
Abstract
Chronic kidney disease (CKD) reflects life events that range from maternal-fetal influences to geriatric exposures. The global direct and indirect costs of CKD are high and include maternal-neonatal hospitalization and treatment, acute kidney injury, dialysis and transplant, missed work, and medications, to name a few. The impact of poor diet, adverse childhood experiences, medication use, and failure to follow consistent public health standards are increasingly appreciated as key influences in the development of CKD. Socioeconomic factors can significantly influence the timing and phenotypic expression in people at risk for developing CKD, although more research is needed to understand these mechanisms. In general, biomedicine has been focused on treating well-established CKD morbidity. This strategy has been short sighted and costly. A more cost-effective approach would focus on early life interventions that hold the potential for mitigating CKD risk and its sequelae. This chapter applies the life course health development principles to review determinants and pathways for CKD evolution and identifies of the gaps in our knowledgebase. We also discuss several research strategies for evaluating the life course health development of CKD.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Chronic kidney disease (CKD)
- Acute kidney injury
- Renal development
- Socioeconomic status
- Nephron mass
- Transplant
- Dialysis
- Maternal
- Research
1 Introduction
Chronic kidney disease (CKD) impacts approximately 650,000 Americans, and health-related costs exceed 28 billion dollars per year (USRDS 2003; Wyld et al. 2015; Feldman et al. 2003; Furth et al. 2006). Given that many patients receive kidney transplants between 20 and 71, it is likely that some of these individuals (particularly the young adults) developed the initial stages of CKD early in life (USRDS 2003; Wyld et al. 2015; Feldman et al. 2003; Furth et al. 2006). Infants and children who develop CKD are at significant risk for associated health problems beyond those directly attributable to kidney disease because of the manifold effects of CKD on health development (Furth et al. 2006).
The risk factors and natural history for CKD progression in infants and children are not well understood (Furth et al. 2006). Currently, North American and European investigators are monitoring childhood cohorts of patients with CKD in order to better understand the natural progression and treatment of CKD and to identify significant risk factors for developing progressive CKD (Furth et al. 2006; Wong et al. 2012; Querfeld et al. 2010; ESCAPE Trial Group et al. 2009). In the same manner, the Chronic Renal Insufficiency Cohort Study (Lash et al. 2011) is identifying characteristics of patients associated with CKD among patients 21–74 years old. These population-based studies are providing data that will enable research to elucidate variables associated with progression of CKD, thereby permitting development of early intervention strategies. Given that the risk of mortality from cardiovascular diseases increases with a declining renal function status in individuals, any insights on reducing the risks or rates of CKD may have also influence cardiac morbidity and mortality in this population (Thompson et al. 2015).
Longitudinal studies of CKD progression and consequences are critical. Renal functional plasticity is substantial in infants, children, and even adults, although it peaks during fetal development. This means that it is possible to alter the negative effects of environmental and other inimical factors that threaten kidney development and function. In adult patients, early identification of modifiable risk factors (including diet) for CKD progression may have profound effects on their outcomes.
The life course health development principles (see Halfon and Forrest 2017) when applied to CKD suggest a set of mechanisms of disease progression and health development , which we will explore more fully throughout this chapter . These include:
-
1.
The need to develop measures of kidney health and developmental plasticity to provide outcome measures that can be evaluated with respect to prevention efforts (Principle 1)
-
2.
The importance of evaluating how kidney health and chronic kidney disease change over the life span (Principle 2)
-
3.
The relationships between gene regulation, environmental exposures, and change in CKD risk over time, as they dynamically interact with one another (Principle 3)
-
4.
Studying how social factors, particularly those related to risk for premature delivery and low birth rate, influence kidney health and chronic kidney disease risk (Principle 4)
-
5.
Understanding the constraints that evolution places on the range of developmental pathways for kidney development and plasticity (Principle 5)
-
6.
The effects of optimal kidney health development on long-term well-being and quality of life for individuals and populations (Principle 6)
-
7.
Elucidating how the synchronization of biological “clocks” with cultural and environmental exposures influences the health development of kidney function (Principle 7)
Life course health development points to the need to study the dynamic interactions between individuals and their physical and social environmental exposures from gestation through to adult life. The principles also suggest that studies on cross-generational as well as individual biological, behavioral, psychosocial, and environmental determinants are important in understanding the development of kidney health and kidney disease (Ben-Shlomo and Kuh 2002).
The CKD framework as defined by the Kidney Disease Outcomes Quality Initiative (KDOQI) provides evidence-based guidelines for management of CKD (National Kidney Foundation; KDOQI (Kidney Disease Outcomes Quality Initiative) 2012). Given the already significant healthcare and societal costs of CKD, there is a need to reframe how we investigate the life course health development determinants of this chronic disease and the ways in which we deliver care in terms of prevention as well as management of extant disease. A clear understanding of these determinants can inform prevention and disease management programs which will have beneficial effects on patient health outcomes and costs of care.
Although diabetes is the single most important cause of CKD and end-stage renal disease in the adult population in the USA, congenital abnormalities of the kidney and urinary tract and reduced nephron endowment secondary to prematurity, intrauterine growth retardation, or early renal injury are the biggest risk factors for CKD that emerges during childhood (NAPRTCS 2011). The following sections of this chapter employ a life course health development framework to guide future research and public policy regarding CKD. The ability to use health services and outcomes research approaches to test public policy effects on health determinants along with the traditional basic, clinical, and translational science methods including the “omics” approach (Chen et al. 2012) provides a path forward within the innovative framework of the life course health development approach (Fig. 1).

Chronic kidney disease life course omics map
2 Preconceptional and Gestational Determinants of CKD
2.1 Preconception
The influence of the intrauterine environment in supporting optimal fetal development, including the developing kidney and metabolic regulatory processes , has lifelong implications for hypertension risk and renal health in later life (Bagby 2015). The impact of nutritional state appears to be multigenerational. In fact, the nutritional state of a woman from the time of her own conception (hence her mother’s nutritional status) can influence how well she can nourish a fetus. Animal studies demonstrate that poor preconceptual nutrition and certain medication exposures both can alter allocation of kidney cell mass and yield reduced nephron number in offspring (Bagby 2015). Key considerations for prevention include developing ways to motivate young girls and women to adopt nutritional supplementation in the preconception period when there are clear benefits for doing so. There is increasing evidence (human and animal studies) that even modest levels of vitamin A deficiency can reduce nephron number in developing fetuses (Puddu et al. 2009). These simple and easily treated risks should not be overlooked. In fact, a recently published hospital-based, case-control study found an association among plasma lycopene, analgesic use, and CKD. This study showed that the higher the plasma lycopene was, the lower the CKD risk, whereas elevated retinol levels were associated with a higher risk of CKD in the context of analgesic use. Thus, while there may be clear benefits in preconception and early fetal life of adequate vitamin A levels , these same micronutrient levels may have differential effects in adult patients at risk for CKD development or progression. Further research on micronutrients and how and when they are delivered (either enterally or parenterally) is an important and cost-effective method of understanding the development of CKD (Chiang et al. 2010).
In a recent landmark study, a group of investigators hypothesized that the risk of CKD may be influenced prenatally. These investigators conducted a population-based, case-control study with 1994 patients with childhood CKD (<21 years of age at diagnosis) and 20,032 controls in order to examine the association of childhood CKD with prenatal risk factors, including fetal growth, maternal diabetes mellitus , and maternal overweight/obesity . Maternal and infant hospital discharge records were linked from 1987 to 2008. Results showed that CKD prevalence in this cohort was 127 cases per 100,000 births. The adjusted odds ratios for CKD associated with prenatal factors were 2.9 (95% CI, 2.3–3.6) for low birth weight, 1.5 (95% CI, 1.1–2.1) for maternal gestational diabetes mellitus , 1.2 (95% CI, 1.1–1.5) for maternal overweight/obesity, and 1.3 (95%CI, 1.05–1.52) for maternal obesity. CKD subtype analysis revealed low birth weight and maternal pregestational diabetes mellitus were associated with increased risk of renal dysplasia/aplasia, whereas poor fetal growth, maternal gestational diabetes mellitus, and maternal overweight/obesity were associated with obstructive uropathy. These findings suggest that targeted modification of these risk factors may reduce the risk of childhood CKD. Such interventions should be a focus of future investigations (Hsu et al. 2014).
Theoretical, experimental, and observational data suggest that there is an increased risk of CKD for infants born prematurely without specific renal birth defects. Indeed, a recent study evaluated 426 participants of the Chronic Kidney Disease in Children study cohort to determine whether low birth weight (<2500 grams), prematurity (gestational age < 36 weeks), small for gestational age (birth weight < 10th percentile for gestational age), or requirement for intensive care unit (ICU) care at birth were risk factors for poor growth outcomes in children identified with subsequent CKD – defined as a median baseline glomerular filtration rate (GFR) of 42.9 ml/min per 1.73 m2. The study found higher than expected prevalences of these risk factors: small for gestational age (14%), low birth weight (17%), prematurity (12%), and neonatal intensive care unit stays (40%) in patients from this CKD cohort. These results led investigators to conclude that small for gestational age and low birth weight are novel risk factors for CKD (Greenbaum et al. 2011).
2.2 Evidence from Animal Models
In rodents, in which nephrogenesis normally continues for 5–7 days postnatally, premature birth has a profound effect on kidney development. Mice born 1–2 days prematurely develop a CKD phenotype by the time they are 5 weeks old, exhibiting hypertension, albuminuria, and reduced nephron number (Stelloh et al. 2012). In other mouse studies, prenatal exposure to low protein and low vitamin A supply, perinatal exposure to gentamicin, and antenatal exposure to steroids all reduce nephron number in offspring (Gilbert et al. 1990; Lelievre-Pegorier et al. 1998; Sutherland et al. 2012a; Ortiz et al. 2001). Rats with 5/6 nephrectomy have been used as a model system to study the mechanisms by which low nephron number results in glomerular hyperfiltration, hypertension, albuminuria, and glomerulosclerosis (Brenner and Anderson 1987). Glomerular hyperfiltration and mechanical stress may contribute to podocyte loss and apoptosis (Reidy and Kaskel 2007; Fogo 2007). Activation of the renin-angiotensin system and TGF-beta has been implicated in podocyte loss and progressive glomerulosclerosis (Reidy and Kaskel 2007; Lopez-Hernandez and Lopez-Novoa 2012).
Other intriguing evidence comes from the baboon model of prematurity in which animals are delivered prematurely and maintained under conditions that very closely approximate the neonatal intensive care unit – including mechanical ventilation and treatment with nephrotoxins such as gentamicin for infections (Gubhaju et al. 2009) and ibuprofen for patent ductus arteriosus (PDA) closure (Sutherland et al. 2012b). Analogous to human studies, the nephrons of the premature baboons continue to develop postnatally, but with a high percentage of histological abnormalities (Gubhaju et al. 2009).
2.3 Prematurity : An Under-recognized Risk Factor for CKD
To date, there have been no prospective, longitudinal population-based prospective cohort studies to confirm the association between prematurity and CKD. However, evidence from well-designed retrospective, longitudinal studies has become increasingly abundant. A systematic review of 31 retrospective cohort or case-control studies found a 70% increase in the risk of CKD by adulthood for infants with a birth weight of <2.5 kg (White et al. 2009).
Clinical signs of oligonephropathy (i.e., low nephron number and mass) among patients born prematurely can often be detected in childhood. Two case series (each including 50 infants born at less than 30–32 weeks gestation) found that children born prematurely had smaller kidneys and higher blood pressure compared with full-term controls, even though their GFR remained normal (Bacchetta et al. 2009; Keijzer-Veen et al. 2010). Microalbuminuria – an early and sensitive indicator of kidney disease and a marker for future cardiovascular morbidity (Rademacher and Sinaiko 2009) – is also common among children 8–11 years old who were born prematurely or with low birth weight (Salgado et al. 2009) (Centers for Disease Control and Prevention (CDC) 2007).
A single case series of six patients with a history of prematurity who developed secondary focal glomerulosclerosis at an average age of 32 years old has been published (Hodgin et al. 2009a), providing an example of prematurity’s association with CKD later in life. These individuals were all noted to be born prematurely (gestational ages of 22–30 weeks) and at the time of their presentation had nephrotic-range proteinuria, hypoalbuminemia, and lack of edema without any other risk factors for secondary focal glomerulosclerosis , suggesting that the premature infants are at higher risk for developing focal glomerulosclerosis later in life and raising the possibility of closer renal follow-up for such patients.
