
Abstract
Electronic spectroscopy has been instrumental in demonstrating the properties of helium droplets as a cryogenic matrix for molecules. The electronic spectrum of glyoxal, which was one of the first molecules investigated in helium droplets by means of electronic spectroscopy, showed two features that provided convincing evidence that the droplets were superfluid. These were free rotation and the distinct shape of the phonon side band which could be directly assigned to the characteristic dispersion curve of a superfluid. On closer examination, however, details such as increased moments of inertia and a spectral response on the droplet size distribution revealed unexpected features of microsolvation in the superfluid helium. In the course of studying many different molecules, it has become clear that electronic spectroscopy in helium droplets provides insight into the detailed effects of microsolvation. These in turn lead to numerous questions regarding the interaction with the superfluid which are discussed in this chapter. In addition, the influence of microsolvation in helium droplets on van der Waals clusters generated inside helium droplets are discussed. Finally, the effect of helium solvation on unimolecular or bimolecular elementary chemical reactions is evaluated in comparison with corresponding experiments in the gas phase. Particular focus of this article lies on the spectral features related to helium solvation which are not yet fully understood.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others
5.1 Introduction
This chapter is devoted to electronic spectroscopy of molecules in superfluid helium droplets. Compared to other spectroscopic techniques such as MW or IR spectroscopy that are discussed in Chap. 3 by Gary Douberly electronic excitation of molecules in the helium droplet environment shows two prominent features in the spectra. These are a phonon wing (PW) reflecting the excitation of the helium environment coupled to the electronic excitation of the dopant. The second feature is a spectral splitting into multiplets that occurs at the pure molecular excitation called zero phonon line (ZPL). This splitting results from inhomogeneities in the solvation of the dopant inside the helium droplet [1–4] as shall be discussed below. Both features do not occur in IR or MW spectroscopy.
In the IR spectra the line resolved rotational bands prove that the molecules rotate freely inside the superfluid droplets. The helium environment has however a strong effect on the moments of inertia of the rotating dopant [1, 3–5]. As shown for the first time in the rotationally resolved IR spectrum of sulfur hexafluoride, the effective moments of inertia reveal a significant increase of the rotating mass in helium droplets [6]. In contrast to electronic excitation the coupling to phonons of the helium droplet has no noticeable effect on the rotations and/or the vibrations of the dopant. Molecular vibrations proceed mostly inside the dopant’s electron cloud and, thereby, are shielded from the influence of the helium environment so that vibrational frequencies are almost identical to the corresponding gas phase values [5]. In contrast, the electron density distribution is that part of the dopant species which is in direct contact with the helium environment and upon electronic excitation is therefore coupled to the phonons of the helium environment. The shape of the dopant molecule as experienced by the helium environment is therefore defined by the outer electron density distribution. Besides an increase of the energy deposited into the dopant’s electrons, electronic excitation is accompanied by a rearrangement of the electron density distribution.
The forces responsible for the dopant to helium interaction are dispersion forces and/or van der Waals forces which exceed dispersion forces among helium atoms. Thus, the dopant species attracts a layer of helium atoms [2–4]. Instead of an isolated and cold molecule doped into a non-viscous cryogenic environment one has to consider a dopant-helium solvation complex which rotates freely inside the superfluid droplet [3–5].
As the dopant enforces the formation of a non-superfluid helium solvation layer, the layer enforces modification of the electron density distribution of the dopant which leads to an energetic shift of electronic states compared to the isolated molecule. This modification is not simply dopant specific but in addition specific for different electronic states of the same dopant species. Depending on the relation of the corrugation of the dopant’s electron density distribution to the size or the van der Waals radius of helium atoms, the solvation complex may exhibit different configurations which differ energetically. Thus, electronic spectra of molecules in helium droplets provide particularly insight into the solvation of molecules in helium and its influence on intramolecular dynamics. In order to make use of the twofold information namely about the dopant as well as about its solvation, it is necessary to decipher the helium induced spectral features. In other words, intramolecular and intermolecular contributions need to be disentangled which is quite a challenge.
Beyond molecular spectroscopy, the formation of a helium solvation complex has certainly some influence on steering, for example, the formation of weakly bound clusters. Since the cluster forming subunits are picked up consecutively, each subunit may attract a helium solvation layer prior to cluster formation and again helium solvation complexes approach each other instead of bare dopant units. Moreover, the ultra-low temperature conditions can promote cluster configurations which are absent at elevated temperatures. Thus, cluster formation in helium droplets provides a larger variety of metastable configurations and in addition those that incorporate helium atoms. So far it is an issue of microsolvation [1, 2, 6, 7].
Besides microsolvation of either single molecules or molecular compounds as for example van der Waals clusters, helium solvation can have a strong impact on molecular dynamics. Electronic excitation accompanying photophysical and photochemical processes are certainly affected by the low temperatures of the droplets. In combination with vanishing viscosity, superfluid helium droplets were expected to be an ideal host for the investigation of intramolecular and intermolecular photochemistry. We will have a critical view on this.
This article on electronic spectroscopy of molecules in helium droplets focusses on helium induced spectral features as revealed by comparison of helium droplet experiments with corresponding gas phase data. Out of the myriads of publications on electronic spectroscopy in helium droplets only selected studies will be discussed in order to highlight microsolvation expressed by helium induced spectral features. Besides several details readily explained by empirical conclusions or chemical intuition, there are many observations that elude empirical interpretation. The quest on modeling microsolvation in superfluid helium and its impact on chemical dynamics needs to consider all such peculiarities reported so far. Any progress in a quantitative understanding of these features is of fundamental importance for quantum chemical modeling.
5.2 Electronic Spectroscopy
In the past the following two techniques are most common in preparing samples of isolated molecules at low temperature. These are adiabatic expansion of a molecular gas into vacuum [8] or matrix isolation of molecules in solid crystals in many cases rare gas crystals or Spolskii matrices [9]. Molecular spectroscopy in superfluid helium droplets can be seen as a kind of hybrid of molecular beam and matrix isolation. Helium droplets are generated via expansion of helium and are provided as a beam of droplets propagating along a well-defined axis inside a vacuum machine. This is the molecular beam aspect. Afterwards, the molecule of interest is doped into or onto the helium droplet by a pick-up process. The solvated dopant resembles matrix isolation with the major difference or rather advantage that the helium matrix is superfluid instead of solid. The doped droplets propagate along a defined axis and, thus, resemble a transient sample similar as molecules do in a molecular beam. Doping of molecules into a helium droplet provides a temperature of only 0.38(1) K for all degrees of freedom of the dopant species except of spin states [1]. This temperature is much lower than can be reached by standard molecular beams. Since helium does not solidify upon cooling at pressures below 25 bar and instead undergoes a transition to a superfluid with vanishing viscosity below 2.17 K, it allows for free rotation of the dopant species. Molecular rotation subject to spectroscopic investigations is of high value for the analysis of the molecular structure. In a solid matrix the rotational degree of freedom is frozen.Footnote 1 In a standard molecular beam, however, rotation is cooled down to a temperature of roughly 1–10 K, however, often with non-thermal state population with a surplus at higher rotational states [8]. A Boltzmann ensemble at a temperature of 0.38 K represents perfect thermal conditions for making use of the rotational degree of freedom for structural analysis of molecular compounds as outlined in Chap. 3 of this monograph. A temperature of only 0.38 K warrants for eliminating vibrational hot bands entirely. Under these conditions an easy reading of vibrational modes from electronic spectra is warranted even selective for electronic states. At appropriate spectral resolution the rotational fine structure can also be resolved in electronic spectra of molecules in helium droplets as shall be discussed below.
Experimentally, electronic spectroscopy can be performed using different detection schemes which provide different information. The basic processes in electronic spectroscopy are absorption or emission of electromagnetic radiation in order to switch electronic states of atoms or molecules. Most of the spectroscopic data discussed in this article are based on these two fundamental processes, whereby absorption is recorded in two variants, namely depletion spectroscopy and fluorescence excitation spectroscopy [1]. Depletion spectroscopy makes use of energy dissipation from the excited dopant species into the helium droplet. The energy transfer initiates evaporative cooling whereby the droplet loses mass as well as volume. The reduction of mass can be monitored as a reduction of the energy flux into a bolometer placed on the droplet beam axis whereas the shrunk volume becomes effective in a reduced ionization cross section when using a quadrupole mass spectrometer as monitor detector. In both cases resonant absorption by the dopant generates a depletion of an intense signal, an effect which bore the term depletion spectroscopy. This technique allows for recording absorption spectra of highly diluted samples and is therefore the method of choice for recording IR spectra of molecules in helium droplets (cf. Chap. 3).
Instead of depletion spectroscopy, the fluorescence as response to electronic excitation of a molecule can be recorded. In general, it requires a radiative step on the decay path of the electronically excited molecule, a precondition which is not necessarily fulfilled. Thus, in contrast to absorption or depletion spectroscopy, resonances to non-radiating states, so-called dark states, are missing in fluorescence excitation spectra. The advantage of recording fluorescence is a zero-background signal which exceeds depletion in the sensitivity by orders of magnitude.
Starting always from the vibronic ground state, as is guaranteed by the droplet temperature of only 0.38 K, the frequency of a laser is tuned across the series of resonances when recording fluorescence excitation spectra or depletion spectra. A spectrum starts with a purely electronic transition, the so-called electronic band origin followed to the blue by vibronic transitions into the multitude of vibrational levels of electronically excited states. At appropriate spectral resolution the rotational band structure can additionally be resolved for each vibronic transition. Thus, at least the normal mode frequencies of electronically excited states if not in addition the moments of inertia of the dopant species are readily obtained when monitoring the dopant’s fluorescence or the depletion signal as shown in the left half of Fig. 5.1.

Schematic of electronic spectroscopy for a closed shell organic molecule indicating vibronic transitions. Left side: excitation spectroscopy starting with the electronic band origin. Left bottom: experimental setup for fluorescence detection for a view along the droplet beam axis. Right side: dispersed emission spectroscopy upon excitation at the electronic band origin. Right bottom: experimental setup for dispersed emission detection for a view along the droplet beam axis. Arrows resemble photons for excitation (up) or spontaneous emission (down)
Instead of recording the fluorescence in dependence on the excitation frequency, another variant of electronic spectroscopy records the fluorescence, however, upon excitation fixed at a certain resonance and dispersed by means of a grating spectrograph as shown in the right half of Fig. 5.1. Upon excitation at the electronic band origin, radiative transitions extend to the multitude of vibrational states of the electronic ground state. Dispersed emission spectra reveal information on the normal mode frequencies of the electronic ground state as complement to the corresponding information on the electronically excited state from excitation spectra. Upon vibronic excitation or excitation to higher electronic states, the efficiency of dissipation of energy in excess to the electronic band origin into the helium droplet, the process depletion spectroscopy is based on, guarantees for radiative decay exclusively from the ground level of the first electronically excited state. As a consequence, dispersed emission spectra recorded for molecules in helium droplets are basically independent of the excitation frequency and start with the electronic band origin followed to the red by vibronic transitions as described above. Accordingly, the fluorescence excitation spectrum and dispersed emission spectra of a molecule in helium droplets should coincide in a single resonance line which represents the electronic band origin. A spectral gap instead of an overlap is indicative for dynamic processes in the electronically excited state.
Dissipation of excess excitation energy beyond the electronic band origin holds for rotational, vibrational, electronic, and in addition phonon energy. This process allows for easily identifying the resonance frequency of the electronic band origin of molecules in helium droplets by simply exciting into the quasi continuum of electronically excited states somewhere in the blue or near UV while recording the dispersed emission in the vicinity of the electronic band origin of the molecule. In general, the resonance with the maximum frequency in the corresponding dispersed emission spectrum represents the electronic band origin.
So far, selected variants of electronic spectroscopy were introduced which are relevant for what shall follow on electronic spectroscopy of molecules in superfluid helium droplets. Before going into the specific details, some general spectral features of helium solvation are mentioned, which are also present in electronic spectra of molecules isolated in solid state matrices. First, due to the weak nevertheless finite polarizability of the helium environment, electronic resonance frequencies are shifted compared to the isolated molecule either to the red or to the blue by rule of thumb about 1% of the transition frequency in the gas phase. Whether to the blue or to the red depends on the difference in the helium induced stabilization energy of the corresponding electronic states. Secondly, a ZPL is accompanied by a PW, representing the excitation of the helium environment coupled to the electronic excitation of the dopant species [11]. Third, the ZPL may exhibit a multiplet splitting [12]. In the case of a solid matrix this is an expression of different sites of the dopant within the solid. The correspondence to helium droplets will be discussed below. Finally, and in contrast to solid matrices, a ZPL exhibits a rotational band structure. In case of appropriate experimental conditions, molecular rotation can even be line resolved in electronic spectra of molecules in helium droplets.
As will be shown in the following, one of the strengths of electronic spectroscopy in helium droplets lies in obtaining vibrational frequencies of the dopant species specific for electronic states. Compared to the accuracy of corresponding theoretical values the helium induced shift of vibrational frequencies is rather small. Other experimental observables such as moments of inertia likewise rotational constants, electronic transition energies, and intramolecular dynamics induced by electronic excitation reveal the influence of helium solvation which becomes most evident in comparison to corresponding data from gas phase experiments. Thus, a key issue of electronic spectroscopy in helium droplets is the investigation of microsolvation which reveals information on both, the dopant as well as the helium droplet.
This chapter focusses on microsolvation of molecules and molecular compounds as well as its influence on molecular dynamics as revealed by electronic spectroscopy. This focus is highlighted by a comparative discussion of experiments made in helium droplets as well as under gas phase conditions. The influence of the helium environment is omnipresent for electronic spectra in helium droplets. We start with electronic spectroscopy of various dopant species in most cases closed shell organic molecules which are heliophilic and, therefore, reside fully solvated inside the helium droplet. Thereby, the focus will be on the spectral structure of the ZPL and the PW. Moreover, microsolvation of van der Waals clusters consisting of a single chromophore molecule and additional noble gas atoms generated inside helium droplets will be discussed. Finally, the influence of solvation on elementary chemical reactions involving electronic excitation or relaxation will be presented. Besides numerous helium induced spectral features which fit at least to empirical explanations, there are other helium induced spectral features which are counterintuitive to the current understanding of superfluid helium as host. Understanding the origin of such features is the challenge for future activities.
5.3 Electronic Spectra of Molecules in Helium Droplets
The discussion of electronic spectroscopy of molecules in superfluid helium droplets starts with an examination mainly of the ZPL and accompanying PW at the electronic band origin of various dopant species. It starts with glyoxal which behaves in many aspects as expected for a molecule solvated in a cryogenic superfluid. It will be continued with tetracene and related polycyclic aromatic hydrocarbons (PAH) before coming to larger organic compounds such as phthalocyanines and porphyrins. Along the line of dopant species, helium induced spectral features become more and more complex and require additional conceptions for an empirical explanation. Up to now, not all of the helium induced spectral features can be rationalized. