
Abstract
Heterotaxy syndrome is characterized by a wide variety of cardiac and extra-cardiac congenital malformations that are primarily induced by disorders of the left-right axis determination during early embryonic development. Prognosis of the disease remains unsatisfactory because the syndrome is often associated with complicated congenital heart diseases. Long-term follow-up of heterotaxy patients, particularly those who underwent Fontan procedure, is now one of the most important issues in pediatric and adult congenital heart disease clinics. Collaborative studies between pediatric cardiologists and basic scientists are essential for improving the prognosis of heterotaxy syndrome.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Heterotaxy syndrome is a rare but serious congenital disease that occurs approximately 1 to 5,000–7,000 of live birth [1]. Patients are generally subdivided into “bilateral right sided” (right isomerism) or “bilateral left sided” (left isomerism) according to the characteristic morphology of atrial appendages of the heart. However, there is a wide spectrum of pathology with considerable overlap of the anatomical features.
2 Molecular and Cellular Mechanisms of Left-Right Determination
The left-right axis determinatio n initiates in the primitive node at E7.5 in mice and develops through the following pathways [2, 3]:
-
1.
Breaking of symmetry as a result of leftward “nodal flow”
-
2.
Transmission of asymmetric signals to the lateral plate mesoderm (LMP)
-
3.
Asymmetric expression of nodal and lefty2 in the LMP
-
4.
Situs-specific morphogenesis mediated by asymmetric expression of Pitx2
2.1 Node Cell Monocilia Create Leftward “Nodal Flow” and Activate Asymmetry Signaling Around the Node
The determination of the left-right asymmetry starts as leftward nodal flow generated by rotational movement of monocilia in the primitive node [4, 5]. Clockwise rotation of motile cilia creates unidirectional leftward flow because the rotational axes of cilia tilt caudal direction of the embryos [6, 7].
There are two models why nodal flow is perceived by nodal and perinodal cells. One hypothesis (chemosensory model) is that the nodal flow produces a gradient of left determinant particles (node vesicular parcels) containing hedgehog proteins and nodal [8, 9], which activate downstream signaling of nodal i n the left-side perinodal cells.
Alternative hypo thesis (mechanosensory model) is that the leftward nodal flow provokes an asymmetrical increase influx of Ca2+ ion in the sensory cilia cells through PKD2, a causative gene for human polycystic kidney disease [9, 10]. This Ca2+ influx is linked to the activation of nodal in the left-side perinodal cells, which is consequently transferred to the left LPM.
2.2 Asymmetry Signaling Transmits to the Left Lateral Plate Mesoderm
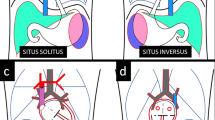
Transmission of nodal to the left LMP followed by lefty2 and Pitx2 activation [11] and the consequent heart morphogenesis in the normal subjects, right/left isomerism, and situs inversus is summarized in Fig. 6.1a.

Signal transmission of nodal to the left lateral plate mesoderm followed by Pitx2 activation and the consequent heart morphogenesis in normal and heterotaxy embryos (Adapted and modified from Ref. [1] with permission)
2.3 Genes Associated with the Human Heterotaxy Syndrome
Recent human and animal model studies have provided insights into the genetic and developmental etiology of the heterotaxy syndrome. In human, genes that are associated with heterotaxy syndrome are ZIC3, NODAL, CFC1, ACVR2B, LEFTY2, CITED2, and GDF1 [12–14].
3 Clinical Manifestation of the Heterotaxy Syndrome
Factors that deteriorate prognosis of the heterotaxy syndrome are complications with pulmonary venous obstruction, pulmonary arterial distortion, regurgitation of atrioventricular valve, elevated pulmonary vascular resistance, and impaired ventricular function [15].
3.1 Right Isomerism
Neonates with right isomerism typically show single atrium, single right ventricle, and univentricular atrioventricular connection often associated with atrioventricular valve regurgitation. First-stage palliation (2–4 weeks after birth) is a control of pulmonary blood flow with pulmonary banding or systemic-pulmonary shunt. Pulmonary venous obstruction due to total anomalous pulmonary venous drainage should be precisely diagnosed and repaired by surgical operation.
The second-stage palliation is the bidirectional Glenn shunt, where the right and/or left superior vena cava is isolated and is connected to the pulmonary artery. This operation is, in general, performed around 6 months after birth.
The third-stage palliation is Fontan procedure. Recently, a modification using extra-cardiac artificial conduit-type total cavo-pulmonary connection (TCPC) is most often employed, because the long-term prognosis of the conventional atrio-pulmonary connection is proved to be unsatisfactory characterized by enlargement of the atrium, intractable atrial tachyarrhythmias, and thromboembolisms.