In infancy/childhood, the most overt cause of CKD and end-stage renal disease is birth defects or congenital anomalies of the kidney and urinary tract that result in inadequate renal mass to provide internal physiologic homeostasis for the provision of growth and development (NAPRTCS 2011; Chen et al. 2012; Bagby 2015; Puddu et al. 2009; Chiang et al. 2010; Hsu et al. 2014; Greenbaum et al. 2011; Horbar et al. 2012; Hack et al. 2002; Stelloh et al. 2012; Gilbert et al. 1990; Lelievre-Pegorier et al. 1998; Sutherland et al. 2012a; Ortiz et al. 2001; Brenner and Anderson 1987; Reidy and Kaskel 2007; Fogo 2007; Lopez-Hernandez and Lopez-Novoa 2012; Gubhaju et al. 2009; Sutherland et al. 2012b; White et al. 2009; Bacchetta et al. 2009; Keijzer-Veen et al. 2010; Rademacher and Sinaiko 2009; Salgado et al. 2009; Centers for Disease Control and Prevention (CDC) 2007; Hodgin et al. 2009a; Harambat et al. 2012; Warady and Chadha 2007; Tabel et al. 2010; Sanna-Cherchi et al. 2009). The long-term consequences of prematurity on renal mass are more subtle but, as mentioned above, are becoming increasingly clear. The advances in neonatal intensive care in the past 50 years have been nothing less than remarkable: according to the most recent data from the Vermont Oxford Network , nearly 90% of infants born weighing 501–1500 grams survive to neonatal intensive care unit (NICU) discharge, and nearly 60% of these very low birth weight survivors leave the NICU without any major neonatal morbidity (Horbar et al. 2012). It is clear that today, premature infants who in another era would have died within a matter of hours are now surviving to adulthood. While there has been a great deal of research into the neurodevelopmental outcomes of premature infants (Hack et al. 2002), the impact of premature birth on other organ systems is less well understood. There is emerging evidence from basic science and clinical research to suggest that both prematurity and medical treatment for its consequences may have serious long-term effects on kidney health development (Greenbaum et al. 2011). The implications of these studies are that the development of CKD and subsequent growth are influenced by maternal preconceptual nutritional status , premature birth, low birth weight, and small for gestational age. These early risk factors merit much more animal and human subject research to understand the specific mechanisms by which they influence the health development of the kidney. Indeed, the conceptualization of gestational renal mass development and its impact on later renal function and sequelae of CKD development continues to be an area of some controversy as well as active investigation.
2.4 Renal Development and Genetic Abnormalities
The congenital anomalies of the kidney and urinary tract spectrum represent relatively common birth defects, present in up to 1 in 500 live births, and are the leading causes of chronic kidney and end-stage renal disease in childhood. Hypoplastic or dysplastic kidneys , obstructive uropathy , and vesicoureteral reflux (VUR) account for 30% of children on dialysis in the USA (NAPRTCS 2011; Chen et al. 2012; Bagby 2015; Puddu et al. 2009; Chiang et al. 2010; Hsu et al. 2014; Greenbaum et al. 2011; Horbar et al. 2012; Hack et al. 2002; Stelloh et al. 2012; Gilbert et al. 1990; Lelievre-Pegorier et al. 1998; Sutherland et al. 2012a; Ortiz et al. 2001; Brenner and Anderson 1987; Reidy and Kaskel 2007; Fogo2007; Lopez-Hernandez and Lopez-Novoa 2012; Gubhaju et al. 2009; Sutherland et al. 2012b; White et al. 2009; Bacchetta et al. 2009; Keijzer-Veen et al. 2010; Rademacher and Sinaiko 2009; Salgado et al. 2009; Centers for Disease Control and Prevention (CDC) 2007; Hodgin et al. 2009a; Harambat et al. 2012; Warady and Chadha 2007; Tabel et al. 2010; Sanna-Cherchi et al. 2009). The development of end-stage renal disease in infancy/childhood results in poor growth and altered health development. Mortality rates among children with end-stage renal disease are 30–150 times higher than age-matched children in the general population (Warady and Chadha 2007). Moreover, kidney and urinary tract malformations that do not result in end-stage renal disease during childhood are risk factors for CKD during adulthood and contribute to significant healthcare and societal costs (Sanna-Cherchi et al. 2009; Westland et al. 2011; Spencer et al. 2011). Genetic studies of children with congenital anomalies of the kidney and urinary tract and animal studies have enabled investigators to identify multiple molecular signaling pathways that are required for normal kidney and urinary tract development. These include Wnts , bone morphogenetic proteins (BMPs) , fibroblast growth factor (FGF) , sonic hedgehog , RET/glial cell-derived neurotrophic factor , and Notch pathways (Horbar et al. 2012; Hack et al. 2002; Stelloh et al. 2012; Gilbert et al. 1990; Lelievre-Pegorier et al. 1998; Sutherland et al. 2012a; Ortiz et al. 2001; Brenner and Anderson 1987; Reidy and Kaskel 2007; Fogo 2007; Lopez-Hernandez and Lopez-Novoa 2012; Gubhaju et al. 2009; Sutherland et al. 2012b; White et al. 2009; Bacchetta et al. 2009; Keijzer-Veen et al. 2010; Rademacher and Sinaiko 2009; Salgado et al. 2009; Centers for Disease Control and Prevention (CDC) 2007; Hodgin et al. 2009a; Harambat et al. 2012; Warady and Chadha 2007; Tabel et al. 2010; Sanna-Cherchi et al. 2009; Westland et al. 2011; Spencer et al. 2011; Bertoli-Avella et al. 2008; Nishimura et al. 1999; Song and Yosypiv 2011; Paces-Fessy et al. 2012; Reidy and Rosenblum 2009). These signaling pathways regulate the outgrowth of the ureteric bud (from the Wolffian duct), the subsequent ureteric bud branching, and the reciprocal interactions between the tips of the ureteric bud and metanephric mesenchyme that lead to nephron and hence kidney formation. Abnormalities in these pathways lead to the spectrum of congenital anomalies of the kidney and urinary tract (Reidy and Rosenblum 2009). While the majority of over 70 genes identified are associated with syndromes involving multiple congenital anomalies, human genetic studies have confirmed a role for defects in several genes in non-syndromic congenital anomalies of the kidney and urinary tract , including HNF1, PAX2, ROBO2, GDNF, RET, SPRY1, FREM, FRET, and the angiotensin receptor 2 gene (Horbar et al. 2012; Hack et al. 2002; Stelloh et al. 2012; Gilbert et al. 1990; Lelievre-Pegorier et al. 1998; Sutherland et al. 2012a; Ortiz et al. 2001; Brenner and Anderson 1987; Reidy and Kaskel 2007; Fogo 2007; Lopez-Hernandez and Lopez-Novoa 2012; Gubhaju et al. 2009; Sutherland et al. 2012b; White et al. 2009; Bacchetta et al. 2009; Keijzer-Veen et al. 2010; Rademacher and Sinaiko 2009; Salgado et al. 2009; Centers for Disease Control and Prevention (CDC) 2007; Hodgin et al. 2009a; Harambat et al. 2012; Warady and Chadha 2007; Tabel et al. 2010; Sanna-Cherchi et al. 2009; Westland et al. 2011; Spencer et al. 2011; Bertoli-Avella et al. 2008; Nishimura et al. 1999; Song and Yosypiv 2011; Paces-Fessy et al. 2012; Chatterjee et al. 2012; Nakayama et al. 2010; Saisawat et al. 2012; Weber et al. 2006). In addition to genetic mutations , prenatal environmental influences such as exposures to renin-angiotensin inhibition or antiepileptic medications may lead to congenital anomalies of the kidney and urinary tract (Carta et al. 2007; Serreau et al. 2005). Mouse models have revealed that the interactions between different signaling pathways are complex and the effects of loss of gene function are time and cell-lineage specific (Paces-Fessy et al. 2012; Reidy and Rosenblum 2009; Sims-Lucas et al. 2012; Schedl and Hastie 2000).
Polycystic kidney disease is the most common and best-studied genetic disease associated with CKD. Autosomal recessive polycystic kidney disease, a relatively rare genetic disorder, occurs in approximately 1 in 20,000 individuals, affecting males and females equally; it commonly presents in the perinatal period; and, it has a high morbidity and mortality rate in the first month of life (Cramer and Guay-Woodford 2015). Autosomal-dominant polycystic kidney disease is the most prevalent inherited renal disease and one of the most common life-threatening genetic diseases.
Autosomal-dominant polycystic kidney disease affects approximately 1 in 500 individuals and does not skip generations. It is often characterized by multiple renal cysts that can eventually lead to kidney failure and may present from birth all the way through to old age. This presentation may be impacted by underlying genetic, epigenetic, and environmental modifying factors. Given the variable phenotypic presentation, autosomal-dominant polycystic kidney disease provides a visual framework for the life course health development approach to care including targets for research and associations with cardiovascular disease and hypertension (see Fig. 2 (Takamitsu and Darwin Bell 2015)).

Life course specific pathways to chronic kidney disease
The second leading cause of CKD in childhood is focal glomerulosclerosis, comprising 14% of children with end-stage renal disease in the USA (NAPRTCS 2011). Populations such as Aboriginal Australians that have low nephron numbers at birth have increased susceptibility to glomerular diseases, especially focal glomerulosclerosis, and there are reports of focal glomerulosclerosis in isolated patients with decreased nephron mass due to oligonephronia or traumatic injury (Hoy et al. 1999, 2010; 2012; Mallick 2003; Hodgin et al. 2009b), suggesting that low nephron number may increase the risk for glomerular disease and CKD (Lackland et al. 2000; Furth et al. 2011; Hallan et al. 2008; Zandi-Nejad et al. 2006).
2.5 Fetal Origins of Adult Disease
David Barker is credited with the observation that many “adult” diseases in fact have their origins in fetal life (Barker and Sir Richard Doll Lecture 2012). To survive in a stressful or resource-poor environment, a fetus must make “choices” about how to utilize scarce resources in a way that maximizes the likelihood of survival in early life in order to reproduce, even at the expense of increased susceptibility to chronic illnesses in later life. This kind of developmental programming among low birth weight infants has been suggested to be an important risk factor for adulthood hypertension (Barker et al. 2007), insulin resistance (Barker 2005), and coronary artery disease (Barker 1995). The so-called Barker hypothesis implicates a reduction in nephron mass from either congenital abnormalities or environmental stressors resulting in CKD. Nephrologist Barry Brenner first substantiated Barker’s theory in the development of CKD. Building on the observation that human nephron number is widely variable (with current best estimates showing a range of glomeruli from 210,000 to 1.8 million per kidney (Hoy et al. 2003)), Brenner proposed that either a congenital or developmentally programmed reduction in nephron number might explain why some individuals are more susceptible to hypertension and CKD (Brenner et al. 1988). Brenner suggested that, among persons with a decreased complement of nephrons, a normal GFR can be maintained initially as individual nephrons enlarge to increase the total surface area available for renal work (Luyckx and Brenner 2005). This leads to a further decrease in nephron number and requires even more hyperfiltration, leading to more rapid nephron dropout and perpetuating renal injury in a vicious cycle (Brenner et al. 1982) (see Fig. 3 (Carmody and Charlton 2013; Brenner et al. 1996; Brenner and Chertow 1994).

This figure demonstrates the impact of cumulative insults on final nephron number in a premature infant. A combination of preterm birth (failure to develop a full functional set of nephrons) in addition to treatment and hemodynamic changes often seen in the treatment of premature infants can lead to increasing workload on the remaining functional nephrons and over the course of years these nephrons begin to scar which leads to subsequent high blood pressure, microalbuminuria, progressive nephron loss and ultimately progressive chronic kidney disease.
Indeed, low nephron number may result from defects in some of the same signaling pathways involved in congenital anomalies of the kidney and urinary tract, as nephron number is ultimately determined by the degree of ureteric bud branching, as nephrons are induced by the tips of the ureteric bud (Reidy and Rosenblum 2009).
The mechanisms by which intrauterine growth retardation and low birth weight result in low nephron number have been studied in rats exposed to resource-poor intrauterine environments. Adverse intrauterine environments can be induced experimentally by caloric or protein restriction or uterine artery ligation and placental insufficiency (Schröder 2003). Offspring develop low nephron number with adult-onset hypertension, proteinuria, and glomerulosclerosis, especially in the setting of additional postnatal stressors such as high-protein diet or additional glomerular injury (Schreuder and Nauta 2007; Baserga et al. 2009, 2010; Merlet-Benichou et al. 1994; Schreuder et al. 2005; Shen et al. 2011; Plank et al. 2010). Alterations in expression of ureteric bud branching factors including downregulation of Pax2, GDNF, FGF7, BMP4, WNT4, and WNT11 mRNAs have been identified and may contribute to the decrease in nephron number (Abdel-Hakeem et al. 2008; Buffat et al. 2007). In addition, increased glomerular apoptosis has been observed (Pham et al. 2003). Studies of the mechanism by which intrauterine growth retardation may induce hypertension have suggested a role for the renin-angiotensin system , renal sympathetic activation , maternal glucocorticoid and fetal kidney 11beta-hydroxysteroid dehydrogenase type 2 activation, and increased sodium retention (Baserga et al. 2010; Ojeda et al. 2007; Grigore et al. 2007; Langley-Evans 1997; Sanders et al. 2005; Franco et al. 2012). Several studies have indicated that altered gene expression may be the result of epigenetic regulation (Baserga et al. 2010; Pham et al. 2003; Woroniecki et al. 2011).
