
Abstract
The heart is the first organ to develop in the embryo, and its formation is an exquisitely regulated process. Inherited mutations in genes required for cardiac development may cause congenital heart disease (CHD), manifested in the newborn or in the adult. Notch is an ancient, highly conserved signaling pathway that communicates adjacent cells to regulate cell fate specification, differentiation, and tissue patterning. Mutations in Notch signaling elements result in cardiac abnormalities in mice and humans, demonstrating an essential role for Notch in heart development. Recent work has shown that endocardial Notch activity orchestrates the early events as well as the patterning and morphogenesis of the ventricular chambers in the mouse and that inactivating mutations in the NOTCH pathway regulator MIND BOMB-1 (MIB1) cause left ventricular non-compaction (LVNC), a cardiomyopathy of poorly understood etiology. Here, we review these data that shed some light on the etiology of LVNC that at least in the case of that caused by MIB1 mutations has a developmental basis.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Heart development begins in the mouse at embryonic day 7 (E7) when pre-cardiac precursor cells move forward bilaterally into the lateral plate mesoderm to form the cardiac crescent [14]. By E8, a linear heart tube is formed after folding of the mesodermal layers from both sides of the embryo, and the two main tissues of the heart are present, the endocardium inside and the outer myocardium. At E9.0, the heart loops and grows by addition of cells from both cardiac poles [20]. At E9.5, the heart divides afterward into developmental domains that would allow the formation of two regions, the valve and the chamber regions [24] (Fig. 12.1a).

Overview of early heart development. (a) Ventral views of the developing mouse embryo. At E7.0, cardiac progenitors (yellow) migrate to the center of the embryo to form the cardiac tube and at E7.5, two cardiac fields can be distinguished: the first heart field (FHF; yellow) and the second heart field (SHF; green). By E8.0, the cardiac tube is formed where endocardium and myocardium are already present. At E9.0 the tube loops and at E9.5, has four anatomically distinct regions: atrium, atrioventricular canal (AVC), ventricle (V), and outflow tract (OFT). Endocardial cushions are formed in valvulogenic regions of the heart (green, OFT and blue, AVC) and trabeculae appear in the ventricles (b). At E15.5, trabeculae are undergoing compaction and there is a thick compact myocardium (c) that becomes very prominent in the adult ventricle that has a smooth surface (d)
We will describe in some detail the process of ventricular chamber development because it is the focus of this review. At E9.5, the process of trabeculation begins in the primitive ventricles. Trabeculae are highly organized sheets of cardiomyocytes that protrude toward the light of the ventricles [3]. Trabeculae are the first structural landmark of the developing ventricles [31]. They increase the ventricular surface and allow the myocardium to grow in the absence of coronary vessels that will be formed later. Trabeculae are important for contractibility, formation of the conduction system, and blood flow direction in the ventricle. Although trabeculae start to form first in the left ventricle and later in the right ventricle, there are no differences between these structures in the two ventricles. Formed trabeculae have no free ends and they make the ventricle appear like a sponge [31] (Fig. 12.1b). From E10.5 onward, the ventricular myocardium has two well-defined regions, morphologically and molecularly, the outer compact myocardium and the inner trabecular layer. The compact myocardium is less differentiated and shows a higher proliferation rate than the trabecular myocardium [31]. Differentiated cardiomyocytes will give rise to the working myocardium and the conduction system [31]. After completion of ventricular septation around E14.5, further growth and maturation of the ventricular chambers require a complete trabecular remodeling and reorganization into an apico-basal orientation that determines the shape of the ventricles (Fig. 12.1c). Trabeculae become compacted and as a result there is a significant change in the ratio of compact vs. trabecular myocardium in the ventricle. Trabeculae compaction is accompanied by the hypoxia-dependent invasion of the coronary vasculature from the epicardium into the compact layer of the myocardium [33]. As they compact, the intertrabecular recesses are transformed into capillaries of the rising coronary plexus. This is followed by a reorganization of the muscle fibers of the heart that organize in a three-layered spiral around the heart reflecting the twisting pattern of heart contraction [31]. Trabeculae stop proliferating while the proliferation of the compact zone sustains the growth of the chamber and the contribution of the compact layer to the heart becomes more significant than the one of the luminal trabeculations that will eventually disappear. How exactly this happens and what molecular mechanisms drive this process is unknown.
