to nicotinic acetylcholinergic receptors (nAChRs), both AD and control patients that smoke have been reported to show an increase in 3H-cytisine (an α4β4 nAChR agonist) binding in the temporal cortex. The α7 nAChR is also a key factor in AD pathology, particularly in relation to internalization of Aβs. Furthermore, there are many reports showing the neuroprotective effects of nicotine. The internalization of Aβ may lead to Aβ clearance in the brain.
We hypothesized that an extracorporeal system that rapidly removes Aβ from the blood may accelerate Aβ clearance from the brain. We have reported that (1) several medical materials including hemodialyzers can effectively remove blood Aβ, (2) the concentrations of blood Aβs decreased during hemodialysis, (3) removal of blood Aβ enhanced Aβ influx into the blood (ideally from the brain), resulting in maintenance or improvement of cognitive function, and (4) Aβ deposition in the brain of hemodialysis patients was significantly lower than in controls. Smoking affected blood Aβ removal efficiencies and brain atrophy. We believe this Extracorporeal Blood Aβ Removal Systems (E-BARS) may contribute as a therapy for AD.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
10.1 Introduction: Amyloid β Protein in Alzheimer’s Disease
One of the major pathological changes associated with Alzheimer’s disease (AD) is the deposition of amyloid β protein (Aβ) as senile plaques and an increase in Aβ peptides in the brain (Kuo et al. 1996; Selkoe 2001). There are several Aβ species in the brain and plasma that are approximately 4 kDa in weight such as the 40-amino acid Aβ1–40 and the 42-amino acid Aβ1–42. Aβ1–42 aggregates more easily and is more toxic (Hung et al. 2008), forming soluble Aβ oligomers that can cause synapse loss and affect long-term potentiation in hippocampal neurons (Walsh et al. 2002). One mechanism proposed to underlie the increase in brain Aβ is reduced Aβ clearance rather than enhanced Aβ production, particularly in sporadic AD cases. Aβ production in the brains of AD patients was reported to be similar to that of normal subjects, yet Aβ clearance from AD brains was approximately 30% lower than in controls (Mawuenyega et al. 2010). In other words, it may be possible to treat AD by increasing Aβ clearance from the brain.
Recently, an anti-Ab monoclonal antibody that selectively targets aggregated forms of Aβ, aducanumab, was reported to be effective in improving cognitive function and reducing the brain Aβ burden, as measured by brain Aβ imaging (Sevigny et al. 2016). Similarly to anti-Aβ antibodies (Hock et al. 2003; Sevigny et al. 2016), peripheral administration of albumin, another Aβ-binding substance, was effective in improving cognitive function in AD patients in a Phase 2 study, and is currently undergoing a Phase 3 trial in AD patients (Boada et al. 2009, 2016).
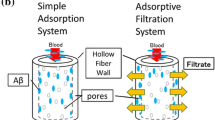
We hypothesized that the rapid removal of Aβ from the blood by an extracorporeal system (E-BARS; extracorporeal blood Aβ removal system) may act as a peripheral Aβ sink from the brain, as shown in Fig. 10.1 (Kawaguchi et al. 2010). Smoking could affect the blood flow in the brain resulting in a change in the excretion of Aβ from the brain into the blood.

Schema of the extracorporeal blood Aβ removal system (E-BARS) for the treatment of Alzheimer’s disease (AD). Our hypothesis: the rapid reduction of Aβ concentrations in the blood by apheresis technology may act as a trigger for enhancing the excretion of Aβ from the brain, resulting in cognitive improvement. (Taken from Kawaguchi et al. 2010 and modified)
10.2 Smoking , Nicotine, and AD
Determining the exact nature of the relationship between smoking and AD or dementia has been controversial. However, a recent meta-analysis revealed that smoking is a risk factor for AD, as described below. These controversial findings may be due to the mixed effects of smoke itself and components of tobacco such as nicotine.