2.6 Late Gestation Is Critical for Nephrogenesis
Although nephrogenesis in humans begins around 9 weeks gestation (Chevalier 1996; Engle 1986), the majority of nephrons (60%) are formed during the third trimester (Hinchliffe et al. 1991). The nephron complement of a human kidney is determined and fixed by 36 weeks gestation (Osathanondh and Potter 1963). Nephrons do not have the ability to regenerate; even in healthy persons, the number of functional nephrons gradually declines over time (leading to the natural age-dependent decline in GFR seen in older and elderly adults (Winearls and Glassock, 2011)).
For infants born prematurely, nephrons continue to form postnatally, but data from an autopsy study of 56 premature infants (birth weight < 1000 g) suggests that postnatal glomerulogenesis ceases after 40 days (Rodriguez et al. 2004). In an ex utero environment, these “potential” and youngest nephrons seem to be particularly vulnerable to maldevelopment and dysfunction: another study showed that up to 13% of postnatal glomeruli are morphologically abnormal (Sutherland et al. 2011) .
3 Acute Kidney Injury as a Risk Factor for CKD
Acute kidney injury in the neonatal intensive care unit is common and often poorly recognized. While its overall incidence is difficult to determine given the lack of multicenter studies and variable definitions of acute kidney injury, one recent study from a single center that used the Acute Kidney Injury Network criteria (Mehta et al. 2007) found that nearly one of every five infants with birth weight < 1500 g who survived to discharge experienced acute kidney injury (Koralkar et al. 2011). Because the majority of infants that develop acute kidney injury are discharged home with a normal creatinine for their gestational age and size, the significance of their renal injury is underappreciated.
Until recently, it was believed that acute kidney injury was completely reversible (Kiley et al. 1960). Although this may be true for prerenal acute kidney injury caused by volume depletion, it is now clear that other forms of acute kidney injury cause irreversible damage and that the more severe the injury, the more likely the progression to CKD. Animal models of acute kidney injury induced by ischemia (Basile et al. 2001), aminoglycoside toxicity (Mingeot-Leclercq and Tulkens 1999), and sepsis (Holly et al. 2006) all exhibit permanent histologic changes even if the serum creatinine returns to normal. Such models of renal injury may be particularly applicable to the premature population, in which hemodynamic instability, antibiotic exposure, and systemic infections are common. As serum creatinine is a poor marker of acute kidney injury risk and represents a better marker for the resulting damage (Askenazi et al. 2012), the emergence of new biomarkers for acute kidney injury has promising potential for predicting risk, damage, progression, and recovery.
Clinical evidence in neonatal and childhood cohorts linking acute kidney injury to the development of CKD is strengthening and well established in adults, leading some authors to even suggest that the rising incidence of acute kidney injury may be partly responsible for the nationwide increase in CKD and end-stage renal disease (Hsu 2007). A recent meta-analysis of adult trials found a substantial and exposure-dependent risk for developing CKD following acute kidney injury, with patients who experienced more severe acute kidney injury developing CKD and end-stage renal disease more frequently (Coca et al. 2012). While there are inherent risks in extrapolating the results of adult studies to pediatric patients, it is worth considering that nephron loss in a newborn infant may have far greater impact on future quality of life than it might for an older adult. Moreover, the limited number of pediatric observational studies undertaken has found similar results. In a single-center pediatric intensive care unit study, approximately 10% of patients with acute kidney injury subsequently developed significant CKD within the next 1–3 years (Mammen et al. 2012a). Alarmingly, nearly 50% of this cohort were deemed “at risk” for CKD based on the presence of hypertension, a reduced GFR (60–90 ml/min/1.73 m2), or hyperfiltration (GFR >150 ml/min/1.73 m2) during the follow-up period (Mammen et al. 2012a).
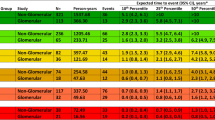
It could be argued that patients who are predisposed to the development of CKD may also be predisposed to the development of acute kidney injury (Rifkin et al. 2012). However, for pediatricians caring for premature infants who have suffered acute kidney injury, this distinction is likely an academic one. Whether acute kidney injury identifies a subgroup of infants most at risk of CKD or in fact leads independently to CKD, the implications for the child are the same, and the long-term implications of acute kidney injury ought not be ignored. Infants born prematurely (or with congenital anomalies of the kidney and urinary tract) begin life in the neonatal intensive care unit with an incomplete complement of nephrons. They are then exposed to a variety of external stressors that can hinder kidney development or cause additional nephron loss (see Fig. 4, (Brophy et al. 2015)). But unlike respiratory or infectious diseases, kidney disease is seldom a proximate cause of life-threatening illness for premature infants while they are in the NICU.

This figure demonstrates some of the possible risks and insults that can occur in utero and ex utero to the renal system in infants, particularly those at higher risk, i.e. premature infants. The in-utero or maternal factors are often major contributors to antenatal renal function and yet may be overlooked during gestation. The impact of ex-utero damage is still not well vetted, but either alone or in combination with in utero insults, may lead to an increased vulnerability to subsequent renal damage as the patient ages and lead to progressive chronic kidney disease
Although general pediatricians are attuned to the short-term risks associated with prematurity such as chronic lung disease, they often miss the implications of acute kidney injury on the long-term renal health of their patients. Indeed, recent analyses have identified the risk factors and repercussions of the use of nonsteroidal anti-inflammatory agents in pediatric patients (Misurac et al. 2013) along with other known nephrotoxins in an inpatient noncritical care environment (Kirkendall et al. 2014; Menon et al. 2014). There is a clear impact of over-the-counter medications alone and in combination with prescription medications on the development of acute kidney injury or worsening of CKD. (Onuigbo and Agbasi 2014) In particular, the geriatric population may be at very high risk for this effect (Mizokami and Mizuno 2015). Across the life span, medications can have a significant impact on the development of acute kidney injury. This in particular may be a very easy target for quality improvement initiatives that target reducing unnecessary use of medications (Goldstein et al. 2013).
4 CKD and Sequelae Beyond the First Year of Life
The interval between the newborn period and the first year of life among individuals with CKD or end-stage renal disease has conventionally been the highest risk period. Recent data has demonstrated improvement in outcomes in this age group in terms of survival (Carey et al. 2007). This appears due to better clinical management and understanding of the underlying renal pathology. The significant impact on families and early child development cannot be underestimated. This is an intense and high-risk period for infants with CKD (Wedekin et al. 2008).
The development of CKD in children ages 1 through 17 years is strongly linked to the presence of congenital anomalies of the kidney and urinary tract diagnosed in infancy, infections, prematurity, glomerular disorders (most often diagnosed in preadolescence), and hereditary causes of renal dysfunction (National Kidney Foundation; KDOQI (Kidney Disease Outcomes Quality Initiative) 2012; Mong Hiep et al. 2010). New emphasis has also been placed on the long-term complications of acute kidney injury in the pediatric intensive care unit population as data suggests as many as 10% of children with kidney injury in this setting will progress to CKD within 1–3 years after injury (Mammen et al. 2012b).
Much of the recent data on the significance of CKD on life course health development outcomes in children springs from the Chronic Kidney Disease in Children (CKiD) prospective cohort study (Feldman et al. 2003). This study has enrolled children ages 1–16 years in a multicenter study aimed at determining the factors associated with progression through the CKD stages and onto end-stage renal disease and to investigate the impact of CKD on growth, cardiovascular complications, and neurodevelopmental outcomes in children.
Growth less than the third percentile has been noted in approximately 35% of children at the point of entry into the North American Pediatric Renal Trials and Collaborative Studies database of children with CKD (Seikaly et al. 2003), with poor growth pervasive in the CKD population despite control of modifiable factors such as nutrition, anemia, and hypoparathyroidism (Mahan and Warady 2006). Initial metabolic evidence of growth failure includes phosphate wasting in early CKD (Ben-Dov et al. 2007) with progression to decreasing calcitriol levels in stages 2–3 CKD resulting later in low 1,25(OH)2 vitamin D levels and hyperparathyroidism in stages 3–4 (Heidbreder et al. 1997), with further hyperphosphatemia and hypocalcemia causing defective mineralization and bony abnormalities as CKD worsens (Wesseling-Perry et al. 2012). Furthermore, early growth and development is an important marker for future growth prediction as children with CKD who are small for gestational age at birth have a higher risk for poor weight gain in childhood (Greenbaum et al. 2011).
With regard to cardiovascular outcomes , diagnoses of hypertension, left ventricular hypertrophy, dyslipidemia, and anemia are notably increased in children with CKD. A majority of children in CKiD are noted to have hypertension with a systolic blood pressure > 95th percentile on at least one occasion with a large number of children with blood pressure >90th percentile not receiving pharmacotherapy (Flynn et al. 2008). A minority of children with CKD have left ventricular hypertrophy as diagnosed by echocardiography. In this study, independent predictors for development of LVH include hypertension, female gender, and lower hemoglobin (Mitsnefes et al. 2010). Dyslipidemia is strongly associated with poor cardiovascular health, and dyslipidemia becomes more notable in those children with a comparatively lower GFR (<30 ml/min per 1.73 m2) and in the presence of nephrotic-range proteinuria (Saland et al. 2010). Even in moderate-stage CKD, anemia is a prevalent finding. Anemia worsens in patients with a GFR less than 43 mL/min per 1.73 m2 and glomerular causes of CKD (Fadrowski et al. 2008; Atkinson et al. 2010a).
Specific effort has been directed toward understanding differences in hemoglobin levels between Caucasian and African-American children. African-American children have been noted to have lower hemoglobin values at diagnosis of CKD, independent of the underlying cause of CKD, compared with both healthy white children and those with CKD. Additionally, racial differences in hemoglobin levels appear to increase at the lower end of the hemoglobin level distribution in a CKD population (Robins and Blum 2007; Atkinson et al. 2010b). Further research utilizing the North American Pediatric Renal Trials and Collaborative Studies CKD registry demonstrated that while the majority of children with stage 3 CKD have anemia, there were no differences between racial groups (Atkinson et al. 2010b).
A 2011 review of CKiD data specific to neurocognition revealed that while neurocognitive functioning was generally within the average range for the cohort, a large percentage of children demonstrated scores at least one standard deviation below the mean in areas such as IQ, academic achievement, and executive functioning with specific predictors for worse outcomes including decline in GFR and proteinuria (Hooper et al. 2011). Additionally, research from outside of the CKiD study has found that young age of disease onset and increasing length of disease and disease severity predict poorer neurocognitive performance (Slickers et al. 2007). Although much has been discovered to date regarding the etiology and pathogenesis of CKD in childhood and adolescence, there remain several areas to expand knowledge in the field. Great strides have been made to improve nutrition and slow worsening of mineral-bone disease in children with CKD. Despite this, there still remains debate regarding therapeutic targets, such as optimal parathyroid levels , as well as differing diagnostic approaches and use of pharmacologic agents in the treatment of mineral-bone disease; thus, much can be gained from research further validating more uniform treatment approaches to optimize patient outcomes.
Much evidence regarding treatment of hypertension utilizing management of volume status via intensive dialysis in CKD progressing to end-stage renal disease is derived from the adult nephrology literature. More research is needed in this domain to further solidify potential benefits on the cardiovascular system in this population using intensified dialysis in the time leading to transplant. Little is known about mechanisms aside from deficient erythropoietin synthesis in the progression of anemia, and further investigation into this topic may yield new clinical interventions to augment the use of erythropoietin-stimulating agents in CKD (Robins and Blum 2007; Atkinson et al. 2010b; Hooper et al. 2011; Slickers et al. 2007; Marciano et al. 2010). Lastly, the field of neurocognition in CKD holds many opportunities for future research – particularly, as survival continues to improve in this population leading to more patients successfully completing college and entering the workforce. In particular, much remains to be gained in understanding the health development course of executive function on both a functional and neuroanatomical basis and the possibility for reversal of dysfunction and neural remodeling following transplantation.
5 Life Course Health Development Origins of Adolescent and Adult CKD
Although the tenets of early-life onset CKD are vitally important to understanding the context of life course impact on this disease process, other pathways likely impact the development of late adolescent and adult CKD. The association of diabetes on the development of CKD is clear. The impact of obesity itself is an area of active investigation (Hall et al. 2015). Other risk factors like hypertension also provide early intervention targets for reducing the incidence and slowing the progression of CKD.
