
Abstract
Spinal cord injury (SCI) resulting in impaired neurological functions creates suffering and economic burden. Directly after the injury, the conservation of tissue at the lesion site and the preservation of the connectivity through the injury epicenter have highest priority. This tissue preservation will not only reduce neurological deficits, but also and allows for more rapid and extensive recovery. Advances in clinical care intended to accomplish this goal include early surgical decompression, support of blood pressure, and the subject of this chapter – experimental neuroprotective therapeutics. The scientific foundation of neuroprotection is that “harmful” secondary injury processes extend and distribute the tissue loss caused by the primary injury event. Existing clinical knowledge together with preclinical experimental evidence has supported phase III clinical trial translation of some neuroprotective agents in the past four decades. Although some have had positive results, the magnitude of improvement was small, and associated complications and controversy surrounding certain therapeutics diminished their role in SCI care. However, these trials generated knowledge valuable to guide current work. Neuroscientists continue to develop new therapeutic approaches by demonstrating neuroprotective efficacy in small and large animal models. A consensus now exists that the preclinical data set to support the expensive process of translation must be very robust and include a well conducted independent replication. Primary endpoints for pivotal clinical trials have been clarified on the basis of aggregate experience and extensive studies on the natural history of SCI. “Secondary injury” consists of numerous mechanisms, and the probability of a robust protective effect of a single agent is small. It is necessary to design rational combinations of therapies to increase efficacy.
Similar content being viewed by others


Keywords
- Spinal Cord Injury
- Ketogenic Diet
- Acute Spinal Cord Injury
- Cervical Spinal Cord Injury
- Spinal Cord Blood Flow
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Scientists often have a naive faith that if only they could discover enough facts about a problem, these facts would somehow arrange themselves in a compelling and true solution. Theodosius Dobzhansky
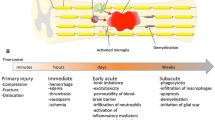
The capability of central nervous system (CNS) tissue for self-repair after severe injury is often insufficient to restore important neurological functions. Due to its small size and axonal concentration, the spinal cord is especially vulnerable to traumatic damage. The primary injury to the spinal cord results from an abrupt physical distortion such as occurs with a diving accident or a military blast injury. Cascades of molecular, cellular, tissue, and organ level events, many of which are pathophysiological, are initiated and propagate the tissue loss caused by the instantaneous primary injury. These events, collectively known as “secondary injury,” are thought to develop in proportion to the primary insult with a consequent progressive loss of residual gray and white matter around the injury epicenter. Secondary injury starts immediately, and its duration depends on which mechanism of pathophysiology is considered (e.g., excitotoxicity, ionic dysregulation, free radical toxicity and lipid peroxidation, mitochondrial and membrane failure, axonal disintegration, apoptosis and cell death, demyelination, inflammation) (see chapter 19), but can last at least weeks. The secondary injury concept is a generalization whose value may be superseded as more specific knowledge of post-injury processes is developed. Neuroprotection is also a broad concept applied to many types of neural injury referring to the attenuation of impending injury. The possibility to provide neuroprotection has been proven in certain settings, e.g., tissue cooling increases the brain and spinal cord’s tolerance to ischemia when initiated prophylactically [34, 267]. In these scenarios, tissue cooling neuroprotects the CNS during ischemia that would otherwise cause neuronal death.
However, more commonly neuroprotection refers to reduction of damage that would otherwise occur after an unpredictable event such as traumatic SCI. The development and testing of neuroprotective agents has largely followed emerging knowledge of pathomechanisms of secondary injury. Thus, early efforts focused on problems such as lowered spinal cord blood flow (SCBF) for which experimental evaluation techniques existed [272]. Current efforts are directed to control molecular events for which newer biomarker measurements are available.
The CNS has unique tissue properties, responses, and vulnerability; neuronal cells extend processes for long distances with high complexity and specificity. Neurons have high metabolic requirements and must maintain membrane function within narrow parameters. CNS glia also have complex functions including regulation of neurotransmitter levels and formation of myelin. The blood-brain barrier prevents the crossing of many classes of drugs, cells, proteins, and other molecules from the blood into the CNS extracellular space. Thus, specialized pharmacokinetic and pharmacodynamic studies are necessary to understand delivery and distribution to the brain and spinal cord. These include the administration route, bioavailability, serum, CSF and tissue levels, half-life, route of elimination, and metabolism to active and inactive products, including the impact of focal blood-brain barrier disruption. Critical issues such as a therapeutic window, dose-response, and adverse or secondary effects must be determined in preclinical animal models and phase I/II human studies. Neuroprotectants range from physical processes that affect the entire body (hypothermia) to drugs, ions, and specially engineered proteins and other biomolecules.
There has been a high failure rate of neuroprotective therapies applied clinically in stroke, traumatic brain injury, and SCI despite many impressive experimental studies in animals. To understand the reasons for this, and to increase efficiency and effectiveness, emphasis has been directed toward both the need for greater rigor in preclinical testing and clinical trial design [123, 196], increased reporting of negative results [229, 304], and the development of quantitative biomarkers.
Another issue to consider is that the distinction between primary and secondary injury is artificial because much of what is called secondary injury is directly triggered by the primary insult, as is a bruise following a blow to an extremity. Thus, the extent to which the consequences of primary injury can be attenuated may be limited. Another key distinction is between secondary injury and secondary insult, the latter being, e.g., an additive insult influenced by therapeutic management such as prolonged hypotension or by concomitant injuries such as hypoxia from pulmonary injuries. This chapter will summarize current knowledge of neuroprotective strategies in SCI discussing the major lessons so far learned and prospects for the near future.
2 Non-pharmacological Strategies for Neuroprotection
2.1 Surgical Decompression
The relief of ongoing pressure on the spinal cord and stabilization of the spine are indicated after SCI when either significant compression or instability is present. Decompression is the most direct neuroprotective procedure. Without spinal cord decompression, many other neuroprotective strategies are futile. If spinal cord compression raises the tissue pressure, ischemia may compromise tissue that might otherwise survive [46, 78]. Early decompression after SCI was assessed in several rodent models. These have shown improved behavioral and anatomical outcomes when decompression was performed earlier, e.g., 6 and even 12 h post-SCI, rather than 24 [284]. There may also be a role for adding dural enlargement to spinal cord decompression. Four-hour post-injury durotomy with allogeneic duraplasty reduced spinal cord cavitation, inflammation, and scar formation when compared to injury or durotomy only control animals [293]. A recent clinical study showed that following SCI, duraplasty improved estimated spinal cord perfusion more than spinal laminectomy decompression alone [248]. Recently, even the acute decompression of necrotic spinal cord tissue has been assessed as a neuroprotectant in large animal models, thus the definition of effective spinal decompression after SCI is in evolution.
Although evidence in animal models shows a benefit to early spinal cord decompression, the optimal clinical timing has been a leading controversy in neurosurgery in recent decades and remains without conclusive evidence. Concerns regarding early surgery have included the safety of acute surgical manipulation of the spinal cord; safety of transportation to specialized care centers; availability of skilled spine surgeons, anesthesiologists, and operative teams at all hours; availability of imaging; and vulnerability of acutely damaged spinal tissue to fluctuations in blood pressure during anesthesia. Substantial published evidence has addressed these concerns supporting the safety of early surgery [101, 112, 134, 331]. The spine surgery culture has shifted to recognize the value of early surgery. An evaluation of the clinical effectiveness of decompressive surgery after SCI took place with the STASCIS Study (Surgical Timing in Acute Spinal Cord Injury). This multicenter, international, prospective, nonrandomized study analyzed the effects of early (<24 h post-SCI) versus late decompression (≥24 h post-SCI) [101]. From 313 patients enrolled, 222 were assessed at the 6-month post-SCI interval; 19.8 % of the early surgery cohort showed a ≥2 grade improvement in AIS grade versus 8.8 % in the late group. There were some imbalances in the relative severity of injury between the groups, but it is evident that early surgery was not harmful in this trial. Thus, surgical decompression should be considered the first significant neuroprotective goal of SCI therapy in concert with other guidelines and potential experimental treatments.
2.2 Support of Blood Pressure
Ischemia is a prominent pathomechanism of SCI secondary injury. Spinal cord blood flow (SCBF) is normally autoregulated so that flow matches metabolic demand over a range of blood pressures (BP). However, after SCI, autoregulation is disrupted, and SCBF becomes more linearly linked to systemic BP [324]. SCI patients frequently have low BP due to neurogenic shock. Inadequate BP is a risk factor for poor neurologic patient outcome [150, 205, 330], presumably by exacerbating ischemic damage. Therefore, support of adequate BP is a key to effective neuroprotection. Recently, it has been shown that intraspinal tissue pressure may be used to guide the mean arterial pressure to optimize spinal cord blood flow [341]. This type of monitoring is well established in brain injury and it is logical to extrapolate to SCI [333].
2.3 Infection
Recent studies have provided evidence that SCI patients with infections such as pneumonia have less neurological recovery [95]. Thus, infection is a “disease-modifying factor.” Schwab and colleagues developed rodent models where an acute immune deficiency state associated with SCI increased vulnerability to infection due to alterations in monocyte function. This has been described as the spinal cord injury-induced immune depression syndrome (SCI-IDS) [42, 265, 266]. Additionally the risk of bacterial infection seemed to be injury-level dependent in an SCI mouse model [42]. A current prospective, multicenter, international clinical protocol, the SCIentinel study, is examining the hypothesis that the severity of immune depression is dependent on injury severity and injury level [180]. Based on this evidence, avoidance of infection is critical in SCI and could be neuroprotective. Recently, it has been learned that the population of bacteria in the gut is altered by SCI [136]. Emerging studies are assessing the impact of the altered microbiome on neurological recovery and complications.
2.4 CSF Drainage
CSF drainage is known to influence SCBF and has achieved prominence as a preemptive strategy to reduce paralysis due to spinal cord ischemia during aortic surgery. The ability of CSF drainage to improve SCBF after SCI is less certain. Spinal cord perfusion pressure (SCPP) is calculated to be proportional to the average pressure head (mean arterial blood pressure – MAP) minus the venous resistance that has been estimated as cerebrospinal fluid pressure (CSFP). Thus, by reducing CSFP, SCBF may be increased. In clinical studies of thoracoabdominal aneurysm repair combined with CSF drainage, an 80 % reduction in the relative risk of postoperative neurological deficits has been reported [63, 65, 316]. After SCI, efforts to maintain SCBF include correction of spinal shock due to lack of sympathetic tone and increased capacitance by keeping MAP >90 mmHg using vasopressors. Pharmacologically increased MAP is thought to compensate for the failure of SCBF autoregulation due to loss of tonic activity of sympathetic neurons. Lowering CSFP may be another alternative to increasing SCBF. A prospective randomized study including 22 patients evaluated the safety and feasibility of CSF drainage and the correlation with neurological recovery at 6 months post-SCI [186]. A CSF drain was placed prior to surgical decompression and maintained for 72 h in all subjects. Subjects were randomized to have CSF drained or not drained, with a drainage threshold of 10 mmHg (the typical value of CSF pressure). After rostral decompressive surgery, there was a marked increase in distal CSFP as well as the onset of a pulsatile waveform. These changes indicated that the CSF measurements prior to surgical decompression (recorded from the lumbar cistern) did not represent the entire CSF space due to rostral occlusion by unreduced bone and disc fragments. Thus, calculations of SCPP prior to decompression were inaccurate, and a reduction in apparent SCBF occurred after decompression when more accurate CSFP values were recorded. In this study, the CSF drainage was conservative; the CSFP remained well above the target value due to restrictions on the rate of drainage per hour and drain closure when intensive patient assessment by nursing was unavailable. Although the study was not powered to assess efficacy nor the patients stratified according to injury level, the authors conclude that there was no neurological detriment or benefit due to the intervention. In a recent study, a pressure recording catheter was inserted into the intrathecal space and then “wedged” between the damaged spinal cord and the dura. The pressure reading was much higher than that of the CSF and considered to represent elevated spinal cord tissue pressure [341]. This finding indicates the weakness of relying on oversimplified formulae for SCPP that assumes spinal cord tissue pressures are reflected by CSFP. A recent porcine SCI study has shown that neither elevating MAP with pressors nor releasing CSF alone improved SCBF other than transiently, but the combination of both manipulations increases SCBF in a more sustained manner [228].
2.5 Therapeutic Hypothermia
The impact of tissue cooling on ischemia in the brain and spinal cord has been studied for more than six decades. Interest in hypothermia partially derived from nature because temperature and metabolic rates are linked. Hibernating mammals dramatically reduce their core body temperature minimizing their metabolic needs. A key idea is that by reducing metabolic requirements, tissue survival during states of low blood flow is improved. Studies in anesthetized dogs by Bigelow and colleagues showed a nearly linear relationship between core temperature and reduced oxygen consumption down to the lowest survivable temperatures (28°C) [29]. Pontius, DeBakey, and colleagues reported that in dogs cooled systemically to 29.4°C, the incidence of post-thoracic aorta occlusion-related paralysis was reduced from 65 % to zero [253]. In the 1960s, hypothermia was employed for neuroprotection to allow temporary circulatory arrest for complicated aneurysm surgery [81]. An important advance was the finding that that mild hypothermia (e.g., 33°C), which is much safer clinically, also had significant neuroprotective effects in rodent stroke models [44]. As of 2016, hypothermia has been studied extensively in several clinical settings. The most compelling current clinical data to support a neuroprotective benefit for hypothermia are from clinical trials in post-cardiac arrest comatose patients [27]. Hypothermia has been tested extensively in traumatic brain injury (TBI) and to a lesser extent in SCI. Despite experimental evidence of reduced tissue loss and improved brain function in animal models [75], multiple clinical trials have failed to show a benefit of post-traumatic hypothermia in adult or pediatric brain trauma [271], other than for transient control of raised intracranial pressure. SCI studies commenced in the 1960s. Maurice Albin and Robert White created one of the first neurocritical care units, located at the Cleveland Metropolitan General Hospital. Numerous experiments examining the impact of hypothermia on the brain and spinal cord were performed. In a study published in 1968 [5], primates were subjected to weight-drop contusion SCI, and after a delay of 4 h, one group underwent local perfusion cooling with a target spinal cord temperature of 10°C maintained for 3 h. The results were impressive, all treated animals recovered, and none of the controls showed substantial recovery. Now, with decades of additional experience, the 1968 results with localized hypothermia seem too good to be true, in part due to increased skepticism about extremely distinct results. In other animal models, hypothermia has been shown to mitigate several aspects of secondary injury cascades. There is modulation of inflammation [76], with reduced neutrophil infiltration [52], less glutamate release [166] and excitotoxicity, and reduced cellular energy metabolism [352]. The preponderance of experimental evidence indicates systemic cooling methods to be more effective [212] than epidural cooling [48].
Several small clinical studies of spinal cooling have been performed, but none has had a sufficiently robust design to provide conclusive evidence of efficacy due to small patient cohort numbers and a lack of suitable controls. The use of hypothermia is greatly underreported based on discussions with senior neurosurgeons who participated in unpublished studies. Since it is not a drug, it is relatively easy to apply hypothermia. Clinical experiments in the 1970s emphasized locally applied cooling due to the influence of Albin and White’s publications. Bricolo and colleagues treated eight neurologically complete acute SCI patients with 2 h of subdural cooling using 5°C saline irrigation. Motor and sensory recovery was reported in four subjects with 2 recovering ambulation [41]. Hansebout and colleagues studied the effect of local epidural cooling in a dog model of spinal cord compression to assess optimal treatment duration [182, 340]. Based on favorable results with 4 h of cooling, they then recruited ten patients with complete SCI to undergo early surgical decompression and local extradural cooling within 8 h of complete SCI. The dural temperature at the injury site was maintained at 6°C for 4 h using a suspended extradural saddle (cord temperature was not recorded). A factor complicating interpretation was the delivery of dexamethasone, 6 mg every 6 h for 11 days, and then tapered until the 18th day. This study had a mean follow-up of 1.9 years. Of six subjects with thoracic SCI, four were unchanged, one became ambulatory, and another had sensory recovery. Of four subjects with a cervical injury, three were unchanged, and one showed motor and sensory recovery [148]. These studies occurred before the publication of the important ASIA impairment scale (AIS) in 1982. Hansebout and Hansebout published a long-term follow-up of the original cohort with ten additional patients enrolled in local cooling and dexamethasone treatment. Results were reported by adapting their previous scoring system to the AIS scale. From the total of twenty treated patients categorized as baseline AIS-A, there was improvement in thoracic injuries of 16.7 % to AIS-B, 33.3 % to AIS-C, 16.7 % to AIS-D, and a mean sensory descent of 2.5 levels per patient. For the cervical cohort, there was improvement of 35.7 % to AIS-B, 21.4 % to AIS-C, and 7.2 % to AIS-D, with a mean motor descent of 0.92 levels per patient [147]. These recovery rates are much better than the published natural history for cervical and thoracic injuries [302, 355], but we know that the reproducibility of small cohorts is poor and robust trial design is needed to obtain valid conclusions regarding therapeutic efficacy [194]. Critique of this study included the contradictory evidence available for the use of steroids, the possibility of improved outcomes as the natural evolution following early decompression surgery, and the interpolation of the clinical data [321]. Given the current information, it would be better not to combine steroids and hypothermia in a clinical trial.
Currently, systemic hypothermia after acute SCI in subjects with motor complete paralysis is under evaluation in an observational prospective cohort study sponsored by the University of Miami/The Miami Project to Cure Paralysis (ClinicalTrials.gov identifier: NCT01739010) at Jackson Memorial Hospital, Miami, FL. Systemic cooling is achieved using an intravascular cooling catheter advanced centrally through the femoral vein. The target temperature is 33°C at a maximum cooling rate of 0.5°C/h, MAP is maintained >90 mmHg, and rewarming begins after 48 h at a rate of 0.1°C/h. A preliminary report of 14 patients scored as AIS-A on admission indicated conversions of three patients to AIS-B, two to AIS-C, and one to AIS-D, with similar rate of complications to the historical control group [204]. In a more recent publication [74], data from 35 subjects with initial AIS-A cervical injury was presented. The rate of conversion to higher AIS grades exceeds than expected from historical controls. Both studies indicate a delay to initiate cooling with an average of 5.8 h for subjects rapidly transferred to the institution, followed by 2.7 h to achieve the target temperature. All patients underwent surgical decompression at an average of 16.5 h after injury. There was no relationship between the timing of cooling onset and neurological outcome. The majority of complications were pulmonary, and the incidence was similar to that in recent series if adjusted for injury severity. Although the data is promising, it remains difficult to isolate the efficacy of hypothermia without a randomized control group. A new study, acute rapid cooling of the traumatically injured cord (ARCTIC), is designed to randomize patients to control and cooling temperature groups.
2.6 Dietary Adjuvants/Restrictions
There has been interest in the influence of dietary factors on CNS inflammation and oxidative stress and diet’s contribution to neurodegenerative diseases such as Alzheimer’s [145, 221] and epilepsy [173]. The positive influence of a ketogenic diet on the incidence of seizures in children has been known for a hundred years [153] and the favorable impact of fasting for millennia. Ketogenic diets reduce circulating glucose and increase ketones as an energy substrate in the CNS. The ability of a ketogenic diet to improve cerebral metabolism after TBI was reviewed by Prins and Matsumoto [260]. In rodents, ketones can reduce free radical generation from mitochondria during stress induced by excess levels of glutamate [218]. Reduction in reactive oxygen species (ROS) has also been observed with caloric restriction. Following unilateral cervical crush hemisection, every-other-day fasting (EODF) leads to reduced injury size, increased gray matter sparing, improved ipsilateral forelimb function, increased functional Tropomyosin receptor kinase B (trkB) receptor levels, and increased plasticity of corticospinal tract (CST) axons [252]. EODF animals weighed 8 % less than controls at the end of the experiment. Blood glucose and β-hydroxybutyrate ketone levels varied inversely throughout the study.
In a rat model of cervical hemi-contusion, Tetzlaff and colleagues mimicked the effects of fasting using a 3:1 ratio regimen of ketogenic diet starting 4 h after injury, with a return to standard diet 12 weeks post-injury. Animals receiving this diet showed improved behavioral recovery, reduced lesion size, increased gray matter sparing, with an increase in the vascular transporters GLUT1 and MCT1 [310]. The post-injury weight gain was similar between the ketogenic and control group. These investigators also studied daily food restriction to 75 %, equivalent to the total food intake of EODF animals, after T10 moderate contusion injury. This continuous caloric restriction did not yield evidence of protection, whereas EODF improved locomotor recovery [168]. When EODF was studied in a mouse model, no behavioral or histological improvement was observed after a T10 crush SCI, and β-hydroxybutyrate levels failed to rise substantially in the first week post-injury despite prominent loss of body weight [311]. These data indicate that the responses in mouse and rat models differ; thus, caution should be exercised in the extrapolation of rodent data to humans.
Creatine is a popular health supplement whose delivery has been shown to maintain ATP homeostasis in TBI models. Rabchevsky and colleagues tested the neuroprotective effect of 2 % creatine supplementation of rat chow given for 4–5 weeks prior to T10 New York University (NYU) or Infinite Horizon device-delivered (IH) contusion injuries on tissue sparing and locomotor recovery (see chapter 25). They also tested the effect of continuation or cessation of the diet after the T10 IH injury. Although no overall effects on locomotor recovery or white matter sparing were found, the creatine supplemented groups in the IH experiment had smaller lesion volumes and more gray matter sparing [261]. IH injuries were found to be more focal in distribution than NYU injuries. These differences provide another example of the variations arising in “standard” injury models and the caution that is needed in extrapolating results for human trial design where injuries are heterogeneous [85].
The neuroprotective effects of long-chain omega-3 polyunsaturated fatty acids (PUFAs) such as docosahexaenoic acid (DHA) are being studied. The effects of acute IV delivery of DHA at 30 min, 1 h, and 3 h post-injury were investigated in a T10 spinal cord compression model. Thirty-minute but not 3 h delivery was associated with substantial neuroprotection with improved neuron and oligodendrocyte survival, reduced inflammation, and significantly improved locomotor scores. Reductions in the oxidation of lipids, proteins, RNA and DNA, and COX-2 expression were found [159], as well as evidence of reduced axonal cytoskeletal damage, demyelination, and preservation of serotonergic innervation below the injury level [338]. A recent study performed in a T6/7 mouse clip compression model found reduced edema, tumor necrosis factor (TNF)-α, nitrotyrosine formation, and glial fibrillary acidic protein (GFAP), FAS ligand, BCL2-associated X protein (BAX), and B-cell lymphoma 2 (BCL2) expression when DHA was administered 30 min after injury [245]. Translation of this therapy has been limited due to lack of cGMP (current good manufacturing practice) grade DHA indicating the difficulty of obtaining clinical grade formulations that affects many agents suitable for clinical testing. The therapeutic window for DHA is brief, apparently true of most neuroprotectants, and its strong safety profile supports acute administration.