10.2.1 Smoking and AD Prevalence
Sabia et al. (2008) reported that ex-smokers had a 30% lower risk of poor vocabulary and low verbal fluency. However, the correlation between smoking history and cognitive decline was inconsistent in longitudinal analysis. Despite this ameliorative effect of smoking on memory (Sabia et al. 2008), the risk of AD was reported to be unaffected by any measure of tobacco consumption (Garcia et al. 2010). Contrary to these favorable or neutral effects of smoking on dementia, there are many reports showing that smoking has a deleterious influence on AD risk. Lower AD risk was observed in alcohol drinkers of both genders who had never smoked (OR = 0.37, 95% CI: 0.21, 0.65), regardless of the presence of apolipoprotein E4 (APOε4). Ott et al. (1998) showed that smokers had an increased risk of dementia (relative risk 2.2 [95% CI: 1.3–3.6]) and AD (relative risk 2.3 [95% CI: 1.3–4.1]) compared with never smokers, based on a study of 6870 people aged 55 years and older. Smoking was a strong risk factor for AD in individuals without the APOε4 allele (relative risk 4.6 [95% CI: 1.5–14.2]), but had no effect in participants with this allele (relative risk 0.6 [95% CI: 0.1–4.8]). By meta-analysis of 19 prospective studies with at least 12 months of follow-up, Anstey et al. (2007) concluded that elderly smokers had increased risks of dementia and cognitive decline. Current smokers at baseline, relative to never smokers, had risks of 1.79 (95% CI: 1.43, 2.23) for AD and 1.78 (95% CI: 1.28, 2.47) for vascular dementia. Compared to those who had never smoked, current smokers at baseline also showed greater yearly declines in Mini-Mental State Examination scores over the follow-up period. Compared to former smokers, current smokers at baseline showed an increased risk of AD and an increased decline in cognitive ability (Anstey et al. 2007). Furthermore, Barnes and Yaffe (2011) reported that smoking was associated with a higher risk of AD (relative risk 1.59 [95% CI: 1.15, 2.20]), and that a 10% reduction in smoking prevalence could potentially lower AD prevalence by about 412,000 cases worldwide and by almost 51,000 cases in the USA, while a 25% reduction in smoking prevalence could potentially prevent more than 1 million AD cases worldwide and 130,000 cases in the USA.
10.2.2 AD Pathology and Smoking
Recently, an interesting animal study on AD pathology was reported that used cigarette smoke rather than administration of some components of tobacco such as nicotine. When APP/PS1 transgenic mice were exposed to smoke from cigarettes, AD pathology, such as Aβ deposition and the Iba1-labeled area indicating an inflammatory response, was enhanced in the cortex and hippocampus. This enhancement was observed in the high-dose smoking group but not in the low-dose group (Moreno-Gonzalez et al. 2013).
Contrary to the animal study, it has been reported that smoking reduces both soluble and insoluble Aβ1–40 and Aβ1–42 in the frontal cortex and Aβ1–40 in the temporal cortex and hippocampus in AD patients (Hellström-Lindahl et al. 2004).
10.2.3 Nicotinic Acetylcholinergic Receptors and Aβs
Regarding nicotinic acetylcholinergic receptors (nAChRs), both AD and control patients that smoked showed increased 3H-cytisine (an agonist of the α4β4 nAChR) binding in the temporal cortex (Hellström-Lindahl et al. 2004). Further, Aβ levels in the brain was reduced in this study. Therefore, these authors proposed that a selective nAChR agonist could be a novel protective therapy for AD.
The α7 nAChR is also a key factor in AD pathology, particularly in relation to internalization of Aβs. Soluble Aβ is known to bind to the α7 nAChR with high affinity (Wang et al. 2000). By in vitro experimentation with SH-SY5Y cells, Yang et al. (2014) revealed that extracellular Aβ1–42 was internalized by the cells and accumulated in endosomes/lysosomes and mitochondria. This internalization was mediated through an α7 nAChR-dependent pathway related to the activation of p38 MAPK and ERK1/2. The authors proposed that blockade of the α7 nAChR may have a beneficial effect by limiting intracellular accumulation of amyloid in the AD brain, thereby representing a potential therapeutic target for AD.
However, there are many articles showing the neuroprotective effects of nicotine. The internalization of Aβ may lead to Aβ clearance from the brain. Akaike and Shimohama’s research group first demonstrated the neuroprotective effect of nicotine on Aβ toxicity (Kihara et al. 1997). Concomitant administration of nicotine with Aβ25–35 ameliorated the death of rat cortical neurons induced by Aβ toxicity. In addition, the selective α7 nAChR antagonist, α-bungarotoxin, blocked this neuroprotective effect of nicotine. This group also revealed that stimulation of the α7 nAChR protected neurons against Aβ-enhanced glutamate neurotoxicity via PI3K (Kihara et al. 2001). Shimohama’s research group reported that treatment of rat microglia with galantamine, an acetylcholinesterase inhibitor, significantly enhanced microglial Aβ phagocytosis via the nAChR pathway (Takata et al. 2010). This group also revealed early accumulation of CD68-positive microglia at Aβ deposition sites and gradual reduction of Aβ in an Aβ-injected AD mouse model, which indicates the importance of the α7 nAChR in microglia as a therapeutic target in AD (Matsumura et al. 2015).
10.3 Our Hypothesis of a Therapeutic System for AD by Removal of Blood Aβ
As described earlier, one mechanism proposed to underlie increased brain Aβ in AD is reduced Aβ clearance rather than an increase in Aβ production, particularly in sporadic AD cases. Therefore, it may be possible to treat AD by enhancing Aβ clearance from the brain. There are several known Aβ transporters such as those involved in the Aβ influx pathway from the brain into the blood; e.g., LRP1 or APOE (Donahue et al. 2006; Bell et al. 2007), and RAGE (Silverberg et al. 2010), which is also known to mediate an Aβ influx pathway into the brain. In addition, perivascular elimination of Aβ in brain capillaries has been proposed (e.g., Morris et al. 2014).