Numerous large clinical databases have been developed over the years that have allowed investigators to better understand the sociodemographic diversity associated with CKD development in adulthood. The Chronic Renal Insufficiency Cohort Study has published multiple articles (Ricardo et al. 2015; Sandsmark et al. 2015), and there are multiple excellent reviews available to better appreciate the relationships among risk factors for adult-onset CKD. Notably, the relationship between CKD and cardiovascular disease is complex and reciprocal. The origins of cardiovascular diseases (including hypertension and obesity) are rooted in the genetic makeup of the individual and, like CKD, are impacted by preconception maternal status, intrauterine environment, and manifold other person ⇔ environmental interactions. Although there are many determinants of CKD progression in late adolescence and adults, an example of one of the emerging prototypical diseases that clearly reflects the early environmental impact on longer-term CKD is metabolic syndrome. The metabolic syndrome is known to be associated with cardiovascular risk, mortality, type 2 diabetes mellitus, and stroke, but its relationship to CKD is an emerging area of research. Patients with metabolic syndrome have a 2.5-fold higher risk of developing CKD, compared to individuals without the syndrome. Renal dysfunction, manifesting as such signs as early microalbuminuria, becomes apparent long before the appearance of hypertension or diabetes. Compared with healthy controls, individuals with metabolic syndrome have increased microvascular disease-tubular atrophy, interstitial fibrosis, arterial sclerosis, and global and segmental sclerosis. The etiology of the renal fibrosis associated with individuals with metabolic syndrome might be caused through a combination of inflammation, insulin resistance, hypertension, and dyslipidemia (Thomas et al. 2011a). The resulting overexpression of inflammatory cytokines, adipocytokines, and hormones such as angiotensin moves the balance from intrarenal homeostasis toward fibrosis pathways. The impact of early dietary intervention and regular exercise are unclear. What is clear is that the requirement for earlier evaluation of these patients by pediatricians speaks to the “epidemic” this has become .
6 Life Course Health Development Origins of Geriatric CKD
Given the aging of developed nations and the ever-increasing expenditures on renal care, estimated to be in the billions worldwide, understanding the opportunities for early recognition and prevention of renal disease development among the aging adult population is imperative (Nitta et al. 2013). Although it is clear that diabetes and hypertension are risk factors for development of CKD, in general CKD is a “silent” disease often going unrecognized until patients are well beyond possible modifiable stages of disease using dietary of medical intervention (Zhang and Rothenbacher 2008).
7 Socioeconomic Status and Race
A variety of socioeconomic determinants of CKD have been identified, including parental social class, living in a resource constrained neighborhoods, and membership in a marginalized or minority group (Shoham et al. 2005). These findings have been replicated in several populations in the USA (Shoham et al. 2005), Canada (Zacharias et al. 2012), and Australia (Cass et al. 2004), although socioeconomically disadvantaged groups in the USA may be at greatest risk (White et al. 2008). Although numerous studies have examined the association between adult socioeconomic status and CKD (Crews et al. 2012; McClellan et al. 2010; Volkova et al. 2008; Cass et al. 2002; Fored et al. 2003; Martins et al. 2006; Merkin et al. 2005; Merkin et al. 2007; Patzer and McClellan 2012; Perneger et al. 1995), few have examined the contribution of early-life socioeconomic determinants of CKD (Shoham et al. 2007; Shoham et al. 2008).
Premature birth often occurs in the context of poverty and social marginalization (Messer et al. 2008a). In the USA, preterm delivery is more common among racial minorities (Messer et al. 2008b). Although racial segregation explains some of the disparity, neighborhoods with a greater proportion of African-Americans show increased premature delivery among both black and white residents (Mason et al. 2009), suggesting that socioeconomic factors may be causative. The understanding of these dynamics is poor, given the complexity of the variables at play. The opportunity for research into better understanding these variables is rich, and the potential return on investment is high, yet targeted funding for research into these socioeconomic factors has not been a major national priority to date. One particular area they may have almost immediate dividends may be a better understanding of the role of maternal drug abuse in offspring risk for renal and hypertensive diseases. This line of investigation does not appear to have been widely addressed but has potential for significant impact via immediate returns on improved understanding of fetal development and longer-term population studies and their effects in the adult population.
Recently, Vart and colleagues performed a meta-regression analysis to identify study-level characteristics related to the strength of the socioeconomic status (SES) associations with CKD. They found that low SES was associated with low estimated glomerular filtration rate (eGFR) (odds ratio (OR) = 1.4, 95% CI = 1.2–1.6), high albuminuria (OR = 1.5, 95% CI = 1.2–1.8), low eGFR/high albuminuria (OR = 1.4, 95% CI = 1.0–1.7), and renal failure (OR = 1.6, 95% CI = 1.4, 1.7) and that variations in the strength of the associations were related to the covariate adjustment, particularly for low eGFR and high albuminuria. Overall, they noted that SES disparities in CKD were robust, irrespective of how SES was measured (Vart et al. 2015).
8 Quality-of-Life Effects of CKD
The social implications, medical regimen adherence, and impact on quality of life for children and families of children with CKD are enormous. The general improvement in survival of children with CKD has also been accompanied by an increased awareness of the effects of this disease on the quality of life of children and their families (Marciano et al. 2010; Soliday et al. 2000). Dietary and fluid management, complex hospital treatments and investigations, and disruption of normative school attendance place the children at risk for significant behavioral and psychiatric disorders, risk that is present into adulthood (Marciano et al. 2010). Childhood CKD also significantly affects family functioning (Mong Hiep et al. 2010). The development of a variety of quality-of-life assessment instruments has improved our understanding of some of the issues facing children with CKD, their parents, and siblings. Despite the availability of these types of assessment tools, there remains a paucity of data across the life course, and this area is rich in research opportunity (Marciano et al. 2010).
9 Implications for Practice
Worldwide, almost 13 million infants are born prematurely each year (Beck et al. 2010). It is unlikely that this entire population is at significant risk for developing CKD due to congenital or acquired nephropenia, but there is currently no test or biomarker for identifying the infants most at risk. No specific clinical management guidelines exist on how to follow low birth weight and intrauterine growth-retarded infants in order to identify those at risk for CKD. One in eight infants is born prematurely in the USA, and there is evidence that ex utero renal growth may be insufficient. Yet, it is not well understood how modifiable factors in postnatal care , such as nutrition and activation of retinoic acid signaling pathways , could optimize renal growth (Thomas and Kaskel 2009). The clinician must therefore rely on historical factors and subtle clinical signs of a low nephron endowment when evaluating infants with a history of prematurity.
A careful review of the neonatal history is the most important procedure to identify infants who deserve the closest follow-up. Note should be made of the infant’s birth weight and gestational age as well as any history of acute kidney injury. Given the long-term implications of the latter, it is important that appropriate follow-up is in place (the specific timing requirements for this are also unclear).
Current American Academy of Pediatrics recommendations call for measuring blood pressure at health maintenance examinations beginning at 3 years among healthy children (National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents 2004). All premature infants should be considered a high-risk group in which blood pressure screening should be performed routinely at earlier visits. Hypertension may be the earliest clinical indicator of nephropenia, as the overworked nephrons require more forceful filtration to maintain an effective GFR. Careful assessment of linear growth at every well child check is also important, as children with CKD have poor linear growth, even when the GFR is only mildly impaired (Kleinknecht et al. 1983).
It is essential that these assessments continue into adolescence, a time when many children see their pediatrician less frequently. Rapid growth in puberty often unmasks renal dysfunction, as vulnerable kidneys are less able to clear an increasing burden of waste products. It is also critical that adolescent patients – particularly those nearing transition to adult practitioners – be made aware of their history of prematurity, the increased long-term risk for CKD, and modifiable risk factors for CKD progression (such as smoking, hypertension, or obesity).
Although an abnormal creatinine at NICU discharge or in childhood carries an ominous prognosis, the serum creatinine is not a sensitive indicator of long-term CKD risk. Clinicians must therefore not be falsely reassured by normal values. The functional redundancy of the kidneys and the ability of individual nephrons to hyperfilter mean that significant disease can be hidden beneath a normal or near-normal creatinine. Routine measurement of serum creatinine should be considered especially if other signs of kidney disease are present.
Microalbuminuria (urine albumin/creatinine of 30–300 mg/g) is also an early indicator of CKD and represents a therapeutic target for CKD progression (Taal and Brenner 2008). Standard urine dipsticks detect only overt albuminuria, but targeted screening of infants and children could result in earlier detection of CKD. Such screening is already standard of care in certain other high-risk groups, such as children with insulin-dependent diabetes mellitus and sickle cell disease.
Any recommendation for increased population-based screening must be tempered with the acknowledgment that presently there is no specific therapy to slow or arrest the progression of CKD. However, risk-based screening appears justified. There are modifiable therapeutic targets for CKD progression (such as hypertension (ESCAPE Trial Group et al. 2009), microalbuminuria (Taal and Brenner 2008), and dyslipidemia (Taal and Brenner 2008)). Additionally, because patients with stage 1 and stage 2 CKD are asymptomatic, there is an opportunity to intervene before complications of CKD have developed. At the very least, identification of patients with early CKD could facilitate education for the child and parents about the treatments for CKD and allow the opportunity to provide counseling on avoiding risk factors that may accelerate its progression (dehydration, nephrotoxic medications, recurrent urinary tract infections, coexisting urologic issues).
10 Implications for Research
Our scientific understanding of the significance of nephron number is advancing rapidly, but there remain significant gaps in our knowledge. We need a better understanding, on the molecular level, of the mechanism of nephrogenesis and its disruptors. Are there strategies that can be used in the NICU to optimize postnatal nephrogenesis for infants born prematurely? Would the clinical use of more sensitive proteomic/metabolomic biomarkers of renal injury result in earlier identification of infants with acute kidney injury and improve their care? Indeed, among low birth weight infants, we lack biomarkers to quantify risk of CKD. Furthermore, once those at risk are identified, we lack knowledge of effective interventions to combat the fetal environment: adult environment imbalance with the goal of preventing CKD. Could a targeted screening program for CKD among NICU graduates improve outcomes or be cost-effective?
Currently, the only direct means of assessing nephron number in humans is through autopsy. Perhaps the most useful research development would be a mechanism to measure the number of nephrons in a living patient. Recently, Bennett et al. have successfully used cationic ferritin to label the glomerular basement membrane , allowing an accurate count of glomeruli in rodents using magnetic resonance imaging (MRI) (Beeman et al. 2011). Though not ready for in vivo use in humans, such techniques would allow the real-time measurement of glomerular counts in the clinical arena in the future and serve as a gold standard for research studies.
Children with CKD secondary to congenital anomalies of the kidney and urinary tract are at risk for hypertension, including masked hypertension. The ESCAPE trial has shown that strict blood pressure control in conjunction with ACE inhibition is reno-protective for these children (ESCAPE Trial Group et al. 2009; Mitsnefes et al. 2010). Beyond this, there is limited data on interventions to delay progression of disease.
Screens for known kidney and urinary gene mutations in populations with non-syndromic and nonfamilial congenital anomalies of the kidney and urinary tract were able to identify mutations in only 10–20% of the sample population. This indicates that the majority of patients with isolated congenital anomalies of the kidney and urinary tract either have mutations in genes not yet identified or have other causes such as epigenetic changes (Weber et al. 2006; Thomas et al. 2011b; Weber et al. 2011). It is also unclear to what extent congenital anomalies of the kidney and urinary tract is oligogenic or the result of interactions between multiple genetic abnormalities (Weber et al. 2006). Studies in mice and humans suggest that gene mutations can result in diverse renal/urologic phenotypes, suggesting there are modifier genes or gene-environment interactions that contribute to congenital anomalies of the kidney and urinary tract but remain incompletely understood (Weber 2012). The genetics of the formation of the lower urinary tract and the potential contribution of epigenetic or genetic abnormalities to the development of posterior urethral valves, one of the leading causes of childhood end-stage renal disease, remains unknown. The study of genetics of congenital anomalies of the kidney and urinary tract may be complicated in that it can result from difficult-to-detect microdeletions and genome-wide association studies may not be able to detect defects if the mutations involved are rare or are of a low effect size (Weber et al. 2011; Hoshi et al. 2012; van Eerde et al. 2012). Exome sequencing may be helpful to identify novel genes. One of the major challenges to understanding the mechanisms of congenital anomalies of the kidney and urinary tract is the interaction between gene networks and a need to better understand time and cell specificity of gene mutations. The GenitoUrinary Development Molecular Anatomy Project (GUDMAP ) has developed a molecular atlas of gene expression in the genitourinary tract (Yu et al. 2012) and is now developing tools to delete genes from specific compartments of the developing kidney and urinary tract using Cre-recombinase technology . This will allow investigators to dissect out functions of genes in these distinct cell compartments. In addition, system-based analysis may be necessary to understand the interactions between gene networks (Gallegos et al. 2012).
Epigenetic modifications are likely mechanisms by which the prenatal environment and genetic factors may interact to engender the increased risk of CKD. Limited data is available on the role of epigenetics in the development of CKD.
