
Abstract
Recent advances of genomic technologies have opened unprecedented possibilities in building high-quality ultra-dense genetic maps. However, with very large numbers of markers available for a mapping population, most of the markers will remain inseparable by recombination. Real situations are also complicated by genotyping errors, which “diversify” a certain part of the markers that would be identical in error-free situations. The higher the error rate the more difficult is the problem of building a reliable map. In our algorithm, we assume that error-free markers can be selected based on the presence of “twins”. There is also a probability of an opposite effect, when non-identical markers may become “twins” because of genotyping errors. Thus, a certain threshold is introduced for the selection of markers with a sufficient number of twins. The developed algorithm (implemented in MultiPoint software) enables mapping big sets of markers (~105–106). Unlike some other algorithms used in building ultra-dense genetic maps, the proposed “twins” approach does not need any prior information (e.g., anchor markers), and hence can be applied to genetically poorly studied organisms.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Recent advances of genomic technologies have opened unprecedented possibilities in building high-quality ultra-dense genetic maps. However, with very large numbers of markers available for a mapping population, most of the markers will remain inseparable by recombination and will represent groups of co-segregating, or absolutely linked markers (AL markers). In such cases, only one marker from each group could be placed on the map that can be referred to as a framework, skeleton, or bin map; the remaining markers can then be attached to the skeleton map (Mester et al. 2003; Korol et al. 2009; Ronin et al. 2010). The real situation is significantly complicated by genotyping errors, which “diversify” a certain part of markers that would be identical in the ideal situation of no errors. The higher the error rate and the number of markers, the more difficult it is to build a reliable map (Buetow 1991). An additional complication is when a part of data points is missing, which is common in the genotyping-by-sequencing (GBS) approach and cannot always be compensated for by the imputation of missing scores.
Several approaches have been suggested for constructing high-density genetic maps aimed at overcoming the aforementioned difficulties. The dominating strategy includes various ways of building hierarchical framework maps (Isidore et al. 2003), e.g. by combining the irresolvable markers of a linkage group into bins (groups of “bound together markers”) in the first phase followed by joint ordering of the representatives of these groups and singleton markers. Our approach to the ordering problem is based on reducing it to the traveler salesperson problem (TSP) and employing Guided Evolutionary Strategy heuristics for building the framework or skeleton map (Mester et al. 2003, 2010; Ronin et al. 2010). An interesting alternative possibility of phasing the mapping analysis is by constructing a minimum spanning tree of a graph followed by improvement of the initial solution based on TSP-inspired heuristics (Wu et al. 2008). For situations of ultra-dense mapping, with thousands and dozens of thousands of markers per chromosome “contaminated” by typing errors, we propose a simple “twins” approach for selecting reliable skeletal markers. Combined with our powerful discrete optimization heuristics, this approach enables the mapping of very big sets of markers (e.g. 105), i.e., suitable to wheat genotyping with the 90 K iSelect chip as well as with the GBS approach. The corresponding algorithms implemented in MultiPoint software were intensively tested using simulated data and a set of 420,000 SNP and GBS markers of a wheat DH population.
Geometry of Genotyping Space in the Presence of Marker Typing Errors
The sample size (N) of mapping populations limits the marker density in the map. Thus, for a DH population with N = 200, the minimal non-zero recombination rate between two adjacent markers cannot be less than 0.5 %. In the absence of errors, all markers should appear in AL groups, with the distance between the groups ≥0.5 cM. Typing errors will lead to the erosion of these groups into “clouds” of falsely different markers. Figure 14.1 illustrates the formation of such a cloud from a set L of 11 AL markers in a multi-dimensional space of markers scored for a sub-sample of 16 individuals from the mapping population. In an ideal error-free situation, all 11 markers would vary identically across the shown 16 genotypes: in the 16-dimensional space these markers are in the same state (aababbbaaaabbaba) and belong to the set L (represented as dots within the grey circle). Due to typing errors, some of the markers change their 16-dimensional states and leave the set L (white holes); corresponding genotypes will be erroneously recorded as “recombinants”. The problem is how to select markers for building a reliable genetic map in a challenging situation when the data set includes thousands of markers per chromosome while a certain proportion of markers are contaminated by erroneous data points and a part of the data points are missing.