Aβ concentrations in the cerebrospinal fluid (CSF) of AD patients are almost 100 times higher than those in plasma. Aβ concentrations in the CSF in cases of AD are reported to be 7.4–42.7 ng/ml for Aβ1–40 and 0.12–0.67 ng/ml for Aβ1–42 (Schoonenboom et al. 2005). Concentrations in the plasma of AD patients are reported to be 190.1 ± 61.7 pg/ml for Aβ1–40 and 23.0 ± 15.5 pg/ml for Aβ1–42 (Lopez et al. 2008). In brief, there are large gradients with respect to Aβ concentrations between the brain and plasma. Therefore, removing Aβ from the blood could accelerate Aβ transfer from the brain, thereby reducing the Aβ burden in the brain.
Peripheral administration of Aβ-binding substances, such as anti-Aβ antibodies, non-immunogenic substances, and albumin, can reduce the Aβ burden in the brain. However, attempts to use Aβ-binding substances in the blood in a therapeutic context resulted in the formation of Aβ complexes with the binding substances inside the body, which were sometimes retained in the plasma for a long period of time (DeMattos et al. 2001). Aβ antibodies generated by passive immunization or by active immunization using synthetic Aβ peptides reduced the occurrence of senile plaques and somewhat improved cognitive impairment in AD patients (Schenk et al. 1999; Hock et al. 2003). Furthermore, non-immunogenic Aβ-binding substances, such as GM1 ganglioside or gelsolin, also decreased the Aβ burden in the brain when they were peripherally injected into mouse models of AD (Matsuoka et al. 2003). Currently, a clinical trial is in progress where AD patients are being treated using intravenous administration of albumin, an Aβ-binding substance (Boada et al. 2009). In this Phase 2 trial, plasma exchange (discard) removes the plasma of AD patients, which contains Aβ–albumin complexes, and a new albumin solution is introduced into the blood as a replacement solution; the results thus far suggest that this therapy has improved cognitive function in AD subjects. The Phase 3 trial is now also underway (Boada et al. 2016).
Based on these observations, the removal of Aβ from the blood could act as peripheral drainage and an Aβ sink from the brain. We proposed that the E-BARS, which transfers Aβ out of the body, may be useful as a therapy for AD (Kawaguchi et al. 2010) (Fig. 10.1). The rapid reduction of Aβ concentrations in the blood could act as a trigger to enhance Aβ excretion from the brain, resulting in cognitive improvement.
10.4 Definition of Aβ Removal Activities of the Devices
The Aβ removal activities assessed in our study were: (1) the removal rate for batch analysis in vitro, (2–1) the removal efficiency based on the concentration change at pre-/post-application of the Aβ removal device, (2–2) the reduction rate of Aβ in the whole blood circulation, and (2–3) the filtration rate. The definitions were as follows:
-
1.
Batch analysis in vitro:
Adsorptive materials were mixed with Aβ solutions or plasma and shaken for the designated time.
-
2.
Flow analysis in vitro and the hemodialysis session
-
2-1
The Aβ removal efficiency of a dialyzer was defined as follows:
-
2-1
\( {\displaystyle \begin{array}{l}\mathrm{Removal}\ \mathrm{efficiency}\left(\%\right)=\\ {}100\times \\ {}\left\{\ 1-\frac{\ \mathrm{concentration}\ \mathrm{of}\ A\beta\ \mathrm{a}\mathrm{fter}\ \mathrm{leaving}\ \mathrm{the}\ \mathrm{dialyzer}\ \left(\mathrm{device}\right)\ \mathrm{a}\mathrm{t}\ \mathrm{a}\ \mathrm{designated}\ \mathrm{time}\ }{\mathrm{concentration}\ \mathrm{of}\ A\beta\ \mathrm{before}\ \mathrm{entering}\ \mathrm{the}\ \mathrm{dialyzer}\ \left(\mathrm{device}\right)\mathrm{at}\ \mathrm{t}\mathrm{hat}\ \mathrm{time}}\right\}\end{array}} \)
-
2-2
The Aβ reduction rate for the experimental pool solution or the whole blood circulation was defined as follows:
\( {\displaystyle \begin{array}{l}\mathrm{Reduction}\ \mathrm{rate}\left(\%\right)=\\ {}100\times \left\{\ 1-\frac{A\beta\ \mathrm{concentration}\ \mathrm{in}\ \mathrm{the}\ \mathrm{pool}\ \mathrm{solution}\ \mathrm{or}\ \mathrm{whole}\ \mathrm{blood}\ \mathrm{circulation}\ \mathrm{a}\mathrm{t}\ \mathrm{a}\ \mathrm{designated}\ \mathrm{time}}{\mathrm{Initial}\ A\beta\ \mathrm{concentration}\ \mathrm{in}\ \mathrm{the}\ \mathrm{pool}\ \mathrm{solution}\ \mathrm{or}\ \mathrm{whole}\ \mathrm{blood}\ \mathrm{circulation}}\right\}\end{array}} \)
-
2-3
The Aβ filtration rate of a dialyzer was defined as follows:
