
Abstract
The Hippo pathway has the unique capacity to sense tissue architecture and the external forces that shape it, and dysregulation of this pathway leads to tumorigenesis. The study of mice bearing systemic or tissue-specific mutations of Hippo elements has driven huge progress in understanding this pathway’s role in normal physiology and disease. Here, we summarize how disruption of Hippo signaling relates to cancer, and we highlight the importance of this pathway as a new drug target for malignant tumors.
The authors declare that no conflicts of interest exist.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others
Keywords
Introduction
The Hippo signaling pathway was first identified as a regulator of organ size in Drosophila [1]. In mammals, canonical Hippo signaling is mediated by mammalian sterile 20-like (MST) kinases, large tumor suppressor (LATS) kinases, the adaptor proteins Salvador homolog 1 (SAV1) and Mps one binder kinase activator protein (MOB1), and the downstream transcription cofactors Yes-associated protein 1 (YAP1) and its paralog, transcriptional coactivator with PDZ-binding motif (TAZ).
In a cell exposed to cell–cell contact, mechanical force, or a stress stimulus, MST phosphorylates LATS in a reaction facilitated by SAV1 and MOB1. LATS in turn phosphorylates YAP1 (S127)/TAZ (S89), which then binds to 14-3-3 protein. This binding prevents phosphorylated YAP1/TAZ from accessing the nucleus and activating the transcription of a broad range of growth factors and anti-apoptotic genes (Fig. 1). Additional control is exerted by the binding of phosphorylated YAP1(S397)/TAZ(S311) to the E3 ligase complex SCFβ-TrCP, which facilitates YAP1/TAZ ubiquitination and proteasomal degradation [2–5] (Fig. 2). Thus, the promotion of cell proliferation by YAP1/TAZ is negatively regulated by Hippo signaling.

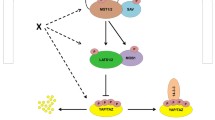
Mammalian canonical and non-canonical Hippo signaling pathways. The core components of the mammalian Hippo pathway are the MST and LATS kinases, and the SAV1 and MOB adaptor proteins. The Hippo pathway is activated in response to increased cell density, cell tension, mechanical forces, stress stimuli, or G-protein-coupled receptor (GPCR) antagonists, and inhibits cell proliferation and other processes potentially contributing to tumorigenesis. Candidate sensory components upstream of the Hippo core include FAT4, DCHS1, ITGB1, CD44, NF2, FRMD6, KIBRA, TAO, and RASSF. Upon Hippo core activation, SAV1 binding to MST allows this kinase to phosphorylate MOB1. Phospho-MOB1 binding to LATS both enhances LATS catalytic activity and allows it to be phosphorylated by MST. Phospho-LATS then phosphorylates and inactivates YAP (or its paralog, TAZ), promoting their cytoplasmic retention through binding to 14-3-3 protein. LATS-phosphorylated YAP1 (or TAZ) is also degraded so transcription factors (such as the TEADs) promoting cell survival are not activated. In contrast, under conditions of low cell density or minimal stress, YAP dissociates from 14-3-3, translocates into the nucleus, and activates transcription factors that induce the expression of pro-survival genes. Solid lines indicate known direct interactions; dashed lines indicate unknown mechanisms. The non-canonical Hippo pathway operates in the tight and adherens junction complexes and involves AMOT in the Crumb complex in the tight junction, ZO-2 in the tight junction, and α-catenin, β-catenin and PTPN14 in the adherence junction. Each of these molecules can bind to phosphorylated YAP1 to control its localization and activity

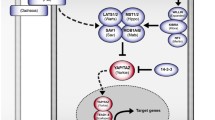
Mechanisms of YAP1/TAZ inactivation. Activation of the Hippo core, as described in Fig. 1, leads to LATS-mediated phosphorylation of YAP1 (S127) and TAZ (S89) and the cytoplasmic retention of these proteins via 14-3-3 binding. Alternatively, LATS-mediated phosphorylation of YAP1 (S397)/TAZ (S311) triggers the binding of the E3 ligase complex SCFβ-TRCP, which facilitates YAP1⁄TAZ ubiquitination and proteasomal degradation. Phosphorylation of YAP1 (Y407)/TAZ (Y321) by SRC family kinases (YES1, SRC, ABL) excludes YAP1/TAZ from the nucleus, whereas methylation of YAP1(K494) by SET7 promotes its nuclear localization. Phosphorylation of TAZ (Ser58 and Ser62) by glycogen synthase kinase 3β (GSK3β) recruits β-TrCP to target TAZ for degradation. RASSF1A activates MST1 by preventing PP2A-mediated dephosphorylation of MST1⁄2 and YAP1. NDR1⁄2 are alternative MST substrates
The upstream sensors activating Hippo signaling in mammals are not clearly understood. With respect to extracellular triggers, E-cadherin (CDH1), CD44, β1-integrin (ITGB1)–Rho–GTPase, β1-integrin–ILK–MYPT1–PP1, G-protein-coupled receptors (GPCRs), and protease-activated receptors (F2Rs) Rho–GTPases–ROCK have all been linked to control of NF2, LATS, or YAP1/TAZ activation, as illustrated in Figs. 1 and 2 [2, 6–15], but the precise sequence of events remains obscure. The roles of intracellular triggers are even less well defined but include KIBRA binding to NF2 and FRMD6 and the activities of TAOK1, PP2A, and RASSFs [16–23].
Establishment of epithelial apical–basal polarity requires the Crumbs, Par, and Scribble complexes [24, 25]. In addition to the canonical Hippo signaling pathway, a non-canonical Hippo pathway exists that involves AMOT in the Crumb complex in the tight junction, ZO-2 in the tight junction, and α-catenin, β-catenin, and PTPN14 in the adherence junction [26–32]. These molecules all bind to phosphorylated YAP1 to control its localization and activity. The Scribble and Par3 polarity complexes also influence Hippo signaling.