While glomerular hypertrophy is thought to be maladaptive, there are conflicting data on the benefit of interventions designed to block renal hypertrophy, such as with mTOR inhibition (Vogelbacher et al. 2007; Kurdian et al. 2012; Fukuda et al. 2012). While the ESCAPE trial (which included children with congenital anomalies of the kidney and urinary tract) showed strict blood pressure control in conjunction with ACE inhibition was beneficial, retrospective analyses were unable to identify a benefit from ACE inhibition in children with congenital anomalies of the kidney and urinary tract (Ardissino et al. 2007; Neild 2009). Interestingly, recent studies have suggested that an imbalance between podocyte and nephron growth can induce glomerulosclerosis (Fukuda et al. 2012). Further studies are needed to better understand the developmental determinants of nephron and podocyte growth and whether imbalance of podocyte and glomerular growth contributes to glomerulosclerosis in genetic forms of focal glomerulosclerosis.
A key question for cases of both congenital anomalies of the kidney and urinary tract and low nephron number is whether the advent of stem cell technologies and gene delivery techniques can result in interventions that can alter or rescue abnormal renal development . Reports of improvement in renal function in rat remnant kidney by injections of the mesenchyme make pursuit of stem cell technologies promising (Cavaglieri et al. 2009; Semedo et al. 2009). However, better understanding of the epigenetic and genetic factors regulating cell types may be necessary to program and tailor stem cells to renal compartments (Hendry and Little 2012).
One of the challenges of congenital anomalies of the kidney and urinary tract is how to use increasing knowledge of genotypes to optimize care of patients. One issue is a need to correlate genotypes with phenotypes and to identify modifiers. In particular, this may be relevant for potential prenatal diagnosis or screening of siblings/relatives. Despite our increased understanding of the genetics of congenital anomalies of the kidney and urinary tract, a major concern is whether there is a potential for intervention. Identification of genetic and epigenetic causes of congenital anomalies of the kidney and urinary tract may improve patient care by improving screening for extrarenal manifestations.
The identification and consistent monitoring of individuals at risk for CKD should not stop after the childhood period. Indeed, it is precisely the aging process and the lifestyle and SES effects on aging populations and particularly females of childbearing age that necessitate the educational, population health and nutritional efforts of our society if we are to impact significantly the incidence and prevalence of CKD.
10.1 Specific Research Recommendations
Clinical Research
-
1.
Nutritional-based studies to better understand the impact and optimization of maternal nutrition and the possible impact on fetal/neonatal renal health.
-
2.
Evaluation of the impact of public education and maternal intervention for nutrition, medication, and lifestyle impact on fetal/neonatal renal health.
-
3.
Examine maternal/environmental exposures that may contribute to congenital anomalies of the kidney and urinary tract and low nephron number.
-
4.
Research on factors that will optimize renal growth in premature infants.
-
5.
Develop research networks of patients with congenital anomalies of the kidney and urinary tract and CKD using global registries to facilitate both cohort studies and interventional studies and to develop bio-repositories.
-
6.
In the USA, facilitate broad-based collaboration between pediatric nephrology centers to enable implementation of clinical and translational studies on congenital anomalies of the kidney and urinary tract, low nephron number, and CKD. Utilize and expand upon existing resources, such as the networks of North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS), Pediatric Clinical and Translation Science Award (CTSA) sites, and the PCORI-funded clinical data research networks, such as PEDSnet.
-
7.
Collate the data from major adult and geriatric CKD and cardiovascular trials, and drive to identify improved biomarkers for progression and associations with CKD.
-
8.
Interventional trials to test optimal timing and efficacy of angiotensin-converting enzyme inhibition in the setting of CKD secondary to congenital anomalies of the kidney and urinary tract and in the adult and geriatric population.
-
9.
Develop national research strategies on the utilization of social media and eHealth to improve adherence and social acceptance of children with CKD .
Translational Research
-
1.
Examine genetic and epigenetic factors that may contribute to nephropenia/congenital anomalies of the kidney and urinary tract using exome sequencing, DNA copy number variant analyses, and histone methylation/acetylation patterns.
-
2.
Examine epigenetic and genetic factors necessary to program kidney stem cells.
-
3.
Develop technologies to deliver stem cells to kidneys and possible gene therapy.
-
4.
Use existing pediatric CKD cohorts, such as those in the CKiD and focal glomerulosclerosis studies, to develop genotype/phenotype correlations and to examine modifiers of renal outcomes.
-
5.
Develop metabolomic and proteomic biomarkers to identify who is at higher risk among those born with intrauterine growth retardation or prematurely.
-
6.
Research on determining nephron number using noninvasive technology.
-
7.
Define best healthcare delivery practices in pediatric and transitional settings.
-
8.
Define acute kidney injury in neonates, and develop strategies to identify high-risk patients whereby premature intervention may prevent development of CKD .
Basic Science Research
-
1.
Time and cell compartment-specific effects of developmental renal gene mutations.
-
2.
Develop system-based (“omics”) approaches to integrate data on multiple gene interactions, incorporating time and cell compartment specificity – identify pathways that contribute to CKD progression, and examine potential drug repurposing.
-
3.
Examine epigenetic mechanisms by which renal developmental programming may lead to risk of CKD.
-
4.
Assess factors that contribute to glomerular and podocyte development and growth.
-
5.
Assemble the necessary tools to begin to build and/or regenerate a kidney.
Translational Priorities: Recommendations for Translating Existing Research into Practice and Policy
-
1.
Develop recommendations for renal follow-up of premature/IUGR infants, and consider recommendations to monitor renal growth and for screening urinalyses in addition to blood pressure assessments.
-
2.
Develop recommendations for use of ambulatory blood pressure monitoring in diagnosis of masked hypertension and optimization of blood pressure control in children with congenital anomalies of the kidney and urinary tract and CKD.
-
3.
Develop innovative healthcare delivery models that improve patient health while reducing health system costs.
-
4.
Focus on improved surveillance systems and access to healthcare providers across regional boundaries.
-
5.
Employ telemedicine for Medicaid and Medicare patients for improved healthcare accessibility .
References
Abdel-Hakeem, A. K., Henry, T. Q., Magee, T. R., Desai, M., Ross, M. G., Mansano, R. Z., et al. (2008). Mechanisms of impaired nephrogenesis with fetal growth restriction: Altered rena transcription and growth factor expression. American Journal of Obstetrics and Gynecology, 199(3), 252 e1–252 e7.
Ardissino, G., Vigano, S., Testa, S., Dacco, V., Paglialonga, F., Leoni, A., et al. (2007). No clear evidence of ACEi efficacy on the progression of chronic kidney disease in children with hypodysplastic nephropathy--report from the ItalKid project database. Nephrology, Dialysis, Transplantation : Official Publication of the European Dialysis and Transplant Association - European Renal Association., 22(9), 2525–2530.
Askenazi, D. J., Koralkar, R., Hundley, H. E., Montesanti, A., Parwar, P., Sonjara, S., & Ambalavanan, N. (2012). Urine biomarkers predict acute kidney injury in newborns. The Journal of Pediatrics, 161(2), 270–275.
Atkinson, M. A., Pierce, C. B., Zack, R. M., et al. (2010a). Hemoglobin differences by race in children with CKD. American Journal of Kidney Diseases, 55(6), 1009–1017.
Atkinson, M. A., Martz, K., Warady, B. A., & Neu, A. M. (2010b). Risk for anemia in pediatric chronic kidney disease patients: A report of NAPRTCS. Pediatric Nephrology, 25(9), 1699–1706.
Bacchetta, J., Harambat, J., Guy, B., Putet, G., Cochat, P., & Dubourg, L. (2009). Long term renal outcome of children born preterm: A regular follow-up is needed. Archives of Pediatrics, 16(Suppl 1), S42–S48.
Bagby, S. P. (2015). Prenatal origins of chronic kidney disease. In P. Kimmel & M. Rosenberg (Eds.), Chronic renal disease (pp. 783–801). New York: Elsevier.
Barker, D. J. (1995). Fetal origins of coronary heart disease. BMJ, 311, 171–174.
Barker, D. J. (2005). The developmental origins of insulin resistance. Hormone Research, 64(Suppl 3), 2–7.
Barker, D. J., & Sir Richard Doll Lecture. (2012). Developmental origins of chronic disease. Public Health, 126, 185–189.
Barker, D. J., Osmond, C., Forsen, T. J., Kajantie, E., & Eriksson, J. G. (2007). Maternal and social origins of hypertension. Hypertension, 50, 565–571.
Baserga, M., Bares, A. L., Hale, M. A., Callaway, C. W., McKnight, R. A., Lane, P. H., et al. (2009). Uteroplacental insufficiency affects kidney VEGF expression in a model of IUGR with compensatory glomerular hypertrophy and hypertension. Early Human Development, 85(6), 361–367.
Baserga, M., Kaur, R., Hale, M. A., Bares, A., Yu, X., Callaway, C. W., et al. (2010). Fetal growth restriction alters transcription factor binding and epigenetic mechanisms of renal 11beta-hydroxysteroid dehydrogenase type 2 in a sex-specific manner. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 299(1), R334–R342.
Basile, D. P., Donohoe, D., Roethe, K., & Osborn, J. L. (2001). Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. American Journal of Physiology. Renal Physiology, 281, F887–F899.
Beck, S., Wojdyla, D., Say, L., Betran, A. P., Merialdi, M., Requejo, J. H., et al. (2010). The worldwide incidence of preterm birth: A systematic review of maternal mortality and morbidity. Bulletin of the World Health Organization, 88, 31–38.
Beeman, S. C., Zhang, M., Gubhaju, L., Wu, T., Bertram, J. F., Frakes, D. H., et al. (2011). Measuring glomerular number and size in perfused kidneys using MRI. American Journal of Physiology. Renal Physiology, 300, F1454–F1457.
Ben-Dov, I. Z., Galitzer, H., Lavi-Moshayoff, V., et al. (2007). The parathyroid is a target organ for FGF23 in rats. The Journal of Clinical Investigation, 117(12), 4003–4008.
Ben-Shlomo, Y., & Kuh, D. (2002). A life course approach to chronic disease epidemiology: Conceptual models, empirical challenges and interdisciplinary perspectives. International Journal of Epidemiology, 31, 285–293.
Bertoli-Avella, A. M., Conte, M. L., Punzo, F., de Graaf, B. M., Lama, G., La Manna, A., et al. (2008). ROBO2 gene variants are associated with familial vesicoureteral reflux. Journal of the American Society of Nephrology, 19(4), 825–831.
Brenner, B. M., & Anderson, S. (1987). The Gordon Wilson lecture. Why kidneys fail: An unifying hypothesis. Transactions of the American Clinical and Climatological Association, 98, 59–70.
Brenner, B. M., & Chertow, G. M. (1994). Congenital oligonephropathy and the etiology of adult hypertension and progressive renal injury. American Journal of Kidney Diseases, 23, 171–175.
Brenner, B. M., Meyer, T. W., & Hostetter, T. H. (1982). Dietary protein intake and the progressive nature of kidney disease: The role of hemodynamically mediated glomerular injury in the pathogenesis of progressive glomerular sclerosis in aging, renal ablation, and intrinsic renal disease. The New England Journal of Medicine, 307, 652–659.
Brenner, B. M., Garcia, D. L., & Anderson, S. (1988). Glomeruli and blood pressure. Less of one, more the other? American Journal of Hypertension, 1, 335–347.
Brenner, B. M., Lawler, E. V., & Mackenzie, H. S. (1996). The hyperfiltration theory: A paradigm shift in nephrology. Kidney International, 49, 1774–1777.
Brophy, P. D., Shoham, D. A., CKD Life Course Group, Charlton, J. R., Carmody, J., Reidy, K. J., Harshman, L., Segar, J., & Askenazi, D. (2015). Early-life course socioeconomic factors and chronic kidney disease. Advances in Chronic Kidney Disease, 22(1), 16–23.
Buffat, C., Boubred, F., Mondon, F., Chelbi, S. T., Feuerstein, J. M., Lelievre-Pegorier, M., et al. (2007). Kidney gene expression analysis in a rat model of intrauterine growth restriction reveals massive alterations of coagulation genes. Endocrinology, 148(11), 5549–5557.
Carey, W. A., Talley, L. I., Sehring, S. A., Jaskula, J. M., & Mathias, R. S. (2007). Outcomes of dialysis initiated during the neonatal period for treatment of end-stage renal disease: A North American pediatric renal trials and collaborative studies special analysis. Pediatrics, 119(2), e468.
Carmody, J. B., & Charlton, J. R. (2013). Short-term gestation, long-term risk: Prematurity and chronic kidney disease. Pediatrics, 131(6), 1168–1179.
Carta, M., Cimador, M., Giuffre, M., Sergio, M., Di Pace, M. R., De Grazia, E., et al. (2007). Unilateral multicystic dysplastic kidney in infants exposed to antiepileptic drugs during pregnancy. Pediatric Nephrology, 22(7), 1054–1057.
Cass, A., Cunningham, J., & Hoy, W. (2002). The relationship between the incidence of end-stage renal disease and markers of socioeconomic disadvantage. New South Wales Public Health Bulletin, 13(7), 147–151.
Cass, A., et al. (2004). Exploring the pathways leading from disadvantage to end-stage renal disease for indigenous Australians. Social Science & Medicine, 58(4), 767–785.