After successful completion of the TCPC, cyanosis disappears and the general conditions of the patients improve. However, number of patients who underwent successful Fontan procedure is approximately 50 % because right isomerism often accompanied with combination of severe and complicated congenital heart diseases [16].
3.2 Left Isomerism
Left isomerism i s typically associated with atrioventricular septal defect, persistent left superior vena cava, interrupted hepatic portion of the inferior vena cava, and atrioventricular conduction disturbance. In left isomerism, sinus node and atrioventricular nodes are usually hypoplastic, and sinus bradycardia or complete atrioventricular block is frequently accompanied.
4 Long-Term Prognosis of Heterotaxy Patients
Although the medical and surgical treatments of the heterotaxy syndrome have remarkably advanced, long-term prognosis of the patients remains unsatisfactory. Right isomerism has been recognized as one of the worst forms of CHD with overall 5-year survival ranging from 30 to 74 %. The results are better in left isomerism with 5-year survival rates ranging between 65 and 84 %, which is still considerably lower than survival for most other forms of CHD [17]. The main reason is that the nature of the Fontan single ventricle physiology is fundamentally imperfect. Representative long-term complications of the Fontan operation are illustrated in Fig. 6.2.

Possible complications of mid- to long term after completion of Fontan procedure for heterotaxy patients (Adapted an d modified from Ref. [1] with permission)
4.1 Protein-Losing Enteropathy
Protein-losing enteropathy (PLE), one of the most severe manifestations of the failing Fontan circulation, occurs in 5–10 % of the total postoperative cases [15]. Chronic loss of serum proteins into the gastrointestinal tracts results in systemic edema, ascites, pleural effusion, diarrhea, gastrointestinal bleeding, susceptibility to infections, and ultimately cachexia. The underlying mechanism of PLE remains uncertain. Elevated inflammatory reactions such as TNF-α or IFN-α, dilatation of intestinal lymphatic vessels, and widening between intestinal epithelial cells may be involved in the protein and fluid losses [18]. Steroids, high molecular weight heparin, sildenafil, surgical interventions, for instan ce, fenestration of atrial-level communications or conversion of the Fontan circuit, are effective. To date, cardiac transplantation is considered as the only and complete resolution of PLE pathophysiology.
4.2 Arrhythmias
Reentrant atrial tachyarrhythmias are the commonest in patients after Fontan procedure and are often associated with deterioration of hemodynamics, either causally or as a result [15]. Hemodynamic abnormalities such as valve regurgitation or outflow obstruction, if present, should be aggressively treated by surgery.
4.3 Heart Failure
Initial feature of heart failure long after the Fontan procedure is characterized by worsening of ventricular relaxation and compliance [15]. These abnormalities may be caused by exposure of hypoxia and volume/pressure overload preceding the Fontan procedure, repetitive surgical operations, and hemodynamic disadvantages of the Fontan circuit. These changes are primarily progressive and consequently lead to failure of the Fontan circuit. Late after the Fontan procedure, systolic dysfunction becomes apparent. Administration of angiotensin converting enzyme inhibitors or β-blockades may be beneficial for particular patients, although the clinical evidence and cellular mechanisms remain to be elucidated.
4.4 Hepatic Dysfunction
Hepatic dysfunction, liver fibrosis, and cirrhosis are common complication of patients long after Fontan operation. Recently, cas es with hepatocellular carcinoma after Fontan operation have been reported [19, 20]. Careful observation should be necessary to detect the hepatic changes long after Fontan operation.
4.5 Management of Failing Fontan Patients
Patients who underwent atrio-pulmonary connection or lateral tunnel procedure are likely to be complicated with thromboembolism or intractable arrhythmias due to enlargement of the right atrium [14]. Surgical intervention with conversion to TCPC is required before such complications become irreversible. Cardiac transplantation may be the only option for patients with severe heart failure, intractable arrhythmias, or recurrent PLE.
5 Future Direction and Clinical Implications
In the basic science field, embryonic development of left-right asymmetry has been uncovered by means of mouse genetic engineering. In addition, advanced human genetics have uncovered many responsible genes for heterotaxy syndrome. By means of innovative technologies such as whole genome sequencing or patient-based human inducible pluripotent stem cells, novel genes will be clarified and analyzed. In the clinical field, anatomical and physiological diagnosis from the fetal period, better clinical managements after birth, and tailor-made surgical operations will improve the prognosis. Cell- or tissue-based regeneration therapies and a new ventricular assist device could improve cardiac function of failing Fontan patients. Multiple approaches including basic and clinical science are necessary to improve the prognosis and quality of life of heterotaxy patients.
References
Shiraishi I, Ichikawa H. Human heterotaxy syndrome – from molecular genetics to clinical features, managements, and prognosis. Circ J. 2012;76:2066–75.
