
Abstract
Reduced plasma levels of HDL-C are associated with an increased risk of CAD and myocardial infarction, as shown in various prospective population studies. However, recent clinical trials on lipid-modifying drugs that increase plasma levels of HDL-C have not shown significant clinical benefit. Notably, in some recent clinical studies, there is no clear association of higher HDL-C levels with a reduced risk of cardiovascular events observed in patients with existing CAD. These observations have prompted researchers to shift from a cholesterol-centric view of HDL towards assessing the function and composition of HDL particles.
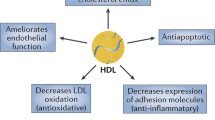
Of importance, experimental and translational studies have further demonstrated various potential antiatherogenic effects of HDL. HDL has been proposed to promote macrophage reverse cholesterol transport and to protect endothelial cell functions by prevention of oxidation of LDL and its adverse endothelial effects. Furthermore, HDL from healthy subjects can directly stimulate endothelial cell production of nitric oxide and exert anti-inflammatory and antiapoptotic effects. Of note, increasing evidence suggests that the vascular effects of HDL can be highly heterogeneous and HDL may lose important anti-atherosclerotic properties and turn dysfunctional in patients with chronic inflammatory disorders. A greater understanding of mechanisms of action of HDL and its altered vascular effects is therefore critical within the context of HDL-targeted therapies.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
It was shown in multiple large prospective studies of cardiovascular risk factors that reduced plasma levels of HDL-cholesterol (HDL-C) are associated with an increased risk of coronary artery disease (CAD) (Castelli et al. 1986; Cullen et al. 1997; Di Angelantonio et al. 2009; Gordon et al. 1977; Sharrett et al. 2001). Multiple biological functions of HDL have been identified, whereby HDL may exert antiatherogenic effects (Annema and von Eckardstein 2013; Barter et al. 2004; Mineo et al. 2006; Rader 2006; Riwanto and Landmesser 2013), e.g., HDL from healthy subjects has been shown to directly promote endothelial antiapoptotic, anti-inflammatory, and antithrombotic effects (Mineo et al. 2006; Nofer et al. 2004; Rye and Barter 2008; Tall et al. 2008; Yuhanna et al. 2001). Accordingly, interventions to improve HDL-C levels and/or HDL function are being intensely evaluated as a potential therapeutic strategy to reduce cardiovascular risk. However, increasing evidence suggests that the endothelial and vascular effects of HDL are highly heterogeneous and vasoprotective properties of HDL are impaired in patients with diabetes, CAD, or chronic kidney dysfunction (Besler et al. 2011; Khera et al. 2011; Riwanto et al. 2013; Sorrentino et al. 2010).
Intriguingly, several recent clinical trials testing the effects of HDL-C-raising therapies have failed to demonstrate cardiovascular risk reduction in patients with CAD. The Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) trial testing the impact of the CETP inhibitor torcetrapib on clinical outcome showed increased risk of mortality and morbidity in patients at high risk for coronary events, despite a substantial increase of HDL-C levels (Barter et al. 2007). Dalcetrapib, another CETP inhibitor, modestly increased HDL-C levels, but the phase 3 trial dal-OUTCOMES study was terminated before completion to a lack of efficacy (Schwartz et al. 2012). More recently, the HPS2-THRIVE trial results showed that adding extended-release niacin/laropiprant, another HDL-cholesterol-raising agent, to statins did not reduce the risk of cardiovascular event (Haynes et al. 2013). Taken together, these observations strongly suggest that plasma HDL-C levels per se are not an optimal therapeutic target.
Of note, accumulating evidence suggests that the vascular effects of HDL can be highly heterogeneous and may turn dysfunctional. HDL loses potential anti-atherosclerotic properties in patients with chronic inflammatory disorders, such as the antiphospholipid syndrome (Charakida et al. 2009), systemic lupus erythematosus and rheumatoid arthritis (McMahon et al. 2006), scleroderma (Weihrauch et al. 2007), metabolic syndrome (de Souza et al. 2008), diabetes (Persegol et al. 2006; Sorrentino et al. 2010), and CAD (Ansell et al. 2003; Besler et al. 2011; Riwanto et al. 2013). In a study of 189 patients with chronic kidney disease on hemodialysis, an impaired anti-inflammatory capacity of HDL was correlated with a poor clinical outcome (Kalantar-Zadeh et al. 2007). Furthermore, HDL isolated from subjects with type 1 or type 2 diabetes mellitus or abdominal obesity had reduced capacity to reverse the inhibition of aortic ring endothelium-dependent relaxation by oxLDL as compared to HDL from healthy control subjects (Persegol et al. 2006, 2007). Importantly, the heterogeneity of the vascular effects of HDL may be attributed to changes in the HDL-associated proteome and lipidome, i.e., changes in the amount and type of proteins and lipids bound to the HDL particle and also posttranslational modifications. In particular, HDL is known to be susceptible to modification in vitro by a variety of oxidants, such as metal ions, peroxyl and hydroxyl radicals, aldehydes, various myeloperoxidase (MPO)-generated oxidants, lipoxygenase, phospholipase A2, elastase, nonenzymatic glycation, and homocysteinylation (Ferretti et al. 2006). These oxidative modifications may contribute to the generation of dysfunctional proinflammatory HDL.
2 HDL and Reverse Cholesterol Transport
2.1 Mechanisms Under Physiological Conditions
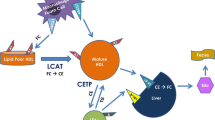
One of the antiatherogenic effects of HDL has been attributed to its function in macrophage reverse cholesterol transport (RCT), i.e., the removal of excess cholesterol from lipid-laden macrophage foam cells in the atherosclerotic plaque and its transport to the liver for excretion in the bile (Rader 2006). The first step of macrophage RCT involves the hydrolysis of cytoplasmic cholesteryl ester into free cholesterol followed by the efflux of the free cholesterol to mature HDL or extracellular lipid-poor apoA-I. Macrophage cholesterol efflux is mediated by active transport systems, which include the ATP-binding cassette transporters ABCA1 and ABCG1 (Rader 2006; Tall et al. 2008). Studies in macrophages from ABCA1-knockout or ABCA1-overexpressing mice have shown that ABCA1 primarily mediates the cholesterol efflux to lipid-poor apoA-I (Bortnick et al. 2000; Haghpassand et al. 2001). In contrast, ABCG1 largely mediates cholesterol efflux from macrophages to mature HDL (Tall et al. 2008).
A study by Wang et al. quantitatively assessed the roles of SR-BI, ABCA1, and ABCG1 in macrophage RCT in mice in vivo (Wang et al. 2007a). Using primary macrophages lacking SR-BI, the authors demonstrated that SR-BI did not promote macrophage RCT in vivo after intraperitoneal injection. In contrast, both ABCA1 and ABCG1 contributed to macrophage RCT in vivo. The study also demonstrated that transplantation of bone marrow from ABCA1/ABCG1-deficient mice accelerated atherosclerotic lesion formation in LDL receptor-deficient mice (Wang et al. 2007a). Interestingly, Yvan-Charvet et al. also demonstrated that SR-BI failed to stimulate net cholesterol efflux from HEK293 cells to plasma HDL and inhibited ABCG1-mediated cholesterol efflux, which was at least in part due to the increased uptake of HDL cholesteryl esters into these cells (Yvan-Charvet et al. 2008).
HDL-associated cholesterol is subsequently esterified by LCAT which transfers a fatty acyl residue from phospholipids to the 3-beta-hydroxy group of cholesterol. The final steps of RCT involve the uptake of HDL-C by the liver and its excretion in the bile. Cholesterol esters and free cholesterol in HDL can either be directly taken up by the liver via SR-BI or transferred to apoB containing lipoproteins in a process mediated by CETP (Cuchel et al. 2009). In the latter pathway, cholesterol esters are taken up by the liver via the LDL receptor and then hydrolyzed to free cholesterol which can be excreted directly into the bile or following conversion to bile acid.
2.2 Alterations of the Cholesterol Efflux Capacity of HDL in Cardiovascular Disease
A case-control study by Khera et al. showed that the cholesterol efflux capacity of apoB-depleted serum was inversely related to carotid IMT in healthy volunteers and to the likelihood of angiographic CAD, even after adjustment for HDL-C and apoA-I levels (Khera et al. 2011). In a separate study, enhanced cholesterol efflux activity from ABCA1-stimulated macrophages was associated with reduced risk of prevalent CAD in unadjusted models in two case-control cohorts: an angiographic cohort comprising stable subjects undergoing elective diagnostic coronary angiography and an outpatient cohort (Li et al. 2013). However, the inverse risk relationship remained significant after adjustment for traditional CAD risk factors only within the outpatient cohort (Li et al. 2013). Surprisingly, higher cholesterol efflux activity was associated with an increase in prospective 3-year risk of myocardial infarction/stroke and major adverse cardiovascular events. The authors further observed that the HDL fraction (1.063 < d < 1.21) contained only a minority (≈40 %) of [(14)C] cholesterol released, with the majority found within the lipoprotein particle-depleted fraction, where ≈60 % was recovered after apoA-I immunoprecipitation. This is in agreement with previous findings on plasmas from patients with apoA-I deficiency, such as Tangier disease and LCAT deficiency, whereby the cholesterol efflux capacity amounted to at least 40 and 20 % of total and apoB-depleted plasmas from healthy controls, respectively (von Eckardstein et al. 1995a). However, in these studies the considerable residual cholesterol efflux capacity of HDL-free or HDL-poor plasma was explained by the abundance of apoA-I-free HDL particles containing apoA-IV, apoA-II, or apoE rather than by albumin (Huang et al. 1994, 1995; von Eckardstein et al. 1995a, b).
Importantly, impaired cholesterol efflux capacity of HDL has been associated with structural changes of the HDL components. Two groups independently demonstrated that oxidative modification of apoA-I, particularly by oxidation of methionine, tyrosine, or tryptophan residues through the myeloperoxidase pathway, dramatically reduced the ability of apoA-I to promote cholesterol efflux through the ABCA1 pathway (Bergt et al. 2004; Huang et al. 2014; Shao et al. 2006, 2010). Both groups also demonstrated that apoA-I in patients with cardiovascular disease was oxidatively modified and defective in promoting cholesterol efflux from macrophages via ABCA1 (Bergt et al. 2004; Shao et al. 2012; Zheng et al. 2004).
Of note, the cholesterol efflux capacity as determined by the assay protocol of Khera et al. which is widely used after its prominent publications is potentially confounded by several HDL-independent factors: the 4-h long incubation with cells raises the possibility that serum components deregulate the activity of ABCA1 and ABCG1. For example, free fatty acids and cytokines are known to alter the expression of ABCA1 and ABCG1 on both the transcriptional and posttranslational level. The use of serum instead of plasma implies the activation of thrombin and other proteases which are known to degrade prebeta-HDL (Eckardstein 2012).
3 Effects of HDL on LDL Oxidation
3.1 Mechanisms Under Physiological Conditions
The antioxidative properties of HDL were first reported by Bowry (Bowry et al. 1992). It has been suggested that HDL limits the formation of oxidized LDL (oxLDL) and oxidation of LDL has been proposed for decades to be a key event in atherogenesis (Heinecke 1998; Witztum and Steinberg 1991). LDL accumulates in the subendothelial space where it is oxidized by different pathways such as lipoxygenase, myeloperoxidase, or NADPH oxidase pathways (Diaz et al. 1997). OxLDL induces the expression of various proinflammatory cytokines, chemokines, and adhesion molecules (Navab et al. 1996). In addition, it promotes monocyte chemotaxis, their differentiation into macrophages, and the subsequent oxLDL uptake that convert them into foam cells, a hallmark of atherosclerotic plaques (Chisolm et al. 1999).