Cavaglieri, R. C., Martini, D., Sogayar, M. C., & Noronha, I. L. (2009). Mesenchymal stem cells delivered at the subcapsule of the kidney ameliorate renal disease in the rat remnant kidney model. Transplantation Proceedings, 41(3), 947–951.
Centers for Disease Control and Prevention (CDC). (2007). Prevalence of chronic kidney disease and associated risk factors—United States, 1999–2004. MMWR. Morbidity and Mortality Weekly Report, 56, 161–165.
Chatterjee, R., Ramos, E., Hoffman, M., Vanwinkle, J., Martin, D. R., Davis, T. K., et al. (2012). Traditional and targeted exome sequencing reveals common, rare and novel functional deleterious variants in RET-signaling complex in a cohort of living US patients with urinary tract malformations. Human Genetics, 131(11), 1725–1738.
Chen, R., Mias, G. I., Li-Pook-Than, J., Jiang, L., Lam, H. Y., Chen, R., Miriami, E., et al. (2012). Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell, 148(6), 1293–1307.
Chevalier, R. L. (1996). Developmental renal physiology of the low birth weight pre-term newborn. The Journal of Urology, 156, 714–719.
Chiang, S. S., Tai, C. W., Chung, C. J., Shiue, H. S., Chen, J. B., Su, C. T., & Hsueh, Y. M. (2010). Micronutrients and lifestyles in Taiwanese patients with stage 3 to 5 chronic kidney disease. Nutrition, 26(3), 276–282.
Coca, S. G., Singanamala, S., & Parikh, C. R. (2012). Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney International, 81, 442–448.
Cramer, M. T., & Guay-Woodford, L. M. (2015). Cystic kidney disease: A primer. Advances in Chronic Kidney Disease, 22(4), 297–305.
Crews, D. C., McClellan, W. M., Shoham, D. A., Gao, L., Warnock, D. G., Judd, S., Muntner, P., Miller, E. R., & Powe, N. R. (2012). Low income and albuminuria among REGARDS (reasons for geographic and racial differences in stroke) study participants. American Journal of Kidney Diseases, 60(5), 779–786.
van Eerde, A. M., Duran, K., van Riel, E., de Kovel, C. G., Koeleman, B. P., Knoers, N. V., et al. (2012). Genes in the ureteric budding pathway: Association study on vesico-ureteral reflux patients. PloS One, 7(4), e31327.
Engle, W. D. (1986). Development of fetal and neonatal renal function. Seminars in Perinatology, 10, 113–124.
ESCAPE Trial Group, Wuhl, E., Trivelli, A., Picca, S., Litwin, M., Peco-Antic, A., et al. (2009). Strict blood-pressure control and progression of renal failure in children. The New England Journal of Medicine, 361, 1639–1650.
Fadrowski, J. J., Pierce, C. B., Cole, S. R., Moxey-Mims, M., Warady, B. A., & Furth, S. L. (2008). Hemoglobin decline in children with chronic kidney disease: Baseline results from the chronic kidney disease in children prospective cohort study. Clinical Journal of the American Society of Nephrology : CJASN., 3(2), 457–462.
Feldman, H. I., Appel, L. J., Chertow, G. M., Cifelli, D., Cizman, B., Daugirdas, J., Fink, J. C., Franklin-Becker, E. D., Go, A. S., Hamm, L. L., He, J., Hostetter, T., Hsu, C. Y., Jamerson, K., Joffe, M., Kusek, J. W., Landis, J. R., Lash, J. P., Miller, E. R., Mohler, E. R., III, Muntner, P., Ojo, A. O., Rahman, M., Townsend, R. R., & Wright, J. T. (2003). The chronic renal insufficiency cohort (CRIC) study: Design and methods. Journal of the American Society of Nephrology, 14(Suppl), S148–S153.
Flynn, J. T., Mitsnefes, M., Pierce, C., et al. (2008). Blood pressure in children with chronic kidney disease: A report from the chronic kidney disease in children study. Hypertension, 52(4), 631–637.
Fogo, A. B. (2007). Mechanisms of progression of chronic kidney disease. Pediatric Nephrology, 22(12), 2011–2022.
Fored, C. M., et al. (2003). Socio-economic status and chronic renal failure: A population-based case-control study in Sweden. Nephrology, Dialysis, Transplantation, 18(1), 82–88.
Franco, M. C., Oliveira, V., Ponzio, B., Rangel, M., Palomino, Z., & Gil, F. Z. (2012). Influence of birth weight on the renal development and kidney diseases in adulthood: Experimental and clinical evidence. International Journal of Nephrology, 2012, 608025.
Fukuda, A., Chowdhury, M. A., Venkatareddy, M. P., Wang, S. Q., Nishizono, R., Suzuki, T., et al. (2012). Growth-dependent podocyte failure causes glomerulosclerosis. Journal of the American Society of Nephrology : JASN., 23(8), 1351–1363.
Furth, S. L., Cole, S. R., Moxey-Mims, M., Kaskel, F., Mak, R., Schwartz, G., Wong, C., Munoz, A., & Warady, B. A. (2006). Design and methods of the chronic kidney disease in children (CKiD) prospective cohort study. Clinical Journal of the American Society of Nephrology : CJASN., 1(5), 1006–1015.
Furth, S. L., Abraham, A. G., Jerry-Fluker, J., Schwartz, G. J., Benfield, M., Kaskel, F., et al. (2011). Metabolic abnormalities, cardiovascular disease risk factors, and GFR decline in children with chronic kidney disease. Clinical Journal of the American Society of Nephrology : CJASN., 6(9), 2132–2140.
Gallegos, T. F., Kouznetsova, V., Kudlicka, K., Sweeney, D. E., Bush, K. T., Willert, K., et al. (2012). A protein kinase a and Wnt-dependent network regulating an intermediate stage in epithelial tubulogenesis during kidney development. Developmental Biology, 364(1), 11–21.
Gilbert, T., Lelievre-Pegorier, M., & Merlet-Benichou, C. (1990). Immediate and long-term renal effects of fetal exposure to gentamicin. Pediatric Nephrology, 4, 445–450.
Goldstein, S. L., Kirkendall, E., Nguyen, H., Schaffzin, J. K., Bucuvalas, J., Bracke, T., Seid, M., Ashby, M., Foertmeyer, N., Brunner, L., Lesko, A., Barclay, C., Lannon, C., & Muething, S. (2013). Electronic health record identification of nephrotoxin exposure and associated acute kidney injury. Pediatrics, 132(3), e756–e767.
Greenbaum, L. A., Muñoz, A., Schneider, M. F., Kaskel, F. J., Askenazi, D. J., Jenkins, R., Hotchkiss, H., Moxey-Mims, M., Furth, S. L., & Warady, B. A. (2011). The association between abnormal birth history and growth in children with CKD. Clinical Journal of the American Society of Nephrology, 6, 14–21.
Grigore, D., Ojeda, N. B., Robertson, E. B., Dawson, A. S., Huffman, C. A., Bourassa, E. A., et al. (2007). Placental insufficiency results in temporal alterations in the renin angiotensin system in male hypertensive growth restricted offspring. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 293(2), R804–R811.
Gubhaju, L., Sutherland, M. R., Yoder, B. A., Zulli, A., Bertram, J. F., & Black, M. J. (2009). Is nephrogenesis affected by preterm birth? Studies in a non-human primate model. American Journal of Physiology. Renal Physiology, 297, F1668–F1677.
Hack, M., Flannery, D. J., Schluchter, M., Cartar, L., Borawski, E., & Klein, N. (2002). Outcomes in young adulthood for very-low-birth-weight infants. The New England Journal of Medicine, 346, 149–157.
Halfon, N., & Forrest, C. B. (2017). The emerging theoretical framework of life course health development. In N. Halfon, C. B. Forrest, R. M. Lerner, & E. Faustman (Eds.), Handbook of life course health-development science. Cham: Springer.
Hall, J. E., do Carmo, J. M., da Silva, A. A., Wang, Z., & Hall, M. E. (2015). Obesity-induced hypertension: Interaction of neurohumoral and renal mechanisms. Circulation Research, 116(6), 991–1006.
Hallan, S., Euser, A. M., Irgens, L. M., Finken, M. J., Holmen, J., & Dekker, F. W. (2008). Effect of intrauterine growth restriction on kidney function at young adult age: The Nord Trondelag health (HUNT 2) study. American Journal of Kidney Diseases: The Official Journal of the National Kidney Foundation, 51(1), 10–20.
Harambat, J., van Stralen, K. J., Kim, J. J., & Tizard, E. J. (2012). Epidemiology of chronic kidney disease in children. Pediatric Nephrology, 27(3), 363–373.
Heidbreder, E., Naujoks, H., Brosa, U., & Schramm, L. (1997). The calcium-parathyroid hormone regulation in chronic renal failure investigation of its dynamic secretion pattern. Hormone and Metabolic Research, 29(2), 70–75.
Hendry, C. E., & Little, M. H. (2012). Reprogramming the kidney: A novel approach for regeneration. Kidney International, 82(2), 138–146.
Hinchliffe, S. A., Sargent, P. H., Howard, C. V., Chan, Y. F., & van Velzen, D. (1991). Human intrauterine renal growth expressed in absolute number of glomeruli assessed by the disector method and Cavalieri principle. Laboratory Investigation, 64, 777–784.
Hodgin, J. B., Rasoulpour, M., Markowitz, G. S., & D'Agati, V. D. (2009a). Very low birth weight is a risk factor for secondary focal segmental glomerulosclerosis. Clinical Journal of the American Society of Nephrology, 4, 71–76.
Holly, M. K., Dear, J. W., Hu, X., Schechter, A. N., Gladwin, M. T., Hewitt, S. M., et al. (2006). Biomarker and drug-target discovery using proteomics in a new rat model of sepsis-induced acute renal failure. Kidney International, 70, 496–506.
Hooper, S. R., Gerson, A. C., Butler, R. W., et al. (2011). Neurocognitive functioning of children and adolescents with mild-to-moderate chronic kidney disease. Clinical Journal of the American Society of Nephrology : CJASN., 6(8), 1824–1830.
Horbar, J. D., Carpenter, J. H., Badger, G. J., Kenny, M. J., Soll, R. F., Morrow, K. A., et al. (2012). Mortality and neonatal morbidity among infants 501 to 1500 grams from 2000 to 2009. Pediatrics, 129, 1019–1026.
Hoshi, M., Batourina, E., Mendelsohn, C., & Jain, S. (2012). Novel mechanisms of early upper and lower urinary tract patterning regulated by RetY1015 docking tyrosine in mice. Development, 139(13), 2405–2415.
Hoy, W. E., Rees, M., Kile, E., Mathews, J. D., & Wang, Z. (1999). A new dimension to the Barker hypothesis: Low birthweight and susceptibility to renal disease. Kidney International, 56(3), 1072–1077.
Hoy, W. E., Douglas-Denton, R. N., Hughson, M. D., Cass, A., Johnson, K., & Bertram, J. F. (2003). A stereological study of glomerular number and volume: Preliminary findings in a multiracial study of kidneys at autopsy. Kidney International. Supplement, 83, S31–S37.
Hoy, W. E., Kincaid-Smith, P., Hughson, M. D., Fogo, A. B., Sinniah, R., Dowling, J., et al. (2010). CKD in aboriginal Australians. American Journal of Kidney Diseases the Official Journal of the National Kidney Foundation, 56(5), 983–993.
Hoy, W. E., Samuel, T., Mott, S. A., Kincaid-Smith, P. S., Fogo, A. B., Dowling, J. P., et al. (2012). Renal biopsy findings among indigenous Australians: A nationwide review. Kidney International, 82(12), 1321–1331.
Hsu, C. Y. (2007). Linking the population epidemiology of acute renal failure, chronic kidney disease and end-stage renal disease. Current Opinion in Nephrology and Hypertension, 16, 221–226.
Hsu, C. W., Yamamoto, K. T., Henry, R. K., De Roos, A. J., & Flynn, J. T. (2014). Prenatal risk factors for childhood CKD. American Society of Nephrology, 25, 2105–2111.
Keijzer-Veen, M. G., Dulger, A., Dekker, F. W., Nauta, J., & van der Heijden, B. J. (2010). Very preterm birth is a risk factor for increased systolic blood pressure at a young adult age. Pediatric Nephrology, 25, 509–516.
Kiley, J. E., Powers, S. R., Jr., & Beebe, R. T. (1960). Acute renal failure: Eighty cases of renal tubular necrosis. The New England Journal of Medicine, 262, 481–486.
Kirkendall, E. S., Spires, W. L., Mottes, T. A., Schaffzin, J. K., Barclay, C., & Goldstein, S. L. (2014). Development and performance of electronic acute kidney injury triggers to identify pediatric patients at risk for nephrotoxic medication-associated harm. Applied Clinical Informatics, 5(2), 313–333.
Kleinknecht, C., Broyer, M., Huot, D., Marti-Henneberg, C., & Dartois, A. M. (1983). Growth and development of nondialyzed children with chronic renal failure. Kidney International. Supplement, 15, S40–S47.