2 Left Ventricular Non-compaction (LVNC)
LVNC is a cardiomyopathy of poorly understood etiology that is characterized by prominent and excessive trabeculations with deep recesses in the ventricular wall [18]. LVNC was first diagnosed in 1984, in a 33-year-old woman assessed by echocardiography [10]. Since then, the heterogeneity of its symptoms might have left many LVNC cases undiagnosed [18], giving a lower estimation of its real impact as a cardiomyopathy. The improvement in imaging technologies and the advent of CMRI allow now a better diagnosis of this disease, and 10 years after the first description, LVNC was included in the World Health Organization (WHO) catalogue of cardiomyopathies [30]. In 2006, the American Heart Association included LVNC to its list of genetic cardiomyopathies caused by an arrest of the normal compaction process of the developing myocardium [23]. LVNC can be almost asymptomatic or manifest as depressed systolic function [7], accompanied by systemic embolism, malignant arrhythmias, heart failure, and sudden death [25], although its clinical manifestation and the age of the affected individual at symptom onset are variable. LVNC was shown to be caused by mutations in genes encoding mainly structural proteins of the cardiac muscle, cytoskeleton, nuclear membrane, and chaperone proteins. In general, familial forms of LVNC are transmitted as an autosomal-dominant trait [7]. The underlying molecular mechanisms that produce the structural abnormalities leading to LVNC are still unknown [34].
3 The NOTCH Signaling Pathway
Notch is a conserved signaling pathway that regulates cell fate specification, differentiation, and tissue patterning via local cell interactions [2]. There are four Notch receptors in mammals, Notch1–Notch4, which are expressed in different tissues and at different times during embryonic and adult life. Notch is a large type I transmembrane receptor that has two distinct domains, an extracellular domain (NECD) responsible for the interaction with the ligands and an intracellular domain (NICD) responsible for signal transduction (Fig. 12.2). NECD contains a varied number of tandem arrays of epidermal growth factor (EGF)-like repeats [2]. NICD is composed of the RAM23 domain and 6 ankyrin repetitions responsible for the interaction with RBPJK, the transcriptional effector of the pathway; two nuclear localization signals; a transcriptional activation domain only present in Notch1 and Notch2; and a terminal PEST domain that negatively regulates the stability of the receptor [21]. NECD and NICD are synthesized as a single polypeptide, which is directly cleaved after translation by a furin convertase in the Golgi. This first cleavage [6] is necessary to form the functional heterodimeric receptor that will be translocated to the membrane where the two domains are still associated by Ca2+-dependent non-covalent bonds [28]. In mammals, the Notch ligands belong to the Delta-like (Dll1, Dll3, and Dll4) and Jagged (Jag1 and Jag2) families. The ligands are type I transmembrane proteins and their extracellular domains are composed by a repetition of EGF-like domains responsible for the interaction with the receptor and the DSL (Delta, Serrate, Lin12) domain (Fig. 12.2) [21]. The glycosyltransferase Fringe can modify the EGF-like domain in the NECD of the receptor by adding O-fucose glycans [5] (Fig. 12.2). This modification determines which ligand preferentially binds to the receptor. There is evidence that the presence of Fringe shifts the activation balance toward Delta-like ligands at the expense of Jagged ligands when both molecules are expressed at the same time in the same tissue [4]. The E3 ubiquitin ligase Mind bomb1 (Mib1) regulates the endocytosis of the Notch ligands when bound to NECD, a prerequisite for Notch activation [16]. Mib1 binds to the two families of Notch ligands, and it is a point of regulation of the pathway in the signaling cell. Upon interaction of ligand and receptor, Mib1 targets by ubiquitylation the ligand for endocytosis [16]. The ligands are endocytosed, mechanically pulling NECD producing a conformational change that exposes a second cleavage site [27] allowing a metalloprotease of the ADAM family to cleave NECD [13] that is finally endocytosed into the signaling cell together with the ligand. Immediately after, γ-secretase/Presenilin cleaves the receptor in a third site liberating the NICD in the cytoplasm of the receiving cell [26] (Fig. 12.2). NICD translocates to the nucleus where it binds to the transcription factor CSL or RBPJK via by the RAM23 domain [32]. In the absence of Notch signaling activation, RBPJK is bound to nuclear corepressors that inhibit gene expression. Upon the interaction of NICD with RBPJK, the transcriptional repressors are released and transcriptional activators such as Master mind-like (Maml) [35] are recruited forming a protein complex that activates the expression of target genes (Fig. 12.2).