3 Pharmacological Strategies Tested in Human Clinical Trials
3.1 Methylprednisolone Sodium Succinate (MPSS)
Studies with MPSS have been important in the evolution of SCI clinical trials raising critical questions about the benefit/risk equation for therapeutic neuroprotection, the relative weight to be assigned to robust prospective research designs as compared to other forms of cohort studies [102] and the importance of a substantial body of evidence in justifying an emerging treatment as a treatment standard. The student of neuroprotection is referred both to the many reviews of this topic and also to the primary studies and subsequent critiques [160], many of high quality. As an anti-inflammatory drug, there are numerous indications for methylprednisolone and a long history of clinical use in the CNS [234]. Likewise, complications related to its use have been reported for almost 40 years [312]. After experimental SCI, MPSS administration was shown to inhibit lipid peroxidation [142, 143], facilitate extracellular calcium recovery [348], and reduce inflammatory responses [158], white matter neurofilament degradation [40], and macrophage/microglia infiltration [239]. Given the large data set indicating beneficial effects in preclinical studies and the absence of an effective clinical treatment for acute SCI, there was enthusiasm for clinical translation of MPSS. There were three prospective randomized studies of MPSS in SCI lead by Bracken and colleagues. The First National Acute Spinal Cord Injury Study (NASCIS I) was initiated in 1979 and completed in 1984. No significant difference in improvement was found between subjects that received doses of 1000 mg or 100 mg of MPSS for 11 days [35, 38]. However, the high dose was associated with an increased relative risk for case fatality and wound infection. The NASCIS II study increased the dose and reported neurological improvement for MPSS versus placebo if the treatment was started within 8 h of injury (30 mg/kg loading dose, 5.4 mg/kg/h for infusion continued for 23 h) [36, 37]. A third NASCIS trial was published in 1997, in which a 24-h treatment period was compared to a 48-h treatment course. There was no placebo control, and MPSS was compared to trilizad, a non-glucocorticoid lipid peroxidation inhibitor. The trial included the Functional Independence Measure as well as ASIA motor and sensory scores as principle outcomes. A Japanese multicenter trial reported following NASCIS II, using the same pharmaceutical protocol, also found a benefit in the MPSS group [238]
A decade after publication of the NASCIS II data, the use of MPSS was widespread and in some cases not supported by the clinical trial evidence. A counter opinion framed MPSS as an inappropriate “gold standard” treatment in acute SCI care. Viewed retrospectively, this early assignment of “gold standard” status is considered to have been unwise and damaging to the study of other therapeutics [160]. However, at the time the excitement that something worked in SCI overshadowed a more measured evaluation of the study results. NASCIS III indicated an advantage to prolonged (48 versus 24 h) treatment if drug delivery was started 3–8 h post-injury, but no advantage if initiated within 3 h [39]. When the NASCIS II protocol was tested in France, no significant differences in outcomes between methylprednisolone and nimodipine were found [247]. A small prospective trial in Japan assessed complication rates with the NASCIS II MPSS protocol versus placebo in patients with cervical injury (and associated cervical stenosis) that did not undergo surgical decompression. A higher rate of pneumonia and GI bleeding was observed in the MPSS group. It should be noted that the average patient age was 61 years old [230]. Further, seven retrospective studies took place in the 1990s, six in the USA and one in Ireland; these have included more than 1300 patients and have shown equivalent motor outcomes in between the steroid and non-steroid groups with slight trends or significance toward the increase of complications in the MPSS groups [119, 121, 122, 152, 206, 257, 259].
MPSS is the most extensively tested pharmacological neuroprotective agent in the SCI field, but evidence to support its use in acute SCI for either the 24- or the 48-h regimen remains controversial due to issues in the research designs and the incidence of associated complications. Three multicenter, randomized clinical trials were completed to address the efficacy of MPSS, and only the NASCIS II has clearly indicated a benefit for neurological scores. The potential increase in serious medical complications including infections, sepsis, respiratory complications, pulmonary embolism, gastrointestinal hemorrhage, pancreatitis, delayed wound healing, worsening of head injury outcomes, and death is a concern. Furthermore, access to the study data has been limited, and concerns have been raised about the control group results and the lack of submission of the data to the FDA to obtain an approved indication [60]. Altogether, these critiques seem harsh treatment of large well-intentioned RCTs, but given the high incidence of serious complications in SCI [133], it is unwise to use an agent that may add to this morbidity. In summary, due to the limited evidence of efficacy in clinical trials and the risk of medical complications, the use of MPSS is only recommended by the American Association of Neurological Surgeons/Congress of Neurological Surgeons (AANS/CNS) as an option which may be supported by clinicians balancing the evidence of side effects toward the limited clinical evidence and the circumstance that there might be no other available therapeutic to offer at the moment [336].
3.2 GM-1 (Monosialotetrahexosylganglioside)
GM-1 belongs to a family of glycosphingolipids that contain only one sialic acid residue; gangliosides are abundant in the nervous system [73, 146]. GM-1 is believed to have trophic effects. These include inducing neurotrophin-3 (NT3) release with further activation of the TrKC receptor [262], mimicking nerve growth factor (NGF) activity, and activation of the TrKB receptor [104]. Synthetic derivatives of GM-1 – LIGA4, LIGA20, and PKS3 – have been shown to protect neurons from glutamate-induced death [223]. Clinically, GM-1 was tested in stroke before SCI [9]. Despite several trials in stroke, its efficacy remained unclear [SASS trial] [113], possibly due to methodological issues [7], not unlike those that affect SCI clinical trials.
The first clinical report testing GM-1 in SCI was a small prospective, randomized, placebo-controlled, double-blinded trial [117, 118]. Thirty-seven patients received a loading dose of MPSS 250 mg followed by 125 mg every 6 h for 72 h, together with GM-1 100 mg/day within 72 h post-SCI, continued daily for 18–32 days. Thirty-four patients were available at 1-year follow-up, and the authors reported a significant improvement in AIS motor scores (p = 0.047) and Frankel grade (p = 0.034) with no adverse effects attributed to GM-1. The following year, the Sygen multicenter, prospective, double-blind, randomized, and stratified study was initiated. The Sygen study is one of the most important trials the SCI field has performed and the largest SCI therapeutics trial ever conducted. 797 patients were enrolled from 1992 to 1997. All patients were first treated according to the MPSS NASCIS II protocol and then randomized to placebo, low-dose GM-1 (300 mg loading dose and then 100 mg/day), or high-dose GM-1(600 mg loading dose and then 200 mg/day) for 56 days [116]. Follow-up with AIS and Benzel classification continued until 52 weeks post-injury. The primary endpoint of the study was “marked improvement” at 26 weeks, defined as a two-grade change in the Modified Benzel Classification from the baseline AIS grade. Although there appeared to be a more rapid neurological recovery in the Sygen-treated group, there was no difference in the overall treatment groups at 26 weeks. However, several interesting trends were observed in the data including more improvement in the AIS-B treatment group and efficacy trends in sensory, motor, bowel, and bladder function as well as ceiling effects in subjects that were initially AIS-C and AIS-D. A ceiling effect means that, e.g., a subject enrolled as AIS-D cannot show a 2 grade improvement and should not have been recruited to the trial. Adverse events did not differ between the treatment groups. There was no true placebo in the GM-1 study because MPSS was considered the gold standard at that time. In summary, the pivotal clinical trial for GM-1 failed to meet its prospectively set primary outcomes. The primary efficacy measure of the Sygen study was a high bar, and statistically significant differences were only observed on post hoc analysis. There is no subsequent clinical study available to validate or disprove the previously mentioned clinical evidence nor has much further experimental evaluation been published [47]. Given the logistical investment to conduct a phase 3 RCT in SCI, it is rare that a therapeutic is further tested after the failure of a well-designed study, even if redesign or stratification might lead to a different result. Currently a GM-1 loading dose of 300 mg followed by 100 mg/day for 56 days after the administration of methylprednisolone within the first 8 h of injury (as per NASCIS II protocol) is recommended by the AANS/CNS as an option (if available) for the treatment of acute SCI. Data from the GM-1 study has proven useful as a comparator for other clinical studies and to help define the natural history of SCI recovery [97, 302]. One of the most striking aspects of the GM-1 story is the paucity of published preclinical data in SCI models prior to the initiation of clinical trials. In the only study reporting the combination of MPSS (30 mg/kg) and GM-1 in a rat model of contusions of graded severity, the acute combination of both drugs blocked the neuroprotective effects of MPSS. In that study, no effect of acute administration of GM-1 was noted, and MPSS alone was neuroprotective in mild and moderate contusion but had minimal benefit after severe contusion [61].
3.3 Magnesium
Magnesium salts and polyethylene glycol are used extensively in medicine. Magnesium neuroprotective properties have been evaluated in animal models for 25 years [167]. Magnesium can block N-Methyl-D-aspartate (NMDA)-type glutamate receptors and reduce calcium influx to cells attenuating glutamate excitotoxicity [361]. Other reported effects include decreased apoptosis [200], improved electrophysiological conduction, reduced lipid peroxidation [315], and normalized lactate levels after SCI [240].
After brain and spinal cord injury, local levels of magnesium are depleted contributing to mitochondrial dysfunction. In rodents intraperitoneal high doses of MgSO4 (600 mg/kg) given immediately after SCI have resulted in early functional recovery and preservation of spinal cord ultrastructure when compared to small doses (100 mg/kg) [174]. Although the effects of magnesium are noteworthy, the doses postulated to induce neuroprotection may be too high and could produce respiratory depression or cardiac arrest in the acute clinical injury setting. Additional preclinical studies assessed magnesium formulations with more efficient blood-brain barrier penetration. One strategy is to combine magnesium salts with polyethylene glycol (PEG) to increase penetration. PEG can have several effects on drug delivery, including increasing the serum half-life of nanoparticles, enhancing brain barrier permeation, and directly supporting membrane repair acute SCI [32]. Several patents have described pharmacological properties of Mg-PEG compounds that achieve higher CSF concentrations as compared to Mg alone, optimal IV delivery rates, and calculations of Cmax, AUC0-∞, and T1/2 for different PEG-magnesium formulations (US Patents 8840933, 8852566). A series of animal studies has examined the relative effects of PEG, Mg2+, and their combination compared to saline after SCI on tissue preservation and neurological outcomes. Kwon and colleagues conducted a detailed study to assess the neuroprotective effects of either MgCl2 or MgSO4 combined with PEG at two different doses, 127 μmol/kg or 254 μmol/kg across four time points for initial delivery: 15 min, 2, 4, and 8 h after injury. Two, four, or six infusions spaced 6 or 8 h apart were compared. Using lesion size as a marker of neuroprotection, it was found that early delivery was more protective, the anion (Cl versus SO4) made no difference, the higher concentration was superior, more IV doses were superior to fewer, and a 6-h dose infusion interval was best [186, 188]. Other groups have reported improved tissue sparing with MgSO4 (300 mg/kg) + PEG versus saline controls in a severe T3 clip compression injury, with no significant differences when compared with either treatment alone [80].
Recognizing that IV CNS magnesium therapy could be limited by low CSF and tissue levels, and the previously reported results by Kwon and colleagues, Medtronic Inc. developed a polymer formulation that enhances magnesium accumulation at the injury site. Acorda Therapeutics licensed this formulation as AC105 (Mg/PEG) and designed a phase II double-blinded, randomized, placebo-controlled study of the safety, tolerability, and potential therapeutic activity of a regimen of six sequential AC105 doses in patients with acute SCI. The first dose was to be given in progressively earlier cohorts, 12, 9, and 6 h after injury with all doses delivered over 30 h (ClinicalTrials.gov Identifier: NCT01750684). Although the study was terminated in 2015, no results have been yet disclosed. Multiple other studies of magnesium in various indications have reported mixed results. A recent large-scale trial (1700 subjects) assessing the prehospital use of magnesium sulfate in stroke did not find a therapeutic effect [274].
3.4 Riluzole
Riluzole is an extensively studied off-patent benzothiazole drug. The neuroprotective effects of riluzole were first reported in ischemia [222]. It counteracts glutamate-mediated neurotoxicity by inhibiting the release of glutamate from presynaptic terminals [56, 227], increasing high affinity glutamate uptake [11], and inhibiting sodium channels, by maintaining voltage-regulated sodium channels in the inactivated state [23, 258]. Riluzole crosses the blood-brain barrier [22]. The molecule was developed in France as an antiepileptic, and there have been several clinical trials for psychiatric conditions such as generalized anxiety disorder, major depressive disorder, and obsessive-compulsive disorder [128]. The only FDA-approved indication of riluzole is to prolong the time interval before assisted ventilation is needed in amyotrophic lateral sclerosis (ALS) patients [233]. Given the established safety record and known safe dose levels, the regulatory pathway to test such drugs in SCI can be much simpler than for novel therapeutics.
Experimental studies have assessed the effect of riluzole delivery in a variety of SCI models. Excessive glutamate is neurotoxic by leading to accumulation of intracellular Ca2+ that activates calpain proteases that break down cytoskeletal components such as spectrin, Microtubule-associated protein 2 (MAP-2), and neurofilament. Pretreatment of rats with 8 mg/kg of riluzole 15 min before and 2 h post 25 g/cm NYU impact to the T10 spinal cord substantially reduced activated calpain and MAP-2 loss in neurons [297]. Four or 8 mg/kg before and at the onset of reperfusion in aortic cross-clamping prevented ischemia-induced necrosis and cytoskeletal proteolysis [195]. Rats receiving riluzole (2 mg/kg 30 min post-thoracic compression and then BID for 10 days) recovered somatosensory evoked potentials (SSEPs) and had smaller lesion volumes [313]; 5 mg/kg 15 min after compressive SCI improved gray matter and selective white matter sparing with better hind-limb coordination and strength [278]; 8 mg/kg post S2 transection reduced spasticity [177], but 10 mg/kg caused motor weakness. Only two studies assessed a clinically meaningful therapeutic window, 8 + 30 mg/kg of MPSS, 2 and 4 h post-contusion promoted tissue sparing and recovery in Basso, Beattie, and Bresnahan (BBB) scores [235]. Eight mg/kg BID 1 and 3-h post-C7 cervical clip compression and continued at 6mg/kg BID for 7 days showed improved SSEPs, tissue preservation, reduced apoptosis, and improved locomotor scores [344]. In this study, a striking difference in the benefit between the 1- and 3-h post-injury groups is observed indicating the brief therapeutic window to reduce secondary injury.
In 2010 the North American Clinical Trials Network (NACTN) initiated a prospective multicenter phase I trial to evaluate the pharmacokinetics, safety, and effects of riluzole in acute SCI (ClinicalTrials.gov Identifier: NCT00876889). Thirty-six patients were enrolled; the dose was 50 mg PO within 12 h of injury, continuing BID for 14 additional days. The trial did not have a concurrent control group, but the comparison group was formed by 36 matched patients meeting the eligibility criteria that received standard of care treatment in NACTN centers from October 2005 to November 2012 enrolled to the NACTN data registry. The authors report a significantly higher mean motor score in cervical patients treated with riluzole (p = 0.021, 31.2 points versus 15.7 points in the control group) from admission to 90 days, with no serious adverse events or deaths related to riluzole [132]. A phase 3 study is now underway for riluzole. A well-designed multicenter, double-blind, randomized, placebo-controlled trial will test a dose of 100 mg BID during the first 24 h post-SCI, followed by 50 mg BID (day 2–14 post-SCI) in an international study enrolling subjects with acute cervical SCI [99] (Clinical trials.gov identifier: NCT01597518). The study is aimed to be completed in 2018.
3.5 Minocycline
This broad-spectrum bacteriostatic antibiotic is the most lipid-soluble tetracycline available and has excellent penetration into the CNS. Minocycline has gained interest for its neuroprotective and anti-inflammatory properties in several neurodegenerative diseases including Huntington’s [54], Parkinson’s [163, 164], schizophrenia [53], and multiple sclerosis [360]. In animal models of SCI, minocycline has been shown to reduce caspase-3 activation, attenuate cell death, reduce lesion size [202], decrease activated microglia/macrophage density, oligodendrocyte death and CST dieback [306], inhibit cytochrome-C release and astrogliosis [326], and inhibit lipid peroxidation [295]. Minocycline moved rapidly through preclinical testing to clinical evaluation due to its prior extensive safety record as an antibiotic and established toxicity limits.
From 2004 to 2008, the University of Calgary conducted a single-center, double-blind, randomized, placebo-controlled, phase II study, to test the safety and estimate outcome changes after an IV minocycline infusion started within 12 h of SCI. Surgical decompression occurred during the first 24 h post-SCI, and minocycline was delivered IV BID until 7 days post-SCI (ClinicalTrials.gov identifier: NCT00559494). MPPS was not given to the subjects. Each had a lumbar catheter placed to allow CSF sampling. To achieve serum levels similar to those found to be effective in experimental models, the authors treated the first five subjects with an IV loading dose of 200 mg, followed by 200 mg BID for 7 days, and then assessed serum and CSF minocycline trough and peak levels. Based on this information, the dose was then revised to an 800 mg loading dose, reduced by 100 mg in each subsequent dose until 400 mg, and then maintained BID for the remainder of the 7 days. The authors found no statistically significant difference in the primary outcome (ISNCSCI motor scores up to 1 year) [49]. Only one subject showed a minocycline linked adverse event with a transient elevation of liver enzymes. Although the study was not powered sufficiently to prove therapeutic efficacy, patients with complete cervical SCI showed a 14-point motor increase and patients with incomplete cervical SCI a 22-point improvement over placebo. Post hoc analysis indicated that the higher minocycline doses were associated with a greater degree of motor recovery. These data shows the importance of obtaining pharmacokinetic data from CSF to assess the adequacy of dosing strategies. A related serum biomarker study showed that subjects receiving minocycline had lower levels of the light chain of neurofilament than those receiving placebo [184]. Minocycline is now being assessed in a multicenter, randomized, double-blind, placebo-controlled Canadian phase 3 trial enrolling subjects with acute cervical SCI. Minocycline will be infused centrally BID at 800 and 700 mg on day 1, 600 and 500 mg on day 2, and 400 mg thereafter from day 3 through 7 (Clinical trials.gov identifier: NCT01828203). An estimated enrollment of 248 subjects is aimed to be completed in 2018.
3.6 Cethrin
RhoA is a major G-protein regulator of the cytoskeleton. Rho’s activity state is regulated by whether it’s in the GTP (active) or GDP (inactive) state. Active RhoA promotes growth cone collapse by activating Rho kinase and also activates apoptosis in the injured spinal cord [83]. C3 transferase, a bacterial enzyme, ADP ribosylates asparagine 41 in the effector-binding domain of Rho, irreversibly preventing its activation [72], one result of which is to prevent axonal growth cone collapse when confronted by inhibitory molecules in the spinal cord. Cethrin is a synthetic molecule that consists of C3 transferase with an added cell membrane transport sequence [214, 343]. Key experiments included in vitro studies where primary retinal neurons treated with C3 transferase extended neurites on myelin substrates. After optic nerve crush and C3 transferase treatment, axons crossed the lesion and grew distally [203]. Experiments in a mouse hemisection model of SCI with C3 transferase showed significant differences in the modified BBB analysis and long-distance regeneration of CST fibers [72]. The cell membrane transport sequence enhanced dural penetration in rodent studies. This sequence alteration is claimed to make extradural delivery through the dura feasible [214].
The Rho inactivator BA-210 was trademarked as Cethrin by BioAxone BioSciences Inc., and a phase I/IIa multicenter, nonrandomized, open-label, dose-escalation trial for patients with complete cervical or thoracic SCI (ClinicalTrials.gov Identifier: NCT00500812) was conducted [100]. From the 48 patients enrolled, 32 completed the 12-month follow-up. Treatment was delivered during surgical decompression with Cethrin mixed within a fibrin sealant placed on top of the dura mater. Five doses were tested, 0.3, 1.0, 3.0, 6.0, and 9.0 mg. Most of the subjects were treated between 24 and 72 h after injury [231]. The most compelling data from the study is a dose-dependent effect on recovery of cervical motor scores with the maximum observed recovery with the 3 mg dose (27 points improved from baseline). Anderson and McKerracher analyzed the data from pooled dose groups using the method of last observation carried forward and compared to the Sygen control group, the European Multicenter Study about Spinal Cord Injury (EMSCI) database, the US Models Systems and Waters published recovery data. The analysis showed a conversion rate from AIS-A to AIS-C of 33 % in cervical patients and 7 % in thoracic patients, the cervical conversion rate being higher than reported in the Sygen control group. The cervical total motor score improvement of 18.6 ± 17.3 exceeds the other data sets, although the EMSCI recovery rates are close. More than 40 % of cervical subjects in the pooled Cethrin group recovered 2 or more neurological levels, which is also higher than usual. BioAxone announced a multicenter, randomized, double-blind, placebo-controlled phase IIb/III trial for Cethrin to begin for patients with acute cervical SCI (ClinicalTrials.gov Identifier: NCT02053883), aimed for completion in 2016. Subsequently, BioAxone licensed Cethrin to Vertex Pharmaceuticals Incorporated in late 2014 and withdrew the study. Cethrin appears now under the name of VX-210, and a new phase IIb/III study (randomized, double-blind, placebo-controlled) sponsored by Vertex launched in early 2016 for patients with acute cervical SCI. The trial is expected to be completed in 2018 (ClinicalTrials.gov Identifier: NCT02669849).
3.7 Naloxone
In the late 1970s, the opiate antagonist naloxone was found to reverse the septic shock-like consequences of IV delivery of endotoxin. B-Endorphin, an opiate agonist, was released in response to shock [155]. In 1981, Faden, Jacobs, and Holaday published an experiment in cats where, following a severe C7 cervical contusion, naloxone or saline was delivered 45 min after injury. Animals receiving naloxone recovered blood pressure rapidly and showed improved forelimb and hindlimb function 3 weeks after the injury. The authors inferred that improved SCBF was the therapeutic mechanism [91]. Further animal studies showed a reduction in ischemia, preserving SCBF and SSEPs [108, 349]. Naloxone also reduced superoxide formation by reactive microglia and antagonized dynorphin A, an endogenous opioid capable of inducing hindlimb paralysis [90], and reestablished normal extracellular calcium levels after SCI in dogs [308]. However, other researchers were unable to verify naloxone mediated improvements on SCBF in rodents with clip compression injury [335]. A small human dose-escalation trial recruited 29 patients and did not show statistical significance in recovery, but had a trend toward SSEP improvements and motor recovery in patients receiving high doses of naloxone (200 mg/m2 loading dose followed by 150 mg/m2/h for 23 h infusion) [107]. There were no serious adverse events except increased pain perception in subjects who had additional injuries such as limb fractures and who were neurologically incomplete. In 1990, naloxone was included as one of the NASCIS II trial arms (5.4 mg/kg loading bolus and 4 mg/kg/h infusion for 23 h) along with MPSS and placebo. No statistically significant evidence of improvement in motor or sensory function was detected [36, 37]. As with many other potential neuroprotectants, it’s hard to predict the therapeutic window in humans, when the effective window in animals is brief, e.g. 30–60 min. There is a tendency to extrapolate that the window will be longer in humans, but the arguments for this are not persuasive, and efforts to provide the earliest possible safe delivery are needed if neuroprotection is to be effective.