Koralkar, R., Ambalavanan, N., Levitan, E. B., McGwin, G., Goldstein, S., & Askenazi, D. (2011). Acute kidney injury reduces survival in very low birth weight infants. Pediatric Research, 69, 354–358.
Kurdian, M., Herrero-Fresneda, I., Lloberas, N., Gimenez-Bonafe, P., Coria, V., Grande, M. T., et al. (2012). Delayed mTOR inhibition with low dose of everolimus reduces TGFbeta expression, attenuates proteinuria and renal damage in the renal mass reduction model. PloS One, 7(3), e32516.
Lackland, D. T., Bendall, H. E., Osmond, C., Egan, B. M., & Barker, D. J. (2000). Low birth weights contribute to high rates of early-onset chronic renal failure in the southeastern United States. Archives of Internal Medicine, 160(10), 1472–1476.
Langley-Evans, S. C. (1997). Hypertension induced by foetal exposure to a maternal low-protein diet, in the rat, is prevented by pharmacological blockade of maternal glucocorticoid synthesis. Journal of Hypertension, 15(5), 537–544.
Lash, J. P., Go, A. S., Appel, L. J., He, J., Ojo, A., Rahman, M., Townsend, R. R., Xie, D., Cifelli, D., Cohan, J., Fink, J. C., Fischer, M. J., Gadegbeku, C., Hamm, L. L., Kusek, J. W., Landis, J. R., Narva, A., Robinson, N., Teal, V., Feldman, H. I., & Chronic Renal Insufficiency Cohort (CRIC) Study Group. (2009). Chronic renal insufficiency cohort (CRIC) study: Baseline characteristics and associations with kidney function. Clinical Journal of the American Society of Nephrology, 4(8), 1302–1311. 2011;6(10):2548-53.
Lelievre-Pegorier, M., Vilar, J., Ferrier, M. L., Moreau, E., Freund, N., Gilbert, T., et al. (1998). Mild vitamin a deficiency leads to inborn nephron deficit in the rat. Kidney International, 54, 1455–1462.
Lopez-Hernandez, F. J., & Lopez-Novoa, J. M. (2012). Role of TGF-beta in chronic kidney disease: An integration of tubular, glomerular and vascular effects. Cell and Tissue Research, 347(1), 141–154.
Luyckx, V. A., & Brenner, B. M. (2005). Low birth weight, nephron number, and kidney disease. Kidney International. Supplement, 97, S68–S77.
Mahan, J. D., & Warady, B. A. (2006). Assessment and treatment of short stature in pediatric patients with chronic kidney disease: A consensus statement. Pediatric Nephrology, 21(7), 917–930.
Mallick, N. (2003). Secondary focal glomerulosclerosis not due to HIV. Nephrology, Dialysis, Transplantation, 18(90006), 64vi–64v7.
Mammen, C., Al Abbas, A., Skippen, P., Nadel, H., Levine, D., Collet, J. P., et al. (2012a). Long-term risk of CKD in children surviving episodes of acute kidney injury in the intensive care unit: A prospective cohort study. American Journal of Kidney Diseases, 59, 523–530.
Marciano, R. C., Bouissou Soares, C. M., Santos Diniz, J. S., Lima, E. M., Penido Silva, J. M., Ribeiro Canhestro, M., Gazzinelli Oliveira, A., Duarte Melo, C., Santos Dias, C., Correa, H., & Araujo de Oliveira, E. (2010). Mental disorders and quality of life in pediatric patients with chronic kidney disease. Jornal Brasileiro de Nefrologia, 32(3), 309–315.
Martins, D., et al. (2006). The association of poverty with the prevalence of albuminuria: Data from the third National Health and nutrition examination survey (NHANES III). American Journal of Kidney Diseases, 47(6), 965–971.
Mason, S. M., et al. (2009). Segregation and preterm birth: The effects of neighborhood racial composition in North Carolina. Health & Place, 15(1), 1–9.
McClellan, W. M., Newsome, B. B., McClure, L. A., Howard, G., Volkova, N., Audhya, P., & Warnock, D. G. (2010). Poverty and racial disparities in kidney disease: The REGARDS study. American Journal of Nephrology, 32(1), 38–46.
Mehta, R. L., Kellum, J. A., Shah, S. V., Molitoris, B. A., Ronco, C., Warnock, D. G., et al. (2007). Acute kidney injury network: Report of an initiative to improve outcomes in acute kidney injury. Critical Care, 11, R31.
Menon, S., Kirkendall, E. S., Nguyen, H., & Goldstein, S. L. (2014). Acute kidney injury associated with high nephrotoxic medication exposure leads to chronic kidney disease after 6 months. The Journal of Pediatrics, 165(3), 522–527.
Merkin, S. S., et al. (2005). Area socioeconomic status and progressive CKD: The atherosclerosis risk in communities (ARIC) study. American Journal of Kidney Diseases, 46(2), 203.
Merkin, S. S., et al. (2007). Individual and neighborhood socioeconomic status and progressive chronic kidney disease in an elderly population: The cardiovascular health study. Social Science & Medicine, 65(4), 809–821.
Merlet-Benichou, C., Gilbert, T., Muffat-Joly, M., Lelievre-Pegorier, M., & Leroy, B. (1994). Intrauterine growth retardation leads to a permanent nephron deficit in the rat. Pediatric Nephrology, 8(2), 175–180.
Messer, L. C., et al. (2008a). Socioeconomic domains and associations with preterm birth. Social Science & Medicine, 67(8), 1247–1257.
Messer, L. C., et al. (2008b). Black-white preterm birth disparity: A marker of inequality. Annals of Epidemiology, 18(11), 851–858.
Mingeot-Leclercq, M. P., & Tulkens, P. M. (1999). Aminoglycosides: Nephrotoxicity. Antimicrobial Agents and Chemotherapy, 43, 1003–1012.
Misurac, J. M., Knoderer, C. A., Leiser, J. D., Nailescu, C., Wilson, A. C., & Andreoli, S. P. (2013). Nonsteroidal anti-inflammatory drugs are an important cause of acute kidney injury in children. The Journal of Pediatrics, 162(6), 1153–1159.
Mitsnefes, M., Flynn, J., Cohn, S., et al. (2010). Masked hypertension associates with left ventricular hypertrophy in children with CKD. Journal of American Society of Nephrology, 21(1), 137–144.
Mizokami, F., & Mizuno, T. (2015). Acute kidney injury induced by antimicrobial agents in the elderly: Awareness and mitigation strategies. Drugs & Aging, 32(1), 1–12.
Mong Hiep, T. T., Ismaili, K., Collart, F., et al. (2010). Clinical characteristics and outcomes of children with stage 3-5 chronic kidney disease. Pediatric Nephrology, 25(5), 935–940.
Nakayama, M., Nozu, K., Goto, Y., Kamei, K., Ito, S., Sato, H., et al. (2010). HNF1B alterations associated with congenital anomalies of the kidney and urinary tract. Pediatric Nephrology, 25(6), 1073–1079.
NAPRTCS. Annual Report. 2011. http://www.kidney.org/professionals/kdoqi/guidelines_ckd/toc.htm
National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents. (2004). The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics, 114, 555–576.
National Kidney Foundation; KDOQI (Kidney Disease Outcomes Quality Initiative) (2012). Clinical Practice Guidelines for Chronic Kidney Disease: Evaluation, Classification and Stratification. Part 4. definition and classification of stages of chronic kidney disease. https://www.kidney.org/professionals/guidelines
Neild, G. H. (2009). What do we know about chronic renal failure in young adults? I. Primary renal disease. Pediatric Nephrology, 24(10), 1913–1919.
Nishimura, H., Yerkes, E., Hohenfellner, K., Miyazaki, Y., Ma, J., Hunley, T. E., et al. (1999). Role of the angiotensin type 2 receptor gene in congenital anomalies of the kidney and urinary tract, CAKUT, of mice and men. Molecular Cell, 3(1), 1–10.
Nitta K, Okada K, Yanai M,· Takahashi S. (2013). Aging and chronic kidney disease. Kidney & Blood Pressure Research 38:109-120.
Ojeda, N. B., Johnson, W. R., Dwyer, T. M., & Alexander, B. T. (2007). Early renal denervation prevents development of hypertension in growth-restricted offspring. Clinical and Experimental Pharmacology & Physiology, 34(11), 1212–1216.
Onuigbo, M. A., & Agbasi, N. (2014). “Quadruple whammy”- a preventable newly described syndrome of post-operative AKI in CKD II and CKD III patients on combination “triple whammy” medications: A Mayo Clinic health system, Eau Claire, Wisconsin experience. Nigerian Journal of Clinical Practice, 17(5), 649–654.
Ortiz, L. A., Quan, A., Weinberg, A., & Baum, M. (2001). Effect of prenatal dexamethasone on rat renal development. Kidney International, 59, 1663–1669.
Osathanondh, V., & Potter, E. (1963). Development of human kidney as shown by microdissection. III. Formation and interrelationship of collecting tubules and nephrons. Archives of Pathology, 76, 290–302.
Paces-Fessy, M., Fabre, M., Lesaulnier, C., & Cereghini, S. (2012). Hnf1b and Pax2 cooperate to control different pathways in kidney and ureter morphogenesis. Human Molecular Genetics, 21(14), 3143–3155.
Patzer, R. E., & McClellan, W. M. (2012). Influence of race, ethnicity and socioeconomic status on kidney disease. Nature Reviews. Nephrology, 8(9), 533–541.
Perneger, T. V., Whelton, P. K., & Klag, M. J. (1995). Race and end-stage renal disease. Socioeconomic status and access to health care as mediating factors. Archives of Internal Medicine, 155(11), 1201–1208.
Pham, T. D., MacLennan, N. K., Chiu, C. T., Laksana, G. S., Hsu, J. L., & Lane, R. H. (2003). Uteroplacental insufficiency increases apoptosis and alters p53 gene methylation in the full-term IUGR rat kidney. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 285(5), R962–R970.
Plank, C., Nusken, K. D., Menendez-Castro, C., Hartner, A., Ostreicher, I., Amann, K., et al. (2010). Intrauterine growth restriction following ligation of the uterine arteries leads to more severe glomerulosclerosis after mesangioproliferative glomerulonephritis in the offspring. American Journal of Nephrology, 32(4), 287–295.
Puddu, M., Fanos, V., Podda, F., & Zaffanello, M. (2009). The kidney from prenatal to adult life: Perinatal programming and reduction of number of nephrons during development. American Journal of Nephrology, 30(2), 162–170.
Querfeld, U., Anarat, A., Bayazit, A. K., Bakkaloglu, A. S., Bilginer, Y., Caliskan, S., Civilibal, M., Doyon, A., Duzova, A., Kracht, D., Litwin, M., Melk, A., Mir, S., Sözeri, B., Shroff, R., Zeller, R., Wühl, E., Schaefer, F., & 4C Study Group. (2010). The cardiovascular comorbidity in children with chronic kidney disease (4C) study: Objectives, design, and methodology. Clinical Journal of the American Society of Nephrology, 5(9), 1642–1648.
Rademacher, E. R., & Sinaiko, A. R. (2009). Albuminuria in children. Current Opinion in Nephrology and Hypertension, 18, 246–251.
Reidy, K., & Kaskel, F. J. (2007). Pathophysiology of focal segmental glomerulosclerosis. Pediatric Nephrology, 22(3), 350–354.
Reidy, K. J., & Rosenblum, N. D. (2009). Cell and molecular biology of kidney development. Seminars in Nephrology, 29(4), 321–337.
Rengo, F., Parisi, V., Rengo, G., Femminella, G. D., Rengo, C., Zincarelli, C., Pagano, G., Festa, G., De Lucia, C., & Leosco, D. (2012). Instruments for geriatric assessment: New multidimensional assessment approaches. Journal of Nephrology, 25 Suppl 19, S73–8.
Ricardo, A. C., Roy, J. A., Tao, K., Alper, A., Chen, J., Drawz, P. E., Fink, J. C., Hsu, C. Y., Kusek, J. W., Ojo, A., Schreiber, M., Fischer, M. J., & CRIC Study Investigators. (2015). Influence of nephrologist care on management and outcomes in adults with chronic kidney disease. Journal of General Internal Medicine, 31, 22. [Epub ahead of print].
Rifkin, D. E., Coca, S. G., & Kalantar-Zadeh, K. (2012). Does AKI truly lead to CKD? Journal of The American Society of Nephrology, 23, 979–984.
Robins, E. B., & Blum, S. (2007). Hematologic reference values for African American children and adolescents. American Journal of Hematology, 82(7), 611–614.
Rodriguez, M. M., Gomez, A. H., Abitbol, C. L., Chandar, J. J., Duara, S., & Zilleruelo, G. E. (2004). Histomorphometric analysis of postnatal glomerulogenesis in extremely preterm infants. Pediatric and Developmental Pathology, 7, 17–25.