The Notch signaling pathway. The Notch ligands Delta and Jagged are tethered to the membrane of the signaling cell. On their way to the plasmatic membrane, the Notch receptors are modified in their extracellular domains (NECD) by Fringe. Upon ligand-receptor interaction, Mib1 ubiquitylates the ligand, targeting it for endocytosis. Pulling of NECD allows the cleavage and release of NICD by γ-secretase. NICD translocates to the nucleus and forms a transcriptional activation complex together with RBPJK and MAML that activates target genes transcription, including Hes and Hey that encode transcriptional repressors
4 NOTCH in Ventricular Chamber Development
Notch pathway elements show a complex expression pattern in the developing mouse chambers. At E9.5, Mib1 is expressed in endocardium and myocardium, Dll4 in the endocardium especially at the base of the forming trabeculae, Jag1 in the myocardium, and N1ICD is only active at discrete sites where the trabeculae form [9]. Analysis of standard and endothelial-specific Notch1 and RBPJk mutants shows that trabeculation is severely impaired and only primitive, poorly organized trabeculae can be observed in these mutants [12]. Molecular analysis indicates that during trabeculation, Notch modulates three different signals: EphrinB2 [1], expressed in the endocardium, is a direct Notch target. Nrg1 [15], also expressed in the endocardium, is an indirect Notch target, and its expression depends both of Notch and EphrinB2 [12]. Nrg1 induces the formation of the ventricular conduction system, a feature of ventricular maturation [29]. A third signaling pathway is Bmp10 [8], expressed in trabecular myocardium and whose expression is severely downregulated in Notch pathway mutants, suggesting that Notch is required to produce a signal that activates Bmp10 in the myocardium [12]. During these early stages, the ligand Dll4 is the main Notch activator in the endocardium [12] (Fig. 12.3).

Proposed mechanism of Notch function in trabeculation. (a, b detail) Dll4 activates Notch1 at the base of the developing trabeculae. N1ICD/RBPJK activates EphrinB2(Efnb2)/EphB4 signaling in the endocardium, which in turn is required for Nrg1 expression in this tissue. Nrg1 activates ErbB2/ErbB4 signaling in myocardium to promote cardiomyocyte differentiation. In addition, Notch1 signaling in endocardium is required for Bmp10 expression and therefore proliferation of trabecular cardiomyocytes (Modified from [12])
To precisely determine the contribution of Notch ligands to ventricular chamber development, we bred mice bearing a conditional allele of Mib1 with myocardium-specific Cre driver line cTnT-Cre [19]. Histological examination at E16.5 revealed that Mib1flox;cTnT-Cre mutants had a dilated heart with a thin compact myocardium and large, non-compacted trabeculae, protruding toward the ventricular lumen. Morphological and functional analysis using echocardiography and cardiac magnetic resonance imaging (CMRI) showed prominent trabeculations, deep intertrabecular recesses, and a significantly reduced ejection fraction in mutant mice [22]. The hearts of these mice had a ratio of non-compacted to compacted myocardium (non-compaction index, NC) of 2.0, a feature diagnostic of LVNC in humans [11, 17]. These features are all strongly reminiscent of LVNC, establishing Mib1flox;cTnT-Cre mice as the first animal model of LVNC [22]. Analysis of chamber markers in E15.5 Mib1flox;cTnT-Cre mice revealed expansion of various compact myocardium markers (Hey2, Tbx20 and n-myc) to the trabeculae and reduced expression of the trabecular markers Anf, Bmp10, and Cx40. These markers were normally expressed at earlier stages, suggesting that maintenance of trabecular maturation and patterning is impaired in Mib1flox;cTnT-Cre mutants. Likewise, lost or reduced Hey1, Hey3, and EphrinB2 expression in the vessels of the compact myocardium suggested that coronary artery development was defective. Proliferation analysis revealed increased proliferation of trabecular cardiomyocytes in the hearts of E15.5 Mib1flox;cTnT-Cre embryos, suggesting this as the cause of the enlarged, non-compacted trabeculae in these mutants. RNA sequencing analysis of E14.5 Mib1flox;cTnT-Cre mutant ventricles identified altered expression of 315 genes (132 upregulated and 183 downregulated). The expression of genes involved in the differentiation of cardiac endothelium/endocardium and cardiomyocytes and coronary vasculogenesis was altered. RNA-Seq data also confirmed the in situ hybridization analysis and demonstrated that Mib1 inactivation in the myocardium disrupts the differentiation and maturation of cardiac endothelial cells and cardiomyocytes. This may impact in turn the process of ventricular maturation and compaction. Genetic ablation of Mib1 in the myocardium thus leads to LVNC cardiomyopathy in mice by arresting ventricular trabeculae maturation and compaction and increasing cardiomyocyte proliferation during fetal development [22].