\( {\displaystyle \begin{array}{l}\mathrm{Filtration}\ \mathrm{rate}\left(\%\right)=\\ {}100\times \left\{\ \frac{\ \mathrm{concentration}\ \mathrm{of}\ \mathrm{filtrated}\ A\beta\ \mathrm{solution}\ \mathrm{at}\ \mathrm{the}\ \mathrm{designated}\ \mathrm{time}\ }{\mathrm{concentration}\ \mathrm{of}\ A\beta\ \mathrm{before}\ \mathrm{the}\ \mathrm{dialyzer}\ \mathrm{at}\ \mathrm{the}\ \mathrm{same}\ \mathrm{time}}\right\}\end{array}} \)
10.5 Adsorption Devices for Blood Aβ Removal
To obtain suitable materials for the removal of blood Aβ, we firstly investigated adsorptive materials for therapeutic blood purification (apheresis). We employed six materials: hexadecyl-alkylated cellulose particles (HDC), used to remove β2-microglobulin in carpal tunnel syndrome; cellulose particles ligated with dextran sulfate (CLD); charcoal (CHA), which is commonly used therapeutically, for example, in hepatic failure; tryptophan-ligated polyvinyl alcohol gel (TRV), used in Guillain–Barré syndrome; and cellulose acetate particles and non-woven polyethylene terephthalate filter, used in ulcerative colitis. Among these materials, HDC and CHA demonstrated a removal rate of almost 99% for both Aβ1–40 and Aβ1–42 in batch analysis using synthetic Aβ peptides (Fig. 10.2) (Kawaguchi et al. 2010).

Aβ removal rate in batch analysis with various adsorptives in a batch reaction for 16 h. HDC hexadecyl-alkylated cellulose particles, CHA charcoal, TRV tryptophan-ligated polyvinyl alcohol gel, CAP cellulose acetate particles, CLD cellulose particles ligated with dextran sulfate, NPT non-woven polyethylene terephthalate filter. HDC and CHA showed significantly higher rates than TRV (p < 0.05) for Aβ1–40 removal and a higher tendency than CAP (p < 0.1) for Aβ1–42 removal. (Taken from Kawaguchi et al. 2010)
HDC is used in cases where there are complications associated with hemodialysis and, therefore, we were able to investigate Aβ concentrations before (pre, inlet of) and after (post, outlet of) HDC column in hemodialysis sessions. The high removal efficiency of HDC was maintained at approximately 50% for both Aβ1–40 and Aβ1–42 during a 4-h hemodialysis session, as shown in Table 10.1.
10.6 Blood Aβ Removal by Hemodialyzers in Hemodialysis
We previously reported that hemodialyzers showed high Aβ removal activity based on analyses of hemodialysis patients (Kitaguchi et al. 2011, 2015; Kato et al. 2012). Measurements of Aβ concentrations at pre (inlet of) and post (outlet of) dialyzers during hemodialysis sessions revealed that the hemodialyzers effectively removed both Aβ1–40 and Aβ1–42 from the plasma of non-diabetic patients. Figure 10.3 shows the Aβ concentrations at the inlet of the dialyzers (Pre) and the outlet of the dialyzers (Post) for each dialysis session (n = 57). The average removal efficiencies for Aβ1–40 were 66.0% at the 1-h point and 52.0% at the 4-h point of the hemodialysis sessions. Those for Aβ1–42 were 61.1% and 49.2%, as shown in Fig. 10.3. The removal efficiency in for Aβ1–40 was significantly higher than for Aβ1–42 both at 1 h and at 4 h of each dialysis session (p < 0.0001 for both time points). Each dialyzer maintained its removal efficiency during the entire dialysis session. This indicates that the dialyzers had sufficient capacity for Aβ removal during the 4-h treatment.

Aβ concentrations measured at pre-/post-dialyzers at 1 and 4 h in the hemodialysis sessions. Aβ removal efficiencies for both Aβ1–40 and Aβ1–42 were quite high, with both being approximately 50% or greater. (a, b) Aβ1–40.; (c, d) Aβ1–42; (a, c) at the 1-h point of the dialysis sessions; (b, d) at the 4-h point of the dialysis sessions. (Taken from Kato et al. 2012 and modified)
10.7 Removal of Blood Aβs Evoked Influx of Aβs into the Blood
Due to the effective removal activity of the dialyzers during the hemodialysis sessions (Fig. 10.3), the concentrations of blood Aβs after 4-h hemodialysis would have been approximately 10% of the concentrations at the starting point if there had been no Aβ influx into the blood (“Calcd” in Fig. 10.4). However, observed concentrations of blood Aβs (“Obsd” in Fig. 10.4) were not decreased compared to “Calcd.” The differences between “Obsd” and “Calcd” were attributed to Aβ influx into the blood. We calculated the influx based on the differential equation described previously (Kitaguchi et al. 2011). The results of this simulation of 37 non-diabetic hemodialysis patients are shown in Fig. 10.4.