3.8 Gacyclidine
Gacyclidine (GK-11 Beaufour-Ipsen Pharmaceutical, Les Ulis, France) is a noncompetitive NMDA receptor antagonist with neuroprotective properties including prevention of glutamate-induced neuronal cell death [82] and stabilization of synaptic currents [332]. Efficacy in SCI animal models was shown in dose-response studies indicating increased functional recovery, reduction in cystic cavitation and astrogliosis [103], higher SSEP amplitudes [115], and an optimal therapeutic window when administered within 30 min of injury [154]. In 1999 a phase II randomized, double-blind, placebo-controlled clinical trial in France was completed; 280 patients were enrolled to receive escalating doses of 0.005, 0.01, and 0.02 mg/kg of GK-11 via IV infusion within 2 h of injury with a second dose within the next 4 h. The analysis showed a nonsignificant trend for a benefit at 30 days in incomplete subjects with cervical injury receiving 0.2 mg/kg. However, this was not evident over placebo at the 1-year follow-up. This study remarkably initiated a neuroprotective strategy within 2 h of injury [319, 320]. Further SCI studies with GK-11 have not apparently been pursued.
3.9 Thyrotropin-Releasing Hormone (TRH)
TRH, a tripeptide produced by the hypothalamus, was found to have anti-inflammatory, antioxidant, and membrane-stabilizing properties following SCI [236]. It also became of interest due to its action as an opiate antagonist [156]. Faden and colleagues first tested the effects of TRH compared to dexamethasone in a cat contusion model as an alternative to naloxone. TRH is a partial opiate antagonist and thus has less analgesic blocking properties than naloxone. Blockade of analgesia was a concern in the clinical use of naloxone in acute SCI. Higher scores in forelimb, hindlimb, and total functional outcomes were found in animals treated with TRH (p < 0.01) [92]. Other properties attributed to TRH from animal experiments include a dose-dependent reduction of leukotriene D4 [105], decreased intracellular pH, tissue cations and edema after trauma [94], increased CNS blood flow [181], excitotoxin antagonizing properties [263], and greater efficacy versus MPSS in tissue sparing [21]. In 1995, results of a small clinical trial for TRH were published by Pitts and colleagues; 20 patients were included in the trial and admitted within 12 h of injury to be randomized to receive TRH (0.2 mg/kg bolus continued by a 0.2 mg/kg/h infusion for additional 6 h) or placebo; both groups included complete and incomplete patients. The analysis took place using NASCIS and Sunnybrook scores at 1, 3, and 7 days and 1, 4, and 12 months post-SCI. The authors report higher motor (58.8 ± 11.9 versus 36.7 ± 23.5 placebo) and sensory (84.5 ± 4.1 versus 55.7 ± 22.7 placebo) scores in incomplete patients at the 4-month evaluation. Although the differences are statistically significant, the data was interpreted as not definitive due to the small sample size. Further, an important number of patients were lost for the 12-month follow-up, and data could not be considered to be informative for this timepoint. Additionally, there were no discernible effects in the complete SCI cohorts [251]. TRH as a neuroprotectant remains an open question.
3.10 Nimodipine
The dihydropyridine calcium channel blocker nimodipine has established clinical efficacy for the prevention of vasospasm after subarachnoid hemorrhage ([209, 249]). As raised intracellular calcium levels are damaging to cells affected by SCI [346], it is logical to test drugs that can block calcium channels. Following SCI in experimental animal models, it was found that administration of nimodipine lead to vascular dilation and reduction of blood pressure [140] unless combined with adrenaline [135] to increase adrenergic tone, in which case SCBF could be improved at the site of injury. Further experiments could not verify improvement in SCBF or electrophysiological conduction for either nimodipine or MPSS monotherapy [162, 268]; nimodipine was tested in a human clinical trial in France [247], involving 48 paraplegic and 58 tetraplegic patients randomized to receive: nimodipine (0.015 mg/kg/h for 2 h and 0.03 mg/kg/h infusion for 7 days), MPSS (as per NASCIS II protocol), both, or placebo. One hundred patients were available for the 1-year evaluation. Neurologic improvements were attributed to the natural evolution of the injury with no differences found in any group as compared to the placebo-treated group; the authors additionally reported no benefit from early decompression (8 h post-SCI) in a cohort of 49 patients.
4 Experimental Pharmacological Strategies
We now consider therapeutics that have considerable experimental data from animal studies but have not been tested clinically in SCI.
4.1 Erythropoietin
Erythropoietin (EPO) is a cytokine glycoprotein hormone produced by the kidney of adults that regulates red cell formation. EPO and its receptor (EPO-R) are expressed in neural tissues, especially neurons and glial cells [68, 77]. In the 1990s it was reported that EPO could reduce apoptosis in a stroke model [269] and that recombinant EPO improved endothelial survival and repair in the ischemic penumbra [208]. EPO and EPO-R are upregulated in the ischemic penumbra by a hypoxemia-inducible factor (HIF) [292]. After further preclinical testing, EPO was translated in clinical trials of ischemia due to stroke [86, 87] and vasospasm after subarachnoid hemorrhage [329]. Clinical efficacy in cerebral ischemia remains unclear [296], and concerns have been raised about the safety of coadministration of t-PA and EPO [356]. In a model of spinal cord ischemia, the immediate administration of EPO, during reperfusion, after 20 min of aortic occlusion, spared ventral motor neurons in a dose-dependent manner [50]. Injection of single doses of intraperitoneal EPO, 1-h post-contusion or clip SCI, was associated with motor recovery and reduced injury sizes [127]. In an NIH-sponsored independent replication study of these reported EPO effects, no differences in locomotor recovery were found [250], although there was a nonsignificant trend for epicenter tissue sparing. A difference in apoptosis was not assessed, but reported in the index study and other experimental studies of EPO, and could have provided a signal of biological effect. An additional study using the EPO derivative, darbepoetin, could not reproduce the therapeutic effects reported by Gorio et al. [225]. Numerous mechanisms of activity of EPO have been reported such as reduced inflammation by attenuation of IL-6 [4] release and attenuation of astrogliosis with reduction of phosphacan/RPTPζ/β [334]. Recent studies have shown that methylprednisolone prevents oligodendrocyte (but not neuronal) excitotoxin-mediated apoptotic cell death by causing glucocorticoid receptor and HIF-1-α/1-β to bind to the EPO promotor leading to its expression and upregulation of antiapoptotic Bcl-xL gene expression. This level of complexity indicates why it is easy for therapeutics trials to fail without robust iterative preclinical support as well as biomarkers of therapeutics activity. Failures to replicate preclinical studies do not necessarily mean that the therapeutic is ineffective but do suggest that the efficacy is lower than the index study. An endogenously active molecule such as EPO can be predicted to have multiple actions. Recently, two small clinical studies have been reported in which EPO was compared to MPSS. A modest superiority of EPO was reported [6, 64]. It is important that in larger studies EPO will be compared to placebo as there is increasing information that MPSS may worsen the transient immune deficiency associated with SCI [180].
4.2 Anti-CD11d
Inflammation is a prominent cause of secondary injury; SCI evokes substantially more potent neuroinflammation than brain injury [357] and elicits a multi-organ systemic reaction [45]. Post-SCI systemic inflammation includes damage to the lung, kidney [129], and liver [109, 273]. The extent of liver inflammation was greater for T4 as compared to T12 injuries. Other data supports that post-SCI immune dysfunction is spinal-level dependent and corresponds to the degree of loss of sympathetic regulation of the immune system [277]. The term “leukocyte” refers to all circulating white blood cells. Those known to be important in SCI are neutrophils, monocytes, and lymphocytes. Both leukocytes and monocytes independently contribute to injury propagation [201]. SCI leads to endothelial injury and the presentation of selectins and vascular cell adhesion protein-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) which regulate monocyte rolling, stabilization, and diapedesis from the blood vessel into the perivascular space. Leukocytes express cell surface integrins that bind to the cell adhesion molecules (CAMs) and have roles both in endothelial adhesion and migration through vessel walls and tissue. This process mainly occurs in postcapillary venules where the shear forces are lower, fostering adhesion. Proof-of-concept studies have sought to deplete circulating white cells or to block their exit from the vascular space into spinal cord tissue. Depletion of circulating macrophages was associated with tissue preservation [30], improved locomotor outcomes [254], and reductions in fibrotic scar formation [362]. However, other studies that depleted neutrophils in mice found a worse outcome [307] indicating the complexity of the effects of inflammation after SCI and the limitations of simplistic approaches such as generalized blocking. Another study found that certain populations of macrophages that migrate into the injury site after SCI are neuroprotective [282] and that depletion of CD11-positive monocytes worsened neurological outcome. Others have shown that the persistence of macrophages is associated with axonal dieback [157]. Thus, the associated biology is complex.
Antibodies have been tested as therapeutics; those against P-selectin partially blocked neutrophil spinal tissue entry with reduced tissue myeloperoxidase activity and improved early locomotor function post-SCI [323]. CD11d (αDβ2 integrin) is expressed by monocytes and leukocytes and is involved in leukocyte activation and endothelial adhesion through interaction with its ligands ICAM-1 and VCAM-1 [28]. Mabon and colleagues developed a monoclonal antibody against αD to be administered intravenously in a transection rat model: 24 h before transection and 2 and 24 after transection (1 mg/kg dose). Two of the three anti-αD tested antibodies (228H and 236L) caused a 65 % reduction of macrophages in the injury site and one (236L) a 43 % reduction in neutrophils [219]. Later studies using a clip compression model assessed the mechanisms by which anti-CD11d was beneficial after SCI finding a reduction in myeloperoxidase (MPO) activity, thiobarbituric acid reactive substances (TBARS), and attenuated inducible nitric oxide synthase (iNOS) expression [16]. In another study, the reduction of reactive oxygen and nitrogen species was assessed demonstrating a decrease in COX-2 and 8-OHdG (a marker of RNA and DNA oxidation) expression, a reduction in protein carbonyl formation, and an increase in APE/Ref-1 (involved in repair of apurinic/apyrimidinic sites) formation [15]. Studies comparing anti-CD11d to MPSS showed both treatments increased neurofilament sparing, but myelin sparing was less with MPSS, with no differences in the neurological outcomes of the anti-CD11d + MPSS versus the MPSS group [339], but with significant differences in BBB scores when plotted against an anti-CD11d cohort reported by the researchers group in a previous publication [131]. There was no additive effect of MPSS; the anti-CD11d group had smaller lesion areas [130], increased serotoninergic fiber density below the injuries, and reduced allodynia in the dorsal trunk and hind paws [237].
Regarding therapeutic window, the treatment could be delayed until 6 h post-injury with improved BBB scores and decreased autonomic dysreflexia [79]. A subsequent study examined the relative expression of the key ligands on monocytes in normal adult humans versus those with general trauma and those with SCI. Higher densities of α4 and CD11d were found on neutrophils and monocytes after SCI but not after general trauma [13]. Anti-CD11d therapy also attenuated the systemic inflammatory response, with reduced inflammatory neutrophil and macrophage infiltration in lungs and kidneys [14]; this response was also evident after treatment with anti-α4β1 antibody [17]. Neuroprotective effects of anti-CD11d were found after SCI in mice [120]. An independent replication of the results previously reported was sponsored by the NIH to validate the results, but only nonsignificant trends on motor recovery and tissue sparing with the anti-CD11d treatments were found [161]. Another recent study used a different strategy to deplete both monocytes and neutrophils and found that the optimum protection required both be reduced [201]. Although there is substantial data in support of anti-CD11d antibody treatment post-SCI, no clinical trials have yet been announced. In stroke, an extensive study of an anti-ICAM antibody indicated the treatment worsened outcome [88] possibly due to the immunological effects of the use of murine antibody. Leukarrest, a humanized, anti- CD11b/18 antibody, reduced infarct size in animal models. However, no benefit was found in a phase III clinical trial [20].
4.3 Polyethylene Glycol
Polyethylene glycol (PEG) is a simple polymer with established safety in the pharmaceutical industry. It is a membrane fusogen, used, for example, in the generation of hybridoma cells to produce monoclonal antibodies [179]. Experiments conducted in a guinea pig severe spinal cord compression model showed that a solution of PEG in distilled water applied by pressure injection in the injury site repaired axons such that compound action potential conduction through the lesion recovered [283] and there was progressive recovery of the cutaneous trunci muscle reflex (CMT) [33]. Subcutaneous delivery of PEG was assessed and apparently selectively targeted the contused areas within the spinal cord with beneficial effects at a 6-h therapeutic window [32]. Histological three-dimensional reconstructions showed higher volumes of intact parenchyma and reduction of cystic cavitation in animals receiving PEG treatment directly over the spinal cord, immediately, or with a delay of 7 h after compression [84]. ROS elevation and lipid peroxidation also were inhibited by PEG [215]. A pilot study in dogs with SCI tested the intravenous delivery of the polymer at 30 % w/w in saline at 2 ml/kg within 72 h post-injury with a second delivery 4–6 h later. The authors reported no adverse events and an ambulation recovery in 68 % of treated dogs versus 25 % of controls and proprioception recovery in 42 % of treated versus 4 % of controls [197]. Mechanistic studies have indicated that PEG stabilizes mitochondrial calcium flux and reduces cytochrome-C release [211, 216, 217]. Additional rodent studies showed that animals treated within 15 min or 6 h with IV PEG had enhanced BBB scores and reduced mechanical allodynia up to a 6-week evaluation timepoint. When PEG and MgSO4 were combined, the additive effect was no greater than either single treatment [80]. An independent study conducted by Baptiste, Fehlings, and colleagues confirmed tissue-preserving effects of IV PEG 30 % delivered 15 min after 35-g clip compression. An increase in retrogradely labeled brainstem axons and a reduction in apoptosis of epicenter cells were observed. The small effects on locomotor scores left the authors to question if the therapeutic impact justified translation as a single treatment for SCI trials [18]. In a rodent contusion model, Kwon and colleagues assessed combinations of PEG with magnesium and found that PEG alone did not significantly improve open field locomotion up to 6 weeks posttreatment [186, 188]. More recent experiments performed by Borgens and colleagues include the incorporation of PEG with silica nanoparticles [55, 57] or micelles such as monomethoxy poly(ethylene glycol)-poly(d,L-lactic acid) di-block copolymer. Apparently, these formulations allow a more precise targeting to damaged neural tissue and a greater repair effect. It has been reported that PEG is superior to Matrigel, or alginate hydrogel in supporting regeneration after complete spinal transection [89]. So far PEG has not been studied in a human SCI clinical trial other than in combination with magnesium.
4.4 Estrogen
Clinical and experimental studies indicate that females tend to exhibit greater neurological recovery than males after equivalent spinal cord injuries [69, 96, 291], implicating a possible role for estrogen as a neuroprotectant. Estrogen has been shown to reduce leukocyte adhesion and infiltration [327] and attenuate microglial superoxide release and phagocytic activity [43]. In vitro experiments showed protection of hippocampal neurons against glucose deprivation, glutamate, FeSO4, and amyloid beta-peptide (Aβ) toxicity [126]. In 2004, Yune and colleagues reported neurological outcomes after a mild NYU contusion impact (12.5 g/cm) in male rats. 17β-estradiol was administered IV at 3, 100, or 300 μg/kg2 hours prior to injury or 100 μg/kg immediately post-injury. For the pre-administered doses, 100 μg/kg had the largest effect. After 30 days the BBB scores for animals in the 100 μg/kg cohort were 18.3 ± 0.6 (p < 0.001) versus 15 ± 1 in vehicle only. The lesion area was reduced, 2.06 ± 0.2 mm2 (p < 0.05) versus 2.5 ± 0.3 mm2, as was apoptosis 50 ± 5 versus 134 ± 8 (p < 0.001) TUNEL + ve, caspase-3 activation 5.7 ± 0.81 versus 9.8 ± 0.95, p < 0.001, and increased BCL-2 and BCL-X labeling. In another study cohort, the steroid was administered immediately after injury, and a substantial effect on BBB at 30 days was observed [351]. These data strongly supported a neuroprotective potential of 17β-estradiol in SCI but lacked clinical relevance since a therapeutic window was not assessed. Sribnick and colleagues reported in 2005 a male rat contusion model using higher doses of 17β-estradiol (4 mg/kg administered 15 min and 24 h post-injury IV). With a short survival of 48 h, there was reduction in tissue edema, infiltration of microglia and macrophages in the injury site and caudal penumbra, and attenuation of myelin loss in the injury site [300]. Further experiments by the group using the same methodology demonstrated attenuation in degradation of 68 kD neurofilament, reduction in calpain protein levels, decrease in cytochrome-C levels, and neuronal apoptosis in estrogen-treated animals [298, 299].
The issue of a neuroprotective effect of estrogen replacement in an SCI model of pre- and postmenopause tested the effect of preimplantation of a 17β-estradiol capsule (180 g/ml) 1 week before injury followed by a 0.5 mm forceps crush at T8/9. Neuroprotection was observed in both pre- and postmenopausal (ovariectomized) rats with improved white matter sparing, anterior horn cell preservation, reduced apoptosis, and higher BBB scores, although premenopausal rats (non-ovariectomized) had higher scores than estrogen treated. There was no effect on the manually voided urine volumes by 21 days post-injury [51]. Testing in the mouse species using a T10-controlled compression confirmed that females had substantially superior outcomes to males on the mouse BBB score [96].
A well-designed study tackled the issue of sex differences by comparing males to ovariectomized females with both implanted with a control, low-dose or high-dose estrogen-releasing capsule (low-dose 180 μg/ml, high-dose 1 mg/ml) 7 days prior to a moderate 150-kilodyne T10 injury. The serum estrogen levels were measured and reflected the dose delivery from the implanted capsule. The 21-day post-injury BBB was not significantly improved over the non-estrogen control in male or female rats. Injury length and tissue sparing favored females over males, but this effect was deemed not to be attributable to estrogen delivery. The authors concluded that no effect of estrogen delivery on SCI outcome was evident [318].
A mouse study used a 24-g clip compression at T6/7 with the subcutaneous delivery of 300–500 μg/kg of E2 before and after injury. They reported that estrogen reduced neutrophil infiltration, expression of myeloperoxidase, iNOS, nitrotyrosine, cyclooxygenase-2 (COX2), BAX, and apoptosis demonstrated by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), TNF-α, IL-6, and IL-1β. The effects were abolished by a coadministered estrogen receptor antagonist [67]. Neuronal antiapoptotic effects and reduction in microglial activation were found in a rat contusion model even at small doses of 5 or 10 μg/kg [270]. A recent study showed that immediate post-T10 NYU 25 g/cm contusion delivery of 300 μg/kg of E2 protected the blood-brain barrier, with reduced hemorrhage, matrix metallopeptidase 9 (MMP9) and sulfonylurea receptor1 (Sur1) Transient receptor potential cation channel subfamily M member 4 (TrpM4) expression, reduced neutrophil and macrophage migration, and reduced inflammatory cytokine expression in male rats [199]. If estrogen is to be considered a potential neuroprotectant, there is a need for studies to establish a therapeutic window and rigorously evaluate safety.
4.5 Progesterone
Progesterone (PRO) is primarily known as female sex hormone produced by the ovary. However, PRO, like estrogen, has pleiotropic effects and is an important neurosteroid that is locally produced in the brain and has a key role in myelination [275]. PRO has been demonstrated to reduce inflammation and attenuate neuronal and glial loss [66, 303] in traumatic brain injury (TBI). Thomas and colleagues first reported testing progesterone therapy for SCI in a standard T9 25 g/cm NYU, contusion model. Adult male rats received 4 mg/kg of PRO intraperitoneally for 5 days beginning 30 min after injury, and significant improvements in BBB scores and white matter preservation were reported at the 6-week timepoint [328]. Fee et al. attempted to replicate some elements of the Thomas study but used a milder injury, 150-kilodyne T10 Infinite Horizon, and both male and female rats. PRO 4 mg/kg or 8 mg/kg was delivered at 30 min and 6 h post-injury and continued for either 5 or 14 days. As with the Kentucky group’s parallel study of estrogen [318], no gender differences were observed with PRO treatment after SCI. Regarding tissue sparing and BBB testing, there were no apparent differences effects between PRO and control at the 21-day assessment timepoint [98].
A recently published study evaluated PRO in male rats with a T8 200-kilodyne Infinite Horizon injury. Animals were randomized to receive subcutaneous injections of 16 mg/kg/day PRO or castor oil, starting 1 h post-injury and continued for 60 days until sacrifice. Outcome assessments included Catwalk automated gait analysis and stereological histological techniques. The authors found a benefit to PRO with improved BBB scores (mean 14 versus 11 in controls), subscores, and gait elements such as a reduced base of support and improved stride length and swing phase. Ex vivo MRI analysis found significant differences in injury volumes and spared tissue indicating a positive effect of PRO. There was a substantial sparing of white matter above and below the lesion epicenter. The number of CC1 immunopositive oligodendroglia, myelin basic protein (MBP)-positive segments, and neurofilament-labeled axonal profiles was increased in PRO-treated rats [114].
Other studies using a variety of injury models have identified several putative mechanisms of PRO effect. Reactive astrocytes in SCI were reduced in the injury site by doses of 4 mg/kg at 1, 24, 48, and 72 h post-T10 transection in male rats [193]. The same treatment paradigm restored the expression of the α3 and β1 regulatory subunits of NA-K-ATPase with further upregulation of growth-associated protein (GAP-43) in ventral horn motoneurons [190], produced upregulation of NG2-positive cells [70], enhanced brain derived neurotrophic factor (BDNF) immunoreactivity, and prevented chromatolytic degeneration in motoneurons with preservation of the cytoskeletal component identified by MAP2 [124, 125]. Regarding potential effects on myelination, a dose of 16 mg/kg started 3 h post-injury and continued until sacrifice increased the expression of mRNA and MBP and increased mRNA and protein expression for oligodendrocyte differentiation factors Olig2 and Nkx2.2 at 3 days. If treatment was continued until 21 days, markers of mature myelin recovery included reduced oligodendrocyte precursor cells and increased oligodendrocyte mRNA expression of proteolipid protein (PLP) and Olig1 [191]. In a different study, inhibition of astrocyte and microglia/macrophage proliferation and activation was found [192]. Currently, there is no indication for the use of progesterone in the clinic as a neuroprotective for SCI, and further study of a therapeutic window in contusion injury would be important.
4.6 Nonsteroidal Anti-inflammatory Drugs (NSAIDs)
Another important class of anti-inflammatory drugs for which there is extensive clinical experience are NSAIDs that inhibit cyclooxygenase 1 and 2 and thus the production of eicosanoids from arachidonic acid. Their activity reduces the production of prostaglandins, thromboxanes, and leukotrienes. Pre-injury delivery of indomethacin substantially reduced tissue prostaglandin levels in a feline SCI model [172]. In another feline SCI study, a triple combination treatment of indomethacin, heparin, and prostacyclin (PGI2) one and three hours post-weight-drop contusion was compared to naloxone and saline control. The combination including indomethacin showed similar Tarlov locomotor scores to those animals receiving naloxone, with both superior to saline control [144]. Fujita and colleagues found that aspirin pretreatment (5–50 mg/kg) also reduced lipid peroxidation and decreased prostaglandin production [111]. A dual lipoxygenase/cyclooxygenase inhibitor BW755C administered prior to trauma decreased the accumulation of thromboxane B2, improving neurological recovery in rat contusion model, but the same effect was not achieved when applied post-injury [93].