HDL is a major carrier of lipid oxidation products and specifically apoA-I, the main protein constituent of HDL, binds to and removes lipid hydroperoxides of LDL in vitro and in vivo (Navab et al. 2000b). In addition, apoA-I may directly reduce cholesteryl ester hydroperoxides and phosphatidylcholine hydroperoxides by oxidation of specific Met residues in the protein (Garner et al. 1998). Interestingly, the resulting oxidized HDL is more rapidly and selectively removed by hepatocytes than native HDL (Garner et al. 1998). Treatment of human artery wall cells with apoA-I but not apoA-II, or by addition of native HDL, apoA-I peptide mimetics, or paraoxonase, prevents the cells from oxidizing LDL in vitro (Navab et al. 2000a). The antioxidative function of apoA-I was corroborated by the finding that recombinant HDL containing only apoA-I and 1-palmitoyl-2-oleoyl-phosphatidylcholine (POPC) was as effective as native HDL in preventing LDL oxidation. In addition, in vivo studies have demonstrated that apoA-I can act as an antioxidative, anti-inflammatory, and anti-atherosclerotic agent (Nicholls et al. 2005a, b; Paszty et al. 1994). This further underlines a key antioxidant role for the HDL major protein apoA-I (Zerrad-Saadi et al. 2009).
Several other HDL-associated apolipoproteins have also been shown to contribute to its antioxidant effects. ApoE has been demonstrated to show isoform-dependent antioxidant activity (Miyata and Smith 1996). ApoE2 stimulates endothelial NO release and exerts anti-inflammatory effects (Sacre et al. 2003). In contrast, apoE4 has been suggested to be proinflammatory (Ophir et al. 2005). Another HDL-associated protein, apoA-IV, showed anti-atherosclerotic, anti-inflammatory, and antioxidant actions in vivo (Ostos et al. 2001; Recalde et al. 2004; Vowinkel et al. 2004). In addition, apoJ, also called clusterin, has been reported to block LDL oxidation by artery wall cells (Navab et al. 1997).
Furthermore, ApoA-II-enriched HDL isolated from transgenic human apoA-II mice protected VLDL from oxidation more efficiently than control HDL (Boisfer et al. 2002). In contrast, in other studies, dyslipidemic mice overexpressing human apoA-II showed accelerated atherosclerosis and reduced antioxidative activity of HDL, which may be attributed to the displacement of apoA-I and PON1 by apoA-II in HDL particles (Ribas et al. 2004; Rotllan et al. 2005). Notably, in apparently healthy subjects from the prospective EPIC-Norfolk (European Prospective Investigation into Cancer and Nutrition-Norfolk) cohort, apoA-II was found to be associated with a decreased risk of future CAD in a nested case-control study (Birjmohun et al. 2007).
Importantly, HDL carries antioxidant enzymes that have the capacity to prevent lipid oxidation or degrade lipid hydroperoxides such as serum paraoxonase/arylesterase 1 (PON1), lecithin/cholesterol acyltransferase (LCAT), and platelet-activating factor acetylhydrolase (PAF-AH). Specifically PON1 has been suggested to be an important regulator of the potential antiatherogenic capacity of HDL (Shih et al. 1998; Tward et al. 2002). It is secreted from the liver and associates with HDL in the plasma. Many studies revealed that PON1 degrades oxidized proinflammatory lipids, measured as a reduction in lipid peroxides (Mackness et al. 1991; Shih et al. 2000; Watson et al. 1995). Higher PON1 activity has been reported to be associated with a lower incidence of major cardiovascular events in human. Conversely, reduced activity of PON1 has been associated with pathological conditions such as diabetes, chronic renal failure rheumatoid arthritis, and various dementia (Soran et al. 2009).
LCAT is another HDL-associated enzyme with antioxidative properties. The antioxidant role of LCAT is thought to be its capacity to hydrolyze oxidized acyl chains from phosphatidylcholine-based oxidized phospholipids and oxidized free fatty acids (Goyal et al. 1997; Subramanian et al. 1999). Genetics and biochemical characterizations of HDLs from patients with low HDL disorders demonstrated that HDL-LCAT activity was reduced in all LCAT mutation carriers as well as in patients with HDL deficiency due to mutation in APOA1 or ABCA1 (Soran et al. 2009; von Eckardstein et al. 1995a). In vivo study in mice deficient for LDL receptor and leptin revealed that LCAT overexpression decreased autoantibodies to oxLDL (Mertens et al. 2003). Besides LCAT and PON1, PAF-AH, another HDL-associated enzyme, nowadays also known as lipoprotein-associated phospholipase A2 (LpPLA2), is able to hydrolyze oxidized phospholipids (Marathe et al. 2003; Noto et al. 2003). The local expression of PAF-AH in arteries of non-hyperlipidemic rabbits reduced the accumulation of oxidatively modified LDL without changing plasma levels of PAF-AH and reduced the expression of endothelial cell adhesion molecules (Arakawa et al. 2005). PAF-AH deficiency caused by a missense mutation in the gene has been indicated to be an independent risk factor for CAD in Japanese men (McIntyre et al. 2009). In contrast to this both activity and mass concentration of LpPLA2 in total serum has been associated with increased rather than decreased cardiovascular risk (Lp et al. 2010). However, recently HDL-associated LpPLA2 was found to be inversely related to risk of coronary events in stable CAD patients (Rallidis et al. 2012). Hence, the lipoprotein distribution of LpPLA2 may determine the pro- or antiatherogenic role of PAF-AH/LpPLA2. Furthermore, plasma levels of PAF-AH are also shown to be an independent risk marker of CAD (Garza et al. 2007). However, a study by Holleboom et al. showed that the reduction in LCAT and PAF-AH activites due to LCAT mutations was not associated with increased plasma lipid peroxidation (Holleboom et al. 2012).
3.2 Impairment of the Anti-Oxidative Effects of HDL in Patients After Surgery and With Cardiovascular Disease
In the acute phase response to surgery, van Lenten et al. demonstrated that the serum amyloid A (SAA) levels in HDL was increased and the activities of PON1 and PAF-AH were reduced (Van Lenten et al. 1995). Concomitant with these changes the anti-inflammatory capacity of HDL was lost in both humans and rabbits (Van Lenten et al. 1995). Isolated HDL from patients before and immediately after surgery were compared for the effects of HDL on LDL-induced monocyte transmigration and lipid hydroperoxide formation (Van Lenten et al. 1995). Prior to surgery, HDL completely inhibited the LDL-induced increase in monocyte transmigration and lipid hydroperoxide formation. In marked contrast, the “acute phase” HDL obtained from the same patients 2–3 days after surgery revealed a significant LDL-induced monocyte transmigration and was less effective in inhibiting lipid hydroperoxide formation. In other words, HDL in the same patient had been transformed from anti-inflammatory towards proinflammatory particles (Van Lenten et al. 1995). Similarly findings were reported in mice infected with influenza A or Chlamydia pneumonia. HDL isolated from virus-infected wild-type mice had impaired capacity to block LDL oxidation as well as LDL-induced monocyte chemotactic activity in human artery wall cell co-cultures (Van Lenten et al. 2001). The mice infected with C. pneumoniae had decreased PON1 activity and reduced HDL capacity to prevent LDL oxidation at days 2 and 3 postinfection (Campbell et al. 2010). Of note, studies by Navab et al. showed that HDL isolated from patients with CAD failed to prevent LDL oxidation by human artery wall cells (Navab et al. 2000a) and had impaired capacity to inhibit LDL-induced monocyte chemotactiy activity (Navab et al. 2001b). Similar results were obtained with HDL isolated from mice genetically predisposed to diet-induced atherosclerosis; it became proinflammatory when the mice were fed an atherogenic diet (Navab et al. 2001a). A subsequent small study by Ansell et al. suggested that the capacity of HDL to alter LDL-induced monocyte chemotactic activity in patients with CAD was somewhat improved after 6 weeks of simvastatin therapy (Ansell et al. 2003). However, HDL from patients with CAD on statin therapy remained proinflammatory when compared to HDL from healthy subjects, despite a significant improvement in plasma lipid levels.
Interestingly, impaired antioxidant properties and increased oxidized fatty acids content were found in HDL from patients with type 2 diabetes (Morgantini et al. 2011). The authors speculated that the elevated levels of oxidized fatty acids in HDL from these patients may account for the impaired antioxidant properties of the lipoprotein (Morgantini et al. 2011). Moreover, in a different study, the ability of HDL to prevent LDL oxidation was found to be reduced in patients with acute coronary syndrome but not in patients with stable coronary artery syndrome (Patel et al. 2011). The antioxidative and cholesterol efflux capacities of HDL were also found to be reduced in ischemic cardiomyopathy (Patel et al. 2013).
4 Effects of HDL on Endothelial Nitric Oxide Bioavailability
4.1 Mechanisms Under Physiological Conditions
Endothelial nitric oxide plays a crucial role in the regulation of vascular tone, platelet aggregation, and angiogenesis. Endothelial nitric oxide synthase (eNOS)-derived nitric oxide (NO) has been shown to exert a variety of atheroprotective effects in the vasculature, such as anti-inflammatory and antithrombotic effects (Landmesser et al. 2004). Consequently, reduced endothelial NO bioavailability has been proposed to promote initiation and progression of atherosclerosis (Landmesser et al. 2004).
It was shown that HDL can directly stimulate eNOS-mediated NO production (Yuhanna et al. 2001). There have been several studies that consistently demonstrated the capacity of HDL to modulate eNOS expression and to stimulate endothelial NO production in vitro and in vivo (Besler et al. 2011; Kuvin et al. 2002; Mineo et al. 2003; Nofer et al. 2004; Ramet et al. 2003; Sorrentino et al. 2010). Furthermore, administration of reconstituted HDL (rHDL) has been shown to improve endothelial function in patients with hypercholesterolemia or in subjects with isolated low HDL due to heterozygous loss-of-function mutations in the ABCA1 gene locus (Bisoendial et al. 2003; Spieker et al. 2002). Several mechanisms have been proposed to account for the endothelial NO-stimulating capacity of HDL. Early studies have demonstrated that HDL prevents oxLDL-mediated eNOS displacements from caveolae and restores enzyme stimulation (Uittenbogaard et al. 2000). Yuhanna et al. demonstrated that HDL binding to endothelial SR-BI is required for eNOS activation (Yuhanna et al. 2001). This binding activates the phosphatidylinositol-3-kinase/Akt signaling pathway and the MAP kinase/extracellular signal-regulated kinase pathway (Mineo et al. 2003). Activation of endothelial Akt by HDL stimulates phosphorylation of eNOS at serine residue 1177 (Mineo et al. 2003; Nofer et al. 2004), which is known to be an important regulatory mechanism leading to eNOS activation (Dimmeler et al. 1999).
There are many different components of HDL known to play a role in the endothelial NO-stimulating capacity. The potential interaction of apoA-I with eNOS in cultured endothelial cells has been previously reported (Ramet et al. 2003). However, lipid-free apoA-I failed to activate eNOS despite being the ligand for SR-BI, suggesting that other HDL components may be important or are required to support the conformation of apoA-I to allow its interaction with SR-BI and to stimulate eNOS (de Beer et al. 2001). In isolated endothelial cell plasma membranes, anti-apoA-I antibody blocks eNOS activation by HDL in vitro (Yuhanna et al. 2001). In contrast anti-apoA-II antibody further enhances eNOS stimulation by HDL (Yuhanna et al. 2001). Furthermore, lysophospholipids may play a role in eNOS activation as demonstrated in several studies. A study by Nofer et al. suggested that HDL-associated sphingolipids such as sphingosylphosphorylcholine, sphingosine-1-phosphate, and lysosulfatide caused eNOS-dependent relaxation of precontracted aortic rings from mice by binding to the lysophospholipid receptor S1P3 expressed in endothelial cells (Nofer et al. 2004). However, the vasodilatory response of HDL was not completely abolished in S1P3-deficient mice (Nofer et al. 2004).