Change in the observed plasma Aβ concentrations in the whole body circulation during hemodialysis sessions (Obsd), and, the calculated plasma Aβ concentrations based on the Aβ removal efficiencies of the dialyzers assuming no Aβ influx into the blood (Calcd). The arrows indicate Aβ influx during the hemodialysis sessions. (Taken from Kitaguchi et al. 2011 and modified)
Table 10.2 shows more detailed results of the simulation of Aβ influx with 30 non-diabetic hemodialysis patients (Kitaguchi et al. 2015). The average removal efficiencies at the 1-hr point of the hemodialysis sessions were 67.3% and 51.3% for Aβ1–40 and Aβ1–42, respectively. Aβ influxes during 4-hr hemodialysis were calculated as 9243 ng and 719 ng for Aβ1–40 and Aβ1–42, respectively, which were around five times the level of pre-existing Aβs in the blood, that is, 1952 ng and 165 ng, just before hemodialysis.
A similar Aβ influx into the blood was also observed in a rat study using HDC.
10.8 Are the Influxes of Aβs into the Blood from the Brain?
Recently, we reported that Aβ accumulation in the brains of hemodialysis (HD) patients was significantly lower than that in age-matched non-hemodialysis controls, as assessed by histopathological studies (Sakai et al. 2016). Senile plaques stained with anti-Aβ antibodies were observed more frequently in non-HD subjects and were either sparse or not seen at all in HD patients (Fig. 10.5). Regarding the ratio of senile plaques (plaque-positive/-negative subjects), there were significantly fewer neuritic and cored plaques in HD patients; only 5 of 17 HD patients showed neuritic plaques stained with 4G8 anti- Aβ antibody, whereas 12 out of 16 non-HD subjects exhibited these plaques. These findings suggest that the brain may be one origin of the Aβ influx during the hemodialysis sessions.

Comparison of senile plaques in patients who had undergone hemodialysis (HD) with those who had not undergone HD (non-HD). (a) Stained with the anti-Aβ17–24 antibody 4G8; (b) stained with the anti-Aβ1–16 antibody DE2. The numbers of all types of Aβ deposition (diffuse, cored, and neuritic plaques) were significantly lower in HD patients. HD, n = 17; non-HD, n = 16. (Taken from Sakai et al. 2016 and modified)
10.9 Effects of Hemodialysis, One of the Blood Aβ Removal Methods, on Cognitive Function
Renal failure is well known to cause cognitive decline. In our cross-sectional study, cognitive function as measured by the MMSE was impaired in renal failure patients who did not receive hemodialysis compared to age-matched healthy controls. However, MMSE scores of hemodialysis patients were similar to those of controls (Fig. 10.6) (Kato et al. 2012).

Cognitive function deteriorated in renal failure; however, hemodialysis appeared to promote recovery or maintenance of this. AMC, age-matched healthy controls (n = 17) (66.6 ± 4.1 years old, 5 male, 12 female); non-HDRF, renal failure patients without hemodialysis (n = 26) (66.6 ± 14.7 years old, 18 male, 8 female); HDRF, renal failure patients who received hemodialysis three times a week (n = 57) (69.4 ± 3.8 years old, 29 male, 28 female). MMSE Mini-Mental State Examination. (Taken from Kato et al. 2012)
Figure 10.7 shows the relationship between plasma Aβ concentrations, cognitive function, renal function, and hemodialysis vintage (the duration of hemodialysis) before and after initiation of hemodialysis. Before initiation of hemodialysis, plasma concentrations of both Aβ1–40 and Aβ1–42 increased along with a concomitant decline in renal function. However, when patients were introduced to hemodialysis (after initiation of hemodialysis), an increase in plasma Aβ concentrations was no longer apparent, but there was instead a slight tendency toward a decrease. Although the cognitive function declined along with the decline in renal function, this was maintained following initiation of hemodialysis (bottom of Fig. 10.7).

Summary of cross-sectional study of renal failure patients before/after initiation of hemodialysis (HD). The central box indicates initiation of hemodialysis. Left of the central box, data from renal failure patients without hemodialysis (non-HDRF) are shown. Right of the central box, data from hemodialysis patients (with-HDRF) are shown. Vertical axis: upper, plasma Aβ1–40 concentrations; middle, plasma Aβ1–42 concentrations; lower, the Mini-Mental State Examination (MMSE) score (30 indicates no mistakes). Plasma for measuring Aβ concentrations after the initiation of hemodialysis was sampled at the beginning of each hemodialysis session. Horizontal axis: before initiation of hemodialysis, plasma creatinine concentrations (CRN), which indicate decline of renal function; after initiation of hemodialysis, the vintage (duration) of hemodialysis. (Data from Kato et al. 2012)
In the prospective study with 18 and 36 months follow-up, average MMSE scores did not significantly change, as shown in Fig. 10.8a, b. However, analysis of the change in individual subjects revealed that most hemodialysis patients maintained or improved their cognitive function, with the exception of patients that showed white matter ischemia at baseline (Fig. 10.8c). This suggests that hemodialysis, with Aβ removal from the blood three times a week, may have a positive effect on cognitive function but has almost no influence on the cognitive effects of brain ischemia.