A geometric model of erosion of AL marker groups due to scoring errors (only 11 markers scored for 16 genotypes are shown)
The Proposed Method and Algorithm
We propose a method of addressing these problems based on a simple idea that with very large numbers of scored markers (e.g., thousands or dozens of thousands per chromosome) and small-to-moderate population size, many markers will be irresolvable by recombination and should appear as groups of AL markers. But some of AL markers will appear as “recombinants” if even a small proportion of scores per marker are erroneous. Thus, we can trust more markers from groups of absolutely linked markers compared to singleton markers. For sample size N and a proportion of genotyping errors p per marker, the probability that in all individuals both alleles of a marker will be unmistakably identified can be estimated under the assumption that the typing errors are independent, as P = (1-p)N ≈ e −Np. Assuming 1 % error rate within a group of AL markers, about a third will still remain error-free. In a DH population of N = 100 individuals, for a chromosome length of 100 cM the minimum interval length will be 1 cM. Consequently, the density of the map cannot be greater than 101 markers. If we genotyped 10,000 markers of this chromosome, only 100 markers (referred to as skeletal markers) can be ordered, whereas the rest will remain absolutely linked to the skeletal markers. Thus, for building a skeleton map one can select presumably error-free markers based on the presence of “twins” in the sample, although there is also a small probability that non-identical markers may become “twins” because of genotyping errors. Therefore, a certain threshold is introduced in our algorithm for the selection of markers with a sufficient number of twins. In regions with a lower density of recombination events (e.g., affected by the centromeric effect on recombination), the map will be less affected by typing errors.
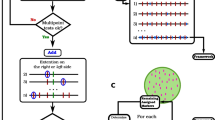
The major steps in our algorithm for building ultra-dense genetic maps (Fig. 14.2) implemented in MultiPoint software (www.multiqtl.com) include: (a) Forming groups of markers with zero distance and selecting a “delegate” from each group containing no fewer twins than the predefined threshold (equal 3 in Fig. 14.2); (b) Except for twins of the candidates, all remaining markers are removed to the Heap; (c) Clustering the delegate markers and ordering the obtained linkage groups (LG); (d) Filling gaps and extending LG ends using markers from Heap; (e) Removal of markers violating map stability and monotonic growth of distance from a marker and its subsequent neighbors.

Scheme of the “twin” algorithm. Illustrated is the marker information flow in the process of map construction
Results and Discussion
Various algorithms have been proposed for building dense genetic maps, including the stepwise increase of the map density (Jansen et al. 2001; Isidore et al. 2003; Mester et al. 2003, 2010; Wu et al. 2008). This problem becomes especially challenging with the current widespread transition from a few hundred to tens or even hundreds of thousands of typed markers per genome. It is well recognized that in such a reality even 1 % of typing errors may lead to a dramatic reduction of map quality, i.e., “more” (markers) may imply “less” (confidence in map quality, at least on a microscale). The problem includes a few aspects: (i) computational complexity, related to the exponential growth of the number of potential marker orders to be tested, (ii) the impossibility to resolve the vast majority of markers by recombination under reasonable population sizes, and (iii) high impact of typing errors on map quality. Our approach is based on the assumption that upon high excess of irresolvable compared to resolvable markers and a low level of typing errors, members of “twin” groups with minimum missing scores can be considered as more credible markers compared to singleton markers.