We next examined whether mutations in the human MIB1 homologue were associated with clinical LVNC and identified two mutations in a cohort of 100 Southern European LVNC cases; one was a heterozygous G to T transversion of nucleotide 2827 in exon 20 (causing a change in amino acid 943 from valine to phenylalanine, the p.Val943Phe mutation). Val943 is located within a region that mediates protein-protein interactions and constitutes the site of MIB1 ubiquitin ligase activity. The second mutation was a heterozygous C to T transition of nucleotide 1587 in MIB1 exon 11 (causing a premature stop codon instead of arginine at position 530 in the MIB1 ankyrin repeats region, the p.Arg530X mutation). These two mutations were tracked back through three and two generations of LVNC-affected individuals, revealing co-segregation with LVNC, confirming the hereditary nature of the disease [22].
In silico modeling suggested that MIB1 is a homodimer where the p.Val943Phe mutant will alter the alignment of the ring finger domains in heterodimers formed by wild-type and mutant p.Val943Phe MIB1, as well as the angle of interaction with JAG1 of the p.Val943Phe wild-type dimers. This possibility was supported by co-immunoprecipitation experiments with tagged wild-type MIB1 and mutant MIB1 isoforms co-transfected in pair-wise combinations into HEK293 cells. The effect of the MIB1 mutations on Jag1 ubiquitylation was tested also in HEK293 cells. Jag1 ubiquitylation was strongly reduced in cells co-transfected with wild-type MIB1 plus p.Val934Phe or wild-type MIB1 plus the p.Arg530X mutant [22].
Lastly, to study the effect of these mutant MIB1 variants in vivo, their mRNAs were microinjected into zebra fish embryos expressing GFP in the developing myocardium. Examination of 72 hpf larvae injected with p.Val943Phe or p.Arg530X mRNAs revealed severely disrupted embryonic development, disrupted cardiac looping, and kinked tail, typical of defective Notch activity. To complement these findings, we cocultured Jag1-HEK293 cells expressing MIB1 variants with Notch1-expressing HEK293 cells co-transfected with a Notch luciferase reporter. Jag1-HEK293 cells expressing wild-type MIB1 increased Notch reporter activity, whereas the p.Val934Phe and p.Arg530X mutant forms reduced reporter activity, indicating that both human mutations disrupt Notch signaling [22].
Accumulated data suggested that both MIB1 mutations result in loss of MIB1 WT function. In the case of p.Arg530X, this would be due to haploinsufficiency caused by insufficient synthesis of WT MIB1 protein; in the case of p.Val934Phe, this would be due to a dominant-negative effect of the mutant protein, titrating down the amount of functional WT MIB1 dimers through heterotypic or homotypic interactions. In both cases, loss of MIB1 function leads to disease inherited in an autosomal-dominant fashion.