Recently, we demonstrated that the HDL-associated enzyme PON1 is participating in HDL’s capacity to stimulate endothelial NO production and to exert NO-dependent endothelial atheroprotective effects (Besler et al. 2011). Inhibition of PON1 in HDL from healthy subjects impaired the capacity of HDL to stimulate endothelial NO production in human aortic endothelial cells. In addition HDL isolated from PON1-deficient mice failed to stimulate NO production in mouse aortic endothelial cells (Besler et al. 2011). Furthermore, inhibition of eNOS-mediated NO production by the pharmacological inhibitor L-NAME prevented the inhibitory effects of HDL from healthy subjects on nuclear factor κB (NF-κB) activity, vascular cell adhesion molecule (VCAM)-1 expression, and endothelial monocyte adhesion. These clearly indicate that the capacity of HDL to stimulate endothelial NO production is important for these endothelial anti-inflammatory effects of HDL (Besler et al. 2011).
4.2 Impaired HDL Capacity to Stimulate NO Production in Patients with Cardiovascular Disease
We and others have recently shown that HDL isolated from patients with stable CAD, diabetes, chronic kidney diseases (CKD), or acute coronary artery syndrome (ACS) displays altered endothelial effects when compared to HDL from healthy subjects. Of note, HDL from patients with diabetes in contrast to HDL from healthy subjects failed to stimulate endothelial cell NO production and to promote endothelial repair in a carotid artery injury model in mice (Sorrentino et al. 2010). Moreover, HDL from patients with either stable CAD or an ACS, in contrast to HDL from age- and gender-matched healthy subjects, inhibited endothelial cell NO production and lost the capacity to limit endothelial inflammatory activation as well as to promote endothelial repair in vivo (Besler et al. 2011). Interestingly, the capacity to stimulate endothelial NO production could be improved upon exercise training (Adams et al. 2013).
HDL from patients with CAD and chronic heart failure (CHF) showed an elevated malondialdehyde (MDA) content as compared to HDL from healthy subjects, which may contribute to the impaired endothelial NO production (Besler et al. 2011). The MDA-lysine adducts in HDL was determined to act via lectin-type oxidized LDL receptor 1 (LOX-1) activation of protein kinase C-βII, which blocks Akt-activating phosphorylation at Ser473 and eNOS-activating phosphorylation at Ser1177. Furthermore, inactivation of PON1 in HDL from healthy subjects resulted in attenuated NO production, in greater protein kinase C-βII activation, decreased activating eNOS-Ser1177 phosphorylation, and increased inactivating eNOS-Thr495 phosphorylation. Furthermore, HDL isolated from PON1-deficient mice failed to stimulate endothelial cell NO production (Besler et al. 2011). These observations likely suggest that alterations of HDL-associated PON1 may severely affect endothelial effects of HDL.
Very recently, we have also shown that MPO and PON1 reciprocally modulate each other’s function in vivo (Huang et al. 2013). MPO may promote site-specific oxidative modification of PON1, therefore limiting its activity. In addition, HDL isolated from patients with an ACS carries enhanced chlorotyrosine content, site-specific PON1 methionine oxidation, and reduced PON1 activity (Huang et al. 2013).
Several groups have reported an inverse relationship between PON1 serum activity and cardiovascular events (Bhattacharyya et al. 2008; Regieli et al. 2009). Interestingly, analysis of SNPs for PON1 identified in genome wide association studies did not reveal a significant association between SNPs, associated with mildly reduced PON1 activity, and the risk of cardiovascular events (Tang et al. 2012). The interpretation of PON1 activity is difficult because it is not known to which extent the paraoxonase and arylesterase activities represent biologically relevant functions. Furthermore, we and others observed important posttranslational modifications of the proteins, which could explain alterations of biological properties of the enzymes (Aviram et al. 1999; Besler et al. 2011; Huang et al. 2013).
5 Endothelial Anti-Inflammatory Effects of HDL
5.1 Mechanisms Under Physiological Conditions
Atherosclerosis is a chronic inflammatory disorder. The cellular inflammatory response is triggered by modified LDL accumulated in the subendothelium and the induction of proinflammatory cytokines and adhesion protein expression. As a consequence monocytes/macrophages infiltrate and accumulate in the arterial wall and express scavenger receptors and toll-like receptors to sequester cholesterol. The cholesterol-loaded macrophages together with T lymphocytes produce a wide array of proinflammatory cytokines and accelerate plague progression (Hansson 2005).
In vitro, HDL has been shown to inhibit the expression of monocyte chemoattractant protein (MCP)-1, an important proinflammatory chemokine in endothelial cells (Mackness et al. 2004; Navab et al. 1991). Furthermore, the potential anti-inflammatory effects of HDL have been demonstrated by several in vivo studies. Infusion of native HDL as well as reconstituted HDL containing apoA-I or apoA-I Milano suppressed cytokine and chemokine expression in animal models of inflammation (Calabresi et al. 1997; Cockerill et al. 1995). Reduced VCAM-1 expression and decreased monocyte/macrophage infiltration were reported after carotid artery cuff injury in apoE-deficient mice (Dimayuga et al. 1999). In the streptozotocin-induced diabetic cardiomyopathy rat model, human apoA-I gene transfer increased HDL-C plasma levels and blocked the diabetes-induced myocardial mRNA expression of VCAM-1 and ICAM-1 (Van Linthout et al. 2008). In contrast, overexpression of human apoA-I in apoE-deficient mice did not result in any change of endothelial VCAM-1 expression and monocyte adherence in early atherosclerotic lesions at the aortic branch sites, despite overall reduction in aortic atherosclerotic lesion formation (Dansky et al. 1999). These studies support the concept that the anti-inflammatory capacity of HDL is heterogeneous, and is dependent on the pathophysiological conditions. In vitro studies showed variations in the capacity of HDL isolated from different human subjects to reduce TNF-α stimulated endothelial VCAM-1 expression (Ashby et al. 1998; Van Lenten et al. 1995). In human studies, the administration of reconstituted HDL increased the anti-inflammatory capacity of HDL from patients with type-2 diabetes (Patel et al. 2009).
One of the proposed mechanisms for the anti-inflammatory effect of HDL is the removal of cholesterol from cell membranes of macrophages and also endothelial cells. The cholesterol depletion and consequently the lipid rafts disruption may downregulate signaling pathways and interfere with the antigen presentation or the expression of toll-like receptor (Anderson et al. 2000; Norata and Catapano 2012; Wang et al. 2012). Additional studies suggest that HDL and apoA-I are able to inhibit the capacity of antigen-presenting cells to stimulate T-cell activation. This inhibition was attributed to cholesterol efflux through ABCA1, an ATP-binding transporter (Tang et al. 2009; Zhu et al. 2010).
The anti-inflammatory capacity of HDL has been mainly attributed to apoA-I, the major protein constituent of HDL. In an in vivo study, apoA-I infusion in rabbits in acute vascular inflammation model reduced neutrophil infiltration and endothelial cell inflammatory activation (Puranik et al. 2008). In addition, apoA-1 mimetic peptides have been shown to reduce vascular inflammation in type I diabetic rats and improve insulin sensitivity in obese mice (Peterson et al. 2007, 2008). It has also been demonstrated that treatment with lipid-free apoA-I and rHDL treatment reduced the expression of chemokines and chemokine receptors in vitro and in vivo by modulating NF-κB and peroxisome proliferator-activated receptor γ (Bursill et al. 2010). Interestingly, apoA-I has also been shown to inhibit palmitate-induced NF-κB activation by reducing Toll-like receptor-4 recruitment into lipid rafts (Cheng et al. 2012). More recently, De Nardo et al. using a systems biology approach identified the transcription factor ATF3 as an HDL-inducible gene to suppress Toll-like receptor-induced proinflammatory cytokines. Mice deficient in ATF3 and apoE revealed enhanced atherosclerotic lesions, and administration of rHDL into apoE-deficient mice resulted in ATF3 upregulation. The authors used rHDL in their study, suggesting that apoA-I is a key mechanistic player, and of note, the effects seem to be unrelated to cholesterol transport (De Nardo et al. 2014).
The lipid moiety of HDL has also been proposed to be important for the anti-inflammatory effects of HDL. In vitro studies using reconstituted HDL containing only apoA-I with a few phospholipid molecules suggested that inhibitory effects of HDL on endothelial cell adhesion molecule expression are also, at least in part, dependent on HDL-associated phospholipid species (Baker et al. 2000). The inhibition of cytokine-induced expression of VCAM-1 by reconstituted HDL varied when different phosphatidylcholine species were used. This suggests that the lipid composition of HDL influences its anti-inflammatory capacity and is likely an important determinant of HDL functionality (Baker et al. 2000; Barter et al. 2004). In addition there are multiple reports suggesting the involvement of sphingosine-1-phosphate (S1P), a biologically active sphingolipid that plays key functions in the immune, inflammatory, and cardiovascular systems. S1P carried by HDL is believed to regulate arterial tone, vascular permeability, and tissue perfusion. During inflammation it induces endothelial adhesion molecules and recruits inflammatory cells, and furthermore it activates a negative feedback loop that consecutively decreases vascular leakage by improving the endothelial barrier function and preventing cytokine-induced leukocyte adhesion (Garcia et al. 2001; Lucke and Levkau 2010; Takeya et al. 2003).
5.2 Impaired Endothelial Anti-Inflammatory Effects of HDL in Patients with CAD, Diabetes, or Chronic Kidney Dysfunction
HDL isolated from patients with inflammatory disease such as CAD, diabetes, or chronic kidney disease display reduced eNOS activation and impaired endothelial repair capacity in a carotid artery injury mouse model. There have been various mechanisms proposed to account for the impaired endothelial anti-inflammatory effects of HDL. Reduced HDL apoA-I levels in inflammatory states has been related to accelerated HDL catabolism and apoA-I substitution in HDL particles by serum amyloid A (SAA) (Esteve et al. 2005; Khovidhunkit et al. 2004). In acute phase, SAA is able to replace apoA-I in HDL, resulting in reduced plasma levels of apoA-I (Parks and Rudel 1985). ApoA-I can be completely replaced by SAA in rabbits and mice, in a subset of small, dense HDL particles and therefore functioning as a structural apolipoprotein (Cabana et al. 1999). HDL isolated from patients with chronic kidney disease is enriched in SAA, which correlated with its reduced anti-inflammatory capacity to inhibit monocyte chemoattractant protein-1 formation in vascular smooth muscle cells (Tolle et al. 2012).
Moreover, HDL carries potential cardioprotective molecules that are significantly altered in compositions in patients with CAD or ACS (Riwanto et al. 2013; Vaisar et al. 2007). Oxidative modifications of HDL, in particular the amino acid residues in apoA-I, have been shown to contribute to the generation of dysfunctional HDL (Bergt et al. 2004; Shao et al. 2006, 2012; Zheng et al. 2004). In vitro study has demonstrated that MPO-catalyzed oxidative modification of HDL or apoA-I converts HDL into a proinflammatory particle which promotes NF-κB activation and endothelial VCAM-1 expression (Undurti et al. 2009). Furthermore, glycation of HDL and apoA-I, a process that is known to occur in diabetes in vivo (Curtiss and Witztum 1985), has been shown to impair the anti-inflammatory capacity of HDL (Nobecourt et al. 2010). Infusion of glycated lipid-free apoA-I failed to decrease adhesion molecule expression following vascular injury (Nobecourt et al. 2010). Moreover, glycation of HDL has also been shown to inhibit the HDL capacity to inhibit oxLDL-induced monocyte adhesion to human aortic endothelial cells in vitro (Hedrick et al. 2000).
6 Effects of HDL on Endothelial Cell Apoptotic Pathways
6.1 Mechanisms Under Physiological Conditions
Endothelial cell dysfunction and injury has been associated with the pathogenesis of atherosclerosis (Landmesser et al. 2004; Nabel and Braunwald 2012; Ross 1999). Studies in pigs have shown that atherosclerotic lesion-prone regions are characterized by a high endothelial cell turnover (Caplan and Schwartz 1973), which has been suggested to be due to increased endothelial cell apoptosis (Caplan and Schwartz 1973). Several studies have indicated an important contribution of endothelial cell apoptosis to the pathophysiology of CAD (Burke et al. 1997; Rossig et al. 2001). Furthermore, it is evident that apoptosis of endothelial cells and smooth muscle cells is detrimental to plaque stability (Bombeli et al. 1997; Kockx and Herman 2000). In the normal arterial wall, endothelial cells may protect themselves against apoptosis using NO-dependent mechanisms. The situation is completely different in atherosclerotic plaques, in a high oxidative stress environment where macrophages produce high amounts of nitric oxide or peroxynitrite that could induce apoptotic cell death (Kockx and Herman 2000). In the aorta the capacity of HDL to quench endothelial cell apoptosis may therefore represent an antiatherogenic property of HDL (de Souza et al. 2010; Nofer et al. 2001; Suc et al. 1997; Sugano et al. 2000).
HDL has been shown to attenuate endothelial cell apoptosis induced by different stimuli such as TNF-α, oxLDL, and growth factor deprivation (de Souza et al. 2010; Nofer et al. 2001; Suc et al. 1997; Sugano et al. 2000). It has been suggested that HDL may inhibit both death-receptor and mitochondrial-mediated apoptotic pathways. Importantly, both the protein and lipid moieties have been reported to contribute to the antiapoptotic capacity of HDL. ApoA-I has been shown to inhibit endothelial cell apoptosis induced by oxLDL, VLDL, and TNF-α (Speidel et al. 1990; Suc et al. 1997; Sugano et al. 2000). In a more detailed study, HDL subpopulations enriched with apoA-I account for approximately 70 % of the antiapoptotic activity of HDL in a cell culture model with human microvascular endothelial cells that were treated with mildly oxidized LDL. Using rHDL consisting of apoA-I, cholesterol and phospholipids potently inhibited oxLDL-induced apoptosis in these cells (de Souza et al. 2010). This suggests that apoA-I plays an important role for the antiapoptotic capacity of HDL in oxLDL-stimulated endothelial cells.
HDL-associated lysosphingolipids have been identified to inhibit endothelial cell apoptosis induced by growth factor deprivation (Kimura et al. 2001, 2003; Nofer et al. 2001). The antiapoptotic capacity of the lipid moiety of HDL was further supported by the findings that the ratio of sphingosine-1-phosphate and sphingomyelin in small dense HDL3 particles was increased and correlated positively with the characteristics of these HDL subpopulations to inhibit endothelial cell apoptosis (Kontush et al. 2007).
Several mechanisms have been suggested for the endothelial antiapoptotic effects of HDL, depending on the stimulus of apoptosis. Endothelial cell death triggered by oxLDL causes a delayed but sustained increase in intracellular calcium which is shown to be prevented by HDL (Suc et al. 1997). HDL inhibits tumor necrosis factor-α-induced endothelial cell death by association as well as suppression of caspase-3, which is a key factor of all primary apoptotic pathways (Sugano et al. 2000). Nofer et al. suggested that sphingosine-1-phosphate (S1P) carried by HDL prevents the growth factor deprived activation of the intrinsic pathway to cell death (Nofer et al. 2001). Of note, lysophospholipids have been shown to enhance endothelial cell survival, but the effects were inhibited by S1P receptor knockdown, as well as by the presence of phosphoinositide 3 (PI3) kinase and Erk pathway antagonists (Kimura et al. 2003).
In addition, HDL activates Akt and causes phosphorylation of the Akt target Bcl-2-associated death promoter (BAD), preventing it from binding to the antiapoptotic protein Bcl-xL (Nofer et al. 2001). HDL also causes phosphoinositide 3 (PI3) kinase-mediated upregulation of the antiapoptotic Bcl-2 family protein Bcl-xL expression (Riwanto et al. 2013). Interestingly, HDL retained its antiapoptotic activity after knockdown of eNOS using specific RNA interference or pharmacological inhibition using L-NAME (Riwanto et al. 2013), indicating that HDL may exert its antiapoptotic activity independently of eNOS activation.
6.2 Impairment of the Endothelial Anti-Apoptotic Effects of HDL in Patients with Cardiovascular Disease
Recently, we reported that HDL isolated from patients with stable CAD or ACS, in contrast to HDL from healthy subjects, failed to inhibit endothelial cell apoptosis in vitro and in apoE-deficient-mice in vivo. Using proteomic analysis of HDL, we observed reduced clusterin and increased apo C-III content in HDL isolated from patients with CAD. Both, supplementing HDL from healthy subjects with apoC-III or pretreatment of healthy HDL with an antibody against clusterin, lead to the activation of proapoptotic signaling pathways in endothelial cells (Riwanto et al. 2013). HDL isolated from these patients stimulated endothelial proapoptotic pathways, in particular p38-MAPK-mediated activation of the proapoptotic Bcl-2-protein tBid. Our study further suggests that differences in the proteome of HDL from patients with CAD, in particular reduced HDL-associated clusterin and increased HDL-associated apoC-III, play an important role for altered activation of endothelial anti- and proapoptotic signaling pathways (Riwanto et al. 2013). In line with this, a nested case-control study found that cholesterol levels in apoC-III-containing HDL particles show a positive association with cardiovascular risk, whereas both total HDL-C and cholesterol in apoC-III-free HDL particles show the expected inverse associations (Jensen et al. 2012). In addition, Undurti et al. demonstrated that MPO-catalyzed oxidation of HDL impaired its capacity to inhibit endothelial apoptosis in vitro (Undurti et al. 2009).
7 HDL Protein Cargo and Prognostic Biomarkers
As highlighted above, alterations in HDL functions are likely, at least in part, attributed to changes in the composition of HDL particles. HDL particles are highly heterogenous in their structure and compositions. They undergo continuous remodeling through apolipoprotein exchanges with other circulating lipoproteins and tissues (Lund-Katz et al. 2003; Saito et al. 2004).
An early study has shown that acute phase response causes significant changes in the HDL-associated proteins and apolipoprotein composition, i.e., enrichment in C-reactive protein (CRP), secretory phospholipase A2-IIa (sPLA2-IIa), serum amyloid A (SAA), and cholesterol ester transfer protein (CETP) (Coetzee et al. 1986). It has been suggested that such remodeling may compromise the antiatherogenic functions of HDL (Jahangiri et al. 2009). These observations have prompted explorative studies into HDL composition. Evaluations of the HDL proteome have been reported with one- or two-dimensional electrophoresis combined with high performance liquid chromatography (HPLC) and tandem mass spectrometry (MS/MS) or matrix-assisted laser desorption/ionization-time flight mass spectrometry (MALDI-TOF-MS) (Heller et al. 2007; Karlsson et al. 2005). More recent studies have used shotgun proteomics, through label-free MS techniques, such as spectral counting and XIC inspection, to analyze HDL protein compositions (Riwanto et al. 2013; Vaisar et al. 2007). Overall, the identification techniques currently available for the analysis of the HDL proteome are semi-quantitative.
Given the complexity of HDL and its multitude of biological functions, it is conceivable to expect that sets of functionally associated proteins can provide information about their participation in the spectrum of atheroprotective actions attributed to HDL. This has further led to studies comparing the HDL proteome of healthy subjects with that of particles from patients with dyslipidemias or CAD (Green et al. 2008; Heller et al. 2007; Riwanto et al. 2013; Vaisar et al. 2007). In the study by Vaisar et al., HDL was shown to carry apolipoproteins and proteins with functions in lipid metabolism, the acute phase response, complement regulation, and blood coagulation (Vaisar et al. 2007). The same group further compared the proteome of HDL3 from 6 CAD patients before and after 1-year treatment with combined statin/niacin therapy (Green et al. 2008). In this study, HDL3 of CAD patients is significantly enriched in apoE and apoCII and carry less apoJ and phospholipid transport protein (PLTP) as compared to HDL from control subjects. Treatment with niacin/statin decreases HDL3 apoE and raises apoJ and PLTP levels (Green et al. 2008). In our recent study, we have observed that reduced clusterin and increased apoC-III content in HDL isolated from patients with CAD lead to activation of proapoptotic signaling pathways in endothelial cells (Riwanto et al. 2013).
Of note, oxidative modifications of HDL-associated proteins have also been demonstrated to contribute to the functional impairment of HDL. In particular, MPO-mediated oxidation of apoA-I has been linked to the generation of dysfunctional forms of HDL (Huang et al. 2014; Shao et al. 2010; Undurti et al. 2009; Wang et al. 2007b; Zheng et al. 2004). Recently, we have shown that MPO and PON1 interact and reciprocally modulate each other’s function in vivo, i.e., PON1 partially inhibits MPO activity, while MPO oxidizes and inactivates PON1 (Huang et al. 2013). Furthermore, HDL isolated from patients with an ACS carries enhanced chlorotyrosine content, site-specific PON1 methionine oxidation, and reduced PON1 activity (Huang et al. 2013).
Taken together, these observations indicate that the HDL proteomic composition may be altered in different disease states which likely have an impact on HDL function. Unraveling the complexities of the HDL through proteomics studies may therefore allow for better understanding of the HDL composition and function. However, proteomics studies of HDL have been limited in size and number due to the labor-intensive nature of the approach. Furthermore, given the diverse concentration range of the proteins in the HDL particles, some proteomics approaches may fail to reflect the actual amount or alteration of low abundant proteins due to signal suppression in the presence of highly abundant proteins. Targeted proteomics using selected reaction monitoring is a powerful tandem mass spectrometry method that can be used to monitor target peptides within a complex protein digest (Picotti and Aebersold 2012; Picotti et al. 2010). The approach will enable absolute quantification of the individual protein contained within HDL particles.
Conclusion and Perspectives
Over the past two decades, many studies have emerged showing various antiatherogenic effects of HDL, including the capacity of HDL to mediate reverse cholesterol transport, and antioxidative, anti-inflammatory, and antiapoptotic effects (Riwanto and Landmesser 2013). Growing evidence has shown that HDL particles are highly heterogeneous and the vasoprotective effects of HDL are altered in patients with CAD, diabetes, and chronic kidney dysfunction, i.e., patients with a high cardiovascular risk profile. Of note, these findings have more recently been supported by observations in cohort studies, suggesting that in patients with advanced CAD, higher plasma levels of HDL-C are no longer associated with reduced risk of cardiovascular events (Angeloni et al. 2013; Schwartz et al. 2012). Importantly, the increase in HDL-C observed with some lipid-modifying drugs has not been uniformly associated with clinical benefit (Barter et al. 2007; Schwartz et al. 2012). These findings have clearly indicated that the association between HDL and cardiovascular disease is far more complex than previously thought and is likely mediated by different HDL functional properties, independent of the cholesterol component of the HDL particle (Heinecke 2011).
It is increasingly evident that the functional heterogeneity of HDL may be attributed to the complexity of the HDL particles, containing proteins and lipids that can be modified or altered in their composition. Of note, rapid advances in the MS proteome technology have allowed tremendous leaps in characterizing the HDL proteome. Studies investigating the lipidome of HDL are still somewhat limited by the available technologies. Importantly, several studies have associated changes in specific HDL protein either due to altered composition or modification to the loss of function of HDL (Besler et al. 2011; Huang et al. 2013; Riwanto et al. 2013; Shao et al. 2006, 2012; Zheng et al. 2004). Nevertheless, it remains to be determined if the alterations are secondary to changes that occur during the disease progression or if the alteration has indeed a causal effect on disease etiology. In this context it is also important to emphasize the problem of confounding between different diseases: for example, many diabetic patients have (sub)clinical chronic kidney disease and/or CAD. As a consequence one must also expect considerable overlap of HDL modifications which were initially identified in patients with either diabetes, CAD, or CKD. It is hence rather unlikely that each disease has its specific footprint of HDL dysfunction and alterations in proteome and lipidome.
Taken together, it is becoming clear that plasma HDL-C levels are not an appropriate marker of vascular effects of high-density lipoproteins and therefore do not represent a reliable therapeutic target. Importantly, HDL-targeted treatment approaches need to take into account the altered vascular effects of HDL in cardiovascular disease. Therefore, targeting HDL-mediated antiatherogenic mechanisms, rather than plasma HDL-C levels, may represent a more promising and interesting therapeutic target. Studies looking into the specific mechanisms leading to a loss of antiatherogenic effects of HDL are of particular importance, in order to develop therapeutic approaches that aim to restore the vasoprotective properties of HDL.
Abbreviations
- ABCA1:
-
ATP-binding cassette transporter A1
- ABCG1:
-
ATP-binding cassette transporter G1
- ACS:
-
Acute coronary syndrome
- apoA-I:
-
Apolipoprotein A1
- apoA-II:
-
Apolipoprotein A2
- apoB:
-
Apolipoprotein B
- apoA-IV:
-
Apolipoprotein A-IV
- apoC-III:
-
Apolipoprotein C-III
- apoE:
-
Apolipoprotein E
- apoJ:
-
Apolipoprotein J
- Bad:
-
Bcl-2-associated death promoter
- Bcl-xL:
-
B-cell lymphoma-extra large
- Bid:
-
BH3 interacting domain death agonist
- CAD:
-
Coronary artery disease
- caspase:
-
Cysteine-aspartic proteases
- CETP:
-
Cholesteryl ester transfer protein
- CHF:
-
Chronic heart failure
- CKD:
-
Chronic kidney disease
- eNOS:
-
Endothelial nitric oxide synthase
- HDL:
-
High-density lipoprotein
- HDL-C:
-
High-density lipoprotein cholesterol
- HPLC:
-
High-performance liquid chromatography
- ICAM-1:
-
Intercellular Adhesion Molecule 1
- LCAT:
-
Lecithin cholesterol acyltransferase
- MS/MS:
-
Tandem mass spectrometry
- LDL:
-
Low density lipoprotein
- L-NAME:
-
L-NG-Nitroarginine Methyl Ester
- LOX-1:
-
Lectin-type oxidized LDL receptor 1
- LpPLA2:
-
Lipoprotein-associated phospholipase A2
- MALDI-TOF-MS:
-
Matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry
- MAPK:
-
Mitogen-activated protein kinase
- MCP:
-
Monocyte chemoattractant protein
- MDA:
-
Malondialdehyde
- MI:
-
Myocardial infarction
- MPO:
-
Myeloperoxidase
- NF-kB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NO:
-
Nitric oxide
- oxLDL:
-
Oxidized low density lipoprotein
- PAF-AH:
-
Platelet-activating factor acetylhydrolase
- PI3K:
-
Phosphoinositide-3-kinase
- PLTP:
-
Phospholipid transport protein
- PON1:
-
Paraoxonase 1
- POPC:
-
1-palmitoyl-2-oleoyl-phosphatidylcholine
- RCT:
-
Reverse cholesterol transport
- rHDL:
-
Reconstituted high-density lipoprotein
- S1P:
-
Sphingosine-1-phosphate
- SAA:
-
Serum amyloid A
- SNP:
-
Single nucleotide polymorphism
- sPLA2-IIa:
-
Secretory phospholipase A2-IIa
- SR-BI:
-
Scavenger receptor type B1
- TNF-α:
-
Tumor necrosis factor-alpha
- VCAM-1:
-
Vascular cell adhesion molecule 1
- VLDL:
-
Very low density lipoprotein
References
Adams V, Besler C, Fischer T, Riwanto M, Noack F, Hollriegel R, Oberbach A, Jehmlich N, Volker U, Winzer EB, Lenk K, Hambrecht R, Schuler G, Linke A, Landmesser U, Erbs S (2013) Exercise training in patients with chronic heart failure promotes restoration of high-density lipoprotein functional properties. Circ Res 113:1345–1355
Anderson HA, Hiltbold EM, Roche PA (2000) Concentration of MHC class II molecules in lipid rafts facilitates antigen presentation. Nat Immunol 1:156–162
Angeloni E, Paneni F, Landmesser U, Benedetto U, Melina G, Luscher TF, Volpe M, Sinatra R, Cosentino F (2013) Lack of protective role of HDL-C in patients with coronary artery disease undergoing elective coronary artery bypass grafting. Eur Heart J 34(46):3557–3562
Annema W, von Eckardstein A (2013) High-density lipoproteins. Multifunctional but vulnerable protections from atherosclerosis. Circ J 77:2432–2448
Ansell BJ, Navab M, Hama S, Kamranpour N, Fonarow G, Hough G, Rahmani S, Mottahedeh R, Dave R, Reddy ST, Fogelman AM (2003) Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation 108:2751–2756
Arakawa H, Qian JY, Baatar D, Karasawa K, Asada Y, Sasaguri Y, Miller ER, Witztum JL, Ueno H (2005) Local expression of platelet-activating factor-acetylhydrolase reduces accumulation of oxidized lipoproteins and inhibits inflammation, shear stress-induced thrombosis, and neointima formation in balloon-injured carotid arteries in nonhyperlipidemic rabbits. Circulation 111:3302–3309
Ashby DT, Rye KA, Clay MA, Vadas MA, Gamble JR, Barter PJ (1998) Factors influencing the ability of HDL to inhibit expression of vascular cell adhesion molecule-1 in endothelial cells. Arterioscler Thromb Vasc Biol 18:1450–1455
Aviram M, Rosenblat M, Billecke S, Erogul J, Sorenson R, Bisgaier CL, Newton RS, La Du B (1999) Human serum paraoxonase (PON 1) is inactivated by oxidized low density lipoprotein and preserved by antioxidants. Free Radic Biol Med 26:892–904
Baker PW, Rye KA, Gamble JR, Vadas MA, Barter PJ (2000) Phospholipid composition of reconstituted high density lipoproteins influences their ability to inhibit endothelial cell adhesion molecule expression. J Lipid Res 41:1261–1267
Barter PJ, Nicholls S, Rye KA, Anantharamaiah GM, Navab M, Fogelman AM (2004) Antiinflammatory properties of HDL. Circ Res 95:764–772
Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B (2007) Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 357:2109–2122
Bergt C, Pennathur S, Fu X, Byun J, O'Brien K, McDonald TO, Singh P, Anantharamaiah GM, Chait A, Brunzell J, Geary RL, Oram JF, Heinecke JW (2004) The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc Natl Acad Sci USA 101:13032–13037
Besler C, Heinrich K, Rohrer L, Doerries C, Riwanto M, Shih DM, Chroni A, Yonekawa K, Stein S, Schaefer N, Mueller M, Akhmedov A, Daniil G, Manes C, Templin C, Wyss C, Maier W, Tanner FC, Matter CM, Corti R, Furlong C, Lusis AJ, von Eckardstein A, Fogelman AM, Luscher TF, Landmesser U (2011) Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J Clin Invest 121:2693–2708
Bhattacharyya T, Nicholls SJ, Topol EJ, Zhang R, Yang X, Schmitt D, Fu X, Shao M, Brennan DM, Ellis SG, Brennan ML, Allayee H, Lusis AJ, Hazen SL (2008) Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA 299:1265–1276
Birjmohun RS, Dallinga-Thie GM, Kuivenhoven JA, Stroes ES, Otvos JD, Wareham NJ, Luben R, Kastelein JJ, Khaw KT, Boekholdt SM (2007) Apolipoprotein A-II is inversely associated with risk of future coronary artery disease. Circulation 116:2029–2035
Bisoendial RJ, Hovingh GK, Levels JH, Lerch PG, Andresen I, Hayden MR, Kastelein JJ, Stroes ES (2003) Restoration of endothelial function by increasing high-density lipoprotein in subjects with isolated low high-density lipoprotein. Circulation 107:2944–2948
Boisfer E, Stengel D, Pastier D, Laplaud PM, Dousset N, Ninio E, Kalopissis AD (2002) Antioxidant properties of HDL in transgenic mice overexpressing human apolipoprotein A-II. J Lipid Res 43:732–741
Bombeli T, Karsan A, Tait JF, Harlan JM (1997) Apoptotic vascular endothelial cells become procoagulant. Blood 89:2429–2442
Bortnick AE, Rothblat GH, Stoudt G, Hoppe KL, Royer LJ, McNeish J, Francone OL (2000) The correlation of ATP-binding cassette 1 mRNA levels with cholesterol efflux from various cell lines. J Biol Chem 275:28634–28640
Bowry VW, Stanley KK, Stocker R (1992) High density lipoprotein is the major carrier of lipid hydroperoxides in human blood plasma from fasting donors. Proc Natl Acad Sci USA 89:10316–10320
Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R (1997) Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med 336:1276–1282
Bursill CA, Castro ML, Beattie DT, Nakhla S, van der Vorst E, Heather AK, Barter PJ, Rye KA (2010) High-density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Arterioscler Thromb Vasc Biol 30:1773–1778
Cabana VG, Reardon CA, Wei B, Lukens JR, Getz GS (1999) SAA-only HDL formed during the acute phase response in apoA-I+/+and apoA-I-/- mice. J Lipid Res 40:1090–1103
Calabresi L, Franceschini G, Sirtori CR, De Palma A, Saresella M, Ferrante P, Taramelli D (1997) Inhibition of VCAM-1 expression in endothelial cells by reconstituted high density lipoproteins. Biochem Biophys Res Commun 238:61–65
Campbell LA, Yaraei K, Van Lenten B, Chait A, Blessing E, Kuo CC, Nosaka T, Ricks J, Rosenfeld ME (2010) The acute phase reactant response to respiratory infection with Chlamydia pneumoniae: implications for the pathogenesis of atherosclerosis. Microbes Infect 12:598–606
Caplan BA, Schwartz CJ (1973) Increased endothelial cell turnover in areas of in vivo Evans Blue uptake in the pig aorta. Atherosclerosis 17:401–417
Castelli WP, Garrison RJ, Wilson PW, Abbott RD, Kalousdian S, Kannel WB (1986) Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham study. JAMA 256:2835–2838
Charakida M, Besler C, Batuca JR, Sangle S, Marques S, Sousa M, Wang G, Tousoulis D, Delgado Alves J, Loukogeorgakis SP, Mackworth-Young C, D'Cruz D, Luscher T, Landmesser U, Deanfield JE (2009) Vascular abnormalities, paraoxonase activity, and dysfunctional HDL in primary antiphospholipid syndrome. JAMA 302:1210–1217
Cheng AM, Handa P, Tateya S, Schwartz J, Tang C, Mitra P, Oram JF, Chait A, Kim F (2012) Apolipoprotein A-I attenuates palmitate-mediated NF-kappaB activation by reducing Toll-like receptor-4 recruitment into lipid rafts. PLoS ONE 7:e33917
Chisolm GM 3rd, Hazen SL, Fox PL, Cathcart MK (1999) The oxidation of lipoproteins by monocytes-macrophages. Biochemical and biological mechanisms. J Biol Chem 274:25959–25962
Cockerill GW, Rye KA, Gamble JR, Vadas MA, Barter PJ (1995) High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler Thromb Vasc Biol 15:1987–1994
Coetzee GA, Strachan AF, van der Westhuyzen DR, Hoppe HC, Jeenah MS, de Beer FC (1986) Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J Biol Chem 261:9644–9651
Cuchel M, Lund-Katz S, de la Llera-Moya M, Millar JS, Chang D, Fuki I, Rothblat GH, Phillips MC, Rader DJ (2009) Pathways by which reconstituted high-density lipoprotein mobilizes free cholesterol from whole body and from macrophages. Arterioscler Thromb Vasc Biol 30:526–532
Cullen P, Schulte H, Assmann G (1997) The Munster heart study (PROCAM): total mortality in middle-aged men is increased at low total and LDL cholesterol concentrations in smokers but not in nonsmokers. Circulation 96:2128–2136
Curtiss LK, Witztum JL (1985) Plasma apolipoproteins AI, AII, B, CI, and E are glucosylated in hyperglycemic diabetic subjects. Diabetes 34:452–461
Dansky HM, Charlton SA, Barlow CB, Tamminen M, Smith JD, Frank JS, Breslow JL (1999) Apo A-I inhibits foam cell formation in Apo E-deficient mice after monocyte adherence to endothelium. J Clin Invest 104:31–39
de Beer MC, Durbin DM, Cai L, Jonas A, de Beer FC, van der Westhuyzen DR (2001) Apolipoprotein A-I conformation markedly influences HDL interaction with scavenger receptor BI. J Lipid Res 42:309–313
De Nardo D, Labzin LI, Kono H, Seki R, Schmidt SV, Beyer M, Xu D, Zimmer S, Lahrmann C, Schildberg FA, Vogelhuber J, Kraut M, Ulas T, Kerksiek A, Krebs W, Bode N, Grebe A, Fitzgerald ML, Hernandez NJ, Williams BR, Knolle P, Kneilling M, Rocken M, Lutjohann D, Wright SD, Schultze JL, Latz E (2014) High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat Immunol 15:152–160
de Souza JA, Vindis C, Hansel B, Negre-Salvayre A, Therond P, Serrano CV Jr, Chantepie S, Salvayre R, Bruckert E, Chapman MJ, Kontush A (2008) Metabolic syndrome features small, apolipoprotein A-I-poor, triglyceride-rich HDL3 particles with defective anti-apoptotic activity. Atherosclerosis 197:84–94
de Souza JA, Vindis C, Negre-Salvayre A, Rye KA, Couturier M, Therond P, Chantepie S, Salvayre R, Chapman MJ, Kontush A (2010) Small, dense HDL 3 particles attenuate apoptosis in endothelial cells: pivotal role of apolipoprotein A-I. J Cell Mol Med 14:608–620
Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, Collins R, Thompson SG, Danesh J (2009) Major lipids, apolipoproteins, and risk of vascular disease. JAMA 302:1993–2000
Diaz MN, Frei B, Vita JA, Keaney JF Jr (1997) Antioxidants and atherosclerotic heart disease. N Engl J Med 337:408–416
Dimayuga P, Zhu J, Oguchi S, Chyu KY, Xu XO, Yano J, Shah PK, Nilsson J, Cercek B (1999) Reconstituted HDL containing human apolipoprotein A-1 reduces VCAM-1 expression and neointima formation following periadventitial cuff-induced carotid injury in apoE null mice. Biochem Biophys Res Commun 264:465–468
Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM (1999) Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399:601–605
Eckardstein A (2012) Tachometer for reverse cholesterol transport? J Am Heart Assoc 1:e003723
Esteve E, Ricart W, Fernandez-Real JM (2005) Dyslipidemia and inflammation: an evolutionary conserved mechanism. Clin Nutr 24:16–31
Ferretti G, Bacchetti T, Negre-Salvayre A, Salvayre R, Dousset N, Curatola G (2006) Structural modifications of HDL and functional consequences. Atherosclerosis 184:1–7
Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D (2001) Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest 108:689–701
Garner B, Waldeck AR, Witting PK, Rye KA, Stocker R (1998) Oxidation of high density lipoproteins. II. Evidence for direct reduction of lipid hydroperoxides by methionine residues of apolipoproteins AI and AII. J Biol Chem 273:6088–6095
Garza CA, Montori VM, McConnell JP, Somers VK, Kullo IJ, Lopez-Jimenez F (2007) Association between lipoprotein-associated phospholipase A2 and cardiovascular disease: a systematic review. Mayo Clin Proc 82:159–165
Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR (1977) High density lipoprotein as a protective factor against coronary heart disease. The Framingham study. Am J Med 62:707–714
Goyal J, Wang K, Liu M, Subbaiah PV (1997) Novel function of lecithin-cholesterol acyltransferase. Hydrolysis of oxidized polar phospholipids generated during lipoprotein oxidation. J Biol Chem 272:16231–16239
Green PS, Vaisar T, Pennathur S, Kulstad JJ, Moore AB, Marcovina S, Brunzell J, Knopp RH, Zhao XQ, Heinecke JW (2008) Combined statin and niacin therapy remodels the high-density lipoprotein proteome. Circulation 118:1259–1267
Haghpassand M, Bourassa PA, Francone OL, Aiello RJ (2001) Monocyte/macrophage expression of ABCA1 has minimal contribution to plasma HDL levels. J Clin Invest 108:1315–1320
Hansson GK (2005) Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 352:1685–1695
Haynes R, Jiang L, Hopewell JC, Li J, Chen F, Parish S, Landray MJ, Collins R, Armitage J, Collins R, Armitage J, Baigent C, Chen Z, Landray M, Chen Y, Jiang L, Pedersen TR, Landray M, Bowman L, Chen F, Hill M, Haynes R, Knott C, Rahimi K, Tobert J, Sleight P, Simpson D, Parish S, Baxter A, Lay M, Bray C, Wincott E, Leijenhorst G, Skattebol A, Moen G, Mitchel Y, Kuznetsova O, Macmahon S, Kjekshus J, Hill C, Lam TH, Sandercock P, Peto R, Hopewell JC (2013) HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J 34:1279–1291
Hedrick CC, Thorpe SR, Fu MX, Harper CM, Yoo J, Kim SM, Wong H, Peters AL (2000) Glycation impairs high-density lipoprotein function. Diabetologia 43:312–320
Heinecke JW (1998) Oxidants and antioxidants in the pathogenesis of atherosclerosis: implications for the oxidized low density lipoprotein hypothesis. Atherosclerosis 141:1–15
Heinecke J (2011) HDL and cardiovascular-disease risk–time for a new approach? N Engl J Med 364:170–171
Heller M, Schlappritzi E, Stalder D, Nuoffer JM, Haeberli A (2007) Compositional protein analysis of high density lipoproteins in hypercholesterolemia by shotgun LC-MS/MS and probabilistic peptide scoring. Mol Cell Proteomics 6:1059–1072
Holleboom AG, Daniil G, Fu X, Zhang R, Hovingh GK, Schimmel AW, Kastelein JJ, Stroes ES, Witztum JL, Hutten BA, Tsimikas S, Hazen SL, Chroni A, Kuivenhoven JA (2012) Lipid oxidation in carriers of lecithin:cholesterol acyltransferase gene mutations. Arterioscler Thromb Vasc Biol 32:3066–3075
Huang Y, von Eckardstein A, Wu S, Maeda N, Assmann G (1994) A plasma lipoprotein containing only apolipoprotein E and with gamma mobility on electrophoresis releases cholesterol from cells. Proc Natl Acad Sci USA 91:1834–1838
Huang Y, von Eckardstein A, Wu S, Assmann G (1995) Cholesterol efflux, cholesterol esterification, and cholesteryl ester transfer by LpA-I and LpA-I/A-II in native plasma. Arterioscler Thromb Vasc Biol 15:1412–1418
Huang Y, Wu Z, Riwanto M, Gao S, Levison BS, Gu X, Fu X, Wagner MA, Besler C, Gerstenecker G, Zhang R, Li XM, DiDonato AJ, Gogonea V, Tang WH, Smith JD, Plow EF, Fox PL, Shih DM, Lusis AJ, Fisher EA, DiDonato JA, Landmesser U, Hazen SL (2013) Myeloperoxidase, paraoxonase-1, and HDL form a functional ternary complex. J Clin Invest 123:3815–3828
Huang Y, DiDonato JA, Levison BS, Schmitt D, Li L, Wu Y, Buffa J, Kim T, Gerstenecker GS, Gu X, Kadiyala CS, Wang Z, Culley MK, Hazen JE, Didonato AJ, Fu X, Berisha SZ, Peng D, Nguyen TT, Liang S, Chuang CC, Cho L, Plow EF, Fox PL, Gogonea V, Tang WH, Parks JS, Fisher EA, Smith JD, Hazen SL (2014) An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat Med 20:193–203
Jahangiri A, de Beer MC, Noffsinger V, Tannock LR, Ramaiah C, Webb NR, van der Westhuyzen DR, de Beer FC (2009) HDL remodeling during the acute phase response. Arterioscler Thromb Vasc Biol 29:261–267
Jensen MK, Rimm EB, Furtado JD, Sacks FM (2012) Apolipoprotein C-III as a potential modulator of the association between HDL-cholesterol and incident coronary heart disease. J Am Heart Assoc 1
Kalantar-Zadeh K, Kopple JD, Kamranpour N, Fogelman AM, Navab M (2007) HDL-inflammatory index correlates with poor outcome in hemodialysis patients. Kidney Int 72:1149–1156
Karlsson H, Leanderson P, Tagesson C, Lindahl M (2005) Lipoproteomics II: mapping of proteins in high-density lipoprotein using two-dimensional gel electrophoresis and mass spectrometry. Proteomics 5:1431–1445
Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ (2011) Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 364:127–135
Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C (2004) Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res 45:1169–1196
Kimura T, Sato K, Kuwabara A, Tomura H, Ishiwara M, Kobayashi I, Ui M, Okajima F (2001) Sphingosine 1-phosphate may be a major component of plasma lipoproteins responsible for the cytoprotective actions in human umbilical vein endothelial cells. J Biol Chem 276:31780–31785
Kimura T, Sato K, Malchinkhuu E, Tomura H, Tamama K, Kuwabara A, Murakami M, Okajima F (2003) High-density lipoprotein stimulates endothelial cell migration and survival through sphingosine 1-phosphate and its receptors. Arterioscler Thromb Vasc Biol 23:1283–1288
Kockx MM, Herman AG (2000) Apoptosis in atherosclerosis: beneficial or detrimental? Cardiovasc Res 45:736–746
Kontush A, Therond P, Zerrad A, Couturier M, Negre-Salvayre A, de Souza JA, Chantepie S, Chapman MJ (2007) Preferential sphingosine-1-phosphate enrichment and sphingomyelin depletion are key features of small dense HDL3 particles: relevance to antiapoptotic and antioxidative activities. Arterioscler Thromb Vasc Biol 27:1843–1849
Kuvin JT, Ramet ME, Patel AR, Pandian NG, Mendelsohn ME, Karas RH (2002) A novel mechanism for the beneficial vascular effects of high-density lipoprotein cholesterol: enhanced vasorelaxation and increased endothelial nitric oxide synthase expression. Am Heart J 144:165–172
Landmesser U, Hornig B, Drexler H (2004) Endothelial function: a critical determinant in atherosclerosis? Circulation 109:II27–II33
Li XM, Tang WH, Mosior MK, Huang Y, Wu Y, Matter W, Gao V, Schmitt D, Didonato JA, Fisher EA, Smith JD, Hazen SL (2013) Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler Thromb Vasc Biol 33:1696–1705
Lp PLASC, Thompson A, Gao P, Orfei L, Watson S, Di Angelantonio E, Kaptoge S, Ballantyne C, Cannon CP, Criqui M, Cushman M, Hofman A, Packard C, Thompson SG, Collins R, Danesh J (2010) Lipoprotein-associated phospholipase A(2) and risk of coronary disease, stroke, and mortality: collaborative analysis of 32 prospective studies. Lancet 375:1536–1544
Lucke S, Levkau B (2010) Endothelial functions of sphingosine-1-phosphate. Cell Physiol Biochem 26:87–96
Lund-Katz S, Liu L, Thuahnai ST, Phillips MC (2003) High density lipoprotein structure. Front Biosci 8:d1044–d1054
Mackness MI, Arrol S, Durrington PN (1991) Paraoxonase prevents accumulation of lipoperoxides in low-density lipoprotein. FEBS Lett 286:152–154
Mackness B, Hine D, Liu Y, Mastorikou M, Mackness M (2004) Paraoxonase-1 inhibits oxidised LDL-induced MCP-1 production by endothelial cells. Biochem Biophys Res Commun 318:680–683
Marathe GK, Zimmerman GA, McIntyre TM (2003) Platelet-activating factor acetylhydrolase, and not paraoxonase-1, is the oxidized phospholipid hydrolase of high density lipoprotein particles. J Biol Chem 278:3937–3947
McIntyre TM, Prescott SM, Stafforini DM (2009) The emerging roles of PAF acetylhydrolase. J Lipid Res 50(Suppl):S255–S259
McMahon M, Grossman J, FitzGerald J, Dahlin-Lee E, Wallace DJ, Thong BY, Badsha H, Kalunian K, Charles C, Navab M, Fogelman AM, Hahn BH (2006) Proinflammatory high-density lipoprotein as a biomarker for atherosclerosis in patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum 54:2541–2549
Mertens A, Verhamme P, Bielicki JK, Phillips MC, Quarck R, Verreth W, Stengel D, Ninio E, Navab M, Mackness B, Mackness M, Holvoet P (2003) Increased low-density lipoprotein oxidation and impaired high-density lipoprotein antioxidant defense are associated with increased macrophage homing and atherosclerosis in dyslipidemic obese mice: LCAT gene transfer decreases atherosclerosis. Circulation 107:1640–1646
Mineo C, Yuhanna IS, Quon MJ, Shaul PW (2003) High density lipoprotein-induced endothelial nitric-oxide synthase activation is mediated by Akt and MAP kinases. J Biol Chem 278:9142–9149
Mineo C, Deguchi H, Griffin JH, Shaul PW (2006) Endothelial and antithrombotic actions of HDL. Circ Res 98:1352–1364
Miyata M, Smith JD (1996) Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat Genet 14:55–61
Morgantini C, Natali A, Boldrini B, Imaizumi S, Navab M, Fogelman AM, Ferrannini E, Reddy ST (2011) Anti-inflammatory and antioxidant properties of HDLs are impaired in type 2 diabetes. Diabetes 60:2617–2623
Nabel EG, Braunwald E (2012) A tale of coronary artery disease and myocardial infarction. N Engl J Med 366:54–63
Navab M, Imes SS, Hama SY, Hough GP, Ross LA, Bork RW, Valente AJ, Berliner JA, Drinkwater DC, Laks H et al (1991) Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. J Clin Invest 88:2039–2046
Navab M, Berliner JA, Watson AD, Hama SY, Territo MC, Lusis AJ, Shih DM, Van Lenten BJ, Frank JS, Demer LL, Edwards PA, Fogelman AM (1996) The Yin and Yang of oxidation in the development of the fatty streak. A review based on the 1994 George Lyman Duff Memorial Lecture. Arterioscler Thromb Vasc Biol 16:831–842
Navab M, Hama-Levy S, Van Lenten BJ, Fonarow GC, Cardinez CJ, Castellani LW, Brennan ML, Lusis AJ, Fogelman AM, La Du BN (1997) Mildly oxidized LDL induces an increased apolipoprotein J/paraoxonase ratio. J Clin Invest 99:2005–2019
Navab M, Hama SY, Anantharamaiah GM, Hassan K, Hough GP, Watson AD, Reddy ST, Sevanian A, Fonarow GC, Fogelman AM (2000a) Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: steps 2 and 3. J Lipid Res 41:1495–1508
Navab M, Hama SY, Cooke CJ, Anantharamaiah GM, Chaddha M, Jin L, Subbanagounder G, Faull KF, Reddy ST, Miller NE, Fogelman AM (2000b) Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: step 1. J Lipid Res 41:1481–1494
Navab M, Berliner JA, Subbanagounder G, Hama S, Lusis AJ, Castellani LW, Reddy S, Shih D, Shi W, Watson AD, Van Lenten BJ, Vora D, Fogelman AM (2001a) HDL and the inflammatory response induced by LDL-derived oxidized phospholipids. Arterioscler Thromb Vasc Biol 21:481–488
Navab M, Hama SY, Hough GP, Subbanagounder G, Reddy ST, Fogelman AM (2001b) A cell-free assay for detecting HDL that is dysfunctional in preventing the formation of or inactivating oxidized phospholipids. J Lipid Res 42:1308–1317
Nicholls SJ, Cutri B, Worthley SG, Kee P, Rye KA, Bao S, Barter PJ (2005a) Impact of short-term administration of high-density lipoproteins and atorvastatin on atherosclerosis in rabbits. Arterioscler Thromb Vasc Biol 25:2416–2421
Nicholls SJ, Dusting GJ, Cutri B, Bao S, Drummond GR, Rye KA, Barter PJ (2005b) Reconstituted high-density lipoproteins inhibit the acute pro-oxidant and proinflammatory vascular changes induced by a periarterial collar in normocholesterolemic rabbits. Circulation 111:1543–1550
Nobecourt E, Tabet F, Lambert G, Puranik R, Bao S, Yan L, Davies MJ, Brown BE, Jenkins AJ, Dusting GJ, Bonnet DJ, Curtiss LK, Barter PJ, Rye KA (2010) Nonenzymatic glycation impairs the antiinflammatory properties of apolipoprotein A-I. Arterioscler Thromb Vasc Biol 30:766–772
Nofer JR, Levkau B, Wolinska I, Junker R, Fobker M, von Eckardstein A, Seedorf U, Assmann G (2001) Suppression of endothelial cell apoptosis by high density lipoproteins (HDL) and HDL-associated lysosphingolipids. J Biol Chem 276:34480–34485
Nofer JR, van der Giet M, Tolle M, Wolinska I, von Wnuck LK, Baba HA, Tietge UJ, Godecke A, Ishii I, Kleuser B, Schafers M, Fobker M, Zidek W, Assmann G, Chun J, Levkau B (2004) HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J Clin Invest 113:569–581
Norata GD, Catapano AL (2012) HDL and adaptive immunity: a tale of lipid rafts. Atherosclerosis 225:34–35
Noto H, Hara M, Karasawa K, Iso ON, Satoh H, Togo M, Hashimoto Y, Yamada Y, Kosaka T, Kawamura M, Kimura S, Tsukamoto K (2003) Human plasma platelet-activating factor acetylhydrolase binds to all the murine lipoproteins, conferring protection against oxidative stress. Arterioscler Thromb Vasc Biol 23:829–835
Ophir G, Amariglio N, Jacob-Hirsch J, Elkon R, Rechavi G, Michaelson DM (2005) Apolipoprotein E4 enhances brain inflammation by modulation of the NF-kappaB signaling cascade. Neurobiol Dis 20:709–718
Ostos MA, Conconi M, Vergnes L, Baroukh N, Ribalta J, Girona J, Caillaud JM, Ochoa A, Zakin MM (2001) Antioxidative and antiatherosclerotic effects of human apolipoprotein A-IV in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 21:1023–1028
Parks JS, Rudel LL (1985) Alteration of high density lipoprotein subfraction distribution with induction of serum amyloid A protein (SAA) in the nonhuman primate. J Lipid Res 26:82–91
Paszty C, Maeda N, Verstuyft J, Rubin EM (1994) Apolipoprotein AI transgene corrects apolipoprotein E deficiency-induced atherosclerosis in mice. J Clin Invest 94:899–903
Patel S, Drew BG, Nakhla S, Duffy SJ, Murphy AJ, Barter PJ, Rye KA, Chin-Dusting J, Hoang A, Sviridov D, Celermajer DS, Kingwell BA (2009) Reconstituted high-density lipoprotein increases plasma high-density lipoprotein anti-inflammatory properties and cholesterol efflux capacity in patients with type 2 diabetes. J Am Coll Cardiol 53:962–971
Patel PJ, Khera AV, Jafri K, Wilensky RL, Rader DJ (2011) The anti-oxidative capacity of high-density lipoprotein is reduced in acute coronary syndrome but not in stable coronary artery disease. J Am Coll Cardiol 58:2068–2075
Patel PJ, Khera AV, Wilensky RL, Rader DJ (2013) Anti-oxidative and cholesterol efflux capacities of high-density lipoprotein are reduced in ischaemic cardiomyopathy. Eur J Heart Fail 15(11):1215–1219
Persegol L, Verges B, Foissac M, Gambert P, Duvillard L (2006) Inability of HDL from type 2 diabetic patients to counteract the inhibitory effect of oxidised LDL on endothelium-dependent vasorelaxation. Diabetologia 49:1380–1386
Persegol L, Verges B, Gambert P, Duvillard L (2007) Inability of HDL from abdominally obese subjects to counteract the inhibitory effect of oxidized LDL on vasorelaxation. J Lipid Res 48:1396–1401
Peterson SJ, Husney D, Kruger AL, Olszanecki R, Ricci F, Rodella LF, Stacchiotti A, Rezzani R, McClung JA, Aronow WS, Ikehara S, Abraham NG (2007) Long-term treatment with the apolipoprotein A1 mimetic peptide increases antioxidants and vascular repair in type I diabetic rats. J Pharmacol Exp Ther 322:514–520
Peterson SJ, Drummond G, Kim DH, Li M, Kruger AL, Ikehara S, Abraham NG (2008) L-4 F treatment reduces adiposity, increases adiponectin levels, and improves insulin sensitivity in obese mice. J Lipid Res 49:1658–1669
Picotti P, Aebersold R (2012) Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods 9:555–566
Picotti P, Rinner O, Stallmach R, Dautel F, Farrah T, Domon B, Wenschuh H, Aebersold R (2010) High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat Methods 7:43–46
Puranik R, Bao S, Nobecourt E, Nicholls SJ, Dusting GJ, Barter PJ, Celermajer DS, Rye KA (2008) Low dose apolipoprotein A-I rescues carotid arteries from inflammation in vivo. Atherosclerosis 196:240–247
Rader DJ (2006) Molecular regulation of HDL metabolism and function: implications for novel therapies. J Clin Invest 116:3090–3100
Rallidis LS, Tellis CC, Lekakis J, Rizos I, Varounis C, Charalampopoulos A, Zolindaki M, Dagres N, Anastasiou-Nana M, Tselepis AD (2012) Lipoprotein-associated phospholipase A(2) bound on high-density lipoprotein is associated with lower risk for cardiac death in stable coronary artery disease patients: a 3-year follow-up. J Am Coll Cardiol 60:2053–2060
Ramet ME, Ramet M, Lu Q, Nickerson M, Savolainen MJ, Malzone A, Karas RH (2003) High-density lipoprotein increases the abundance of eNOS protein in human vascular endothelial cells by increasing its half-life. J Am Coll Cardiol 41:2288–2297
Recalde D, Ostos MA, Badell E, Garcia-Otin AL, Pidoux J, Castro G, Zakin MM, Scott-Algara D (2004) Human apolipoprotein A-IV reduces secretion of proinflammatory cytokines and atherosclerotic effects of a chronic infection mimicked by lipopolysaccharide. Arterioscler Thromb Vasc Biol 24:756–761
Regieli JJ, Jukema JW, Doevendans PA, Zwinderman AH, Kastelein JJ, Grobbee DE, van der Graaf Y (2009) Paraoxonase variants relate to 10-year risk in coronary artery disease: impact of a high-density lipoprotein-bound antioxidant in secondary prevention. J Am Coll Cardiol 54:1238–1245
Ribas V, Sanchez-Quesada JL, Anton R, Camacho M, Julve J, Escola-Gil JC, Vila L, Ordonez-Llanos J, Blanco-Vaca F (2004) Human apolipoprotein A-II enrichment displaces paraoxonase from HDL and impairs its antioxidant properties: a new mechanism linking HDL protein composition and antiatherogenic potential. Circ Res 95:789–797
Riwanto M, Landmesser U (2013) High density lipoproteins and endothelial functions: mechanistic insights and alterations in cardiovascular disease. J Lipid Res 54:3227–3243
Riwanto M, Rohrer L, Roschitzki B, Besler C, Mocharla P, Mueller M, Perisa D, Heinrich K, Altwegg L, von Eckardstein A, Luscher TF, Landmesser U (2013) Altered activation of endothelial anti- and pro-apoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of HDL-proteome remodeling. Circulation 127(8):891–904
Ross R (1999) Atherosclerosis – an inflammatory disease. N Engl J Med 340:115–126
Rossig L, Dimmeler S, Zeiher AM (2001) Apoptosis in the vascular wall and atherosclerosis. Basic Res Cardiol 96:11–22
Rotllan N, Ribas V, Calpe-Berdiel L, Martin-Campos JM, Blanco-Vaca F, Escola-Gil JC (2005) Overexpression of human apolipoprotein A-II in transgenic mice does not impair macrophage-specific reverse cholesterol transport in vivo. Arterioscler Thromb Vasc Biol 25:e128–e132
Rye KA, Barter PJ (2008) Antiinflammatory actions of HDL: a new insight. Arterioscler Thromb Vasc Biol 28:1890–1891
Sacre SM, Stannard AK, Owen JS (2003) Apolipoprotein E (apoE) isoforms differentially induce nitric oxide production in endothelial cells. FEBS Lett 540:181–187
Saito H, Lund-Katz S, Phillips MC (2004) Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Prog Lipid Res 43:350–380
Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Mundl H, Nicholls SJ, Shah PK, Tardif JC, Wright RS (2012) Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med 367:2089–2099
Shao B, Oda MN, Bergt C, Fu X, Green PS, Brot N, Oram JF, Heinecke JW (2006) Myeloperoxidase impairs ABCA1-dependent cholesterol efflux through methionine oxidation and site-specific tyrosine chlorination of apolipoprotein A-I. J Biol Chem 281:9001–9004
Shao B, Tang C, Heinecke JW, Oram JF (2010) Oxidation of apolipoprotein A-I by myeloperoxidase impairs the initial interactions with ABCA1 required for signaling and cholesterol export. J Lipid Res 51:1849–1858
Shao B, Pennathur S, Heinecke JW (2012) Myeloperoxidase targets apolipoprotein A-I, the major high density lipoprotein protein, for site-specific oxidation in human atherosclerotic lesions. J Biol Chem 287:6375–6386
Sharrett AR, Ballantyne CM, Coady SA, Heiss G, Sorlie PD, Catellier D, Patsch W (2001) Coronary heart disease prediction from lipoprotein cholesterol levels, triglycerides, lipoprotein(a), apolipoproteins A-I and B, and HDL density subfractions: the atherosclerosis risk in communities (ARIC) study. Circulation 104:1108–1113
Shih DM, Gu L, Xia YR, Navab M, Li WF, Hama S, Castellani LW, Furlong CE, Costa LG, Fogelman AM, Lusis AJ (1998) Mice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosis. Nature 394:284–287
Shih DM, Xia YR, Wang XP, Miller E, Castellani LW, Subbanagounder G, Cheroutre H, Faull KF, Berliner JA, Witztum JL, Lusis AJ (2000) Combined serum paraoxonase knockout/apolipoprotein E knockout mice exhibit increased lipoprotein oxidation and atherosclerosis. J Biol Chem 275:17527–17535
Soran H, Younis NN, Charlton-Menys V, Durrington P (2009) Variation in paraoxonase-1 activity and atherosclerosis. Curr Opin Lipidol 20:265–274
Sorrentino SA, Besler C, Rohrer L, Meyer M, Heinrich K, Bahlmann FH, Mueller M, Horvath T, Doerries C, Heinemann M, Flemmer S, Markowski A, Manes C, Bahr MJ, Haller H, von Eckardstein A, Drexler H, Landmesser U (2010) Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 121:110–122
Speidel MT, Booyse FM, Abrams A, Moore MA, Chung BH (1990) Lipolyzed hypertriglyceridemic serum and triglyceride-rich lipoprotein cause lipid accumulation in and are cytotoxic to cultured human endothelial cells. High density lipoproteins inhibit this cytotoxicity. Thromb Res 58:251–264
Spieker LE, Sudano I, Hurlimann D, Lerch PG, Lang MG, Binggeli C, Corti R, Ruschitzka F, Luscher TF, Noll G (2002) High-density lipoprotein restores endothelial function in hypercholesterolemic men. Circulation 105:1399–1402
Subramanian VS, Goyal J, Miwa M, Sugatami J, Akiyama M, Liu M, Subbaiah PV (1999) Role of lecithin-cholesterol acyltransferase in the metabolism of oxidized phospholipids in plasma: studies with platelet-activating factor-acetyl hydrolase-deficient plasma. Biochim Biophys Acta 1439:95–109
Suc I, Escargueil-Blanc I, Troly M, Salvayre R, Negre-Salvayre A (1997) HDL and ApoA prevent cell death of endothelial cells induced by oxidized LDL. Arterioscler Thromb Vasc Biol 17:2158–2166
Sugano M, Tsuchida K, Makino N (2000) High-density lipoproteins protect endothelial cells from tumor necrosis factor-alpha-induced apoptosis. Biochem Biophys Res Commun 272:872–876
Takeya H, Gabazza EC, Aoki S, Ueno H, Suzuki K (2003) Synergistic effect of sphingosine 1-phosphate on thrombin-induced tissue factor expression in endothelial cells. Blood 102:1693–1700
Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N (2008) HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab 7:365–375
Tang C, Liu Y, Kessler PS, Vaughan AM, Oram JF (2009) The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J Biol Chem 284:32336–32343
Tang WH, Hartiala J, Fan Y, Wu Y, Stewart AF, Erdmann J, Kathiresan S, Roberts R, McPherson R, Allayee H, Hazen SL (2012) Clinical and genetic association of serum paraoxonase and arylesterase activities with cardiovascular risk. Arterioscler Thromb Vasc Biol 32:2803–2812
Tolle M, Huang T, Schuchardt M, Jankowski V, Prufer N, Jankowski J, Tietge UJ, Zidek W, van der Giet M (2012) High-density lipoprotein loses its anti-inflammatory capacity by accumulation of pro-inflammatory-serum amyloid A. Cardiovasc Res 94:154–162
Tward A, Xia YR, Wang XP, Shi YS, Park C, Castellani LW, Lusis AJ, Shih DM (2002) Decreased atherosclerotic lesion formation in human serum paraoxonase transgenic mice. Circulation 106:484–490
Uittenbogaard A, Shaul PW, Yuhanna IS, Blair A, Smart EJ (2000) High density lipoprotein prevents oxidized low density lipoprotein-induced inhibition of endothelial nitric-oxide synthase localization and activation in caveolae. J Biol Chem 275:11278–11283
Undurti A, Huang Y, Lupica JA, Smith JD, DiDonato JA, Hazen SL (2009) Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J Biol Chem 284:30825–30835
Vaisar T, Pennathur S, Green PS, Gharib SA, Hoofnagle AN, Cheung MC, Byun J, Vuletic S, Kassim S, Singh P, Chea H, Knopp RH, Brunzell J, Geary R, Chait A, Zhao XQ, Elkon K, Marcovina S, Ridker P, Oram JF, Heinecke JW (2007) Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest 117:746–756
Van Lenten BJ, Hama SY, de Beer FC, Stafforini DM, McIntyre TM, Prescott SM, La Du BN, Fogelman AM, Navab M (1995) Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J Clin Invest 96:2758–2767
Van Lenten BJ, Wagner AC, Nayak DP, Hama S, Navab M, Fogelman AM (2001) High-density lipoprotein loses its anti-inflammatory properties during acute influenza a infection. Circulation 103:2283–2288
Van Linthout S, Spillmann F, Riad A, Trimpert C, Lievens J, Meloni M, Escher F, Filenberg E, Demir O, Li J, Shakibaei M, Schimke I, Staudt A, Felix SB, Schultheiss HP, De Geest B, Tschope C (2008) Human apolipoprotein A-I gene transfer reduces the development of experimental diabetic cardiomyopathy. Circulation 117:1563–1573
von Eckardstein A, Huang Y, Wu S, Funke H, Noseda G, Assmann G (1995a) Reverse cholesterol transport in plasma of patients with different forms of familial HDL deficiency. Arterioscler Thromb Vasc Biol 15:691–703
von Eckardstein A, Huang Y, Wu S, Sarmadi AS, Schwarz S, Steinmetz A, Assmann G (1995b) Lipoproteins containing apolipoprotein A-IV but not apolipoprotein A-I take up and esterify cell-derived cholesterol in plasma. Arterioscler Thromb Vasc Biol 15:1755–1763
Vowinkel T, Mori M, Krieglstein CF, Russell J, Saijo F, Bharwani S, Turnage RH, Davidson WS, Tso P, Granger DN, Kalogeris TJ (2004) Apolipoprotein A-IV inhibits experimental colitis. J Clin Invest 114:260–269
Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH, Tall AR, Rader DJ (2007a) Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest 117:2216–2224
Wang Z, Nicholls SJ, Rodriguez ER, Kummu O, Horkko S, Barnard J, Reynolds WF, Topol EJ, DiDonato JA, Hazen SL (2007b) Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat Med 13:1176–1184
Wang SH, Yuan SG, Peng DQ, Zhao SP (2012) HDL and ApoA-I inhibit antigen presentation-mediated T cell activation by disrupting lipid rafts in antigen presenting cells. Atherosclerosis 225:105–114
Watson AD, Berliner JA, Hama SY, La Du BN, Faull KF, Fogelman AM, Navab M (1995) Protective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J Clin Invest 96:2882–2891
Weihrauch D, Xu H, Shi Y, Wang J, Brien J, Jones DW, Kaul S, Komorowski RA, Csuka ME, Oldham KT, Pritchard KA (2007) Effects of D-4 F on vasodilation, oxidative stress, angiostatin, myocardial inflammation, and angiogenic potential in tight-skin mice. Am J Physiol Heart Circ Physiol 293:H1432–H1441
Witztum JL, Steinberg D (1991) Role of oxidized low density lipoprotein in atherogenesis. J Clin Invest 88:1785–1792
Yuhanna IS, Zhu Y, Cox BE, Hahner LD, Osborne-Lawrence S, Lu P, Marcel YL, Anderson RG, Mendelsohn ME, Hobbs HH, Shaul PW (2001) High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat Med 7:853–857
Yvan-Charvet L, Pagler TA, Wang N, Senokuchi T, Brundert M, Li H, Rinninger F, Tall AR (2008) SR-BI inhibits ABCG1-stimulated net cholesterol efflux from cells to plasma HDL. J Lipid Res 49:107–114
Zerrad-Saadi A, Therond P, Chantepie S, Couturier M, Rye KA, Chapman MJ, Kontush A (2009) HDL3-mediated inactivation of LDL-associated phospholipid hydroperoxides is determined by the redox status of apolipoprotein A-I and HDL particle surface lipid rigidity: relevance to inflammation and atherogenesis. Arterioscler Thromb Vasc Biol 29:2169–2175
Zheng L, Nukuna B, Brennan ML, Sun M, Goormastic M, Settle M, Schmitt D, Fu X, Thomson L, Fox PL, Ischiropoulos H, Smith JD, Kinter M, Hazen SL (2004) Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest 114:529–541
Zhu X, Owen JS, Wilson MD, Li H, Griffiths GL, Thomas MJ, Hiltbold EM, Fessler MB, Parks JS (2010) Macrophage ABCA1 reduces MyD88-dependent toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res 51:3196–3206
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this chapter
Cite this chapter
Riwanto, M., Rohrer, L., von Eckardstein, A., Landmesser, U. (2015). Dysfunctional HDL: From Structure-Function-Relationships to Biomarkers. In: von Eckardstein, A., Kardassis, D. (eds) High Density Lipoproteins. Handbook of Experimental Pharmacology, vol 224. Springer, Cham. https://doi.org/10.1007/978-3-319-09665-0_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-09665-0_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-09664-3
Online ISBN: 978-3-319-09665-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)