Further experimental studies conducted during the 1990s assessed the effects of indomethacin after SCI. These showed neurological improvement with reduced tissue injury in rats (2 mg/kg post-injury + every 24 h during 7 days) [290], decreased edema and attenuated motor evoked potential changes (10 mg/kg 30 min pre-injury) [342], and ultrastructural demonstration of decreased edema (10 mg/kg 30 min pre-injury) [279].
A lack of effect of indomethacin was also reported: no locomotor improvement unless combined with pregnenolone and lipopolysaccharide (0.2 mg after injury + every 24h during 21 days) [138], increased lipid peroxidation when compared to saline treatment (3 mg/kg after injury) [139], and increased levels of MPO and TNF with greater motor deficit and histological damage (5, 10, 20 mg/kg 1 h pre-injury) [149]. The study by Guth and colleagues is notable as this was a combination selected to increase monocyte cytokine secretion using lipopolysaccharide, to provide anti-inflammatory steroid activity with pregnenolone, and to block prostaglandin production with indomethacin. Multiple pair-wise and triple combinations were tested that included aspirin and dehydroepiandrosteron (DHEAS) as alternate anti-inflammatory controls. The injury was created at T8 using 2 s of jeweler’s forceps compression. Only indomethacin, pregnenolone, and lipopolysaccharide (LPS) combined were associated with substantial locomotor recovery and reduction in the lesion area. Eighteen years later, an National Institute of Neurological Disorders and Stroke (NINDS) supported replication was reported by the team at Ohio State. An attempt was made to replicate the forceps compression injury, and a sustained release pellet (lipopolysaccharide 0.2 mg, indomethacin 0.16 mg, pregnenolone 150 mg) was inserted into tissue adjacent to the laminectomy, and the animals survived for 28 days. An effect of the triple combination was found on white matter preservation, preserved neurofilament area, and axon density. No effect, however, was detected in the BBB scores, subscores, or imputed Tarlov scores, with assessments up to 28 days post-injury [256]. In a brief rebuttal, Dr. Guth pointed out that the control animals in the replication study showed more spontaneous recovery than in the original study making it difficult to show a difference in locomotor recovery [137].
A very interesting property of some NSAIDs is the ability to reduce RhoA activation after SCI. In the first study, indomethacin was injected at 2 mg/kg/day IP after a T8 dorsal over hemisection. The tissue was examined on day 3 for sources of RhoA activity. The treated rats showed a decrease in RhoA expression in microglia/macrophages, astrocytes, and neutrophils with an increase in GAP-43 positive neurites [276]. In a second experiment, the impact of ibuprofen versus naproxen on RhoA activation, axonal growth, and locomotor recovery was assessed, with drugs delivered for 28 days via an Alzet minipump, ibuprofen (60 mg/kg/day), naproxen (50 mg/kg/day), or saline. The treatment was initiated immediately after an extensive dorsal T6/7 oversection, or 7 days after a T8/9 moderate NYU contusion injury. In vitro assays showed inhibition of RhoA activation by ibuprofen and indomethacin but not naprosyn. Interestingly, the inhibition was of similar potency to C3 transferase (see Cethrin above). At 5–7 days post-contusion, RhoA was markedly reduced in spinal cord tissue by ibuprofen but not naprosyn. In the sharp section, animals with immediate delivery CST axon growth distal to the lesion were found in anterogradely traced animals that received ibuprofen but not naprosyn or control. In the contusion animals with treatment initiated at 7 days post-SCI, improved serotonergic axonal growth below the injury level was observed. In both injury paradigms, the BBB score at 42 days was higher in the ibuprofen-treated animals [110]. A subsequent study further examined these issues using both in vitro assays and rat experiments. A T7/8 25 g/cm NYU contusion was created and 3 days after injury an Alzet minipump implanted to deliver 70 mg/kg/day ibuprofen, 25 mg/kg/day naproxen, or PBS. The rats survived either 2 or 6.5 weeks. In a separate experiment, mice underwent T8 spinal cord transection and were simultaneously implanted with minipumps to deliver either 35 or 70 mg/kg/day of ibuprofen for 28 days. The in vitro studies showed a dose-dependent reduction in MAG-mediated inhibition. Both weight-bearing status and BBB scores were increasing in ibuprofen- but not naprosyn-treated animals up to 50 days post-injury, and Rho activation was reduced in spinal cord tissue. Tissue preservation was significantly better in ibuprofen as compared to naprosyn- and PBS-treated animals at 2- and 4-week timepoints. There was extensive sprouting of the CST rostral to the contusion site in contused rats that received ibuprofen, and the terminations of the tract were not retracted from the injury as is usually seen and was observed in the naprosyn- and PBS-treated animals. No distal CST regeneration, however, was observed. Six weeks post-injury, ibuprofen-treated rats showed substantially more serotonergic fiber density distal to the contusion site, apparently due to regenerative sprouting from preserved processes. In completely transected mice, regenerative growth of serotonergic fibers through the transection and distally was observed together with improvements in the Basso Mouse Scale for Locomotion (BMS) [337]. The Fu et al. study was selected for replication by the Facilities of Research Excellence—Spinal Cord Injury (FORE-SCI) NIH group, conducted at UC Irvine. Despite consultation with the senior author, Dr. Li, of the index study, no aspect of the original results could be confirmed other than the suppression of Rho activation by ibuprofen [280]. A recently reported study assessed ibuprofen in combination with C16, a vascular preserving peptide; ghrelin, a neuroprotective gastric hormone; and ketogenic diet in a C5 hemicontusion paradigm. No effects were apparent using rodent upper extremity functional assessments and light microscopy-based tissue sparing analysis [309].
Selective inhibitors of inducible cyclooxygenase 2 were studied. Following a 25-g/cm NYU impact, celebocid (3 mg/kg) was delivered PO 20 min post-injury. The levels of prostaglandin E2 (PGE2) and thromboxane B2 (TxB2) were reduced by the drug at 4 and 24 h post-injury [264]. A single injection of NS-398 5 mg/kg delivered 15 min prior to a 12.5 g/cm NYU T13 also decreased prostaglandin E2 (PGE2), increased spared tissue, improved BBB scores 14–28 days post-SCI, and reduced mechanical allodynia and thermal hyperalgesia [141]. It is notable that in this injury model, the pain behaviors are found in forelimbs above the injury and the hindlimbs below the injury. This indicates that CNS reorganization after SCI occurs not only at and below the injury level.
4.7 Glibenclamide
Glibenclamide, known as glyburide in the USA, is a well-known sulfonylurea developed in the 1960s for the oral treatment of type 2 diabetes [226]. Due to its ability to close KATP channels (pore formed by subunits SUR1 and Kir6.2), it stimulates the secretion of insulin by β islet cells [10]. Interest in its potential for SCI was due to the identification of an upregulation of the SUR1 subunit in a characteristic phenomenon of TBI and SCI known as progressive hemorrhagic necrosis (PHN) [285], which is described as a progressively confluent petechial hemorrhage adjacent to the injury sites [12]. The mechanism of PHN is not yet known, but it is understood to be a progressive capillary blocking due to mechanical stress after secondary injury [325] or leukocyte infiltration [175].
In a rat cervical hemicontusion model, SUR1 was identified to be upregulated in endothelial cells and neurons surrounding necrotic injuries, beginning its expression in the epicenter at 6 h post-injury and expanding to the injury rim and adjacent tissues within 24 h. The delivery of glibenclamide via infusion pump (200 ng/h) after 2–3 min of injury resulted in a threefold reduction of injury volumes; decreased intraspinal hemorrhage at 6, 12, and 24 h; and significant neurobehavioral improvement in BBB scores and inclined plane testing at 1, 3, and 7 days post-injury [288]. An NIH-sponsored independent replication study was carefully conducted. When initial data failed to replicate the index study of Simard and colleagues, extensive efforts were made to ensure that the same injury methodology was being employed. When this was achieved, an effect of glibenclamide could be verified [255, 256]. This experience underscores the importance of exact attention to detail in replication studies. In a subsequent study, the effect of glibenclamide was assessed in two hemicontusion injury methods. In the more extensive injury, small but significant effects of glibenclamide on behavioral outcomes and tissue sparing were observed [287]. It should be noted, however, that the drug was administered within 5 min of injury. In another study, glibenclamide was compared to riluzole with administration performed either immediately after unilateral C7 injury or at the 3-h timepoint. Unfortunately, nearly all of the control animals died resulting in a comparison between the two drugs but without a vehicle comparison. Glibenclamide resulted in superior tissue preservation, grip strength, rotarod stability, and rearing [289]. A subsequent study assessed the effects of glibenclamide on the progression of hemorrhagic injury volume using MRI [286]. Glibenclamide appears as a promising neuroprotective approach for SCI, but further studies in more severe lesion models and with additional timepoints of drug delivery should be conducted to compare its effects to other agents in rodent models. Clinical trials in stroke and TBI have been initiated.
4.8 Thiazolidinediones
Thiazolidinediones (TZDs) are used in the treatment of type 2 diabetes and are agonists of the peroxisome proliferator-activated receptors gamma (PPARγ). These are “nuclear receptor” DNA-binding compounds that regulate fatty acid storage and hepatic glucose metabolism [170]. A model assessing factors involved in the macrophage-mediated expansion of SCI cavity size included testing of ciglitazone (the prototypical TZD) with an in vitro cavity model of cultured astrocytes and activated macrophages. Ciglitazone potently reduced the increase in the areas of culture cavities mediated by zymogen-activated macrophages [106]. Later with the development of pioglitazone (PGZ) which crosses the blood-brain barrier [317], a rat contusion model compared high (10 mg/kg) versus low (1 mg/kg) doses of the TZD, started 15 min post-injury and maintained BID for 7 days. The differences in BBB scores were not significant with a positive trend for the high-dose group (15.4 ± 1.1 versus 13.3 ± 1.3 in controls), but differences were more evident in subscores (toe clearance, coordination, parallel paw position). The high-dose group had improvements in spared white and gray matter areas rostral to the epicenter and better neuronal counts rostral and caudal to the epicenter; no differences were observed in caspase-3 immunoreactivity or macrophage detection [232]. A concurrent study by a different laboratory compared the treatment effects of PGZ and rosiglitazone (RGZ) (0.5, 1.5, 3 mg/kg at different timepoints) and the PPARγ antagonist GW9662 (2 mg/kg 1 h prior to any TZD injection). Both TZDs decreased lesion areas (57 % RGZ, 64 % PGZ) significantly and increased neuronal survival; decreased astrogliosis, microglial activation, myelin loss, induction of inflammatory genes; and enhanced induction of neuroprotective heat-shock proteins and antioxidants, with improved BBB scores (final scores at day 7 PGZ 12.9 ± 1.4, RGZ 12.2 ± 1.1, vehicle 8.9 ± 0.7); these effects were inhibited by the GW9662 treatment. The therapeutic window of intervention indicated no significant effects of a delay in administration >2 h and similar effects with 3 or 1.5 mg doses with no effects at 0.5 mg; additionally PGZ exhibited a decrease in thermal hyperalgesia [244]. In other studies, intrathecal delivery of RGZ decreased neuropathic pain [59], and similar effects were observed with oral administration of PGZ [220]. Anti-inflammatory effects of RGZ (reduction of TNF-α, IL-β, myeloperoxidase activity) were evident in a clip compression model [358], and a comparison between MPSS and RGZ demonstrated greater increased neurotrophin expression and improved early recovery in the RGZ group [359]. There are no published studies in SCI to disprove the effects of TZD treatment, and having two studies performed by independent groups with similar results within the same year is strong evidence. There are additional reports from Chinese studies [169, 207, 345]. It may be valuable to test TZD in a large animal contusion model.
4.9 Atorvastatin
Atorvastatin and statins, in general, are competitive inhibitors of the 3-hydroxy-3methylglutarylcoenzymeA (HMG-CoA) reductase which catalyzes the reduction of HMG-CoA to mevalonate, inhibiting the synthesis of hepatic cholesterol, increasing the uptake of low density lipoprotein (LDL), reducing triglyceride levels, and increasing high density lipoprotein (HDL) levels. Statins have anti-inflammatory effects, in astrocyte and microglial cell cultures exposed to LPS. Lovastatin blocked the expression of inducible nitric oxide synthase (NOS) and TNF-α, IL-1β, and IL-6 [241]. In MBP-induced experimental models of experimental autoimmune encephalomyelitis (EAE) in Lewis rats, lovastatin was found to reduce the numbers of infiltrating mononuclear cells by downregulating the ICAM ligand Lymphocyte function-associated antigen-1 (LFA-1), to reduce demyelination, and to reverse paralysis [301]. In further EAE studies, atorvastatin was found to increase a Th2 over Th1 cytokine and cellular phenotype [350]. The studies in SCI by Pannu and colleagues used a rat contusion model with an oral atorvastatin dose of 5 mg/kg/day until sacrifice (15 days). All groups in the study received a pretreatment regimen of atorvastatin for 7 days; animals that continued on statins exhibited higher BBB scores that started to deviate significantly from the vehicle group by day 3 (6.57 ± 0.55 vs 0.166 ± 0.33) and showed near-normal scores by day 15 (19.13 ± 0.53 vs 9.04 ± 1.22). Statin-treated animals had reduced post-injury levels of TNF-α and IL-1β, decreased infiltration of ED-1 macrophages, attenuated neuron, and oligodendrocyte apoptosis demonstrated by TUNEL assay, reduced gliosis, tissue necrosis, and demyelination [242]. Evaluation of the therapeutic window was subsequently assessed by not pretreating and randomizing contused animals to start treatment at 2, 4, or 6 h post-injury, with continued drug treatment until sacrifice (42 days). BBB analysis showed much higher scores for all treated groups at 42 days (2h 19.5 ± 1, 4h 19.25 ± 1, 18.5 ± 0.5) versus 10.5 ± 1.5 vehicle control; these results were supported by attenuated necrosis and demyelination; decreased neutrophil and macrophage infiltration; decreased expression of iNOS, TNF-α, and IL-1β; attenuated expression of MMP9 via inhibition of the RhoA/ROCK pathway with preservation of the blood-brain barrier; and reduced neuronal apoptosis [243]. These are remarkable results clearly requiring independent evaluation. A Canadian study assessed the relative effects of simvastatin and atorvastatin as the former is more lipophilic, using oral gavage delivery in saline, fed in chocolate ensure, or purified simvastatin alone administered subcutaneously initiated 2h after injury. The OSU device was used to deliver a 1.5-cm displacement at 300 m/s. In brief, the results failed to show effects that justify clinical translation. There were higher BBB scores in the statin group in one experiment that could not be reproduced in others: white matter tissue sparing showed a nonsignificant trend toward positive effects but did not reach statistical significance (42 ± 6% vs. 34 ± 8% in controls), and the one statistically significant difference was obtained at 3 days comparing the extent of ED1 staining at the injury epicenter [224]. A second Canadian study examined the effect of atorvastatin-administered IP 5 mg/kg 2 h post-SCI for 15d with a 4 week survival. An early reduction in TUNEL labeling and caspase-3 activation was observed, with a statistically significant improvement in BBB score at 4 weeks post-SCI. In conclusion, statins may offer an approach for post-SCI neuroprotection, but additional evidence is needed to resolve inconsistencies in the literature and determine if the cost and effort of a clinical trial is warranted.
4.10 Inosine
Inosine is a purine nucleoside found in tRNAs and is a popular athletic supplement. It’s metabolism to uric acid has antioxidant effects. One of its earliest uses in neurology was as a component of the drug isoprinosine that was found to have beneficial immune-modulating properties during viral diseases such as subacute sclerosing panencephalitis [171]. Inosine stimulated axonal outgrowth in retinal ganglion cell cultures [26]. Subsequently, its impact on CST structural plasticity was studied in a rodent model of unilateral pyramidal section. Twenty-four hours after the injury, animals were implanted with minipumps to continuously infuse inosine (10 mM 0.5 μl/h) for 14 days into the non-axotomized sensorimotor cortex. Inosine stimulated the uninjured pyramidal axons below the lesion to extend new collaterals across the midline into denervated areas of the lesioned tract [24]. An upregulation of GAP-43 expression was observed in axons forming new collaterals [25] as is seen in regenerating axons. Mammalian sterile 20-like kinase-3b (Mst3b) activation was specifically linked to inosine stimulated axon outgrowth [165]. Further studies have indicated that Mst3b is an important regulator of axonal growth after injury in both CNS and peripheral nervous system (PNS) neurons [165, 213]. In models of unilateral stroke, intraventricular delivery of inosine [354] did not reduce infarct size but was associated with improved recovery of affected forepaw function, sprouting of CST axons into the denervated tract, and upregulation of genes associated with axonal growth in the uninjured cortex. In a second stroke study, inosine amplified the effects of a co-administered Nogo blocker and was again associated with increased forelimb function [353]. In a rodent T8 dorsal spinal cord hemisection model, the effects of intraventricular (50 mM 0.5 μl/h) and intravenous (8–27 mg/kg/day) inosine delivery were compared. Both delivery methods were associated with significant increases in BBB scores and ladder walking tests. Inosine treatment did not affect lesion size but increased CST sprouting in the cervical spinal cord rostral to the lesion with new synaptic contacts with long propriospinal interneurons that could form novel relays and also was associated with increased serotoninergic immunoreactivity to the lumbar spinal cord below the cord section [176]. In a Chinese study, a single dose of inosine was administered IP at 2, 12, or 24 h after T9 clip compression injury. Epicenter tissue preservation and a reduction in the number of apoptotic cells were detected at 3 days in the 2- and 12-h treatment groups with the 2-h treatment being the best [210]. In another experiment, inosine effects appeared to be enhanced by oscillating field stimulation with regeneration of ascending and descending projections of spinal cord tracts and recovery of the cutaneous trunci muscle reflex in chronic SCI [31]. Continued subcutaneous delivery of inosine started 15 min post-thoracic contusion injury reduced the number of ED-1 positive profiles at the injury site [62]. In 2012, an NIH-sponsored independent replication study of the methodology published by Benowitz in 1999 was reported from UC Irvine. The authors described difficulties with complications and deaths secondary to the pyramidotomy surgeries; nevertheless, after several trials the results failed to confirm that CST axons sprouted across the midline [304, 305]. Sources of possible error in interpreting the histological appearance of CST axons at the midline were discussed. The most recent study testing inosine in SCI reported neuronal survival, tissue sparing, and improved locomotion after oral administration of inosine (1.2 g/kg/day, beginning 2 h post-injury) [185]. In summary, there is substantial evidence for inosine as a promoter of axonal plasticity after injury. Combined with its established use in drugs and as a health supplement, clinical testing may be reasonable if pivotal preclinical studies to assess the route of administration and pharmacokinetics were supportive.
4.11 Imatinib
Imatinib a tyrosine kinase antagonist, is one of the most successful cancer drugs ever developed and has fundamentally changed the treatment of chronic myelogenous leukemia. As with other “repurposed” drugs, we have discussed such as minocycline, riluzole, statins, and glibenclamide, there is extensive human experience with dosage and side effects. This experience makes the translation pathway less complicated. Imatinib was shown to preserve blood-brain barrier integrity in a stroke model by inhibiting the activity of the platelet-dervied growth factor (PDGF) α receptor [314]. Furthermore, imatinib is a very illuminating example of disparities between index studies and replication attempts. Potent neuroprotective effects were reported by Abrams et al. in 2012 [2]. The dose was 250 mg/kg given orally 30 min after a thoracic 25 g/cm NYU contusion and continued for 5 days. Several significant functional effects were seen, improved BBB scores, contact placing responses, and a substantial reduction in bladder urine retention after voiding. There was epicenter tissue and neurofilament preservation, reduced numbers of macrophages, astrocyte reactivity, and chondroitin sulfate proteoglycan (CSPG) deposition in the injury region. Blood-barrier integrity was improved with a reduction in albumin leakage. Detectable PDGFR-α and PDGFR-β were reduced at 5 days post-injury, and tight junction protein claudin 5 was better preserved. An NIH-supported replication study failed to reproduce the findings other than the significantly reduced post-void residual bladder volume [281]. A potent rebuttal by the index study authors showed that the replication injury severity was significantly less than that of the index study precluding the ability to demonstrate recovery of weight-supported stepping and creating a ceiling effect for contact placing. Other flaws in the replication were noted as well [1]. In a recently published study, Kjell and colleagues further examined the therapeutic window assessing delivery at 4, 8, and 24 h, continuing for 14 days, after a milder contusion injury that facilitated automatic analysis of coordinated stepping. They used the same dose as the original study 250 mg/kg PO. Effects on the BBB, subscores, and coordination were evident with 4- but not 8- or 24-h delivery [178]. Improved bladder emptying was found at all timepoints as compared to placebo control. A specific peripheral blood profile of cytokine activation was consistent in animals receiving imatinib. In an EAE model of spinal multiple sclerosis (MS), imatinib was shown to markedly attenuate disease by protecting blood-brain barrier integrity, by reducing endothelial expression of molecules mediating the entry of lymphocytes and other inflammatory cells to the tissue, and by promoting the Th2 phenotype in the peripheral immune system [3]. Peripheral IL-4 was markedly increased; IL-4 is an important promoter of Th2 and M2 tissue repair macrophage phenotype. One interesting point the authors do not discuss is that the dose administered to the rats appears far higher than that used for human malignancies, typically a total of 400–800 mg/day in adults [246]. For a typical 70-kg male, 250 mg/kg would be a daily dose of 17,500 mg, 40× higher than the standard cancer dose.
5 Conclusions and Final Remarks
Currently, there is no clinically established neuroprotective treatment that has met the standard of showing a prospectively established increment of neurological recovery superior to control, or alternate therapy, in a suitably designed randomized study with acceptable morbidity. There are several reasons for this that are rooted in the preclinical, translational, and pivotal phases of studies, the intransigent biology of SCI, and even in the politics of clinical trials. The daily reality of SCI occurrence and its permanent outcomes drives the machinery of clinical translation. Other factors that impact progress and decision-making in the neuroprotective therapeutics field include the organization of medical care competition, market forces, availability of research funding, the media and public perception, high-status publications, and the energy and leadership of investigators. Clinical factors that complicate therapeutics trials include human variability, the unique context of each injury, variations in management, secondary complications, and type and extent of rehabilitation. Clinical trial methodology to address subject and injury heterogeneity may rely on stratification and partitioning methods [85, 322] as well as responder analyses [294]. Pharmacokinetic studies have rarely been performed in SCI neuroprotection trials, and recent evidence indicates that the systemic exposure to an enterally delivered neuroprotectant is highly variable between subjects [58]. This indicates that serum levels might be needed to guide the dosing schedule in order to achieve uniform exposure to the drug between individuals in a clinical trial. Dosage adjustment may also be required to achieve patient drug levels similar to those employed in key preclinical studies [49]. Unlike diseases such as liver and cardiac injury, there are no validated quantitative biomarkers available to provide insight into the real-time effects of CNS neuroprotectants [189] although serum light chain neurofilament has recently been reported to correlate with injury severity [151] and to be modified by minocycline treatment after cervical SCI [183]. Post-injury imaging is also rather insensitive to subtle neuroprotective effects although the relative rate and extent of T2 signal distribution is under investigation as a marker of injury severity [198]. Thus, clinical examination is the primary tool to assess efficacy. This is sensible when an intervention is potent but risks missing subtle signals that might be important guideposts to address issues of the therapeutic window, dose, pharmacokinetics, and duration of therapy.