For an illustration of the efficiency of our “twins” approach, two examples are provided here: simulated data for one chromosome with 10,000 markers for a DH population with N = 200 (two variants of the same marker set were considered, with and without marker typing errors), and real DH data on ~24,000 markers of wheat chromosome 3B (the whole genome set included ~420,000 markers). In the first example, the map length was 212 cM. For error-free data, the skeleton map included 197 markers. For data with 1 % typing errors, about 1/8 of the markers appear as AL groups, while 7/8 of the markers appear as clouds surrounding AL groups, as explained in Fig. 14.1 and illustrated by Fig. 14.3 (grey dots). Figure 14.3 illustrates the distribution of markers with errors relative to the skeleton map (when it is known, as with simulated data).

The structure of clouds with markers with scoring errors
The analysis of simulated data with 1 % errors (Table 14.1) demonstrates how a meaningful map can be obtained for such data when nothing is known about the order of markers, which is a standard situation with non-model species. Obviously, the result may depend on the threshold size of the AL groups to be represented in the skeleton map. Thus, with threshold = 4, AL groups with two and three markers are excluded from consideration together with singletons (moved to heap) and the first variant of the skeleton map is constructed (stage 1 of the procedure). Stage 2 is cleaning the map. MultiPoint package enables the detection and removal of markers violating the order stability and monotonic growth of distances in the skeleton map (Ronin et al. 2010). After cleaning, markers from the heap can be checked as candidates for filling in the gaps (if gaps are present in the obtained skeleton map). The results in Table 14.1 show a relatively weak dependence on the arbitrarily selected threshold of the AL group size and very good correspondence between the map characteristics (the number of skeletal markers and length of the map) obtained under zero and 1 % marker typing errors. Clearly, each of the remaining >9,800 markers can be attached to the corresponding interval or marker on the skeleton map. Figure 14.4 shows the skeleton map of the second example, on wheat chromosome 3B (DH population, the total set included ~420,000 markers).

Map of wheat chromosome 3B, the largest in the wheat genome (the figure is split into two parts to fit the page size limits)
References
Buetow KH (1991) Influence of aberrant observations on high resolution linkage analysis outcomes. Am J Hum Genet 49:985–994
Isidore E, van Os H, Andrzejewski S et al (2003) Toward a marker-dense meiotic map of the potato genome: lessons from linkage group I. Genetics 165:2107–2116
Jansen J, de Jong AG, van Ooijen JW (2001) Constructing dense genetic linkage maps. Theor Appl Genet 102:1113–1122
Korol AB, Mester D, Frenkel Z, Ronin Y (2009) Methods for genetic analysis in the Triticeae. In: Feuillet C, Muehlbauer GJ (eds) Genetics and genomics of the Triticeae. Springer, Berlin, pp 163–199
Mester D, Ronin Y, Minkov D et al (2003) Constructing large scale genetic maps using an evolutionary strategy algorithm. Genetics 165:2269–2282
Mester D, Ronin Y, Korostishevsky M, et al (2010) Discrete optimization for some TSP-like genome mapping problems. In: Varela J, Acuna S (eds) Handbook of optimization theory. Nova Science, New York, pp 1-40
Ronin Y, Mester D, Minkov D, Korol AB (2010) Building reliable genetic maps: different mapping strategies may result in different maps. Nat Sci 2:576–589
Wu Y, Bhat PR, Close TJ, Lonardi S (2008) Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PLoS Genet 4, e1000212
Acknowledgments
This work was supported by the Israel Science Foundation (ISF grant #800/10), Binational Agricultural Research and Development Fund (BARD grant #3873/06), MultiQTL Ltd, and the Israeli Ministry of Absorption.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this paper
Cite this paper
Ronin, Y., Minkov, D., Mester, D., Akhunov, E., Korol, A. (2015). Building Ultra-Dense Genetic Maps in the Presence of Genotyping Errors and Missing Data. In: Ogihara, Y., Takumi, S., Handa, H. (eds) Advances in Wheat Genetics: From Genome to Field. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55675-6_14
Download citation
DOI: https://doi.org/10.1007/978-4-431-55675-6_14
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55674-9
Online ISBN: 978-4-431-55675-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)