The localization and activity of YAP1/TAZ are also regulated by several other non-Hippo molecular events. For example, the phosphorylation of YAP1 (Y407)/TAZ (Y321) by SRC family kinases (YES1, SRC, ABL) [33] induces nuclear localization of YAP1, while methylation of YAP1 (K494) by SET7 excludes YAP1 from the nucleus [34]. Like Hippo-mediated phosphorylation, phosphorylation of TAZ (Ser58 and Ser62) by glycogen synthase kinase 3β (GSK3β) recruits β-TrCP, driving TAZ degradation [35].
The main transcription factors regulated by YAP1/TAZ are the TEADs, but, as illustrated in Fig. 3, many other transcription factors are affected by YAP1/TAZ. Accordingly, the Hippo pathway regulates cell proliferation, organ size, and tumor suppression. As shown in Fig. 4, the Hippo pathway also engages in crosstalk with several morphogenetic signaling pathways whose disruption is linked to morphogenesis and carcinogenesis [36]. While YAP1 and TAZ share most structural features, TAZ lacks two sequences present in YAP1: a PxxφP motif and an SH3-binding motif. The effect of these differences on specific transcriptional activation patterns is under investigation.

Transcription factors and downstream target genes activated by Hippo signaling. Left: the main transcription factors regulated by YAP1/TAZ are the TEADs. Following interaction with nuclear YAP1/TAZ, TEADs enhance the transcription of anti-apoptotic genes such as CTGF and BIRC5 (survivin). Right: list of other transcription factors controlled either positively or negatively by YAP1 or TAZ. Bottom: cellular processes regulated by YAP1/TAZ

Crosstalk between the Hippo pathway and other signaling pathways. The Hippo pathway is influenced by and interacts with components of the indicated morphogenetic pathways whose dysregulation is related to tumorigenesis
Identifying the roles of mammalian Hippo signaling elements has been a challenge because multiple homologs exist for almost all components. Each isoform’s function has required separate clarification via the use of specific knockout (KO) mice. Strikingly, mutants lacking NF2, MST1/2, or LATS1, as well as YAP1 transgenic animals, all develop some type of tumor. In human cancers, inactivation of the Hippo pathway due to either genetic (NF2, SAV1, MOB1A, LATS2, or YAP1) or epigenetic changes has been documented [37–40]. Indeed, for a variety of tumors, loss of Hippo signaling can predict patient sensitivity to chemotherapy as well as disease-free survival and overall survival [41]. This function of Hippo components as potent tumor suppressors makes them attractive targets for new anti-cancer therapies. To date, certain porphyrin derivatives, such as verteporfin (VP) and protoporphyrin IX (PPIX), have been reported to inhibit YAP1–TEAD association [42]. GPCR (Gα12/13, Gαq/11, Gαi/o) antagonists, GPCR (Gαs) agonists [8], and Src family antagonists (dasatinib) [33] have also been proposed as candidate Hippo modulators for tumor therapy.
In this chapter, we describe gene-targeted mouse models that have been useful in characterizing the physiological and tumor-suppressive properties of the Hippo pathway in various tissues, and relate these models to human cancers.
Loss of Hippo Signaling in the Liver
YAP1 transgenic mice show hepatomegaly due to an increase in liver cell numbers rather than cell size. This hyperplasia was completely reversible in the short term but led to irreversible liver tumorigenesis in the long term [37]. YAP1 hyperactivation caused adult hepatocytes to dedifferentiate into liver progenitor cells that exhibited enhanced self-renewal and engraftment capacities as well as increased Notch2 transcription promoting the cholangiocyte lineage [43]. In contrast, mice deficient for YAP1 specifically showed loss of hepatocytes and cholangiocytes, and resisted the induction of hepatocarcinoma (HCC) [38, 42, 44].
In human HCC, amplification of the YAP1 gene is uncommon (~5–10 %) [37], but loss of YAP1 (Ser127) phosphorylation and elevated nuclear YAP1 are frequently observed [38, 39]. Increased YAP1 is now considered an independent prognostic marker for HCC [41]. Among human liver tumor subtypes, YAP1 elevation is most frequent in combined hepatocellular cholangiocarcinomas (cHC-CC; 67%). YAP1 is also higher in more HCCs exhibiting “stemness” (EpCAM+ and keratin 19+; 56%) than in HCCs lacking “stemness” (17%). In addition, impaired phosphorylation of MOB1 occurs in 81% of human HCCs [40].
Complete knockout of both MST1 and MST2 (DKO) in mice results in embryonic lethality, whereas retention of a single functional copy of either MST1 or MST2 is sufficient for normal embryogenesis. These mutants show enhanced proliferation of liver stem cells and progenitor cells (such as oval cells) that give rise to both hepatocytes and cholangiocytes. Eventually these mutants develop liver tumors due to loss of function of the single MST1 or MST2 allele. These tumors display characteristics of both HCC and CC, and feature the expansion and transformation of a mixed liver progenitor cell population [45]. Liver-specific MST1/2 DKO mice develop similar hepatomegaly and HCC/CC tumors by 5–6 months after birth [46].
It is still unclear whether LATS function is essential for tumor suppression in the liver. One study showed that loss of MST1/2 decreased MOB1 and YAP1 phosphorylation but had no effect on LATS1/2 phosphorylation [45]. However, another study demonstrated reduced levels of phosphorylated YAP1 and LATS1/2 but unexpectedly decreased TAZ protein levels in MST1/2 DKO liver and tumors [40]. These data suggest that LATS1/2 and TAZ may be unlikely to play a major role in this liver phenotype.
Liver-specific deletion of Sav1 in mice enhances oval cell proliferation such that the mutants eventually develop HCC/CC liver tumors. However, levels of phospho-YAP1 and phospho-LATS1/2 are normal in SAV1 KO livers, suggesting that Sav1 promotes oval cell expansion and tumorigenesis in this model but acts independently of LATS1/2 and YAP1 [46, 47].
Liver-specific deletion of NF2 in mice also results in hepatocyte and cholangiocyte proliferation, leading to HCC/CC [42, 48, 49]. In one study, YAP1 and LATS1/2 phosphorylation were reduced in NF2-deficient liver and nuclear YAP1 was increased. Deletion of one copy of YAP1 was sufficient to reverse tumorigenesis in this model, indicating that NF2-mediated tumor suppression is achieved by inactivating YAP1 [48]. However, another group reported that liver-specific deletion of NF2 led to dramatic progenitor cell expansion without altering YAP1 localization or phosphorylation. EGFR inhibition blocked tumorigenesis triggered by NF2 deletion in this model, arguing against a role for YAP1 [49].
Loss of Hippo Signaling in the Pancreas
Pancreas-specific MST1/2 DKO mice (using PDX1-Cre Tg) exhibit a small pancreas in which the exocrine compartment shows increased cell proliferation but also extensive disorganization and pancreatitis-like autodigestion [50]. Pancreatic MST1/2 deficiency also induces postnatal de-differentiation of acinar cells into duct-like cells and thus reduces the total number of acinar cells [51]. However, neither these nor any other pancreas-specific Hippo mutants examined to date develop tumors. Interestingly, doxycycline-dependent YAP1 activation in the mouse pancreas expands its size and increases the number of acinar cells, leading to ductal metaplasia [44]. Thus, the phenotype observed in pancreas-specific MST DKO mice may be YAP1 independent.
In humans, pancreatic ductal adenocarcinoma (PDAC) frequently carries an oncogenic Kras mutation. In an inducible KrasG12D-driven mouse PDAC model, the mutants can develop malignancies in which KrasG12D and downstream MAPK activation are lost, but DNA amplification and/or mRNA overexpression of YAP1 are acquired [52, 53]. Excessive YAP1 may thus be the major driver of invasive and recurrent PDAC.
Loss of Hippo Signaling in the Intestine
The role of Hippo signaling in the intestine is complex. YAP1 is overexpressed and localized in the nucleus in most human colon cancers, but silenced in a subset of highly aggressive and undifferentiated human colorectal carcinomas [54]. In mice, reports differ regarding Hippo element functions in intestinal stem cell (ISC) expansion and intestinal regeneration. In one study, loss of YAP1 in intestinal epithelial cells (Villin-Cre) had little effect on intestinal development but severely impaired DSS-induced intestinal regeneration. In the same study, doxycycline-dependent YAP1 activation expanded multipotent undifferentiated dysplastic progenitor cells in the intestine in a manner partly dependent on Notch activation [44]. However, another study found that intestinal YAP1 deficiency (Villin-Cre) resulted in WNT hypersensitivity leading to hyperplasia, ISC expansion, and formation of ectopic crypts and microadenomas. This report also showed that transgenic YAP1 expression reduced WNT target gene expression and induced rapid loss of intestinal crypts. In this latter case, the authors speculated that cytoplasmic YAP1 can bind to DVL and dampen WNT signaling [54]. The discrepancies between these reports require resolution because both increased nuclear YAP1 and decreased cytoplasmic YAP1 are associated with an increased risk of colon cancer in humans.
Intestine-specific Sav1-deficient mice show accelerated crypt regeneration and DSS-induced colonic polyp formation dependent on nuclear YAP1 but independent of β-catenin [55]. In contrast, intestine-specific MST1/2-deficient mice (Villin-Cre) display YAP1-dependent activation of Notch and β-catenin, as well as ISC expansion associated with colonic adenoma formation [56]. Recent work has shown that phosphorylation of YAP1(Y407) by the SRC family tyrosine kinase YES1 allows YAP1 to enter the nucleus and form a complex with the transcription factor TBX5 and β-catenin. This complex then promotes the transcription of anti-apoptotic genes such as BCL2L1 and BIRC5 [33]. Thus, a SRC–YAP1–β catenin–TBX5 complex may be essential to the transformation and survival of β-catenin-driven cancers.
Loss of Hippo Signaling in the Lung
Lung-specific MST1/2 DKO mice (TTF1(NKX2.1)-Cre) exhibit perinatal mortality due to respiratory failure [57]. The lungs appear immature, with reduced type I pneumocytes and increased immature type II pneumocytes lacking microvilli, lamellar bodies, and surfactant protein expression. These features are reminiscent of human infant respiratory distress syndrome (IRDS). MST normally phosphorylates and stabilizes the transcription factor FOXA2, which regulates alveolar epithelial cell maturation and surfactant protein expression. The IRDS phenotype of MST1/2 DKO mice is independent of YAP1 hyperactivation but dependent on FOXA2 [57].
Whether MST1/2 deficiency in the adult mouse lung would trigger lung tumorigenesis remains unknown. In Sca1+ murine lung tumor–propagating cells (TPC), TAZ is overexpressed by more than twofold and NF2 is downregulated by more than 15-fold compared with non-TPC [58]. Knockdown of YAP1 or TAZ in lung cancer cell lines decreases their cellular migration capacity and ability to metastasize. Conversely, expression of doxycycline-inducible constitutively active YAP1 in Kras Tg mice results in lung tumor progression in vivo [58].
TAZ KO mice display alveolarization defects similar to those seen in human emphysema patients [59–62]. However, although TAZ activates TTF1, a transcription factor essential for lung formation, TAZ KO lungs show a normal pattern of TTF1 target gene expression [63]. Mutant mice heterozygous for TAZ deficiency are resistant to bleomycin-induced lung fibrosis, suggesting that TAZ regulates fibrotic responses in this tissue [61].
Humans exposed to asbestos often develop malignant mesothelioma (MM). Mutation of NF2 occurs in 50 % of human MM and YAP1 activation is frequent [64]. However, no statistically significant MM development has been reported to date in mice bearing mutations of Hippo elements.
Loss of Hippo Signaling in the Skin
YAP1 plays a key role in murine skin morphogenesis. Normal basal epidermal progenitors with nuclear YAP1 localization are driven to proliferate, whereas those with cytoplasmic YAP1 localization differentiate into hair follicles. YAP1 overexpression in mouse skin causes expansion of epidermal stem cells and progenitors, resulting in epidermal thickening, hyperkeratosis, hair follicle evagination, and the development of squamous cell-like carcinomas in skin grafts [65, 66]. Conversely, YAP1 deficiency in mouse epidermis, or disruption of YAP1–TEAD interaction, leads to epidermal hypoplasia and decreased keratinocyte proliferation due to loss of epidermal stem/progenitor cells.
At least one wild-type (WT) MOB1A or MOB1B allele is essential for murine embryogenesis, and any loss of this remaining allele results in trichilemmal carcinomas of the skin [67]. YAP1 activation and MOB1A/1B inactivation are also frequently observed in human trichilemmal carcinomas. Newborn MOB1A/1B DKO mice show hyperplastic keratinocyte progenitors and defective keratinocyte terminal differentiation, and rapidly die of malnutrition. MOB1A/1B-deficient keratinocytes exhibit hyperproliferation, apoptotic resistance, impaired contact inhibition, enhanced progenitor self-renewal, and increased centrosomes. LATS1/2 and YAP1 activities are altered in these cells [67].
SAV1-deficient mice exhibit a thick epidermal skin layer and succumb to early embryonic lethality [68]. SAV1-deficient primary keratinocytes show hyperproliferation, progenitor expansion, decreased apoptosis, and inhibition of terminal differentiation.
Surprisingly, keratinocyte-specific MST1/2 DKO mice are healthy and show normal YAP1 phosphorylation and activation. YAP1 is therefore likely phosphorylated by an alternative mechanism in keratinocytes [22], perhaps by NDR1/2 kinases or GPCR signaling [8].
As noted above, cell adhesion and junction complex proteins can also regulate YAP1. Keratinocyte-specific loss of α-catenin results in keratinocyte hyperplasia and squamous cell carcinomagenesis similar to that observed in YAP1 transgenic mice [22]. In normal keratinocytes, α-catenin interacts directly with YAP1 and restricts it to the cytoplasm [39]. YAP1 localization in the skin is also influenced by β-catenin, PTPN14, AMOT, and ZO-2 [69, 70].
Loss of Hippo Signaling in T Lymphocytes
In humans, inactivating MST1 mutations occur in families with T cell immunodeficiency or autoimmune disorders [71, 72]. In MST1 KO mice, naïve T cells show impaired CCL19/21-induced LFA-1 clustering and reduced cell migration [73–75]. In addition, MST1-deficient T cells exhibit defective FOXO1–IL-7R–BCL2 signaling, FAS upregulation, and a FOXO1/3-dependent decrease in SOD2 and catalase. As a result, these mutant T cells possess increased ROS and undergo apoptosis [76], a mechanism that may also underlie the observed immunodeficiency in humans with MST1 mutations. MOB1 phosphorylation is decreased in MST1 KO T cells but LATS1/2 and YAP1 phosphorylation are essentially normal, suggesting that LATS1/2 and YAP1 are not downstream effectors of MST1 and MOB1 in naïve T cells.
Despite their T cell deficiency, aged MST1 KO mice develop autoimmune disease because MST1 normally enhances FOXP3 expression and thus sustains regulatory T cell development through stabilizing FOXO1/3 [75]. These mutants are also susceptible to chemical induction of T-ALL and undergo accelerated spontaneous lymphomagenesis in a p53-deficient background [77].
The chemokines CCL19/21 normally induce the binding of DOCK8 to phosphorylated MOB1, promoting actin polarization and thymocyte migration from the wild-type thymus. MST1/2 DKO thymocytes show a loss of DOCK8 binding to MOB1 [78]. As a result, T cell–specific MST1/2 DKO thymocytes develop normally but undergo apoptosis before they can exit the thymus, leading to an absence of mature T cells in the circulation and lymphoid tissues [78].
Loss of Hippo Signaling in Salivary Glands
TAZ phosphorylation increases during salivary submandibular gland (SMG) development in mice [79]. In human Sjogren’s syndrome (SS) patients, TAZ is localized in the nucleus rather than in the junctional regions of the SMGs, causing an abnormal accumulation of TAZ downstream targets such as extracellular matrix components, fibronectin, and CTGF [79]. LATS2 deficiency also results in defective SMG morphogenesis and a reduction in junction-localized TAZ in mice. Lastly, 5 % of MOB1 partially deficient mice develop salivary gland adenomas and carcinomas [67].
Loss of Hippo Signaling in Neurons
In humans, mutations of DCHS1 or FAT4 cause Van Maldergem syndrome, which is characterized by a periventricular neuronal heterotopia (the mislocalization of cortical neuron progenitors) accompanied by auditory and renal deficits as well as craniofacial, skeletal, and limb malformations [80]. In mice, reduction of DCHS1 or FAT4 in embryonic neuroepithelium increases neuronal progenitors but reduces their differentiation. These cells are then mislocalized in the neocortex in a YAP1-dependent manner [80], producing a phenotype similar to human Van Maldergem syndrome.
Inactivating mutations of NF2 in humans are associated with sporadic schwannomas, meningiomas, and ependymomas [81]. Heterozygous NF2 mutant mice do not develop neuronal tumors, but homozygous loss of NF2 specifically in Schwann cells results in their hyperplasia and eventually schwannomas [81]. Additional loss of one p53 allele results in the appearance of malignant peripheral nerve sheath tumors [82]. Complete loss of NF2 in mice is embryonic lethal. Telencephalon-specific NF2 KO embryos show neural progenitor expansion associated with increased nuclear YAP1/TAZ [83]. YAP1 or TEAD overexpression promotes cell cycle progression and inhibits neuronal differentiation by inducing the expression of downstream targets such as cyclin D1 and Pax3 [84, 85].
In normal brain, YAP1 is a downstream effector of the SHH pathway and increases neural stem cell proliferation [86, 87]. YAP1 and SHH signaling engage in crosstalk, since GLI2, which participates in SHH signaling, also acts downstream of YAP1 to control neuronal differentiation [88]. YAP1 is often upregulated in both human and mouse SHH-dependent or PATCHED1-mutated medulloblastomas [86, 87].
Loss of Hippo Signaling in Bone
Several Hippo elements have been implicated in controlling tumor suppression in bone. Many human osteosarcoma patients show high levels of nuclear YAP1 in their tumor cells [89], and aged mice bearing heterozygous mutations of NF2 develop osteosarcomas [90]. Some SAV1 heterozygous mouse mutants also develop osteosarcomas [68], as do some MOB1A/1B partially deficient mice [67].
Concluding Remarks
This chapter has summarized evidence showing that heterozygous inactivation of Hippo components leads to tumorigenesis in a wide range of tissues. For more detail on the in vivo functions of the Hippo pathway, please refer to several excellent recent reviews [36, 91, 92]. In general, loss of Hippo signaling or abnormal activation of YAP1/TAZ leads to impaired cell contact inhibition and eventually malignant transformation. Hippo components also control the differentiation/de-differentiation of many stem/progenitor cells. Thus, proper Hippo signaling not only protects against tumor formation but also regulates a vast array of physiological and pathological phenomena.
The studies we have cited demonstrate a strong correlation between findings in human diseases and the phenotypes of Hippo mutant mouse strains. These animals may therefore be of great value for studying the mechanisms underlying various pathological conditions and for determining the efficacy of new therapeutic strategies. Manipulation of Hippo signaling could spur stem cell expansion for regenerative medicine or halt tumor progression to benefit cancer patients. As noted above, porphyrin derivatives [42], GPCR agonists and antagonists [8], and Src family antagonists [33] have already been proposed as candidate Hippo regulators for cancer therapy. Future work will no doubt uncover other Hippo-based approaches worth exploring for their anticancer potential.
References
Harvey K, Tapon N (2007) The Salvador-Warts-Hippo pathway – an emerging tumour-suppressor network. Nat Rev Cancer 7:182–191
Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S (2011) Role of YAP/TAZ in mechanotransduction. Nature 474:179–183
Liu CY, Zha ZY, Zhou X, Zhang H, Huang W, Zhao D, Li T, Chan SW, Lim CJ, Hong W, Zhao S, Xiong Y, Lei QY, Guan KL (2010) The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF-TrCP E3 ligase. J Biol Chem 285:37159–37169
Zhao B, Li L, Tumaneng K, Wang CY, Guan KL (2010) A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev 24:72–85
Zhao B, Li L, Lei Q, Guan KL (2010) The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev 24:862–874
Sainio M, Zhao F, Heiska L, Turunen O, den Bakker M, Zwarthoff E, Lutchman M, Rouleau GA, Jaaskelainen J, Vaheri A, Carpen O (1997) Neurofibromatosis 2 tumor suppressor protein colocalizes with ezrin and CD44 and associates with actin-containing cytoskeleton. J Cell Sci 110(Pt 18):2249–2260
Morrison H, Sherman LS, Legg J, Banine F, Isacke C, Haipek CA, Gutmann DH, Ponta H, Herrlich P (2001) The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev 15:968–980
Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, Fu XD, Mills GB, Guan KL (2012) Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150:780–791
Mo JS, Yu FX, Gong R, Brown JH, Guan KL (2012) Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs). Genes Dev 26:2138–2143
Kim M, Kim M, Lee S, Kuninaka S, Saya H, Lee H, Lee S, Lim DS (2013) cAMP/PKA signalling reinforces the LATS-YAP pathway to fully suppress YAP in response to actin cytoskeletal changes. EMBO J 32:1543–1555
Sansores-Garcia L, Bossuyt W, Wada K, Yonemura S, Tao C, Sasaki H, Halder G (2011) Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J 30:2325–2335
Wada K, Itoga K, Okano T, Yonemura S, Sasaki H (2011) Hippo pathway regulation by cell morphology and stress fibers. Development 138:3907–3914
Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S (2013) A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 154:1047–1059
Tang Y, Rowe RG, Botvinick EL, Kurup A, Putnam AJ, Seiki M, Weaver VM, Keller ET, Goldstein S, Dai J, Begun D, Saunders T, Weiss SJ (2013) MT1-MMP-dependent control of skeletal stem cell commitment via a beta1-integrin/YAP/TAZ signaling axis. Dev Cell 25:402–416
Serrano I, McDonald PC, Lock F, Muller WJ, Dedhar S (2013) Inactivation of the Hippo tumour suppressor pathway by integrin-linked kinase. Nat Commun 4:2976
Xiao L, Chen Y, Ji M, Volle DJ, Lewis RE, Tsai MY, Dong J (2011) KIBRA protein phosphorylation is regulated by mitotic kinase aurora and protein phosphatase 1. J Biol Chem 286:36304–36315
Yu J, Zheng Y, Dong J, Klusza S, Deng WM, Pan D (2010) Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and expanded. Dev Cell 18:288–299
Moleirinho S, Chang N, Sims AH, Tilston-Lunel AM, Angus L, Steele A, Boswell V, Barnett SC, Ormandy C, Faratian D, Gunn-Moore FJ, Reynolds PA (2013) KIBRA exhibits MST-independent functional regulation of the Hippo signaling pathway in mammals. Oncogene 32:1821–1830
Poon CL, Lin JI, Zhang X, Harvey KF (2011) The sterile 20-like kinase Tao-1 controls tissue growth by regulating the Salvador-Warts-Hippo pathway. Dev Cell 21:896–906
Boggiano JC, Vanderzalm PJ, Fehon RG (2011) Tao-1 phosphorylates Hippo/MST kinases to regulate the Hippo-Salvador-Warts tumor suppressor pathway. Dev Cell 21:888–895
Guo C, Zhang X, Pfeifer GP (2011) The tumor suppressor RASSF1A prevents dephosphorylation of the mammalian STE20-like kinases MST1 and MST2. J Biol Chem 286:6253–6261
Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, Camargo FD (2011) Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell 144:782–795
Oh HJ, Lee KK, Song SJ, Jin MS, Song MS, Lee JH, Im CR, Lee JO, Yonehara S, Lim DS (2006) Role of the tumor suppressor RASSF1A in Mst1-mediated apoptosis. Cancer Res 66:2562–2569
Bryant DM, Mostov KE (2008) From cells to organs: building polarized tissue. Nat Rev Mol Cell Biol 9:887–901
Shin K, Fogg VC, Margolis B (2006) Tight junctions and cell polarity. Annu Rev Cell Dev Biol 22:207–235
Varelas X, Samavarchi-Tehrani P, Narimatsu M, Weiss A, Cockburn K, Larsen BG, Rossant J, Wrana JL (2010) The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-beta-SMAD pathway. Dev Cell 19:831–844
Chen CL, Gajewski KM, Hamaratoglu F, Bossuyt W, Sansores-Garcia L, Tao C, Halder G (2010) The apical-basal cell polarity determinant Crumbs regulates Hippo signaling in Drosophila. Proc Natl Acad Sci U S A 107:15810–15815
Robinson BS, Huang J, Hong Y, Moberg KH (2010) Crumbs regulates Salvador/Warts/Hippo signaling in Drosophila via the FERM-domain protein Expanded. Curr Biol 20:582–590
Grzeschik NA, Parsons LM, Allott ML, Harvey KF, Richardson HE (2010) Lgl, aPKC, and Crumbs regulate the Salvador/Warts/Hippo pathway through two distinct mechanisms. Curr Biol 20:573–581
Ling C, Zheng Y, Yin F, Yu J, Huang J, Hong Y, Wu S, Pan D (2010) The apical transmembrane protein Crumbs functions as a tumor suppressor that regulates Hippo signaling by binding to Expanded. Proc Natl Acad Sci U S A 107:10532–10537
Martin-Belmonte F, Perez-Moreno M (2012) Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer 12:23–38
Menendez J, Perez-Garijo A, Calleja M, Morata G (2010) A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc Natl Acad Sci U S A 107:14651–14656
Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, Zack TI, Wang X, Tsherniak A, Schinzel AC, Shao DD, Schumacher SE, Weir BA, Vazquez F, Cowley GS, Root DE, Mesirov JP, Beroukhim R, Kuo CJ, Goessling W, Hahn WC (2012) Beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 151:1457–1473
Oudhoff MJ, Freeman SA, Couzens AL, Antignano F, Kuznetsova E, Min PH, Northrop JP, Lehnertz B, Barsyte-Lovejoy D, Vedadi M, Arrowsmith CH, Nishina H, Gold MR, Rossi FM, Gingras AC, Zaph C (2013) Control of the hippo pathway by Set7-dependent methylation of Yap. Dev Cell 26:188–194
Huang W, Lv X, Liu C, Zha Z, Zhang H, Jiang Y, Xiong Y, Lei QY, Guan KL (2012) The N-terminal phosphodegron targets TAZ/WWTR1 protein for SCFbeta-TrCP-dependent degradation in response to phosphatidylinositol 3-kinase inhibition. J Biol Chem 287:26245–26253
Nishio M, Otsubo K, Maehama T, Mimori K, Suzuki A (2013) Capturing the mammalian Hippo: elucidating its role in cancer. Cancer Sci 104:1271–1277
Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, Mu D, Lucito R, Powers S, Lowe SW (2006) Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125:1253–1267
Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D (2007) Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130:1120–1133
Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, Halder G, Lai ZC, Guan KL (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21:2747–2761
Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, Bardeesy N (2009) Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16:425–438
Xu MZ, Yao TJ, Lee NP, Ng IO, Chan YT, Zender L, Lowe SW, Poon RT, Luk JM (2009) Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer 115:4576–4585
Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D (2012) Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev 26:1300–1305
Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, Shrestha K, Cahan P, Stanger BZ, Camargo FD (2014) Hippo pathway activity influences liver cell fate. Cell 157:1324–1338
Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, Brummelkamp TR (2007) YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol 17:2054–2060
Song H, Mak KK, Topol L, Yun K, Hu J, Garrett L, Chen Y, Park O, Chang J, Simpson RM, Wang CY, Gao B, Jiang J, Yang Y (2010) Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc Natl Acad Sci U S A 107:1431–1436
Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, Wang Y, Halder G, Finegold MJ, Lee JS, Johnson RL (2010) Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci U S A 107:1437–1442
Lee KP, Lee JH, Kim TS, Kim TH, Park HD, Byun JS, Kim MC, Jeong WI, Calvisi DF, Kim JM, Lim DS (2010) The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci U S A 107:8248–8253
Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, Giovannini M, Liu P, Anders RA, Pan D (2010) The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell 19:27–38
Benhamouche S, Curto M, Saotome I, Gladden AB, Liu CH, Giovannini M, McClatchey AI (2010) Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes Dev 24:1718–1730
George NM, Day CE, Boerner BP, Johnson RL, Sarvetnick NE (2012) Hippo signaling regulates pancreas development through inactivation of Yap. Mol Cell Biol 32:5116–5128
Gao T, Zhou D, Yang C, Singh T, Penzo-Mendez A, Maddipati R, Tzatsos A, Bardeesy N, Avruch J, Stanger BZ (2013) Hippo signaling regulates differentiation and maintenance in the exocrine pancreas. Gastroenterology 144:1543–1553, 1553 e1541
Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, Chang Q, Chu GC, Al-Khalil R, Jiang S, Xia H, Fletcher-Sananikone E, Lim C, Horwitz GI, Viale A, Pettazzoni P, Sanchez N, Wang H, Protopopov A, Zhang J, Heffernan T, Johnson RL, Chin L, Wang YA, Draetta G, DePinho RA (2014) Yap1 activation enables bypass of oncogenic kras addiction in pancreatic cancer. Cell 158:185–197
Zhang W, Nandakumar N, Shi Y, Manzano M, Smith A, Graham G, Gupta S, Vietsch EE, Laughlin SZ, Wadhwa M, Chetram M, Joshi M, Wang F, Kallakury B, Toretsky J, Wellstein A, Yi C (2014) Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci Signal 7:ra42
Barry ER, Morikawa T, Butler BL, Shrestha K, de la Rosa R, Yan KS, Fuchs CS, Magness ST, Smits R, Ogino S, Kuo CJ, Camargo FD (2013) Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 493:106–110
Cai J, Zhang N, Zheng Y, de Wilde RF, Maitra A, Pan D (2010) The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev 24:2383–2388
Zhou D, Zhang Y, Wu H, Barry E, Yin Y, Lawrence E, Dawson D, Willis JE, Markowitz SD, Camargo FD, Avruch J (2011) Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc Natl Acad Sci U S A 108:E1312–E1320
Chung C, Kim T, Kim M, Kim M, Song H, Kim TS, Seo E, Lee SH, Kim H, Kim SK, Yoo G, Lee DH, Hwang DS, Kinashi T, Kim JM, Lim DS (2013) Hippo-Foxa2 signaling pathway plays a role in peripheral lung maturation and surfactant homeostasis. Proc Natl Acad Sci U S A 110:7732–7737
Lau AN, Curtis SJ, Fillmore CM, Rowbotham SP, Mohseni M, Wagner DE, Beede AM, Montoro DT, Sinkevicius KW, Walton ZE, Barrios J, Weiss DJ, Camargo FD, Wong KK, Kim CF (2014) Tumor-propagating cells and Yap/Taz activity contribute to lung tumor progression and metastasis. EMBO J 33:468–481
Hossain Z, Ali SM, Ko HL, Xu J, Ng CP, Guo K, Qi Z, Ponniah S, Hong W, Hunziker W (2007) Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1. Proc Natl Acad Sci U S A 104:1631–1636
Makita R, Uchijima Y, Nishiyama K, Amano T, Chen Q, Takeuchi T, Mitani A, Nagase T, Yatomi Y, Aburatani H, Nakagawa O, Small EV, Cobo-Stark P, Igarashi P, Murakami M, Tominaga J, Sato T, Asano T, Kurihara Y, Kurihara H (2008) Multiple renal cysts, urinary concentration defects, and pulmonary emphysematous changes in mice lacking TAZ. Am J Physiol Renal Physiol 294:F542–F553
Mitani A, Nagase T, Fukuchi K, Aburatani H, Makita R, Kurihara H (2009) Transcriptional coactivator with PDZ-binding motif is essential for normal alveolarization in mice. Am J Respir Crit Care Med 180:326–338
Tian Y, Kolb R, Hong JH, Carroll J, Li D, You J, Bronson R, Yaffe MB, Zhou J, Benjamin T (2007) TAZ promotes PC2 degradation through a SCFbeta-Trcp E3 ligase complex. Mol Cell Biol 27:6383–6395
Park KS, Whitsett JA, Di Palma T, Hong JH, Yaffe MB, Zannini M (2004) TAZ interacts with TTF-1 and regulates expression of surfactant protein-C. J Biol Chem 279:17384–17390
Sekido Y (2013) Molecular pathogenesis of malignant mesothelioma. Carcinogenesis 34:1413–1419
Silvis MR, Kreger BT, Lien WH, Klezovitch O, Rudakova GM, Camargo FD, Lantz DM, Seykora JT, Vasioukhin V (2011) Alpha-catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Sci Signal 4:ra33
Zhang H, Pasolli HA, Fuchs E (2011) Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci U S A 108:2270–2275
Nishio M, Hamada K, Kawahara K, Sasaki M, Noguchi F, Chiba S, Mizuno K, Suzuki SO, Dong Y, Tokuda M, Morikawa T, Hikasa H, Eggenschwiler J, Yabuta N, Nojima H, Nakagawa K, Hata Y, Nishina H, Mimori K, Mori M, Sasaki T, Mak TW, Nakano T, Itami S, Suzuki A (2012) Cancer susceptibility and embryonic lethality in Mob1a/1b double-mutant mice. J Clin Invest 122:4505–4518
Lee JH, Kim TS, Yang TH, Koo BK, Oh SP, Lee KP, Oh HJ, Lee SH, Kong YY, Kim JM, Lim DS (2008) A crucial role of WW45 in developing epithelial tissues in the mouse. EMBO J 27:1231–1242
Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei Q, Guan KL (2011) Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev 25:51–63
Hirate Y, Hirahara S, Inoue K, Suzuki A, Alarcon VB, Akimoto K, Hirai T, Hara T, Adachi M, Chida K, Ohno S, Marikawa Y, Nakao K, Shimono A, Sasaki H (2013) Polarity-dependent distribution of angiomotin localizes Hippo signaling in preimplantation embryos. Curr Biol 23:1181–1194
Abdollahpour H, Appaswamy G, Kotlarz D, Diestelhorst J, Beier R, Schaffer AA, Gertz EM, Schambach A, Kreipe HH, Pfeifer D, Engelhardt KR, Rezaei N, Grimbacher B, Lohrmann S, Sherkat R, Klein C (2012) The phenotype of human STK4 deficiency. Blood 119:3450–3457
Nehme NT, Pachlopnik Schmid J, Debeurme F, Andre-Schmutz I, Lim A, Nitschke P, Rieux-Laucat F, Lutz P, Picard C, Mahlaoui N, Fischer A, de Saint BG (2012) MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood 119:3458–3468
Katagiri K, Imamura M, Kinashi T (2006) Spatiotemporal regulation of the kinase Mst1 by binding protein RAPL is critical for lymphocyte polarity and adhesion. Nat Immunol 7:919–928
Dong Y, Du X, Ye J, Han M, Xu T, Zhuang Y, Tao W (2009) A cell-intrinsic role for Mst1 in regulating thymocyte egress. J Immunol 183:3865–3872
Du X, Shi H, Li J, Dong Y, Liang J, Ye J, Kong S, Zhang S, Zhong T, Yuan Z, Xu T, Zhuang Y, Zheng B, Geng JG, Tao W (2014) Mst1/Mst2 regulate development and function of regulatory T cells through modulation of Foxo1/Foxo3 stability in autoimmune disease. J Immunol 192:1525–1535
Choi J, Oh S, Lee D, Oh HJ, Park JY, Lee SB, Lim DS (2009) Mst1-FoxO signaling protects Naive T lymphocytes from cellular oxidative stress in mice. PLoS One 4:e8011
Kim TS, Lee DH, Kim SK, Shin SY, Seo EJ, Lim DS (2012) Mammalian sterile 20-like kinase 1 suppresses lymphoma development by promoting faithful chromosome segregation. Cancer Res 72:5386–5395
Mou F, Praskova M, Xia F, Van Buren D, Hock H, Avruch J, Zhou D (2012) The Mst1 and Mst2 kinases control activation of rho family GTPases and thymic egress of mature thymocytes. J Exp Med 209:741–759
Enger TB, Samad-Zadeh A, Bouchie MP, Skarstein K, Galtung HK, Mera T, Walker J, Menko AS, Varelas X, Faustman DL, Jensen JL, Kukuruzinska MA (2013) The Hippo signaling pathway is required for salivary gland development and its dysregulation is associated with Sjogren’s syndrome. Lab Investig J Tech Method Pathol 93:1203–1218
Cappello S, Gray MJ, Badouel C, Lange S, Einsiedler M, Srour M, Chitayat D, Hamdan FF, Jenkins ZA, Morgan T, Preitner N, Uster T, Thomas J, Shannon P, Morrison V, Di Donato N, Van Maldergem L, Neuhann T, Newbury-Ecob R, Swinkells M, Terhal P, Wilson LC, Zwijnenburg PJ, Sutherland-Smith AJ, Black MA, Markie D, Michaud JL, Simpson MA, Mansour S, McNeill H, Gotz M, Robertson SP (2013) Mutations in genes encoding the cadherin receptor-ligand pair DCHS1 and FAT4 disrupt cerebral cortical development. Nat Genet 45:1300–1308
Giovannini M, Robanus-Maandag E, van der Valk M, Niwa-Kawakita M, Abramowski V, Goutebroze L, Woodruff JM, Berns A, Thomas G (2000) Conditional biallelic Nf2 mutation in the mouse promotes manifestations of human neurofibromatosis type 2. Genes Dev 14:1617–1630
Robanus-Maandag E, Giovannini M, van der Valk M, Niwa-Kawakita M, Abramowski V, Antonescu C, Thomas G, Berns A (2004) Synergy of Nf2 and p53 mutations in development of malignant tumours of neural crest origin. Oncogene 23:6541–6547
Lavado A, He Y, Pare J, Neale G, Olson EN, Giovannini M, Cao X (2013) Tumor suppressor Nf2 limits expansion of the neural progenitor pool by inhibiting Yap/Taz transcriptional coactivators. Development 140:3323–3334
Cao X, Pfaff SL, Gage FH (2008) YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev 22:3320–3334
Milewski RC, Chi NC, Li J, Brown C, Lu MM, Epstein JA (2004) Identification of minimal enhancer elements sufficient for Pax3 expression in neural crest and implication of Tead2 as a regulator of Pax3. Development 131:829–837
Fernandez LA, Northcott PA, Dalton J, Fraga C, Ellison D, Angers S, Taylor MD, Kenney AM (2009) YAP1 is amplified and up-regulated in hedgehog-associated medulloblastomas and mediates Sonic hedgehog-driven neural precursor proliferation. Genes Dev 23:2729–2741
Kagey JD, Brown JA, Moberg KH (2012) Regulation of Yorkie activity in Drosophila imaginal discs by the Hedgehog receptor gene patched. Mech Dev 129:339–349
Lin YT, Ding JY, Li MY, Yeh TS, Wang TW, Yu JY (2012) YAP regulates neuronal differentiation through Sonic hedgehog signaling pathway. Exp Cell Res 318:1877–1888
Chan LH, Wang W, Yeung W, Deng Y, Yuan P, Mak KK (2014) Hedgehog signaling induces osteosarcoma development through Yap1 and H19 overexpression. Oncogene 33(40):4857–4866
McClatchey AI, Saotome I, Mercer K, Crowley D, Gusella JF, Bronson RT, Jacks T (1998) Mice heterozygous for a mutation at the Nf2 tumor suppressor locus develop a range of highly metastatic tumors. Genes Dev 12:1121–1133
Varelas X (2014) The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 141:1614–1626
Mo JS, Park HW, Guan KL (2014) The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep 15:642–656
Acknowledgments
We gratefully acknowledge the support of the Uehara Memorial Foundation and the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this paper
Cite this paper
Nishio, M., Goto, H., Suzuki, M., Fujimoto, A., Mimori, K., Suzuki, A. (2015). The Hippo Signaling Pathway: A Candidate New Drug Target for Malignant Tumors. In: Nakao, K., Minato, N., Uemoto, S. (eds) Innovative Medicine. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55651-0_7
Download citation
DOI: https://doi.org/10.1007/978-4-431-55651-0_7
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55650-3
Online ISBN: 978-4-431-55651-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)