It is considered that there are faults in the preclinical evaluation process [71, 123, 196]. A therapeutic should show a very reproducible and robust effect to have a chance to have an effect in a group of humans with SCI. Preclinical testing is performed in genetically similar animals using protocols that emphasize exact repetition. The intent is to minimize heterogeneity to achieve clarity. Confidence in the value of animal testing for SCI rests largely on success in other disease states such as cancer, diabetes, and organ transplantation. Highly reproducible SCI methods [347] and assessment techniques have been developed [19]. Despite these tools, it is frustratingly difficult for others to reproduce positive findings reported by another research group [304, 305]. The field is struggling with this issue especially after a series of failed replications sponsored by NINDS. There is no easy solution to the replication problem, and apparently minor details can substantially affect experimental outcomes. There may be a role to consolidate testing into highly expert core facilities devoid of vested interest that can further perfect the art and science of replication beyond what has yet been achieved. Another important preclinical question is whether efficacy should be shown in more than one species before clinical translation [187] seems difficult unless an exact replication can be achieved in the rodent species. Often, the failure to replicate is left without a resolution that is unsatisfactory for the advance of our field.
Outcome measures in preclinical experiments should have a correlate to human recovery. For therapeutics to be used to recover function after cervical SCI, recovery of arm and hand function are critical issues. This raises a challenging question in the use of large animal models in so far as prominent models such as pigs are not useful for detailed assessment of hand function. The authors’ opinion is that primate model testing is justified for high-risk therapeutics such as directly implanted cells but not for low-risk pharmacological interventions. Upper extremity assessments are available for rodent models of cervical SCI [8].
Typically, only positive reports are published. It would be valuable to the field to communicate the absence of therapeutic effects and the presence of adverse events and analyze the potential causes of failure (underpowered sample, unspecific outcome measures, underdosing, overdosing, incorrect therapeutic window, improper route of administration, etc.) and communicate these results. Another conclusion, given the complexity of secondary injury, is the expectation that a single agent will provide “remarkable” protection within the realities of a human clinical population is flawed. It may be essential to learn to combine multiple therapeutics that target several secondary injury mechanisms. As shown in this review, neuroprotectant testing has a long history in SCI. The field is continuing to mature and adding knowledge and technology at an impressive rate. The need for collaboration and standardization, data sharing, and data mining is increasingly evident and accepted. It would be beneficial if preclinical testing of future therapeutics could be extremely robust, independently replicated, tested in at least 2 species, and cleared of all reasonable concerns related to method of delivery before clinical testing is initiated. It is apparent that only full honesty and maximum diminution of bias will lead to efficient and meaningful progress in SCI neuroprotection. It may be that the field needs a fundamentally new approach as all of the foregoing suggestions are really methods to improve the current basic model. This model of preclinical prediction in animal models and then staged translation to humans appears to be inherently unreliable. Therefore, in conclusion there is an opportunity and need for insight and innovative approaches to advance this field.
References
Abrams MB, Nilsson I, Kjell J, Lewandowski S, Codeluppi S, Eriksson U, Olson L (2014) Response to the report, “A re-assessment of treatment with a tyrosine kinase inhibitor (imatinib) on tissue sparing and functional recovery after spinal cord injury” by Sharp et al. Exp Neurol 257:182–185. doi:10.1016/j.expneurol.2014.04.025
Abrams MB, Nilsson I, Lewandowski SA, Kjell J, Codeluppi S, Olson L, Eriksson U (2012) Imatinib enhances functional outcome after spinal cord injury. PLoS One 7(6):e38760. doi:10.1371/journal.pone.0038760
Adzemovic MV, Zeitelhofer M, Eriksson U, Olsson T, Nilsson I (2013) Imatinib ameliorates neuroinflammation in a rat model of multiple sclerosis by enhancing blood-brain barrier integrity and by modulating the peripheral immune response. PLoS One 8(2):e56586. doi:10.1371/journal.pone.0056586
Agnello D, Bigini P, Villa P, Mennini T, Cerami A, Brines ML, Ghezzi P (2002) Erythropoietin exerts an anti-inflammatory effect on the CNS in a model of experimental autoimmune encephalomyelitis. Brain Res 952(1):128–134
Albin MS, White RJ, Acosta-Rua G, Yashon D (1968) Study of functional recovery produced by delayed localized cooling after spinal cord injury in primates. J Neurosurg 29(2):113–120. doi:10.3171/jns.1968.29.2.0113
Alibai E, Zand F, Rahimi A, Rezaianzadeh A (2014) Erythropoietin plus methylprednisolone or methylprednisolone in the treatment of acute spinal cord injury: a preliminary report. Acta Med Iran 52(4):275–279
Alter M (1998) GM1 ganglioside for acute ischemic stroke. Trial design issues. Ann N Y Acad Sci 845:391–401
Anderson KD, Gunawan A, Steward O (2005) Quantitative assessment of forelimb motor function after cervical spinal cord injury in rats: relationship to the corticospinal tract. Exp Neurol 194(1):161–174. doi:10.1016/j.expneurol.2005.02.006
Argentino C, Sacchetti ML, Toni D, Savoini G, D’Arcangelo E, Erminio F et al (1989) GM1 ganglioside therapy in acute ischemic stroke. Italian Acute Stroke Study – Hemodilution + Drug. Stroke 20(9):1143–1149
Ashcroft FM (1996) Mechanisms of the glycaemic effects of sulfonylureas. Horm Metab Res 28(9):456–463. doi:10.1055/s-2007-979837
Azbill RD, Mu X, Springer JE (2000) Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res 871(2):175–180
Balentine JD (1978) Pathology of experimental spinal cord trauma. I. The necrotic lesion as a function of vascular injury. Lab Invest 39(3):236–253
Bao F, Bailey CS, Gurr KR, Bailey SI, Rosas-Arellano MP, Brown A et al (2011) Human spinal cord injury causes specific increases in surface expression of beta integrins on leukocytes. J Neurotrauma 28(2):269–280. doi:10.1089/neu.2010.1618
Bao F, Brown A, Dekaban GA, Omana V, Weaver LC (2011) CD11d integrin blockade reduces the systemic inflammatory response syndrome after spinal cord injury. Exp Neurol 231(2):272–283. doi:10.1016/j.expneurol.2011.07.001
Bao F, Chen Y, Dekaban GA, Weaver LC (2004) An anti-CD11d integrin antibody reduces cyclooxygenase-2 expression and protein and DNA oxidation after spinal cord injury in rats. J Neurochem 90(5):1194–1204. doi:10.1111/j.1471-4159.2004.02580.x
Bao F, Chen Y, Dekaban GA, Weaver LC (2004) Early anti-inflammatory treatment reduces lipid peroxidation and protein nitration after spinal cord injury in rats. J Neurochem 88(6):1335–1344
Bao F, Omana V, Brown A, Weaver LC (2012) The systemic inflammatory response after spinal cord injury in the rat is decreased by alpha4beta1 integrin blockade. J Neurotrauma 29(8):1626–1637. doi:10.1089/neu.2011.2190
Baptiste DC, Austin JW, Zhao W, Nahirny A, Sugita S, Fehlings MG (2009) Systemic polyethylene glycol promotes neurological recovery and tissue sparing in rats after cervical spinal cord injury. J Neuropathol Exp Neurol 68(6):661–676. doi:10.1097/NEN.0b013e3181a72605
Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG (2006) Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma 23(5):635–659. doi:10.1089/neu.2006.23.635
Becker KJ (2002) Anti-leukocyte antibodies: LeukArrest (Hu23F2G) and Enlimomab (R6.5) in acute stroke. Curr Med Res Opin 18(Suppl 2):s18–s22
Behrmann DL, Bresnahan JC, Beattie MS (1994) Modeling of acute spinal cord injury in the rat: neuroprotection and enhanced recovery with methylprednisolone, U-74006F and YM-14673. Exp Neurol 126(1):61–75. doi:10.1006/exnr.1994.1042
Benavides J, Camelin JC, Mitrani N, Flamand F, Uzan A, Legrand JJ et al (1985) 2-Amino-6-trifluoromethoxy benzothiazole, a possible antagonist of excitatory amino acid neurotransmission – II. Biochemical properties. Neuropharmacology 24(11):1085–1092
Benoit E, Escande D (1991) Riluzole specifically blocks inactivated Na channels in myelinated nerve fibre. Pflugers Arch 419(6):603–609
Benowitz LI, Goldberg DE, Madsen JR, Soni D, Irwin N (1999) Inosine stimulates extensive axon collateral growth in the rat corticospinal tract after injury. Proc Natl Acad Sci U S A 96(23):13486–13490
Benowitz LI, Goldberg DE, Irwin N (2001) A purine-sensitive mechanism regulates the molecular program for axon growth. Restor Neurol Neurosci 19(1-2):41–49
Benowitz LI, Jing Y, Tabibiazar R, Jo SA, Petrausch B, Stuermer CA, Rosenberg PA, Irwin N (1996) Axon outgrowth is regulated by an intracellular purine-sensitive mechanism in retinal ganglion cells. J Biol Chem 243(45):29626–29634
Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K (2002) Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med 346(8):557–563. doi:10.1056/NEJMoa003289
Bevilacqua MP (1993) Endothelial-leukocyte adhesion molecules. Annu Rev Immunol 11:767–804. doi:10.1146/annurev.iy.11.040193.004003
Bigelow WG, Lindsay WK et al (1950) Oxygen transport and utilization in dogs at low body temperatures. Am J Physiol 160(1):125–137
Blight AR (1994) Effects of silica on the outcome from experimental spinal cord injury: implication of macrophages in secondary tissue damage. Neuroscience 60(1):263–273
Bohnert DM, Purvines S, Shapiro S, Borgens RB (2007) Simultaneous application of two neurotrophic factors after spinal cord injury. J Neurotrauma 24(5):846–863. doi:10.1089/neu.2006.0101
Borgens RB, Bohnert D (2001) Rapid recovery from spinal cord injury after subcutaneously administered polyethylene glycol. J Neurosci Res 66(6):1179–1186
Borgens RB, Shi R (2000) Immediate recovery from spinal cord injury through molecular repair of nerve membranes with polyethylene glycol. FASEB J 14(1):27–35
Botterell EH, Lougheed WM, Scott JW, Vandewater SL (1956) Hypothermia, and interruption of carotid, or carotid and vertebral circulation, in the surgical management of intracranial aneurysms. J Neurosurg 13(1):1–42. doi:10.3171/jns.1956.13.1.0001
Bracken MB, Collins WF, Freeman DF, Shepard MJ, Wagner FW, Silten RM et al (1984) Efficacy of methylprednisolone in acute spinal cord injury. JAMA 251(1):45–52
Bracken MB, Holford TR (1993) Effects of timing of methylprednisolone or naloxone administration on recovery of segmental and long-tract neurological function in NASCIS 2. J Neurosurg 79(4):500–507. doi:10.3171/jns.1993.79.4.0500
Bracken MB, Shepard MJ, Collins WF, Holford TR, Young W, Baskin DS et al (1990) A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the Second National Acute Spinal Cord Injury Study. N Engl J Med 322(20):1405–1411. doi:10.1056/NEJM199005173222001
Bracken MB, Shepard MJ, Hellenbrand KG, Collins WF, Leo LS, Freeman DF et al (1985) Methylprednisolone and neurological function 1 year after spinal cord injury. Results of the National Acute Spinal Cord Injury Study. J Neurosurg 63(5):704–713. doi:10.3171/jns.1985.63.5.0704
Bracken MB, Shepard MJ, Holford TR, Leo-Summers L, Aldrich EF, Fazl M et al (1997) Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. Results of the Third National Acute Spinal Cord Injury Randomized Controlled Trial. National Acute Spinal Cord Injury Study. JAMA 277(20):1597–1604
Braughler JM, Hall ED (1984) Effects of multi-dose methylprednisolone sodium succinate administration on injured cat spinal cord neurofilament degradation and energy metabolism. J Neurosurg 61(2):290–295. doi:10.3171/jns.1984.61.2.0290
Bricolo A, Ore GD, Da Pian R, Faccioli F (1976) Local cooling in spinal cord injury. Surg Neurol 6(2):101–106
Brommer B, Engel O, Kopp MA, Watzlawick R, Muller S, Pruss H et al (2016) Spinal cord injury-induced immune deficiency syndrome enhances infection susceptibility dependent on lesion level. Brain 139(Pt 3):692–707. doi:10.1093/brain/awv375
Bruce-Keller AJ, Keeling JL, Keller JN, Huang FF, Camondola S, Mattson MP (2000) Antiinflammatory effects of estrogen on microglial activation. Endocrinology 141(10):3646–3656. doi:10.1210/endo.141.10.7693
Busto R, Dietrich WD, Globus MY, Ginsberg MD (1989) Postischemic moderate hypothermia inhibits CA1 hippocampal ischemic neuronal injury. Neurosci Lett 101(3):299–304
Campbell SJ, Zahid I, Losey P, Law S, Jiang Y, Bilgen M et al (2008) Liver Kupffer cells control the magnitude of the inflammatory response in the injured brain and spinal cord. Neuropharmacology 55(5):780–787. doi:10.1016/j.neuropharm.2008.06.074
Carlson GD, Minato Y, Okada A, Gorden CD, Warden KE, Barbeau JM et al (1997) Early time-dependent decompression for spinal cord injury: vascular mechanisms of recovery. J Neurotrauma 14(12):951–962
Carvalho MO, Barros Filho TE, Tebet MA (2008) Effects of methylprednisolone and ganglioside GM-1 on a spinal lesion: a functional analysis. Clinics (Sao Paulo) 63(3):375–380
Casas CE, Herrera LP, Prusmack C, Ruenes G, Marcillo A, Guest JD (2005) Effects of epidural hypothermic saline infusion on locomotor outcome and tissue preservation after moderate thoracic spinal cord contusion in rats. J Neurosurg Spine 2(3):308–318. doi:10.3171/spi.2005.2.3.0308
Casha S, Zygun D, McGowan MD, Bains I, Yong VW, Hurlbert RJ (2012) Results of a phase II placebo-controlled randomized trial of minocycline in acute spinal cord injury. Brain 135(Pt 4):1224–1236. doi:10.1093/brain/aws072
Celik M, Gokmen N, Erbayraktar S, Akhisaroglu M, Konakc S, Ulukus C et al (2002) Erythropoietin prevents motor neuron apoptosis and neurologic disability in experimental spinal cord ischemic injury. Proc Natl Acad Sci U S A 99(4):2258–2263. doi:10.1073/pnas.042693799
Chaovipoch P, Jelks KA, Gerhold LM, West EJ, Chongthammakun S, Floyd CL (2006) 17beta-estradiol is protective in spinal cord injury in post- and pre-menopausal rats. J Neurotrauma 23(6):830–852. doi:10.1089/neu.2006.23.830
Chatzipanteli K, Yanagawa Y, Marcillo AE, Kraydieh S, Yezierski RP, Dietrich WD (2000) Posttraumatic hypothermia reduces polymorphonuclear leukocyte accumulation following spinal cord injury in rats. J Neurotrauma 17(4):321–332
Chaudhry IB, Hallak J, Husain N, Minhas F, Stirling J, Richardson P et al (2012) Minocycline benefits negative symptoms in early schizophrenia: a randomised double-blind placebo-controlled clinical trial in patients on standard treatment. J Psychopharmacol 26(9):1185–1193. doi:10.1177/0269881112444941
Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S et al (2000) Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 6(7):797–801. doi:10.1038/77528
Chen B, Zuberi M, Borgens RB, Cho Y (2012) Affinity for, and localization of, PEG-functionalized silica nanoparticles to sites of damage in an ex vivo spinal cord injury model. J Biol Eng 6(1):18. doi:10.1186/1754-1611-6-18
Cheramy A, Barbeito L, Godeheu G, Glowinski J (1992) Riluzole inhibits the release of glutamate in the caudate nucleus of the cat in vivo. Neurosci Lett 147(2):209–212
Cho Y, Shi R, Ivanisevic A, Borgens RB (2010) Functional silica nanoparticle-mediated neuronal membrane sealing following traumatic spinal cord injury. J Neurosci Res 88(7):1433–1444. doi:10.1002/jnr.22309
Chow DS, Teng Y, Toups EG, Aarabi B, Harrop JS, Shaffrey CI et al (2012) Pharmacology of riluzole in acute spinal cord injury. J Neurosurg Spine 17(1 Suppl):129–140. doi:10.3171/2012.5.AOSPINE12112
Churi SB, Abdel-Aleem OS, Tumber KK, Scuderi-Porter H, Taylor BK (2008) Intrathecal rosiglitazone acts at peroxisome proliferator-activated receptor-gamma to rapidly inhibit neuropathic pain in rats. J Pain 9(7):639–649. doi:10.1016/j.jpain.2008.02.002
Coleman WP, Benzel D, Cahill DW, Ducker T, Geisler F, Green B et al (2000) A critical appraisal of the reporting of the National Acute Spinal Cord Injury Studies (II and III) of methylprednisolone in acute spinal cord injury. J Spinal Disord 13(3):185–199
Constantini S, Young W (1994) The effects of methylprednisolone and the ganglioside GM1 on acute spinal cord injury in rats. J Neurosurg 80(1):97–111. doi:10.3171/jns.1994.80.1.0097
Conta AC, Stelzner DJ (2008) Immunomodulatory effect of the purine nucleoside inosine following spinal cord contusion injury in rat. Spinal Cord 46(1):39–44. doi:10.1038/sj.sc.3102057
Coselli JS, LeMaire SA, Koksoy C, Schmittling ZC, Curling PE (2002) Cerebrospinal fluid drainage reduces paraplegia after thoracoabdominal aortic aneurysm repair: results of a randomized clinical trial. J Vasc Surg 35(4):631–639
Costa DD, Beghi E, Carignano P, Pagliacci C, Faccioli F, Pupillo E (2015) Tolerability and efficacy of erythropoietin (EPO) treatment in traumatic spinal cord injury: a preliminary randomized comparative trial vs. methylprednisolone (MP). Neurol Sci. doi:10.1007/s10072-015-2182-5
Crawford ES, Svensson LG, Hess KR, Shenaq SS, Coselli JS, Safi HJ et al (1991) A prospective randomized study of cerebrospinal fluid drainage to prevent paraplegia after high-risk surgery on the thoracoabdominal aorta. J Vasc Surg 13(1):36–45; discussion 45-36
Cutler SM, Cekic M, Miller DM, Wali B, VanLandingham JW, Stein DG (2007) Progesterone improves acute recovery after traumatic brain injury in the aged rat. J Neurotrauma 24(9):1475–1486. doi:10.1089/neu.2007.0294
Cuzzocrea S, Genovese T, Mazzon E, Esposito E, Di Paola R, Muia C et al (2008) Effect of 17beta-estradiol on signal transduction pathways and secondary damage in experimental spinal cord trauma. Shock 29(3):362–371. doi:10.1097/shk.0b013e31814545dc
Dame C, Juul SE, Christensen RD (2001) The biology of erythropoietin in the central nervous system and its neurotrophic and neuroprotective potential. Biol Neonate 79(3-4):228–235, doi:47097
Datto JP, Yang J, Dietrich WD, Pearse DD (2015) Does being female provide a neuroprotective advantage following spinal cord injury? Neural Regen Res 10(10):1533–1536. doi:10.4103/1673-5374.165213
De Nicola AF, Labombarda F, Gonzalez SL, Gonzalez Deniselle MC, Guennoun R, Schumacher M (2003) Steroid effects on glial cells: detrimental or protective for spinal cord function? Ann N Y Acad Sci 1007:317–328
DeGraba TJ, Pettigrew LC (2000) Why do neuroprotective drugs work in animals but not humans? Neurol Clin 18(2):475–493
Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L (2002) Rho signaling pathway targeted to promote spinal cord repair. J Neurosci 22(15):6570–6577, doi:20026637
Derry DM, Wolfe LS (1967) Gangliosides in isolated neurons and glial cells. Science 158(3807):1450–1452
Dididze M, Green BA, Dietrich WD, Vanni S, Wang MY, Levi AD (2013) Systemic hypothermia in acute cervical spinal cord injury: a case-controlled study. Spinal Cord 51(5):395–400. doi:10.1038/sc.2012.161
Dietrich WD, Bramlett HM (2010) The evidence for hypothermia as a neuroprotectant in traumatic brain injury. Neurotherapeutics 7(1):43–50. doi:10.1016/j.nurt.2009.10.015
Dietrich WD, Chatzipanteli K, Vitarbo E, Wada K, Kinoshita K (2004) The role of inflammatory processes in the pathophysiology and treatment of brain and spinal cord trauma. Acta Neurochir Suppl 89:69–74
Digicaylioglu M, Bichet S, Marti HH, Wenger RH, Rivas LA, Bauer C, Gassmann M (1995) Localization of specific erythropoietin binding sites in defined areas of the mouse brain. Proc Natl Acad Sci U S A 92(9):3717–3720
Dimar JR 2nd, Glassman SD, Raque GH, Zhang YP, Shields CB (1999) The influence of spinal canal narrowing and timing of decompression on neurologic recovery after spinal cord contusion in a rat model. Spine (Phila Pa 1976) 24(16):1623–1633
Ditor DS, Bao F, Chen Y, Dekaban GA, Weaver LC (2006) A therapeutic time window for anti-CD 11d monoclonal antibody treatment yielding reduced secondary tissue damage and enhanced behavioral recovery following severe spinal cord injury. J Neurosurg Spine 5(4):343–352. doi:10.3171/spi.2006.5.4.343
Ditor DS, John SM, Roy J, Marx JC, Kittmer C, Weaver LC (2007) Effects of polyethylene glycol and magnesium sulfate administration on clinically relevant neurological outcomes after spinal cord injury in the rat. J Neurosci Res 85(7):1458–1467. doi:10.1002/jnr.21283
Drake CG, Barr HW, Coles JC, Gergely NF (1964) The use of extracorporeal circulation and profound hypothermia in the treatment of ruptured intracranial aneurysm. J Neurosurg 21:575–581. doi:10.3171/jns.1964.21.7.0575
Drian MJ, Kamenka JM, Pirat JL, Privat A (1991) Non-competitive antagonists of N-methyl-D-aspartate prevent spontaneous neuronal death in primary cultures of embryonic rat cortex. J Neurosci Res 29(1):133–138. doi:10.1002/jnr.490290116
Dubreuil CI, Winton MJ, McKerracher L (2003) Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. J Cell Biol 162(2):233–243. doi:10.1083/jcb.200301080
Duerstock BS, Borgens RB (2002) Three-dimensional morphometry of spinal cord injury following polyethylene glycol treatment. J Exp Biol 205(Pt 1):13–24
Dvorak MF, Noonan VK, Fallah N, Fisher CG, Rivers CS, Ahn H et al (2014) Minimizing errors in acute traumatic spinal cord injury trials by acknowledging the heterogeneity of spinal cord anatomy and injury severity: an observational Canadian cohort analysis. J Neurotrauma 31(18):1540–1547. doi:10.1089/neu.2013.3278
Ehrenreich H, Hasselblatt M, Dembowski C, Cepek L, Lewczuk P, Stiefel M et al (2002) Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med 8(8):495–505
Ehrenreich H, Weissenborn K, Prange H, Schneider D, Weimar C, Wartenberg K, Schellinger PD, Bohn M, Becker H, Wegrzyn M, Jähnig P, Herrmann M, Knauth M, Bähr M, Heide W, Wagner A, Schwab S, Reichmann H, Schwendemann G, Dengler R, Kastrup A, Bartels C; EPO Stroke Trial Group (2009) Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke. 40(12):e647–656. doi:10.1161/STROKEAHA.109.564872
Enlimomab Acute Stroke Trial Investigators (2001) Use of anti-ICAM-1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology 57(8):1428–1434
Estrada V, Brazda N, Schmitz C, Heller S, Blazyca H, Martini R, Muller HW (2014) Long-lasting significant functional improvement in chronic severe spinal cord injury following scar resection and polyethylene glycol implantation. Neurobiol Dis 67:165–179. doi:10.1016/j.nbd.2014.03.018
Faden AI, Jacobs TP (1984) Dynorphin-related peptides cause motor dysfunction in the rat through a non-opiate action. Br J Pharmacol 81(2):271–276
Faden AI, Jacobs TP, Holaday JW (1981) Opiate antagonist improves neurologic recovery after spinal injury. Science 211(4481):493–494
Faden AI, Jacobs TP, Holaday JW (1981) Thyrotropin-releasing hormone improves neurologic recovery after spinal trauma in cats. N Engl J Med 305(18):1063–1067. doi:10.1056/NEJM198110293051806
Faden AI, Lemke M, Demediuk P (1988) Effects of BW755C, a mixed cyclo-oxygenase-lipoxygenase inhibitor, following traumatic spinal cord injury in rats. Brain Res 463(1):63–68
Faden AI, Yum SW, Lemke M, Vink R (1990) Effects of TRH-analog treatment on tissue cations, phospholipids and energy metabolism after spinal cord injury. J Pharmacol Exp Ther 255(2):608–614
Failli V, Kopp MA, Gericke C, Martus P, Klingbeil S, Brommer B et al (2012) Functional neurological recovery after spinal cord injury is impaired in patients with infections. Brain 135(Pt 11):3238–3250. doi:10.1093/brain/aws267
Farooque M, Suo Z, Arnold PM, Wulser MJ, Chou CT, Vancura RW et al (2006) Gender-related differences in recovery of locomotor function after spinal cord injury in mice. Spinal Cord 44(3):182–187. doi:10.1038/sj.sc.3101816
Fawcett JW, Curt A, Steeves JD, Coleman WP, Tuszynski MH, Lammertse D et al (2007) Guidelines for the conduct of clinical trials for spinal cord injury as developed by the ICCP panel: spontaneous recovery after spinal cord injury and statistical power needed for therapeutic clinical trials. Spinal Cord 45(3):190–205. doi:10.1038/sj.sc.3102007
Fee DB, Swartz KR, Joy KM, Roberts KN, Scheff NN, Scheff SW (2007) Effects of progesterone on experimental spinal cord injury. Brain Res 1137(1):146–152. doi:10.1016/j.brainres.2006.12.024
Fehlings MG, Nakashima H, Nagoshi N, Chow DS, Grossman RG, Kopjar B (2016) Rationale, design and critical end points for the Riluzole in Acute Spinal Cord Injury Study (RISCIS): a randomized, double-blinded, placebo-controlled parallel multi-center trial. Spinal Cord 54(1):8–15. doi:10.1038/sc.2015.95
Fehlings MG, Theodore N, Harrop J, Maurais G, Kuntz C, Shaffrey CI et al (2011) A phase I/IIa clinical trial of a recombinant Rho protein antagonist in acute spinal cord injury. J Neurotrauma 28(5):787–796. doi:10.1089/neu.2011.1765
Fehlings MG, Vaccaro A, Wilson JR, Singh A, W. Cadotte D, Harrop JS (2012) Early versus delayed decompression for traumatic cervical spinal cord injury: results of the Surgical Timing in Acute Spinal Cord Injury Study (STASCIS). PLoS One 7(2):e32037. doi:10.1371/journal.pone.0032037
Fehlings MG, Wilson JR, Cho N (2014) Methylprednisolone for the treatment of acute spinal cord injury: counterpoint. Neurosurgery 61(Suppl 1):36–42. doi:10.1227/NEU.0000000000000412
Feldblum S, Arnaud S, Simon M, Rabin O, D’Arbigny P (2000) Efficacy of a new neuroprotective agent, gacyclidine, in a model of rat spinal cord injury. J Neurotrauma 17(11):1079–1093
Ferrari G, Anderson BL, Stephens RM, Kaplan DR, Greene LA (1995) Prevention of apoptotic neuronal death by GM1 ganglioside. Involvement of Trk neurotrophin receptors. J Biol Chem 270(7):3074–3080
Feuerstein G, Lux WE Jr, Ezra D, Faden AI (1984) Reversal of leukotriene D4 hypotension by thyrotropin-releasing hormone. Neurosci Res 2(1-2):121–124
Fitch MT, Doller C, Combs CK, Landreth GE, Silver J (1999) Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci 19(19):8182–8198
Flamm ES, Young W, Collins WF, Piepmeier J, Clifton GL, Fischer B (1985) A phase I trial of naloxone treatment in acute spinal cord injury. J Neurosurg 63(3):390–397. doi:10.3171/jns.1985.63.3.0390
Flamm ES, Young W, Demopoulos HB, DeCrescito V, Tomasula JJ (1982) Experimental spinal cord injury: treatment with naloxone. Neurosurgery 10(2):227–231
Fleming JC, Bailey CS, Hundt H, Gurr KR, Bailey SI, Cepinskas G et al (2012) Remote inflammatory response in liver is dependent on the segmental level of spinal cord injury. J Trauma Acute Care Surg 72(5):1194–1201. doi:10.1097/TA.0b013e31824d68bd; discussion 1202
Fu Q, Hue J, Li S (2007) Nonsteroidal anti-inflammatory drugs promote axon regeneration via RhoA inhibition. J Neurosci 27(15):4154–4164. doi:10.1523/JNEUROSCI.4353-06.2007
Fujita Y, Shingu T, Kurihara M, Miyake H, Kono T, Tsujimura M, Mori K (1985) Evaluation of a low dose administration of aspirin, dipyridamol and steroid. Therapeutic effects on motor function and protective effects on Na + -K + -activated ATPase activity against lipid peroxidation in an experimental model of spinal cord injury. Paraplegia 23(1):56–57. doi:10.1038/sc.1985.9
Furlan JC, Noonan V, Cadotte DW, Fehlings MG (2011) Timing of decompressive surgery of spinal cord after traumatic spinal cord injury: an evidence-based examination of pre-clinical and clinical studies. J Neurotrauma 28(8):1371–1399. doi:10.1089/neu.2009.1147
Ganglioside GM1 in acute ischemic stroke. The SASS trial (1994) Stroke 25(6):1141–1148
Garcia-Ovejero D, Gonzalez S, Paniagua-Torija B, Lima A, Molina-Holgado E, De Nicola AF, Labombarda F (2014) Progesterone reduces secondary damage, preserves white matter, and improves locomotor outcome after spinal cord contusion. J Neurotrauma 31(9):857–871. doi:10.1089/neu.2013.3162
Gaviria M, Privat A, d’Arbigny P, Kamenka J, Haton H, Ohanna F (2000) Neuroprotective effects of a novel NMDA antagonist, Gacyclidine, after experimental contusive spinal cord injury in adult rats. Brain Res 874(2):200–209
Geisler FH, Coleman WP, Grieco G, Poonian D; Sygen Study Group (2001) The Sygen multicenter acute spinal cord injury study. Spine (Phila Pa 1976) 26(24 Suppl):S87–98
Geisler FH, Dorsey FC, Coleman WP (1991) Correction: recovery of motor function after spinal-cord injury – a randomized, placebo-controlled trial with GM-1 ganglioside. N Engl J Med 325(23):1659–1660
Geisler FH, Dorsey FC, Coleman WP (1991) Recovery of motor function after spinal-cord injury – a randomized, placebo-controlled trial with GM-1 ganglioside. N Engl J Med 324(26):1829–1838. doi:10.1056/NEJM199106273242601
George ER, Scholten DJ, Buechler CM, Jordan-Tibbs J, Mattice C, Albrecht RM (1995) Failure of methylprednisolone to improve the outcome of spinal cord injuries. Am Surg 61(8):659–663; discussion 663-654
Geremia NM, Bao F, Rosenzweig TE, Hryciw T, Weaver L, Dekaban GA, Brown A (2012) CD11d antibody treatment improves recovery in spinal cord-injured mice. J Neurotrauma 29(3):539–550. doi:10.1089/neu.2011.1976
Gerhart KA, Johnson RL, Menconi J, Hoffman RE, Lammertse DP (1995) Utilization and effectiveness of methylprednisolone in a population-based sample of spinal cord injured persons. Paraplegia 33(6):316–321. doi:10.1038/sc.1995.71
Gerndt SJ, Rodriguez JL, Pawlik JW, Taheri PA, Wahl WL, Micheals AJ, Papadopoulos SM (1997) Consequences of high-dose steroid therapy for acute spinal cord injury. J Trauma 42(2):279–284
Gladstone DJ, Black SE, Hakim AM; Heart and Stroke Foundation of Ontario Centre of Excellence in Stroke, Recovery (2002) Toward wisdom from failure: lessons from neuroprotective stroke trials and new therapeutic directions. Stroke 33(8):2123–2136
Gonzalez SL, Labombarda F, Gonzalez Deniselle MC, Guennoun R, Schumacher M, De Nicola AF (2004) Progesterone up-regulates neuronal brain-derived neurotrophic factor expression in the injured spinal cord. Neuroscience 125(3):605–614. doi:10.1016/j.neuroscience.2004.02.024
Gonzalez SL, Lopez-Costa JJ, Labombarda F, Gonzalez Deniselle MC, Guennoun R, Schumacher M, De Nicola AF (2009) Progesterone effects on neuronal ultrastructure and expression of microtubule-associated protein 2 (MAP2) in rats with acute spinal cord injury. Cell Mol Neurobiol 29(1):27–39. doi:10.1007/s10571-008-9291-0
Goodman Y, Bruce AJ, Cheng B, Mattson MP (1996) Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J Neurochem 66(5):1836–1844
Gorio A, Gokmen N, Erbayraktar S, Yilmaz O, Madaschi L, Cichetti C et al (2002) Recombinant human erythropoietin counteracts secondary injury and markedly enhances neurological recovery from experimental spinal cord trauma. Proc Natl Acad Sci U S A 99(14):9450–9455. doi:10.1073/pnas.142287899
Grant P, Song JY, Swedo SE (2010) Review of the use of the glutamate antagonist riluzole in psychiatric disorders and a description of recent use in childhood obsessive-compulsive disorder. J Child Adolesc Psychopharmacol 20(4):309–315. doi:10.1089/cap.2010.0009
Gris D, Hamilton EF, Weaver LC (2008) The systemic inflammatory response after spinal cord injury damages lungs and kidneys. Exp Neurol 211(1):259–270. doi:10.1016/j.expneurol.2008.01.033
Gris D, Marsh DR, Dekaban GA, Weaver LC (2005) Comparison of effects of methylprednisolone and anti-CD11d antibody treatments on autonomic dysreflexia after spinal cord injury. Exp Neurol 194(2):541–549. doi:10.1016/j.expneurol.2005.03.016
Gris D, Marsh DR, Oatway MA, Chen Y, Hamilton EF, Dekaban GA, Weaver LC (2004) Transient blockade of the CD11d/CD18 integrin reduces secondary damage after spinal cord injury, improving sensory, autonomic, and motor function. J Neurosci 24(16):4043–4051. doi:10.1523/JNEUROSCI.5343-03.2004
Grossman RG, Fehlings MG, Frankowski RF, Burau KD, Chow DS, Tator C et al (2014) A prospective, multicenter, phase I matched-comparison group trial of safety, pharmacokinetics, and preliminary efficacy of riluzole in patients with traumatic spinal cord injury. J Neurotrauma 31(3):239–255. doi:10.1089/neu.2013.2969
Grossman RG, Frankowski RF, Burau KD, Toups EG, Crommett JW, Johnson MM et al (2012) Incidence and severity of acute complications after spinal cord injury. J Neurosurg Spine 17(1 Suppl):119–128. doi:10.3171/2012.5.AOSPINE12127
Guest J, Eleraky MA, Apostolides PJ, Dickman CA, Sonntag VK (2002) Traumatic central cord syndrome: results of surgical management. J Neurosurg 97(1 Suppl):25–32
Guha A, Tator CH, Smith CR, Piper I (1989) Improvement in post-traumatic spinal cord blood flow with a combination of a calcium channel blocker and a vasopressor. J Trauma 29(10):1440–1447
Gungor B, Adiguzel E, Gursel I, Yilmaz B, Gursel M (2016) Intestinal microbiota in patients with spinal cord injury. PLoS One 11(1):e0145878. doi:10.1371/journal.pone.0145878
Guth L (2012) A reassessment of LPS/indomethacin/pregnenolone combination therapy after spinal cord injury in rats. Exp Neurol 233(2):686. doi:10.1016/j.expneurol.2011.11.024
Guth L, Zhang Z, DiProspero NA, Joubin K, Fitch MT (1994) Spinal cord injury in the rat: treatment with bacterial lipopolysaccharide and indomethacin enhances cellular repair and locomotor function. Exp Neurol 126(1):76–87. doi:10.1006/exnr.1994.1043
Guven MB, Cirak B, Yuceer N, Ozveren F (1999) Is indomethacin harmful in spinal cord injury treatment? An experimental study. Pediatr Neurosurg 31(4):189–193
Haghighi SS, Chehrazi BB, Wagner FC Jr (1988) Effect of nimodipine-associated hypotension on recovery from acute spinal cord injury in cats. Surg Neurol 29(4):293–297
Hains BC, Yucra JA, Hulsebosch CE (2001) Reduction of pathological and behavioral deficits following spinal cord contusion injury with the selective cyclooxygenase-2 inhibitor NS-398. J Neurotrauma 18(4):409–423. doi:10.1089/089771501750170994
Hall ED (1993) The effects of glucocorticoid and nonglucocorticoid steroids on acute neuronal degeneration. Adv Neurol 59:241–248
Hall ED, Yonkers PA, Andrus PK, Cox JW, Anderson DK (1992) Biochemistry and pharmacology of lipid antioxidants in acute brain and spinal cord injury. J Neurotrauma 9(Suppl 2):S425–S442
Hallenbeck JM, Jacobs TP, Faden AI (1983) Combined PGI2, indomethacin, and heparin improves neurological recovery after spinal trauma in cats. J Neurosurg 58(5):749–754. doi:10.3171/jns.1983.58.5.0749
Hamaguchi T, Ono K, Yamada M (2010) REVIEW: curcumin and Alzheimer’s disease. CNS Neurosci Ther 16(5):285–297. doi:10.1111/j.1755-5949.2010.00147.x
Hamberger A, Svennerholm L (1971) Composition of gangliosides and phospholipids of neuronal and glial cell enriched fractions. J Neurochem 18(10):1821–1829
Hansebout RR, Hansebout CR (2014) Local cooling for traumatic spinal cord injury: outcomes in 20 patients and review of the literature. J Neurosurg Spine 20(5):550–561. doi:10.3171/2014.2.SPINE13318
Hansebout RR, Tanner JA, Romero-Sierra C (1984) Current status of spinal cord cooling in the treatment of acute spinal cord injury. Spine (Phila Pa 1976) 9(5):508–511
Harada N, Taoka Y, Okajima K (2006) Role of prostacyclin in the development of compression trauma-induced spinal cord injury in rats. J Neurotrauma 23(12):1739–1749. doi:10.1089/neu.2006.23.1739
Hawryluk GW, Whetstone WD, Saigal R, Ferguson AR, Talbott JF, Bresnahan JC et al (2015) Mean arterial blood pressure correlates with neurological recovery following human spinal cord injury: analysis of high frequency physiologic data. J Neurotrauma. doi:10.1089/neu.2014.3778
Hayakawa K, Okazaki R, Ishii K, Ueno T, Izawa N, Tanaka Y et al (2012) Phosphorylated neurofilament subunit NF-H as a biomarker for evaluating the severity of spinal cord injury patients, a pilot study. Spinal Cord 50(7):493–496. doi:10.1038/sc.2011.184
Heary RF, Vaccaro AR, Mesa JJ, Northrup BE, Albert TJ, Balderston RA, Cotler JM (1997) Steroids and gunshot wounds to the spine. Neurosurgery 41(3):576–583; discussion 583-574
Helmholz HF, Haddow MK (1930) Eight years experience with the ketogenic diet in the treatment of epilepsy. JAMA 95(10):707–709
Hirbec H, Gaviria M, Vignon J (2001) Gacyclidine: a new neuroprotective agent acting at the N-methyl-D-aspartate receptor. CNS Drug Rev 7(2):172–198
Holaday JW, Faden AI (1978) Naloxone reversal of endotoxin hypotension suggests role of endorphins in shock. Nature 275(5679):450–451
Holaday JW, Tseng LF, Loh HH, Li CH (1978) Thyrotropin releasing hormone antagonizes beta endorphin hypothermia and catalepsy. Life Sci 22(17):1537–1544
Horn KP, Busch SA, Hawthorne AL, van Rooijen N, Silver J (2008) Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci 28(38):9330–9341. doi:10.1523/JNEUROSCI.2488-08.2008
Hsu CY, Dimitrijevic MR (1990) Methylprednisolone in spinal cord injury: the possible mechanism of action. J Neurotrauma 7(3):115–119
Huang WL, King VR, Curran OE, Dyall SC, Ward RE, Lal N et al (2007) A combination of intravenous and dietary docosahexaenoic acid significantly improves outcome after spinal cord injury. Brain 130(Pt 11):3004–3019. doi:10.1093/brain/awm223
Hurlbert RJ (2000) Methylprednisolone for acute spinal cord injury: an inappropriate standard of care. J Neurosurg 93(1 Suppl):1–7
Hurtado A, Marcillo A, Frydel B, Bunge MB, Bramlett HM, Dietrich WD (2012) Anti-CD11d monoclonal antibody treatment for rat spinal cord compression injury. Exp Neurol 233(2):606–611. doi:10.1016/j.expneurol.2010.11.015
Imamura H, Tator CH (1998) Effect of intrathecal nimodipine on spinal cord blood flow and evoked potentials in the normal or injured cord. Spinal Cord 36(7):497–506
Investigators Ninds Net-Pd (2006) A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology 66(5):664–671. doi:10.1212/01.wnl.0000201252.57661.e1
Investigators Ninds Net-Pd (2008) A pilot clinical trial of creatine and minocycline in early Parkinson disease: 18-month results. Clin Neuropharmacol 31(3):141–150. doi:10.1097/WNF.0b013e3181342f32
Irwin N, Li YM, O’Toole JE, Benowitz LI (2006) Mst3b, a purine-sensitive Ste20-like protein kinase, regulates axon outgrowth. Proc Natl Acad Sci U S A 103(48):18320–18325. doi:10.1073/pnas.0605135103
Ishikawa T, Marsala M (1999) Hypothermia prevents biphasic glutamate release and corresponding neuronal degeneration after transient spinal cord ischemia in the rat. Cell Mol Neurobiol 19(2):199–208
Izumi Y, Roussel S, Pinard E, Seylaz J (1991) Reduction of infarct volume by magnesium after middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab 11(6):1025–1030. doi:10.1038/jcbfm.1991.170
Jeong MA, Plunet W, Streijger F, Lee JH, Plemel JR, Park S et al (2011) Intermittent fasting improves functional recovery after rat thoracic contusion spinal cord injury. J Neurotrauma 28(3):479–492. doi:10.1089/neu.2010.1609
Jia HB, Wang XM, Qiu LL, Liu XY, Shen JC, Ji Q, Yang JJ (2013) Spinal neuroimmune activation inhibited by repeated administration of pioglitazone in rats after L5 spinal nerve transection. Neurosci Lett 543:130–135. doi:10.1016/j.neulet.2013.03.046
Jones JR, Barrick C, Kim KA, Lindner J, Blondeau B, Fujimoto Y et al (2005) Deletion of PPARgamma in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc Natl Acad Sci U S A 102(17):6207–6212. doi:10.1073/pnas.0306743102
Jones CE, Dyken PR, Huttenlocher PR, Jabbour JT, Maxwell KW (1982) Inosiplex therapy in subacute sclerosing panencephalitis. A multicentre, non-randomised study in 98 patients. Lancet 1(8280):1034–1037
Jonsson HT Jr, Daniell HB (1976) Altered levels of PGF in cat spinal cord tissue following traumatic injury. Prostaglandins 11(1):51–61
Jyoti A, Sethi P, Sharma D (2009) Curcumin protects against electrobehavioral progression of seizures in the iron-induced experimental model of epileptogenesis. Epilepsy Behav 14(2):300–308. doi:10.1016/j.yebeh.2008.11.011
Kaptanoglu E, Beskonakli E, Okutan O, Selcuk Surucu H, Taskin Y (2003) Effect of magnesium sulphate in experimental spinal cord injury: evaluation with ultrastructural findings and early clinical results. J Clin Neurosci 10(3):329–334
Kawata K, Morimoto T, Ohashi T, Tsujimoto S, Hoshida T, Tsunoda S, Sakaki T (1993) Experimental study of acute spinal cord injury: a histopathological study. No Shinkei Geka 21(1):45–51
Kim D, Zai L, Liang P, Schaffling C, Ahlborn D, Benowitz LI (2013) Inosine enhances axon sprouting and motor recovery after spinal cord injury. PLoS One 8(12):e81948. doi:10.1371/journal.pone.0081948
Kitzman PH (2009) Effectiveness of riluzole in suppressing spasticity in the spinal cord injured rat. Neurosci Lett 455(2):150–153. doi:10.1016/j.neulet.2009.03.016
Kjell J, Finn A, Hao J, Wellfelt K, Josephson A, Svensson CI (2015) Delayed imatinib treatment for acute spinal cord injury: functional recovery and serum biomarkers. J Neurotrauma. doi:10.1089/neu.2014.3863
Kohler G, Milstein C (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256(5517):495–497
Kopp MA, Druschel C, Meisel C, Liebscher T, Prilipp E, Watzlawick R et al (2013) The SCIentinel study – prospective multicenter study to define the spinal cord injury-induced immune depression syndrome (SCI-IDS) – study protocol and interim feasibility data. BMC Neurol 13:168. doi:10.1186/1471-2377-13-168
Koskinen LO (1989) Effects of TRH on blood flow and the microcirculation. Ann N Y Acad Sci 553:353–369
Kuchner EF, Hansebout RR (1976) Combined steroid and hypothermia treatment of experimental spinal cord injury. Surg Neurol 6(6):371–376
Kuhle J, Gaiottino J, Leppert D, Petzold A, Bestwick JP, Malaspina A et al (2014) Serum neurofilament light chain is a biomarker of human spinal cord injury severity and outcome. J Neurol Neurosurg Psychiatry. doi:10.1136/jnnp-2013-307454
Kuhle J, Gaiottino J, Leppert D, Petzold A, Bestwick JP, Malaspina A et al (2015) Serum neurofilament light chain is a biomarker of human spinal cord injury severity and outcome. J Neurol Neurosurg Psychiatry 86(3):273–279. doi:10.1136/jnnp-2013-307454
Kuricova M, Ledecky V, Liptak T, Madari A, Grulova I, Slovinska L et al (2014) Oral administration of inosine promotes recovery after experimental spinal cord injury in rat. Neurol Sci 35(11):1785–1791. doi:10.1007/s10072-014-1840-3
Kwon BK, Curt A, Belanger LM, Bernardo A, Chan D, Markez JA et al (2009) Intrathecal pressure monitoring and cerebrospinal fluid drainage in acute spinal cord injury: a prospective randomized trial. J Neurosurg Spine 10(3):181–193. doi:10.3171/2008.10.SPINE08217
Kwon BK, Okon EB, Tsai E, Beattie MS, Bresnahan JC, Magnuson DK et al (2011) A grading system to evaluate objectively the strength of pre-clinical data of acute neuroprotective therapies for clinical translation in spinal cord injury. J Neurotrauma 28(8):1525–1543. doi:10.1089/neu.2010.1296
Kwon BK, Roy J, Lee JH, Okon E, Zhang H, Marx JC, Kindy MS (2009) Magnesium chloride in a polyethylene glycol formulation as a neuroprotective therapy for acute spinal cord injury: preclinical refinement and optimization. J Neurotrauma 26(8):1379–1393. doi:10.1089/neu.2009-0884
Kwon BK, Stammers AM, Belanger LM, Bernardo A, Chan D, Bishop CM et al (2010) Cerebrospinal fluid inflammatory cytokines and biomarkers of injury severity in acute human spinal cord injury. J Neurotrauma 27(4):669–682. doi:10.1089/neu.2009.1080
Labombarda F, Gonzalez SL, Gonzalez DM, Guennoun R, Schumacher M, de Nicola AF (2002) Cellular basis for progesterone neuroprotection in the injured spinal cord. J Neurotrauma 19(3):343–355. doi:10.1089/089771502753594918
Labombarda F, Gonzalez SL, Lima A, Roig P, Guennoun R, Schumacher M, de Nicola AF (2009) Effects of progesterone on oligodendrocyte progenitors, oligodendrocyte transcription factors, and myelin proteins following spinal cord injury. Glia 57(8):884–897. doi:10.1002/glia.20814
Labombarda F, Gonzalez S, Lima A, Roig P, Guennoun R, Schumacher M, De Nicola AF (2011) Progesterone attenuates astro- and microgliosis and enhances oligodendrocyte differentiation following spinal cord injury. Exp Neurol 231(1):135–146. doi:10.1016/j.expneurol.2011.06.001
Labombarda F, Gonzalez S, Roig P, Lima A, Guennoun R, Schumacher M, De Nicola AF (2000) Modulation of NADPH-diaphorase and glial fibrillary acidic protein by progesterone in astrocytes from normal and injured rat spinal cord. J Steroid Biochem Mol Biol 73(3-4):159–169
Lammertse D, Tuszynski MH, Steeves JD, Curt A, Fawcett JW, Rask C, Ditunno JF, Fehlings MG, Guest JD, Ellaway PH, Kleitman N, Blight AR, Dobkin BH, Grossman R, Katoh H, Privat A, Kalichman M; International Campaign for Cures of Spinal Cord Injury Paralysis (2007) Guidelines for the conduct of clinical trials for spinal cord injury as developed by the ICCP panel: clinical trial design. Spinal Cord 45(3):232–242. doi:10.1038/sj.sc.3102010
Lang-Lazdunski L, Heurteaux C, Vaillant N, Widmann C, Lazdunski M (1999) Riluzole prevents ischemic spinal cord injury caused by aortic crossclamping. J Thorac Cardiovasc Surg 117(5):881–889
Lapchak PA, Zhang JH, Noble-Haeusslein LJ (2013) RIGOR guidelines: escalating STAIR and STEPS for effective translational research. Transl Stroke Res 4(3):279–285. doi:10.1007/s12975-012-0209-2
Laverty PH, Leskovar A, Breur GJ, Coates JR, Bergman RL, Widmer WR et al (2004) A preliminary study of intravenous surfactants in paraplegic dogs: polymer therapy in canine clinical SCI. J Neurotrauma 21(12):1767–1777. doi:10.1089/neu.2004.21.1767
Le E, Aarabi B, Hersh DS, Shanmuganathan K, Diaz C, Massetti J, Akhtar-Danesh N (2015) Predictors of intramedullary lesion expansion rate on MR images of patients with subaxial spinal cord injury. J Neurosurg Spine 22(6):611–621. doi:10.3171/2014.10.SPINE14576
Lee JY, Choi HY, Na WH, Ju BG, Yune TY (2015) 17beta-estradiol inhibits MMP-9 and SUR1/TrpM4 expression and activation and thereby attenuates BSCB disruption/hemorrhage after spinal cord injury in male rats. Endocrinology 156(5):1838–1850. doi:10.1210/en.2014-1832
Lee JS, Han YM, Yoo DS, Choi SJ, Choi BH, Kim JH et al (2004) A molecular basis for the efficacy of magnesium treatment following traumatic brain injury in rats. J Neurotrauma 21(5):549–561. doi:10.1089/089771504774129883
Lee SM, Rosen S, Weinstein P, van Rooijen N, Noble-Haeusslein LJ (2011) Prevention of both neutrophil and monocyte recruitment promotes recovery after spinal cord injury. J Neurotrauma 28(9):1893–1907. doi:10.1089/neu.2011.1860
Lee SM, Yune TY, Kim SJ, Park DW, Lee YK, Kim YC et al (2003) Minocycline reduces cell death and improves functional recovery after traumatic spinal cord injury in the rat. J Neurotrauma 20(10):1017–1027. doi:10.1089/089771503770195867
Lehmann M, Fournier A, Selles-Navarro I, Dergham P, Sebok A, Leclerc N et al (1999) Inactivation of Rho signaling pathway promotes CNS axon regeneration. J Neurosci 19(17):7537–7547
Levi AD, Casella G, Green BA, Dietrich WD, Vanni S, Jagid J, Wang MY (2010) Clinical outcomes using modest intravascular hypothermia after acute cervical spinal cord injury. Neurosurgery 66(4):670–677. doi:10.1227/01.NEU.0000367557.77973.5F
Levi L, Wolf A, Belzberg H (1993) Hemodynamic parameters in patients with acute cervical cord trauma: description, intervention, and prediction of outcome. Neurosurgery 33(6):1007–1016; discussion 1016-1007
Levy ML, Gans W, Wijesinghe HS, SooHoo WE, Adkins RH, Stillerman CB (1996) Use of methylprednisolone as an adjunct in the management of patients with penetrating spinal cord injury: outcome analysis. Neurosurgery 39(6):1141–1149
Li X, Du J, Xu S, Lin X, Ling Z (2013) Peroxisome proliferator-activated receptor-gamma agonist rosiglitazone reduces secondary damage in experimental spinal cord injury. J Int Med Res 41(1):153–161. doi:10.1177/0300060513476601
Li Y, Lu Z, Keogh CL, Yu SP, Wei L (2007) Erythropoietin-induced neurovascular protection, angiogenesis, and cerebral blood flow restoration after focal ischemia in mice. J Cereb Blood Flow Metab 27(5):1043–1054. doi:10.1038/sj.jcbfm.9600417
Liu GJ, Luo J, Zhang LP, Wang ZJ, Xu LL, He GH et al (2011) Meta-analysis of the effectiveness and safety of prophylactic use of nimodipine in patients with an aneurysmal subarachnoid haemorrhage. CNS Neurol Disord Drug Targets 10(7):834–844
Liu F, You SW, Yao LP, Liu HL, Jiao XY, Shi M et al (2006) Secondary degeneration reduced by inosine after spinal cord injury in rats. Spinal Cord 44(7):421–426. doi:10.1038/sj.sc.3101878
Liu-Snyder P, Logan MP, Shi R, Smith DT, Borgens RB (2007) Neuroprotection from secondary injury by polyethylene glycol requires its internalization. J Exp Biol 210(Pt 8):1455–1462. doi:10.1242/jeb.02756
Lo TP Jr, Cho KS, Garg MS, Lynch MP, Marcillo AE, Koivisto DL et al (2009) Systemic hypothermia improves histological and functional outcome after cervical spinal cord contusion in rats. J Comp Neurol 514(5):433–448. doi:10.1002/cne.22014
Lorber B, Howe ML, Benowitz LI, Irwin N (2009) Mst3b, an Ste20-like kinase, regulates axon regeneration in mature CNS and PNS pathways. Nat Neurosci 12(11):1407–1414. doi:10.1038/nn.2414
Lord-Fontaine S, Yang F, Diep Q, Dergham P, Munzer S, Tremblay P, McKerracher L (2008) Local inhibition of Rho signaling by cell-permeable recombinant protein BA-210 prevents secondary damage and promotes functional recovery following acute spinal cord injury. J Neurotrauma 25(11):1309–1322. doi:10.1089/neu.2008.0613
Luo J, Borgens R, Shi R (2002) Polyethylene glycol immediately repairs neuronal membranes and inhibits free radical production after acute spinal cord injury. J Neurochem 83(2):471–480
Luo J, Borgens R, Shi R (2004) Polyethylene glycol improves function and reduces oxidative stress in synaptosomal preparations following spinal cord injury. J Neurotrauma 21(8):994–1007. doi:10.1089/0897715041651097
Luo J, Shi R (2007) Polyethylene glycol inhibits apoptotic cell death following traumatic spinal cord injury. Brain Res 1155:10–16. doi:10.1016/j.brainres.2007.03.091
Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM (2007) Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 145(1):256–264. doi:10.1016/j.neuroscience.2006.11.065
Mabon PJ, Weaver LC, Dekaban GA (2000) Inhibition of monocyte/macrophage migration to a spinal cord injury site by an antibody to the integrin alphaD: a potential new anti-inflammatory treatment. Exp Neurol 166(1):52–64. doi:10.1006/exnr.2000.7488
Maeda T, Kiguchi N, Kobayashi Y, Ozaki M, Kishioka S (2008) Pioglitazone attenuates tactile allodynia and thermal hyperalgesia in mice subjected to peripheral nerve injury. J Pharmacol Sci 108(3):341–347
Maiti P, Manna J, Veleri S, Frautschy S (2014) Molecular chaperone dysfunction in neurodegenerative diseases and effects of curcumin. Biomed Res Int 2014:495091. doi:10.1155/2014/495091
Malgouris C, Bardot F, Daniel M, Pellis F, Rataud J, Uzan A et al (1989) Riluzole, a novel antiglutamate, prevents memory loss and hippocampal neuronal damage in ischemic gerbils. J Neurosci 9(11):3720–3727
Manev H, Favaron M, Vicini S, Guidotti A, Costa E (1990) Glutamate-induced neuronal death in primary cultures of cerebellar granule cells: protection by synthetic derivatives of endogenous sphingolipids. J Pharmacol Exp Ther 252(1):419–427
Mann CM, Lee JH, Hillyer J, Stammers AM, Tetzlaff W, Kwon BK (2010) Lack of robust neurologic benefits with simvastatin or atorvastatin treatment after acute thoracic spinal cord contusion injury. Exp Neurol 221(2):285–295. doi:10.1016/j.expneurol.2009.11.006
Mann C, Lee JH, Liu J, Stammers AM, Sohn HM, Tetzlaff W, Kwon BK (2008) Delayed treatment of spinal cord injury with erythropoietin or darbepoetin – a lack of neuroprotective efficacy in a contusion model of cord injury. Exp Neurol 211(1):34–40. doi:10.1016/j.expneurol.2007.12.013
Marble A (1971) Glibenclamide, a new sulphonylurea: whither oral hypoglycaemic agents? Drugs 1(2):109–115
Martin D, Thompson MA, Nadler JV (1993) The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur J Pharmacol 250(3):473–476
Martirosyan NL, Kalani MY, Bichard WD, Baaj AA, Gonzalez FL, Preul MC, Theodore N (2015) Cerebrospinal fluid drainage and induced hypertension improve spinal cord perfusion after acute spinal cord injury in pigs. Neurosurgery. doi:10.1227/NEU.0000000000000638
Matosin N, Frank E, Engel M, Lum JS, Newell KA (2014) Negativity towards negative results: a discussion of the disconnect between scientific worth and scientific culture. Dis Model Mech 7(2):171–173. doi:10.1242/dmm.015123
Matsumoto T, Tamaki T, Kawakami M, Yoshida M, Ando M, Yamada H (2001) Early complications of high-dose methylprednisolone sodium succinate treatment in the follow-up of acute cervical spinal cord injury. Spine (Phila Pa 1976) 26(4):426–430
McKerracher L, Anderson KD (2013) Analysis of recruitment and outcomes in the phase I/IIa Cethrin clinical trial for acute spinal cord injury. J Neurotrauma 30(21):1795–1804. doi:10.1089/neu.2013.2909
McTigue DM, Tripathi R, Wei P, Lash AT (2007) The PPAR gamma agonist Pioglitazone improves anatomical and locomotor recovery after rodent spinal cord injury. Exp Neurol 205(2):396–406. doi:10.1016/j.expneurol.2007.02.009
Miller RG, Mitchell JD, Moore DH (2012) Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev (3):CD001447. doi:10.1002/14651858.CD001447.pub3
Miller JD, Sakalas R, Ward JD, Young HF, Adams WE, Vries JK, Becker DP (1977) Methylprednisolone treatment in patients with brain tumors. Neurosurgery 1(2):114–117
Mu X, Azbill RD, Springer JE (2000) Riluzole and methylprednisolone combined treatment improves functional recovery in traumatic spinal cord injury. J Neurotrauma 17(9):773–780
Naftchi NE (1982) Prevention of damage in acute spinal cord injury by peptides and pharmacologic agents. Peptides 3(3):235–247
Oatway MA, Chen Y, Bruce JC, Dekaban GA, Weaver LC (2005) Anti-CD11d integrin antibody treatment restores normal serotonergic projections to the dorsal, intermediate, and ventral horns of the injured spinal cord. J Neurosci 25(3):637–647. doi:10.1523/JNEUROSCI.3960-04.2005
Otani K, Abe H, Kadoya S et al (1994) Beneficial effect of methylprednisolone sodium succinate in the treatment of acute spinal cord injury (translation of Japanese). Sekitsui Sekizui J 7:633–647
Oudega M, Vargas CG, Weber AB, Kleitman N, Bunge MB (1999) Long-term effects of methylprednisolone following transection of adult rat spinal cord. Eur J Neurosci 11(7):2453–2464
Ozdemir M, Cengiz SL, Gurbilek M, Ogun TC, Ustun ME (2005) Effects of magnesium sulfate on spinal cord tissue lactate and malondialdehyde levels after spinal cord trauma. Magnes Res 18(3):170–174
Pahan K, Sheikh FG, Namboodiri AM, Singh I (1997) Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. J Clin Invest 100(11):2671–2679. doi:10.1172/JCI119812
Pannu R, Barbosa E, Singh AK, Singh I (2005) Attenuation of acute inflammatory response by atorvastatin after spinal cord injury in rats. J Neurosci Res 79(3):340–350. doi:10.1002/jnr.20345
Pannu R, Christie DK, Barbosa E, Singh I, Singh AK (2007) Post-trauma Lipitor treatment prevents endothelial dysfunction, facilitates neuroprotection, and promotes locomotor recovery following spinal cord injury. J Neurochem 101(1):182–200. doi:10.1111/j.1471-4159.2006.04354.x
Park SW, Yi JH, Miranpuri G, Satriotomo I, Bowen K, Resnick DK, Vemuganti R (2007) Thiazolidinedione class of peroxisome proliferator-activated receptor gamma agonists prevents neuronal damage, motor dysfunction, myelin loss, neuropathic pain, and inflammation after spinal cord injury in adult rats. J Pharmacol Exp Ther 320(3):1002–1012. doi:10.1124/jpet.106.113472
Paterniti I, Impellizzeri D, Di Paola R, Esposito E, Gladman S, Yip P et al (2014) Docosahexaenoic acid attenuates the early inflammatory response following spinal cord injury in mice: in-vivo and in-vitro studies. J Neuroinflammation 11:6. doi:10.1186/1742-2094-11-6
Petain A, Kattygnarath D, Azard J, Chatelut E, Delbaldo C, Geoerger B, Barrois M, Séronie-Vivien S, LeCesne A, Vassal G; Innovative Therapies with Children with Cancer European consortium (2008) Population pharmacokinetics and pharmacogenetics of imatinib in children and adults. Clin Cancer Res 14(21):7102–7109. doi:10.1158/1078-0432.CCR-08-0950
Petitjean ME, Pointillart V, Dixmerias F, Wiart L, Sztark F, Lassie P et al (1998) Medical treatment of spinal cord injury in the acute stage. Ann Fr Anesth Reanim 17(2):114–122
Phang I, Werndle MC, Saadoun S, Varsos GV, Czosnyka M, Zoumprouli A, Papadopoulos MC (2015) Expansion duroplasty improves intraspinal pressure, spinal cord perfusion pressure and vascular pressure reactivity index in patients with traumatic spinal cord injury. J Neurotrauma. doi:10.1089/neu.2014.3668
Philippon J, Grob R, Dagreou F, Guggiari M, Rivierez M, Viars P (1986) Prevention of vasospasm in subarachnoid haemorrhage. A controlled study with nimodipine. Acta Neurochir (Wien) 82(3-4):110–114
Pinzon A, Marcillo A, Pabon D, Bramlett HM, Bunge MB, Dietrich WD (2008) A re-assessment of erythropoietin as a neuroprotective agent following rat spinal cord compression or contusion injury. Exp Neurol 213(1):129–136. doi:10.1016/j.expneurol.2008.05.018
Pitts LH, Ross A, Chase GA, Faden AI (1995) Treatment with thyrotropin-releasing hormone (TRH) in patients with traumatic spinal cord injuries. J Neurotrauma 12(3):235–243
Plunet WT, Streijger F, Lam CK, Lee JH, Liu J, Tetzlaff W (2008) Dietary restriction started after spinal cord injury improves functional recovery. Exp Neurol 213(1):28–35. doi:10.1016/j.expneurol.2008.04.011
Pontius RG, Brockman HL, Hardy EG, Cooley DA, Debakey ME (1954) The use of hypothermia in the prevention of paraplegia following temporary aortic occlusion: experimental observations. Surgery 36(1):33–38
Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT (1999) Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol 158(2):351–365. doi:10.1006/exnr.1999.7118
Popovich PG, Lemeshow S, Gensel JC, Tovar CA (2012) Independent evaluation of the effects of glibenclamide on reducing progressive hemorrhagic necrosis after cervical spinal cord injury. Exp Neurol 233(2):615–622. doi:10.1016/j.expneurol.2010.11.016
Popovich PG, Tovar CA, Wei P, Fisher L, Jakeman LB, Basso DM (2012) A reassessment of a classic neuroprotective combination therapy for spinal cord injured rats: LPS/pregnenolone/indomethacin. Exp Neurol 233(2):677–685. doi:10.1016/j.expneurol.2011.11.045
Poynton AR, O’Farrell DA, Shannon F, Murray P, McManus F, Walsh MG (1997) An evaluation of the factors affecting neurological recovery following spinal cord injury. Injury 28(8):545–548
Pratt J, Rataud J, Bardot F, Roux M, Blanchard JC, Laduron PM, Stutzmann JM (1992) Neuroprotective actions of riluzole in rodent models of global and focal cerebral ischaemia. Neurosci Lett 140(2):225–230
Prendergast MR, Saxe JM, Ledgerwood AM, Lucas CE, Lucas WF (1994) Massive steroids do not reduce the zone of injury after penetrating spinal cord injury. J Trauma 37(4):576–579; discussion 579-580
Prins ML, Matsumoto JH (2014) The collective therapeutic potential of cerebral ketone metabolism in traumatic brain injury. J Lipid Res 55(12):2450–2457. doi:10.1194/jlr.R046706
Rabchevsky AG, Sullivan PG, Fugaccia I, Scheff SW (2003) Creatine diet supplement for spinal cord injury: influences on functional recovery and tissue sparing in rats. J Neurotrauma 20(7):659–669. doi:10.1089/089771503322144572
Rabin SJ, Bachis A, Mocchetti I (2002) Gangliosides activate Trk receptors by inducing the release of neurotrophins. J Biol Chem 277(51):49466–49472. doi:10.1074/jbc.M203240200
Renaud LP, Blume HW, Pittman QJ, Lamour Y, Tan AT (1979) Thyrotropin-releasing hormone selectively depresses glutamate excitation of cerebral cortical neurons. Science 205(4412):1275–1277
Resnick DK, Nguyen P, Cechvala CF (2001) Selective cyclooxygenase 2 inhibition lowers spinal cord prostaglandin concentrations after injury. Spine J 1(6):437–441
Riegger T, Conrad S, Liu K, Schluesener HJ, Adibzahdeh M, Schwab JM (2007) Spinal cord injury-induced immune depression syndrome (SCI-IDS). Eur J Neurosci 25(6):1743–1747. doi:10.1111/j.1460-9568.2007.05447.x
Riegger T, Conrad S, Schluesener HJ, Kaps HP, Badke A, Baron C et al (2009) Immune depression syndrome following human spinal cord injury (SCI): a pilot study. Neuroscience 158(3):1194–1199. doi:10.1016/j.neuroscience.2008.08.021
Robertson CS, Foltz R, Grossman RG, Goodman JC (1986) Protection against experimental ischemic spinal cord injury. J Neurosurg 64(4):633–642. doi:10.3171/jns.1986.64.4.0633
Ross IB, Tator CH (1993) Spinal cord blood flow and evoked potential responses after treatment with nimodipine or methylprednisolone in spinal cord-injured rats. Neurosurgery 33(3):470–476; discussion 476-477
Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, Sasaki R (1998) In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci U S A 95(8):4635–4640
Samantaray S, Smith JA, Das A, Matzelle DD, Varma AK, Ray SK, Banik NL (2011) Low dose estrogen prevents neuronal degeneration and microglial reactivity in an acute model of spinal cord injury: effect of dosing, route of administration, and therapy delay. Neurochem Res 36(10):1809–1816. doi:10.1007/s11064-011-0498-y
Sandestig A, Romner B, Grande PO (2014) Therapeutic hypothermia in children and adults with severe traumatic brain injury. Ther Hypothermia Temp Manag 4(1):10–20. doi:10.1089/ther.2013.0024
Sandler AN, Tator CH (1976) Regional spinal cord blood flow in primates. J Neurosurg 45(6):647–659. doi:10.3171/jns.1976.45.6.0647
Sauerbeck AD, Laws JL, Bandaru VV, Popovich PG, Haughey NJ, McTigue DM (2015) Spinal cord injury causes chronic liver pathology in rats. J Neurotrauma 32(3):159–169. doi:10.1089/neu.2014.3497
Saver JL, Starkman S, Eckstein M, Stratton SJ, Pratt FD, Hamilton S et al (2015) Prehospital use of magnesium sulfate as neuroprotection in acute stroke. N Engl J Med 372(6):528–536. doi:10.1056/NEJMoa1408827
Schumacher M, Hussain R, Gago N, Oudinet JP, Mattern C, Ghoumari AM (2012) Progesterone synthesis in the nervous system: implications for myelination and myelin repair. Front Neurosci 6:10. doi:10.3389/fnins.2012.00010
Schwab JM, Conrad S, Elbert T, Trautmann K, Meyermann R, Schluesener HJ (2004) Lesional RhoA+ cell numbers are suppressed by anti-inflammatory, cyclooxygenase-inhibiting treatment following subacute spinal cord injury. Glia 47(4):377–386. doi:10.1002/glia.20031
Schwab JM, Zhang Y, Kopp MA, Brommer B, Popovich PG (2014) The paradox of chronic neuroinflammation, systemic immune suppression, autoimmunity after traumatic chronic spinal cord injury. Exp Neurol 258:121–129. doi:10.1016/j.expneurol.2014.04.023
Schwartz G, Fehlings MG (2001) Evaluation of the neuroprotective effects of sodium channel blockers after spinal cord injury: improved behavioral and neuroanatomical recovery with riluzole. J Neurosurg 94(2 Suppl):245–256
Sharma HS, Olsson Y, Cervos-Navarro J (1993) Early perifocal cell changes and edema in traumatic injury of the spinal cord are reduced by indomethacin, an inhibitor of prostaglandin synthesis. Experimental study in the rat. Acta Neuropathol 85(2):145–153
Sharp KG, Yee KM, Stiles TL, Aguilar RM, Steward O (2013) A re-assessment of the effects of treatment with a non-steroidal anti-inflammatory (ibuprofen) on promoting axon regeneration via RhoA inhibition after spinal cord injury. Exp Neurol 248:321–337. doi:10.1016/j.expneurol.2013.06.023
Sharp KG, Yee KM, Steward O (2014) A re-assessment of treatment with a tyrosine kinase inhibitor (imatinib) on tissue sparing and functional recovery after spinal cord injury. Exp Neurol 254:1–11. doi:10.1016/j.expneurol.2013.12.019
Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G et al (2009) Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med 6(7):e1000113. doi:10.1371/journal.pmed.1000113
Shi R, Borgens RB (1999) Acute repair of crushed guinea pig spinal cord by polyethylene glycol. J Neurophysiol 81(5):2406–2414
Shields CB, Zhang YP, Shields LB, Han Y, Burke DA, Mayer NW (2005) The therapeutic window for spinal cord decompression in a rat spinal cord injury model. J Neurosurg Spine 3(4):302–307. doi:10.3171/spi.2005.3.4.0302
Simard JM, Kilbourne M, Tsymbalyuk O, Tosun C, Caridi J, Ivanova S et al (2009) Key role of sulfonylurea receptor 1 in progressive secondary hemorrhage after brain contusion. J Neurotrauma 26(12):2257–2267. doi:10.1089/neu.2009.1021
Simard JM, Popovich PG, Tsymbalyuk O, Caridi J, Gullapalli RP, Kilbourne MJ, Gerzanich V (2013) MRI evidence that glibenclamide reduces acute lesion expansion in a rat model of spinal cord injury. Spinal Cord 51(11):823–827. doi:10.1038/sc.2013.99
Simard JM, Popovich PG, Tsymbalyuk O, Gerzanich V (2012) Spinal cord injury with unilateral versus bilateral primary hemorrhage – effects of glibenclamide. Exp Neurol 233(2):829–835. doi:10.1016/j.expneurol.2011.11.048
Simard JM, Tsymbalyuk O, Ivanov A, Ivanova S, Bhatta S, Geng Z et al (2007) Endothelial sulfonylurea receptor 1-regulated NC Ca-ATP channels mediate progressive hemorrhagic necrosis following spinal cord injury. J Clin Invest 117(8):2105–2113. doi:10.1172/JCI32041
Simard JM, Tsymbalyuk O, Keledjian K, Ivanov A, Ivanova S, Gerzanich V (2012) Comparative effects of glibenclamide and riluzole in a rat model of severe cervical spinal cord injury. Exp Neurol 233(1):566–574. doi:10.1016/j.expneurol.2011.11.044
Simpson RK Jr, Baskin DS, Dudley AW, Bogue L, Rothenberg F (1991) The influence of long-term nifedipine or indomethacin therapy on neurologic recovery from experimental spinal cord injury. J Spinal Disord 4(4):420–427
Sipski ML, Jackson AB, Gomez-Marin O, Estores I, Stein A (2004) Effects of gender on neurologic and functional recovery after spinal cord injury. Arch Phys Med Rehabil 85(11):1826–1836
Siren AL, Knerlich F, Poser W, Gleiter CH, Bruck W, Ehrenreich H (2001) Erythropoietin and erythropoietin receptor in human ischemic/hypoxic brain. Acta Neuropathol 101(3):271–276
Smith JS, Anderson R, Pham T, Bhatia N, Steward O, Gupta R (2010) Role of early surgical decompression of the intradural space after cervical spinal cord injury in an animal model. J Bone Joint Surg Am 92(5):1206–1214. doi:10.2106/JBJS.I.00740
Snapinn SM, Jiang Q (2007) Responder analyses and the assessment of a clinically relevant treatment effect. Trials 8:31. doi:10.1186/1745-6215-8-31
Sonmez E, Kabatas S, Ozen O, Karabay G, Turkoglu S, Ogus E et al (2013) Minocycline treatment inhibits lipid peroxidation, preserves spinal cord ultrastructure, and improves functional outcome after traumatic spinal cord injury in the rat. Spine (Phila Pa 1976) 38(15):1253–1259. doi:10.1097/BRS.0b013e3182895587
Souvenir R, Doycheva D, Zhang JH, Tang J (2015) Erythropoietin in stroke therapy: friend or foe. Curr Med Chem 22(10):1205–1213
Springer JE, Azbill RD, Kennedy SE, George J, Geddes JW (1997) Rapid calpain I activation and cytoskeletal protein degradation following traumatic spinal cord injury: attenuation with riluzole pretreatment. J Neurochem 69(4):1592–1600
Sribnick EA, Matzelle DD, Ray SK, Banik NL (2006) Estrogen treatment of spinal cord injury attenuates calpain activation and apoptosis. J Neurosci Res 84(5):1064–1075. doi:10.1002/jnr.21016
Sribnick EA, Samantaray S, Das A, Smith J, Matzelle DD, Ray SK, Banik NL (2010) Postinjury estrogen treatment of chronic spinal cord injury improves locomotor function in rats. J Neurosci Res 88(8):1738–1750. doi:10.1002/jnr.22337
Sribnick EA, Wingrave JM, Matzelle DD, Wilford GG, Ray SK, Banik NL (2005) Estrogen attenuated markers of inflammation and decreased lesion volume in acute spinal cord injury in rats. J Neurosci Res 82(2):283–293. doi:10.1002/jnr.20622
Stanislaus R, Singh AK, Singh I (2001) Lovastatin treatment decreases mononuclear cell infiltration into the CNS of Lewis rats with experimental allergic encephalomyelitis. J Neurosci Res 66(2):155–162
Steeves JD, Kramer JK, Fawcett JW, Cragg J, Lammertse DP, Blight AR, Marino RJ, Ditunno JF Jr, Coleman WP, Geisler FH, Guest J, Jones L, Burns S, Schubert M, van Hedel HJ, Curt A; EMSCI Study Group (2011) Extent of spontaneous motor recovery after traumatic cervical sensorimotor complete spinal cord injury. Spinal Cord 49(2):257–265. doi:10.1038/sc.2010.99
Stein DG (2008) Progesterone exerts neuroprotective effects after brain injury. Brain Res Rev 57(2):386–397. doi:10.1016/j.brainresrev.2007.06.012
Steward O, Popovich PG, Dietrich WD, Kleitman N (2012) Replication and reproducibility in spinal cord injury research. Exp Neurol 233(2):597–605. doi:10.1016/j.expneurol.2011.06.017
Steward O, Sharp K, Yee KM (2012) A re-assessment of the effects of intracortical delivery of inosine on transmidline growth of corticospinal tract axons after unilateral lesions of the medullary pyramid. Exp Neurol 233(2):662–673. doi:10.1016/j.expneurol.2011.09.019
Stirling DP, Khodarahmi K, Liu J, McPhail LT, McBride CB, Steeves JD et al (2004) Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci 24(9):2182–2190. doi:10.1523/JNEUROSCI.5275-03.2004
Stirling DP, Liu S, Kubes P, Yong VW (2009) Depletion of Ly6G/Gr-1 leukocytes after spinal cord injury in mice alters wound healing and worsens neurological outcome. J Neurosci 29(3):753–764. doi:10.1523/JNEUROSCI.4918-08.2009
Stokes BT, Hollinden G, Fox P (1984) Improvement in injury induced hypocalcia by high-dose naloxone intervention. Brain Res 290(1):187–190
Streijger F, Lee JH, Duncan GJ, Ng MT, Assinck P, Bhatnagar T et al (2014) Combinatorial treatment of acute spinal cord injury with ghrelin, ibuprofen, C16, and ketogenic diet does not result in improved histologic or functional outcome. J Neurosci Res 92(7):870–883. doi:10.1002/jnr.23372
Streijger F, Plunet WT, Lee JH, Liu J, Lam CK, Park S et al (2013) Ketogenic diet improves forelimb motor function after spinal cord injury in rodents. PLoS One 8(11):e78765. doi:10.1371/journal.pone.0078765
Streijger F, Plunet WT, Plemel JR, Lam CK, Liu J, Tetzlaff W (2011) Intermittent fasting in mice does not improve hindlimb motor performance after spinal cord injury. J Neurotrauma 28(6):1051–1061. doi:10.1089/neu.2010.1715
Stubbs SS, Morrell RM (1973) Intravenous methylprednisolone sodium succinate: adverse reactions reported in association with immunosuppresive therapy. Transplant Proc 5(2):1145–1146
Stutzmann JM, Pratt J, Boraud T, Gross C (1996) The effect of riluzole on post-traumatic spinal cord injury in the rat. Neuroreport 7(2):387–392
Su EJ, Fredriksson L, Geyer M, Folestad E, Cale J, Andrae J et al (2008) Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat Med 14(7):731–737. doi:10.1038/nm1787
Suzer T, Coskun E, Islekel H, Tahta K (1999) Neuroprotective effect of magnesium on lipid peroxidation and axonal function after experimental spinal cord injury. Spinal Cord 37(7):480–484
Svensson LG, Hess KR, D’Agostino RS, Entrup MH, Hreib K, Kimmel WA et al (1998) Reduction of neurologic injury after high-risk thoracoabdominal aortic operation. Ann Thorac Surg 66(1):132–138
Swanson CR, Joers V, Bondarenko V, Brunner K, Simmons HA, Ziegler TE et al (2011) The PPAR-gamma agonist pioglitazone modulates inflammation and induces neuroprotection in parkinsonian monkeys. J Neuroinflammation 8:91. doi:10.1186/1742-2094-8-91
Swartz KR, Fee DB, Joy KM, Roberts KN, Sun S, Scheff NN et al (2007) Gender differences in spinal cord injury are not estrogen-dependent. J Neurotrauma 24(3):473–480. doi:10.1089/neu.2006.0167
Tadie M, D’Arbigny P, Mathé J, Loubert G, Saint-Marc C, Menthonnex P (1999) Acute spinal cord injury: early care and treatment in a multicenter study with gacyclidine. Soc Neuroscience 25(1090):b138-0250014
Tadie M, Gaviria M, Mathé J-F, Menthonnex PH, Loubert G, Lagarrigue J, Saint-Marc C, Argenson C, Kempf CH, D’Arbigny P, Kamenca J-M, Privat A, Carli P (2003) Early care and treatment with a neuroprotective drug, Gacyclidine, in patients with acute spinal cord injury. Le Rachis 15(6):363–376
Tan LA, Kasliwal MK, Fontes RB, Fessler RG (2014) Local cooling for traumatic spinal cord injury. J Neurosurg Spine 21(5):845–847. doi:10.3171/2014.5.SPINE14472
Tanadini LG, Hothorn T, Jones LA, Lammertse DP, Abel R, Maier D (2015) Toward inclusive trial protocols in heterogeneous neurological disorders: prediction-based stratification of participants with incomplete cervical spinal cord injury. Neurorehabil Neural Repair. doi:10.1177/1545968315570322
Taoka Y, Okajima K, Uchiba M, Murakami K, Kushimoto S, Johno M et al (1997) Role of neutrophils in spinal cord injury in the rat. Neuroscience 79(4):1177–1182
Tator CH, Fehlings MG (1991) Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg 75(1):15–26. doi:10.3171/jns.1991.75.1.0015
Tator CH, Koyanagi I (1997) Vascular mechanisms in the pathophysiology of human spinal cord injury. J Neurosurg 86(3):483–492. doi:10.3171/jns.1997.86.3.0483
Teng YD, Choi H, Onario RC, Zhu S, Desilets FC, Lan S et al (2004) Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc Natl Acad Sci U S A 101(9):3071–3076. doi:10.1073/pnas.0306239101
Thomas T, Bryant M, Clark L, Garces A, Rhodin J (2001) Estrogen and raloxifene activities on amyloid-beta-induced inflammatory reaction. Microvasc Res 61(1):28–39. doi:10.1006/mvre.2000.2267
Thomas AJ, Nockels RP, Pan HQ, Shaffrey CI, Chopp M (1999) Progesterone is neuroprotective after acute experimental spinal cord trauma in rats. Spine (Phila Pa 1976) 24(20):2134–2138
Tseng MY, Hutchinson PJ, Richards HK, Czosnyka M, Pickard JD, Erber WN et al (2009) Acute systemic erythropoietin therapy to reduce delayed ischemic deficits following aneurysmal subarachnoid hemorrhage: a Phase II randomized, double-blind, placebo-controlled trial. Clinical article. J Neurosurg 111(1):171–180. doi:10.3171/2009.3.JNS081332
Vale FL, Burns J, Jackson AB, Hadley MN (1997) Combined medical and surgical treatment after acute spinal cord injury: results of a prospective pilot study to assess the merits of aggressive medical resuscitation and blood pressure management. J Neurosurg 87(2):239–246. doi:10.3171/jns.1997.87.2.0239
van Middendorp JJ, Hosman AJ, Doi SA (2013) The effects of the timing of spinal surgery after traumatic spinal cord injury: a systematic review and meta-analysis. J Neurotrauma 30(21):1781–1794. doi:10.1089/neu.2013.2932
Vandame D, Desmadryl G, Becerril Ortega J, Teigell M, Crouzin N, Buisson A et al (2007) Comparison of the pharmacological properties of GK11 and MK801, two NMDA receptor antagonists: towards an explanation for the lack of intrinsic neurotoxicity of GK11. J Neurochem 103(4):1682–1696. doi:10.1111/j.1471-4159.2007.04925.x
Varsos GV, Werndle MC, Czosnyka ZH, Smielewski P, Kolias AG, Phang I et al (2015) Intraspinal pressure and spinal cord perfusion pressure after spinal cord injury: an observational study. J Neurosurg Spine 23(6):763–771. doi:10.3171/2015.3.SPINE14870
Vitellaro-Zuccarello L, Mazzetti S, Madaschi L, Bosisio P, Fontana E, Gorio A, De Biasi S (2008) Chronic erythropoietin-mediated effects on the expression of astrocyte markers in a rat model of contusive spinal cord injury. Neuroscience 151(2):452–466. doi:10.1016/j.neuroscience.2007.11.004
Wallace MC, Tator CH (1986) Failure of naloxone to improve spinal cord blood flow and cardiac output after spinal cord injury. Neurosurgery 18(4):428–432
Walters BC, Hadley MN, Hurlbert RJ, Aarabi B, Dhall SS, Gelb DE, Harrigan MR, Rozelle CJ, Ryken TC, Theodore N; American Association of Neurological Surgeons; Congress of Neurological Surgeons (2013) Guidelines for the management of acute cervical spine and spinal cord injuries: 2013 update. Neurosurgery 60(Suppl 1);82–91. doi:10.1227/01.neu.0000430319.32247.7f
Wang X, Budel S, Baughman K, Gould G, Song KH, Strittmatter SM (2009) Ibuprofen enhances recovery from spinal cord injury by limiting tissue loss and stimulating axonal growth. J Neurotrauma 26(1):81–95. doi:10.1089/neu.2007.0464
Ward RE, Huang W, Curran OE, Priestley JV, Michael-Titus AT (2010) Docosahexaenoic acid prevents white matter damage after spinal cord injury. J Neurotrauma 27(10):1769–1780. doi:10.1089/neu.2010.1348
Weaver LC, Gris D, Saville LR, Oatway MA, Chen Y, Marsh DR et al (2005) Methylprednisolone causes minimal improvement after spinal cord injury in rats, contrasting with benefits of an anti-integrin treatment. J Neurotrauma 22(12):1375–1387. doi:10.1089/neu.2005.22.1375
Wells JD, Hansebout RR (1978) Local hypothermia in experimental spinal cord trauma. Surg Neurol 10(3):200–204
Werndle MC, Saadoun S, Phang I, Czosnyka M, Varsos GV, Czosnyka ZH et al (2014) Monitoring of spinal cord perfusion pressure in acute spinal cord injury: initial findings of the injured spinal cord pressure evaluation study*. Crit Care Med 42(3):646–655. doi:10.1097/CCM.0000000000000028
Winkler T, Sharma HS, Stalberg E, Olsson Y (1993) Indomethacin, an inhibitor of prostaglandin synthesis attenuates alteration in spinal cord evoked potentials and edema formation after trauma to the spinal cord: an experimental study in the rat. Neuroscience 52(4):1057–1067
Winton MJ, Dubreuil CI, Lasko D, Leclerc N, McKerracher L (2002) Characterization of new cell permeable C3-like proteins that inactivate Rho and stimulate neurite outgrowth on inhibitory substrates. J Biol Chem 277(36):32820–32829. doi:10.1074/jbc.M201195200
Wu Y, Satkunendrarajah K, Teng Y, Chow DS, Buttigieg J, Fehlings MG (2013) Delayed post-injury administration of riluzole is neuroprotective in a preclinical rodent model of cervical spinal cord injury. J Neurotrauma 30(6):441–452. doi:10.1089/neu.2012.2622
Yan J, Li B, Chen JW, Jiang SD, Jiang LS (2012) Spinal cord injury causes bone loss through peroxisome proliferator-activated receptor-gamma and Wnt signalling. J Cell Mol Med 16(12):2968–2977. doi:10.1111/j.1582-4934.2012.01624.x
Young W (1992) Role of calcium in central nervous system injuries. J Neurotrauma 9(Suppl 1):S9–S25
Young W (2002) Spinal cord contusion models. Prog Brain Res 137:231–255
Young W, Flamm ES (1982) Effect of high-dose corticosteroid therapy on blood flow, evoked potentials, and extracellular calcium in experimental spinal injury. J Neurosurg 57(5):667–673. doi:10.3171/jns.1982.57.5.0667
Young W, Flamm ES, Demopoulos HB, Tomasula JJ, DeCrescito V (1981) Effect of naloxone on posttraumatic ischemia in experimental spinal contusion. J Neurosurg 55(2):209–219. doi:10.3171/jns.1981.55.2.0209
Youssef S, Stuve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM et al (2002) The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature 420(6911):78–84. doi:10.1038/nature01158
Yune TY, Kim SJ, Lee SM, Lee YK, Oh YJ, Kim YC et al (2004) Systemic administration of 17beta-estradiol reduces apoptotic cell death and improves functional recovery following traumatic spinal cord injury in rats. J Neurotrauma 21(3):293–306. doi:10.1089/089771504322972086
Zager EL, Ames A 3rd (1988) Reduction of cellular energy requirements. Screening for agents that may protect against CNS ischemia. J Neurosurg 69(4):568–579. doi:10.3171/jns.1988.69.4.0568
Zai L, Ferrari C, Dice C, Subbaiah S, Havton LA, Coppola G et al (2011) Inosine augments the effects of a Nogo receptor blocker and of environmental enrichment to restore skilled forelimb use after stroke. J Neurosci 31(16):5977–5988. doi:10.1523/JNEUROSCI.4498-10.2011
Zai L, Ferrari C, Subbaiah S, Havton LA, Coppola G, Strittmatter S et al (2009) Inosine alters gene expression and axonal projections in neurons contralateral to a cortical infarct and improves skilled use of the impaired limb. J Neurosci 29(25):8187–8197. doi:10.1523/JNEUROSCI.0414-09.2009
Zariffa J, Kramer JL, Fawcett JW, Lammertse DP, Blight AR, Guest J et al (2011) Characterization of neurological recovery following traumatic sensorimotor complete thoracic spinal cord injury. Spinal Cord 49(3):463–471. doi:10.1038/sc.2010.140
Zechariah A, ElAli A, Hermann DM (2010) Combination of tissue-plasminogen activator with erythropoietin induces blood-brain barrier permeability, extracellular matrix disaggregation, and DNA fragmentation after focal cerebral ischemia in mice. Stroke 41(5):1008–1012. doi:10.1161/STROKEAHA.109.574418
Zhang B, Gensel JC (2014) Is neuroinflammation in the injured spinal cord different than in the brain? Examining intrinsic differences between the brain and spinal cord. Exp Neurol 258:112–120. doi:10.1016/j.expneurol.2014.04.007
Zhang Q, Hu W, Meng B, Tang T (2010) PPARgamma agonist rosiglitazone is neuroprotective after traumatic spinal cord injury via anti-inflammatory in adult rats. Neurol Res 32(8):852–859. doi:10.1179/016164110X12556180206112
Zhang Q, Huang C, Tang T, Shi Q, Yang H (2011) Comparative neuroprotective effects of methylprednisolone and rosiglitazone, a peroxisome proliferator-activated receptor-gamma following spinal cord injury. Neurosciences (Riyadh) 16(1):46–52
Zhang Y, Metz LM, Yong VW, Bell RB, Yeung M, Patry DG, Mitchell JR (2008) Pilot study of minocycline in relapsing-remitting multiple sclerosis. Can J Neurol Sci 35(2):185–191
Zhang L, Rzigalinski BA, Ellis EF, Satin LS (1996) Reduction of voltage-dependent Mg2+ blockade of NMDA current in mechanically injured neurons. Science 274(5294):1921–1923
Zhu Y, Soderblom C, Krishnan V, Ashbaugh J, Bethea JR, Lee JK (2015) Hematogenous macrophage depletion reduces the fibrotic scar and increases axonal growth after spinal cord injury. Neurobiol Dis 74:114–125. doi:10.1016/j.nbd.2014.10.024
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Santamaria, A.J., Guest, J.D. (2017). The Current Status of Neuroprotection for Spinal Cord Injury. In: Weidner, N., Rupp, R., Tansey, K. (eds) Neurological Aspects of Spinal Cord Injury. Springer, Cham. https://doi.org/10.1007/978-3-319-46293-6_20
Download citation
DOI: https://doi.org/10.1007/978-3-319-46293-6_20
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-46291-2
Online ISBN: 978-3-319-46293-6
eBook Packages: MedicineMedicine (R0)