Change in cognitive function of hemodialysis patients in prospective studies. (a) Mini-Mental State Examination (MMSE) changes over 18 months; (b) MMSE changes over 36 months; (c) change in MMSE from baseline for each patient. A change of −1 to 4 is regarded as maintained or improved. Patients whose MMSE declined by −4 and −5 showed white matter ischemia at baseline. (Taken from Kitaguchi et al. 2015 and modified)
Furthermore, using a database of over 200,000 hemodialysis patients in Japan, the risk of dementia was revealed to be significantly lower in the patient subgroup with a longer duration of hemodialysis in subjects without diabetes (Nakai et al. 2018).
10.10 Effects of Smoking on Removal of Blood Aβ
We then investigated the effects of smoking on Aβ removal efficiencies in hemodialysis. Subjects were non-diabetic hemodialysis patients; n = 57, 29 male and 28 female; age, 69.4 ± 3.8 years old (59–76 years old); duration of hemodialysis, 13.9 ± 9.4 years (1–37 years); 28 smokers and 29 non-smokers, with “smoker” defined as a patient who had ever smoked (former smokers and current smokers). Information regarding the duration of smoking, the number of cigarettes per day, and the brands of cigarettes were obtained by interview with each patient. The product of the duration and the number of cigarettes per day was also used for analysis.
Interestingly, removal efficiencies for both Aβ1–40 and Aβ1–42 in smokers significantly decreased during the 4-h hemodialysis sessions (Table 10.3). The efficiencies for non-smokers showed a tendency to increase, which was insignificant, rather than a decrease. The reason for this difference is unclear at present. One possibility is that Aβ species in the blood of smokers may have certain characteristics that cause saturation of Aβ adsorption or clogging of the inner surface of dialyzer membranes. A second possibility is that Aβ species flowing into the blood during hemodialysis may be more difficult to remove using a dialyzer in smokers than in non-smokers.
However, there is a limitation regarding this speculation on the effects of smoking. The ratio of male/female subjects was higher in smokers than in non-smokers. Therefore, the differences between smokers and non-smokers could be partially attributable to gender.
10.11 Effects of Smoking on Cognitive Function and Brain Atrophy in Renal Failure Patients
Figure 10.9 indicates that there appears to be no clear difference between the smoker and non-smoker cognitive function, as measured by the MMSE, in our study with a small sample size. The MMSE scores of smokers were similar to those of non-smokers in all three groups; age-matched healthy controls (AMC, seven smokers, ten non-smokers), renal failure patients who did not need hemodialysis (non-HDRF, seven smokers, seven non-smokers), and severe renal failure patients who received hemodialysis three times a week (HDRF, 28 smokers, 29 non-smokers).

The cognitive function of smokers and non-smokers was similar in our study. The patients were the same as those represented in Fig. 10.6 except that smoking history was obtained from only 16 non-HDRF patients. AMC age-matched healthy controls (seven smokers, ten non-smokers), non-HDRF renal failure patients without hemodialysis (seven smokers, nine non-smokers), HDRF severe renal failure patients who received hemodialysis three times a week (28 smokers, 29 non-smokers). MMSE Mini-Mental State Examination
However, brain CT scans revealed that there were differences in brain atrophy between smokers and non-smokers. Frontal/temporal and temporal/parietal atrophies were more severe in smokers than in non-smokers, as shown in Fig. 10.10 (p = 0.0465 and p = 0.0062, respectively, by the χ2 test). This suggests that the effects of smoking on the brain may not be sufficiently serious to affect cognitive function in our study, or that hemodialysis including Aβ removal from the blood three times a week may maintain cognitive function despite the presence of more severe atrophies in smokers.

Brain atrophy in smokers and non-smokers. Frontal/temporal atrophy and temporal/parietal atrophy was more severe in smokers than in non-smokers, as detected by brain CT scans (p = 0.0465 and p = 0.0062, respectively, by the χ2 test). (Taken from Kitaguchi et al. 2015)
10.12 Closing
As described above, removal of blood Aβ may enhance Aβ influx into the blood from the brain, resulting in maintenance or improvement of cognitive function. We believe that the E-BARS could contribute as a therapy for Alzheimer’s disease. With respect to smoking, the patient’s history in this regard may have some effect on brain atrophy and on the forms of Aβs existing in the blood. Additional study will be necessary in the future to further clarify this.
References
Anstey KJ, von Sanden C, Salim A, O’Kearney R (2007) Smoking as a risk factor for dementia and cognitive decline: a meta-analysis of prospective studies. Am J Epidemiol 166:367–378
Barnes DE, Yaffe K (2011) The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol 10:819–828
Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV (2007) Transport pathways for clearance of human Alzheimer’s amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab 27:909–918
Boada M, Ortiz P, Anaya F, Hernández I, Muñoz J, Núñez L, Olazarán J, Roca I, Cuberas G, Tárraga L, Buendia M, Pla RP, Ferrer I, Páez A (2009) Amyloid-targeted therapeutics in Alzheimer’s disease: use of human albumin in plasma exchange as a novel approach for Abeta mobilization. Drug News Perspect 22:325–239
Boada M, Ramos-Fernández E, Guivernau B, Muñoz FJ, Costa M, Ortiz AM, Jorquera JI, Núñez L, Torres M, Páez A (2016) Treatment of Alzheimer disease using combination therapy with plasma exchange and haemapheresis with albumin and intravenous immunoglobulin: rationale and treatment approach of the AMBAR (Alzheimer Management By Albumin Replacement) study. Neurologia 31:473–481
DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM (2001) Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 98:8850–8855
Donahue JE, Flaherty SL, Johanson CE, Duncan JA 3rd, Silverberg GD, Miller MC, Tavares R, Yang W, Wu Q, Sabo E, Hovanesian V, Stopa EG (2006) RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol 4:405–415
García AM, Ramón-Bou N, Porta M (2010) The effects of tobacco exposure before the age of onset of AD was investigated as a case-control study. Isolated and joint effects of tobacco and alcohol consumption on risk of Alzheimer’s disease. J Alzheimers Dis 20:577–586
Hellström-Lindahl E, Mousavi M, Ravid R, Nordberg A (2004) Reduced levels of Abeta 40 and Abeta 42 in brains of smoking controls and Alzheimer’s patients. Neurobiol Dis 15:351–360
Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Müller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, de Quervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM (2003) Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron 38:547–554
Hung LW, Ciccotosto GD, Giannakis E, Tew DJ, Perez K, Masters CL, Cappai R, Wade JD, Barnham KJ (2008) Amyloid-b peptide (Ab) neurotoxicity is modulated by the rate of peptide aggregation: Ab dimers and trimers correlate with neurotoxicity. J Neurosci 28:11950–11958
Kato M, Kawaguchi K, Nakai S, Murakami K, Hori H, Ohashi A, Hiki Y, Ito S, Shimano Y, Suzuki N, Sugiyama S, Ogawa H, Kusimoto H, Mutoh T, Yuzawa Y, Kitaguchi N (2012) Potential therapeutic system for Alzheimer’s disease: removal of blood Abs by hemodialyzers and its effect on the cognitive functions of renal-failure patients. J Neural Transm 119:1533–1544
Kawaguchi K, Kitaguchi N, Nakai S, Murakami K, Asakura K, Mutoh T, Fujita Y, Sugiyama S (2010) Novel therapeutic approach for Alzheimer’s disease by removing amyloid-β protein from the brain with an extracorporeal removal system. J Artif Organs 13:31–37
Kawaguchi K, Saigusa A, Yamada S, Gotoh T, Nakai S, Hiki Y, Hasegawa M, Yuzawa Y, Kitaguchi N (2016) Toward the treatment for Alzheimer’s disease: adsorption is primary mechanism of removing amyloid β protein with hollow-fiber dialyzers of the suitable materials, Polysulfone and polymethyl methacrylate. J Artif Organs 19:149–158
Kihara T, Shimohama S, Sawada H, Kimura J, Kume T, Kochiyama H, Maeda T, Akaike A (1997) Nicotinic receptor stimulation protects neurons against beta-amyloid toxicity. Ann Neurol 42:159–163
Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, Kume T, Akaike A (2001) Alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J Biol Chem 276:13541–13546
Kitaguchi N, Kawaguchi K, Nakai S, Murakami K, Ito S, Hoshino H, Hori H, Ohashi A, Shimano Y, Suzuki N, Yuzawa Y, Mutoh T, Sugiyama S (2011) Reduction of Alzheimer’s disease amyloid-β in plasma by hemodialysis and its relation to cognitive functions. Blood Purif 32:57–62
Kitaguchi N, Hasegawa M, Ito S, Kawaguchi K, Hiki Y, Nakai S, Suzuki N, Shimano Y, Ishida O, Kushimoto H, Kato M, Koide S, Kanayama K, Kato T, Ito K, Takahashi H, Mutoh T, Sugiyama S, Yuzawa Y (2015) A prospective study on blood Aβ levels and the cognitive function of patients with hemodialysis: a potential therapeutic strategy for Alzheimer’s disease. J Neural Transm 122:1593–1607
Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, Ball MJ, Roher AE (1996) Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem 271:4077–4081