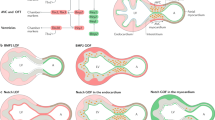
As to the role of Mib1 in ventricular chamber development, we have proposed that myocardial Mib1 activity enables Jag1-mediated activation of Notch1 in the endocardium, to sustain trabeculae patterning, maturation, and compaction [22] and (Fig. 12.4). Abrogation of Mib1-mediated signaling in the myocardium disrupts trabeculae maturation and patterning, arresting the development of chamber myocardium and resulting in LVNC (Fig. 12.4). The expansion of compact zone markers to the trabeculae of E15.5 Mib1flox;cTnT-Cre mutants further suggests that trabeculae patterning and maturation is impaired (Fig. 12.4). In addition, the disruption of coronary vessel markers in Mib1flox;cTnT-Cre mutants indicates that abrogation of myocardial-endocardial Notch signaling indirectly impairs coronary vessel formation.

Proposed mechanism of Notch function in trabecular maturation and compaction. (a) In wild-type (WT) embryos, ubiquitylation of Jag1 (and another ligand?) by Mib1 in the myocardium allows Notch1 activation in the endocardium. N1ICD is required to sustain trabecular patterning, maturation, and compaction. The compact myocardium (expressing Hey2, Tbx20 and n-myc) proliferates actively, unlike the trabecular myocardium (expressing Anf, Bmp10 and Cx40). Notch-dependent chamber maturation leads to compacted ventricular myocardium in the adult mouse. (b) In Mib1flox; cTnT-Cre mutants, Notch1 activity is impaired and compact myocardium markers (Hey2, Tbx20 and n-myc) are consequently expanded to the trabeculae, which remain abnormally proliferative. The resulting disruption of trabecular patterning, maturation, and compaction manifests as LVNC (Modified from [22])
5 Future Directions and Clinical Implications
The data reviewed here establish the causal role of NOTCH dysregulation in LVNC, a congenital cardiomyopathy that results from a developmental arrest in ventricular maturation and myocardial compaction. These findings will lead to a better diagnosis and risk stratification, allowing timely intervention in LVNC-associated complications. In addition, since other NOTCH pathway elements may be involved in LVNC, they could serve as novel diagnostic or therapeutic disease targets. Further research is required to determine the molecular mechanisms and regulatory interactions underlying NOTCH function in ventricular chamber development using genetically modified mouse models and the full implication of altered NOTCH signaling in human LVNC.
References
Adams RH, Wilkinson GA, Weiss C, et al. Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999;13:295–306.
Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–6.
Ben-Shachar G, Arcilla RA, Lucas RV, et al. Ventricular trabeculations in the chick embryo heart and their contribution to ventricular and muscular septal development. Circ Res. 1985;57:759–66.
Benedito R, Roca C, Sorensen I, et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137:1124–35.
Blair SS. Notch signaling: fringe really is a glycosyltransferase. Curr Biol. 2000;10:R608–12.
Blaumueller CM, Qi H, Zagouras P, et al. Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell. 1997;90:281–91.
Captur G, Nihoyannopoulos P. Left ventricular non-compaction: genetic heterogeneity, diagnosis and clinical course. Int J Cardiol. 2010;140:145–53.
Chen H, Shi S, Acosta L, et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development. 2004;131:2219–31.
Del Monte G, Grego-Bessa J, Gonzalez-Rajal A, et al. Monitoring Notch1 activity in development: evidence for a feedback regulatory loop. Dev Dyn. 2007;236:2594–614.
Engberding R, Bender F. Identification of a rare congenital anomaly of the myocardium by two-dimensional echocardiography: persistence of isolated myocardial sinusoids. Am J Cardiol. 1984;53:1733–4.
Finsterer J, Stollberger C. Heterogenous myopathic background of left ventricular hypertrabeculation/noncompaction. Am J Med Genet A. 2004;131:221; author reply 222-223.
Grego-Bessa J, Luna-Zurita L, Del Monte G, et al. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007;12:415–29.
Hartmann D, De Strooper B, Serneels L, et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet. 2002;11:2615–24.
Harvey RP. Patterning the vertebrate heart. Nat Rev Genet. 2002;3:544–56.
Hertig CM, Kubalak SW, Wang Y, et al. Synergistic roles of neuregulin-1 and insulin-like growth factor-I in activation of the phosphatidylinositol 3-kinase pathway and cardiac chamber morphogenesis. J Biol Chem. 1999;274:37362–9.
Itoh M, Kim CH, Palardy G, et al. Mind bomb is a ubiquitin ligase that is essential for efficient activation of Notch signaling by Delta. Dev Cell. 2003;4:67–82.
Jenni R, Oechslin E, Schneider J, et al. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart. 2001;86:666–71.
Jenni R, Oechslin EN, Van Der Loo B. Isolated ventricular non-compaction of the myocardium in adults. Heart. 2007;93:11–5.
Jiao K, Kulessa H, Tompkins K, et al. An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003;17:2362–7.
Kelly RG, Buckingham ME. The anterior heart-forming field: voyage to the arterial pole of the heart. Trends Genet. 2002;18:210–6.
Kopan R. Notch: a membrane-bound transcription factor. J Cell Sci. 2002;115:1095–7.
Luxan G, Casanova JC, Martinez-Poveda B, et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med. 2013;19:193–201.
Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–16.
Moorman AF, Christoffels VM. Cardiac chamber formation: development, genes, and evolution. Physiol Rev. 2003;83:1223–67.
Oechslin EN, Attenhofer Jost CH, Rojas JR, et al. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol. 2000;36:493–500.
Okochi M, Steiner H, Fukumori A, et al. Presenilins mediate a dual intramembranous gamma-secretase cleavage of Notch-1. EMBO J. 2002;21:5408–16.
Parks AL, Klueg KM, Stout JR, et al. Ligand endocytosis drives receptor dissociation and activation in the Notch pathway. Development. 2000;127:1373–85.
Rand MD, Grimm LM, Artavanis-Tsakonas S, et al. Calcium depletion dissociates and activates heterodimeric notch receptors. Mol Cell Biol. 2000;20:1825–35.
Rentschler S, Zander J, Meyers K, et al. Neuregulin-1 promotes formation of the murine cardiac conduction system. Proc Natl Acad Sci U S A. 2002;99:10464–9.
Richardson P, Mckenna W, Bristow M, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation. 1996;93:841–2.
Sedmera D, Pexieder T, Vuillemin M, et al. Developmental patterning of the myocardium. Anat Rec. 2000;258:319–37.
Tamura K, Taniguchi Y, Minoguchi S, et al. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H). Curr Biol. 1995;5:1416–23.
Tao J, Doughman Y, Yang K, et al. Epicardial HIF signaling regulates vascular precursor cell invasion into the myocardium. Dev Biol. 2013;376:136–49.
Towbin JA. Left ventricular noncompaction: a new form of heart failure. Heart Fail Clin. 2010;6:453–69. viii.
Wu L, Aster JC, Blacklow SC, et al. MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat Genet. 2000;26:484–9.
Acknowledgments
We apologize to those authors whose work was not cited. J.L. de la Pompa is funded by grants SAF2013-45543, RD12/0042/0005 (RIC), and RD12/0019/0003 (TERCEL) from the Spanish Ministry of Economy and Competitiveness (MINECO) and grant FP7-ITN 215761 (NotchIT) from the European Commission. G. Luxán had a PhD fellowship from the MINECO (FPI Program, ref. BES-2008-002904) and G. D’Amato holds a PhD fellowship associated with grant FP7-ITN 215761 (NotchIT). The CNIC is supported by the MINECO and the Pro-CNIC Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution-Noncommercial 2.5 License (http://creativecommons.org/licenses/by-nc/2.5/), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited. The images or other third party material in this chapter are included in the work's Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work's Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2016 The Author(s)
About this chapter
Cite this chapter
Luxán, G., D’Amato, G., de la Pompa, J.L. (2016). Intercellular Signaling in Cardiac Development and Disease: The NOTCH pathway. In: Nakanishi, T., Markwald, R., Baldwin, H., Keller, B., Srivastava, D., Yamagishi, H. (eds) Etiology and Morphogenesis of Congenital Heart Disease. Springer, Tokyo. https://doi.org/10.1007/978-4-431-54628-3_12
Download citation
DOI: https://doi.org/10.1007/978-4-431-54628-3_12
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-54627-6
Online ISBN: 978-4-431-54628-3
eBook Packages: MedicineMedicine (R0)