Hamada H. Molecular mechanisms of left-right development. Heart development and regeneration. In: Rosenthal N, Harvey RP, editors. London: Academic Press Inc; 2010. vol 1, p. 297–306.
Kawasumi A, Nakamura T, Iwai N, Yashiro K, Saijoh Y, Belo JA, et al. Left-right asymmetry in the level of active Nodal protein produced in the node is translated into left-right asymmetry in the lateral plate of mouse embryos. Dev Biol. 2011;353:321–30.
Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, et al. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–37.
Hirokawa N, Tanaka Y, Okada Y. Cilia, KIF3 molecular motor and nodal flow. Curr Opin Cell Biol. 2012;24:31–9.
Nonaka S, Yoshiba S, Watanabe D, Ikeuchi S, Goto T, Marshall WF, et al. De novo formation of left-right asymmetry by posterior tilt of nodal cilia. PLoS Biol. 2005;3:e268.
Basu B, Brueckner M. Cilia multifunctional organelles at the center of vertebrate left-right asymmetry. Curr Top Dev Biol. 2008;85:151–74.
Okada Y, Nonaka S, Tanaka Y, Saijoh Y, Hamada H, Hirokawa N. Abnormal nodal flow precedes situs inversus in iv and inv mice. Mol Cell. 1999;4:459–68.
Tanaka Y, Okada Y, Hirokawa N. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature. 2005;435(7039):172–7.
McGrath J, Somlo S, Makova S, Tian X, Brueckner M. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell. 2003;114:61–73.
Martin JF, Amendt BA, Brown NA. Heart development and regeneration. In: Rosenthal N, Harvey RP, editors. London: Academic Press Inc., 2010. vol 1. p. 307–22.
Zhu L, Harutyunyan KG, Peng JL, Wang J, Schwartz RJ, Belmont JW. Identification of a novel role of ZIC3 in regulating cardiac development. Hum Mol Genet. 2007;16:1649–60.
Bamford RN, Roessler E, Burdine RD, Saplakoğlu U, dela Cruz J, Splitt M, et al. Loss-of-function mutations in the EGF-CFC gene CFC1 are associated with human left-right laterality defects. Nat Genet. 2000;26:365–9.
Kaasinen E, Aittomäki K, Eronen M, Vahteristo P, Karhu A, Mecklin JP, et al. Recessively inherited right atrial isomerism caused by mutations in growth/differentiation factor 1 (GDF1). Hum Mol Genet. 2010;19:2747–53.
Hutter D, Redington AN. The principles of Management, and outcomes for, patients with functionally univentricular hearts. In: Anderson RH, Baker EJ, Penny DJ, Redington AN, Rigby ML, Wernovsky G, editors. Paediatric cardiology. 3rd ed. Philadelphia: Churchill Livingstone; 2009. p. 687–96.
Ota N, Fujimoto Y, Murata M, Tosaka Y, Ide Y, Tachi M, et al. Improving outcomes of the surgical management of right atrial isomerism. Ann Thorac Surg. 2012;93:832–8.
Delmo Walter EM, Ewert P, Hetzer R, Hübler M, Alexi-Meskishvili V, et al. Biventricular repair in children with complete atrioventricular septal defect and a small left ventricle. Eur J Cardiothorac Surg. 2008;33:40–7.
Serraf A, Bensari N, Houyel L, Capderou A, Roussin R, Lebret E, Ly M, Belli E. Surgical management of congenital heart defects associated with heterotaxy syndrome. Eur J Cardiothorac Surg. 2010;38:721–7.
Ostrow AM, Freeze H, Rychik J. Protein-losing enteropathy after fontan operation: investigations into possible pathophysiologic mechanisms. Ann Thorac Surg. 2006;82:695–700.
Ghaferi AA, Hutchins GM. Progression of liver pathology in patients undergoing the Fontan procedure: chronic passive congestion, cardiac cirrhosis, hepatic adenoma, and hepatocellular carcinoma. J Thorac Cardiovasc Surg. 2005;129:1348–52.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution-Noncommercial 2.5 License (http://creativecommons.org/licenses/by-nc/2.5/), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited. The images or other third party material in this chapter are included in the work's Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work's Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2016 The Author(s)
About this chapter
Cite this chapter
Shiraishi, I. (2016). Left-Right Asymmetry and Human Heterotaxy Syndrome. In: Nakanishi, T., Markwald, R., Baldwin, H., Keller, B., Srivastava, D., Yamagishi, H. (eds) Etiology and Morphogenesis of Congenital Heart Disease. Springer, Tokyo. https://doi.org/10.1007/978-4-431-54628-3_6
Download citation
DOI: https://doi.org/10.1007/978-4-431-54628-3_6
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-54627-6
Online ISBN: 978-4-431-54628-3
eBook Packages: MedicineMedicine (R0)