Lopez OL, Kuller LH, Mehta PD, Becker JT, Gach HM, Sweet RA, Chang YF, Tracy R, DeKosky ST (2008) Plasma amyloid levels and the risk of AD in normal subjects in the Cardiovascular Health Study. Neurology 70:1664–1671
Matsumura A, Suzuki S, Iwahara N, Hisahara S, Kawamata J, Suzuki H, Yamauchi A, Takata K, Kitamura Y, Shimohama S (2015) Temporal changes of CD68 and α7 nicotinic acetylcholine receptor expression in microglia in Alzheimer’s disease-like mouse models. J Alzheimers Dis 44:409–423
Matsuoka Y, Saito M, LaFrancois J, Saito M, Gaynor K, Olm V, Wang L, Casey E, Lu Y, Shiratori C, Lemere C, Duff K (2003) Novel therapeutic approach for the treatment of Alzheimer’s disease by peripheral administration of agents with an affinity to β-Amyloid. J Neurosci 23:29–33
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ (2010) Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330:1774
Moreno-Gonzalez I, Estrada LD, Sanchez-Mejias E, Soto C (2013) Smoking exacerbates amyloid pathology in a mouse model of Alzheimer’s disease. Nat Commun 4:1495
Morris AWJ, Carare RO, Schreiber S, Hawkes CA (2014) The cerebrovascular basement membrane: role in the clearance of β-amyloid and cerebral amyloid angiopathy. Front Aging Neurosci 6:1–9
Nakai S, Wakai K, Kanda E, Kawaguchi K, Sakai K, Kitaguchi N (2018) Is hemodialysis itself a risk factor for dementia? An analysis of nationwide registry data of patients on maintenance hemodialysis in Japan. Ren Replace Ther 4:12. https://doi.org/10.1186/s41100-018-0154-y
Ott A, Slooter AJ, Hofman A, van Harskamp F, Witteman JC, Van Broeckhoven C, van Duijn CM, Breteler MM (1998) Smoking and risk of dementia and Alzheimer’s disease in a population-based cohort study: the Rotterdam Study. Lancet 351:1840–1843
Sabia S, Marmot M, Dufouil C, Singh-Manoux A (2008) Smoking history and cognitive function in middle age from the Whitehall II study. Arch Intern Med 168:1165–1173
Sakai K, Senda T, Hata R, Kuroda M, Hasegawa M, Kato M, Abe M, Kawaguchi K, Nakai S, Hiki Y, Yuzawa Y, Kitaguchi N (2016) Patients that have undergone hemodialysis exhibit lower amyloid deposition in the brain: evidence supporting a therapeutic strategy for Alzheimer’s disease by removal of blood amyloid. J Alzheimers Dis 51:997–1002
Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P (1999) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400:173–177
Schoonenboom NS, Mulder C, Van Kamp GJ, Mehta SP, Scheltens P, Blankenstein MA, Mehta PD (2005) Amyloid beta 38, 40, and 42 species in cerebrospinal fluid: more of the same? Ann Neurol 58:139–142
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiolo Rev 81:741–766
Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM, Sandrock A (2016) The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537:50–56
Silverberg GD, Miller MC, Messier AA, Majmudar S, Machan JT, Donahue JE, Stopa EG, Johanson CE (2010) Amyloid deposition and influx transporter expression at the blood-brain barrier increase in normal aging. J Neuropathol Exp Neurol 69:98–108
Takata K, Kitamura Y, Saeki M, Terada M, Kagitani S, Kitamura R, Fujikawa Y, Maelicke A, Tomimoto H, Taniguchi T, Shimohama S (2010) Galantamine-induced amyloid-{beta} clearance mediated via stimulation of microglial nicotinic acetylcholine receptors. J Biol Chem 285:40180–40191
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid b protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416:535–539
Wang HY, Lee DHS, D’Andrea MR, Peterson PA, Shank RP, Reitz AB (2000) Beta-amyloid(1–42) binds to α7 nicotinic acetylcholine receptor with high affinity—implications for Alzheimer’s disease pathology. J Biol Chem 275:5626–5632
Yang WN, Ma KG, Chen XL, Shi LL, Bu G, Hu XD, Han H, Liu Y, Qian YH (2014) Mitogen-activated protein kinase signaling pathways are involved in regulating α7 nicotinic acetylcholine receptor-mediated amyloid-β uptake in SH-SY5Y cells. Neuroscience 278:276–290
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
This chapter is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
Copyright information
© 2018 The Author(s)
About this chapter

Cite this chapter
Kitaguchi, N., Kawaguchi, K., Sakai, K. (2018). Removal of Blood Amyloid As a Therapeutic Strategy for Alzheimer’s Disease: The Influence of Smoking and Nicotine. In: Akaike, A., Shimohama, S., Misu, Y. (eds) Nicotinic Acetylcholine Receptor Signaling in Neuroprotection. Springer, Singapore. https://doi.org/10.1007/978-981-10-8488-1_10
Download citation
DOI: https://doi.org/10.1007/978-981-10-8488-1_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-8487-4
Online ISBN: 978-981-10-8488-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)