Saisawat, P., Tasic, V., Vega-Warner, V., Kehinde, E. O., Gunther, B., Airik, R., et al. (2012). Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Kidney International, 81(2), 196–200.
Saland, J. M., Pierce, C. B., Mitsnefes, M. M., et al. (2010). Dyslipidemia in children with chronic kidney disease. Kidney International, 78(11), 1154–1163.
Salgado, C. M., Jardim, P. C., Teles, F. B., & Nunes, M. C. (2009). Influence of low birth weight on microalbuminuria and blood pressure of school children. Clinical Nephrology, 71, 367–374.
Sanders, M. W., Fazzi, G. E., Janssen, G. M., Blanco, C. E., & De Mey, J. G. (2005). High sodium intake increases blood pressure and alters renal function in intrauterine growth-retarded rats. Hypertension, 46(1), 71–75.
Sandsmark, D. K., Messé, S. R., Zhang, X., Roy, J., Nessel, L., Lee Hamm, L., He, J., Horwitz, E. J., Jaar, B. G., Kallem, R. R., Kusek, J. W., Mohler, E. R., 3rd, Porter, A., Seliger, S. L., Sozio, S. M., Townsend, R. R., Feldman, H. I., & Kasner, S. E. (2015). Proteinuria, but not eGFR, predicts stroke risk in chronic kidney disease: Chronic renal insufficiency cohort study. Stroke, 46, 2075. [Epub ahead of print].
Sanna-Cherchi, S., Ravani, P., Corbani, V., Parodi, S., Haupt, R., Piaggio, G., et al. (2009). Renal outcome in patients with congenital anomalies of the kidney and urinary tract. Kidney International, 76(5), 528–533.
Schedl, A., & Hastie, N. D. (2000). Cross-talk in kidney development. Current Opinion in Genetics and Development, 10(5), 543–549.
Schreuder, M. F., & Nauta, J. (2007). Prenatal programming of nephron number and blood pressure. Kidney International, 72(3), 265–268.
Schreuder, M. F., Nyengaard, J. R., Fodor, M., van Wijk, J. A., & Delemarre-van de Waal, H. A. (2005). Glomerular number and function are influenced by spontaneous and induced low birth weight in rats. Journal of the American Society of Nephrology: JASN., 16(10), 2913–2919.
Schröder, H. J. (2003). Models of fetal growth restriction. European Journal of Obstetrics, Gynecology, and Reproductive Biology, 110, S29–S39.
Seikaly, M. G., Ho, P. L., Emmett, L., Fine, R. N., & Tejani, A. (2003). Chronic renal insufficiency in children: The 2001 Annual report of the NAPRTCS. Pediatric Nephrology, 18(8), 796–804.
Semedo, P., Correa-Costa, M., Antonio Cenedeze, M., Maria Avancini Costa Malheiros, D., Antonia dos Reis, M., Shimizu, M. H., et al. (2009). Mesenchymal stem cells attenuate renal fibrosis through immune modulation and remodeling properties in a rat remnant kidney model. Stem Cells, 27(12), 3063–3073.
Serreau, R., Luton, D., Macher, M. A., Delezoide, A. L., Garel, C., & Jacqz-Aigrain, E. (2005). Developmental toxicity of the angiotensin II type 1 receptor antagonists during human pregnancy: A report of 10 cases. BJOG : An International Journal of Obstetrics and Gynaecology, 112(6), 710–712.
Shen, Q., Xu, H., Wei, L. M., Chen, J., & Liu, H. M. (2011). Intrauterine growth restriction and postnatal high-protein diet affect the kidneys in adult rats. Nutrition, 27(3), 364–371.
Shoham, D. A., Vupputuri, S., & Kshirsagar, A. V. (2005). Chronic kidney disease and life course socioeconomic status: A review. Advances in Chronic Kidney Disease, 12(1), 56–63.
Shoham, D. A., et al. (2007). Kidney disease in life-course socioeconomic context: The atherosclerosis risk in communities (ARIC) study. American Journal of Kidney Diseases, 49(2), 217–226.
Shoham, D. A., et al. (2008). Kidney disease and the cumulative burden of life course socioeconomic conditions: The atherosclerosis risk in communities (ARIC) study. Social Science & Medicine, 67(8), 1311–1320.
Sims-Lucas, S., Di Giovanni, V., Schaefer, C., Cusack, B., Eswarakumar, V. P., & Bates, C. M. (2012). Ureteric morphogenesis requires Fgfr1 and Fgfr2/Frs2alpha signaling in the metanephric mesenchyme. Journal of the American Society of Nephrology, 23(4), 607–617.
Slickers, J., Duquette, P., Hooper, S., & Gipson, D. (2007). Clinical predictors of neurocognitive deficits in children with chronic kidney disease. Pediatric Nephrology, 22(4), 565–572.
Soliday, E., Kool, E., & Lande, M. B. (2000). Psychosocial adjustment in children with kidney disease. Journal of Pediatric Psychology, 25(2), 93–103.
Song, R., & Yosypiv, I. V. (2011). Genetics of congenital anomalies of the kidney and urinary tract. Pediatric Nephrology, 26(3), 353–364.
Spencer, J. D., Schwaderer, A., McHugh, K., Vanderbrink, B., Becknell, B., & Hains, D. S. (2011). The demographics and costs of inpatient vesicoureteral reflux management in the USA. Pediatric Nephrology, 26(11), 1995–2001.
Stelloh, C., Allen, K. P., Mattson, D. L., Lerch-Gaggl, A., Reddy, S., & El-Meanawy, A. (2012). Prematurity in mice leads to reduction in nephron number, hypertension, and proteinuria. Translational Research, 159, 80–89.
Sutherland, M. R., Gubhaju, L., Moore, L., Kent, A. L., Dahlstrom, J. E., Horne, R. S., et al. (2011). Accelerated maturation and abnormal morphology in the preterm neonatal kidney. Journal of American Society of Nephrology, 22, 1365–1374.
Sutherland, M. R., Yoder, B. A., McCurnin, D., Seidner, S., Gubhaju, L., Clyman, R. I., et al. (2012a). Effects of ibuprofen treatment on the developing preterm baboon kidney. American Journal of Physiology. Renal Physiology, 302(10), F1286–F1292.
Taal, M. W., & Brenner, B. M. (2008). Renal risk scores: Progress and prospects. Kidney International, 73, 1216–1219.
Tabel, Y., Haskologlu, Z. S., Karakas, H. M., & Yakinci, C. (2010). Ultrasonographic screening of newborns for congenital anomalies of the kidney and the urinary tracts. Urology Journal, 7(3), 161–167.
Takamitsu, S., & Darwin Bell, P. (2015). Molecular pathways and therapies in autosomal-dominant polycystic kidney disease. Physiology, 30(3), 195–207.
Thomas, R., & Kaskel, F. J. (2009). It's not over till the last glomerulus forms. Kidney International, 76(4), 361–363.
Thomas, G., Sehgal, A. R., Kashyap, S. R., Srinivas, T. R., Kirwan, J. P., & Navaneethan, S. D. (2011a). Metabolic syndrome and kidney disease: A systematic review and meta-analysis. Clinical Journal of the American Society of Nephrology, 6(10), 2364–2373.
Thomas, R., Sanna-Cherchi, S., Warady, B. A., Furth, S. L., Kaskel, F. J., & Gharavi, A. G. (2011b). HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatric Nephrology, 26(6), 897–903.
Thompson, S., James, M., Wiebe, N., Hemmelgarn, B., Manns, B., Klarenbach, S., Tonelli, M., & for the Alberta Kidney Disease Network. (2015). Cause of death in patients with reduced kidney function. Journal of the American Society of Nephrology, 26, 2504.
USRDS. (2003). 2003 Annual data report: Atlas of end stage renal disease in the United States. Bethesda: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases.
Vart, P., Gansevoort, R. T., Joosten, M. M., Bültmann, U., & Reijneveld, S. A. (2015). Socioeconomic disparities in chronic kidney disease: A systematic review and meta-analysis. American Journal of Preventive Medicine, 48(5), 580–592.
Vogelbacher, R., Wittmann, S., Braun, A., Daniel, C., & Hugo, C. (2007). The mTOR inhibitor everolimus induces proteinuria and renal deterioration in the remnant kidney model in the rat. Transplantation, 84(11), 1492–1499.
Volkova, N., et al. (2008). Neighborhood poverty and racial differences in ESRD incidence. Journal of the American Society of Nephrology, 19(2), 356–364.
Warady, B. A., & Chadha, V. (2007). Chronic kidney disease in children: The global perspective. Pediatric Nephrology, 22(12), 1999–2009.
Weber, S. (2012). Novel genetic aspects of congenital anomalies of kidney and urinary tract. Current Opinion in Pediatrics, 24(2), 212–218.
Weber, S., Moriniere, V., Knuppel, T., Charbit, M., Dusek, J., Ghiggeri, G. M., et al. (2006). Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: Results of the ESCAPE study. Journal of the American Society of Nephrology, 17(10), 2864–2870.
Weber, S., Landwehr, C., Renkert, M., Hoischen, A., Wuhl, E., Denecke, J., et al. (2011). Mapping candidate regions and genes for congenital anomalies of the kidneys and urinary tract (CAKUT) by array-based comparative genomic hybridization. Nephrology, Dialysis, Transplantation : Official Publication of the European Dialysis and Transplant Association - European Renal Association., 26(1), 136–143.
Wedekin, M., Ehrich, J. H. H., Offner, G., & Pope, L. (2008). Aetiology and outcome of acute and chronic renal failure in infants. Nephrology, Dialysis, Transplantation, 23, 1575–1580.
Wesseling-Perry, K., Pereira, R. C., Tseng, C. H., et al. (2012). Early skeletal and biochemical alterations in pediatric chronic kidney disease. Clinical Journal of the American Society of Nephrology : CJASN., 7(1), 146–152.
Westland, R., Schreuder, M. F., Bokenkamp, A., Spreeuwenberg, M. D., & van Wijk, J. A. (2011). Renal injury in children with a solitary functioning kidney--the KIMONO study. Nephrology, Dialysis, Transplantation: Official Publication of the European Dialysis and Transplant Association - European Renal Association, 26(5), 1533–1541.
White, S. L., et al. (2008). Socioeconomic disadvantage and kidney disease in the United States, Australia, and Thailand. American Journal of Public Health, 98(7), 1306–1313.
White, S. L., Perkovic, V., Cass, A., Chang, C. L., Poulter, N. R., Spector, T., et al. (2009). Is low birth weight an antecedent of CKD in later life?. A systematic review of observational studies. American Journal of Kidney Diseases, 54, 248–261.
Winearls, C. G., & Glassock, R. J. (2011). Classification of chronic kidney disease in the elderly: Pitfalls and errors. Nephron. Clinical Practice, 119(Suppl 1), c2–c4.
Wong, C. J., Moxey-Mims, M., Jerry-Fluker, J., Warady, B. A., & Furth, S. L. (2012). CKiD (CKD in children) prospective cohort study: A review of current findings. American Journal of Kidney Diseases, 60(6), 1002–1011.
Woroniecki, R., Gaikwad, A. B., & Susztak, K. (2011). Fetal environment, epigenetics, and pediatric renal disease. Pediatric Nephrology, 26(5), 705–711.
Wyld, M. L., Lee, C. M., Zhuo, X., White, S., Shaw, J. E., Morton, R. L., Colagiuri, S., & Chadban, S. J. (2015). Cost to government and society of chronic kidney disease stage 1-5: A national cohort study. Internal Medicine Journal, 45(7), 741–747.
Yu, J., Valerius, M. T., Duah, M., Staser, K., Hansard JK, Guo, J. J., et al. (2012). Identification of molecular compartments and genetic circuitry in the developing mammalian kidney. Development, 139(10), 1863–1873.
Zacharias, J. M., et al. (2012). Prevalence, risk factors and awareness of albuminuria on a Canadian first nation: A community-based screening study. BMC Public Health, 12, 290.
Zandi-Nejad, K., Luyckx, V. A., & Brenner, B. M. (2006). Adult hypertension and kidney disease: The role of fetal programming. Hypertension, 47(3), 502–508.
Zhang, Q.-L., & Rothenbacher, D. (2008). Prevalence of chronic kidney disease in population-based studies: Systematic review. BMC Public Health, 8, 117.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
This chapter is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made. The images or other third party material in this chapter are included in the work's Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work's Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2018 The Author(s)
About this chapter

Cite this chapter
Brophy, P.D. et al. (2018). Chronic Kidney Disease: A Life Course Health Development Perspective. In: Halfon, N., Forrest, C., Lerner, R., Faustman, E. (eds) Handbook of Life Course Health Development . Springer, Cham. https://doi.org/10.1007/978-3-319-47143-3_16
Download citation
DOI: https://doi.org/10.1007/978-3-319-47143-3_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-47141-9
Online ISBN: 978-3-319-47143-3
eBook Packages: MedicineMedicine (R0)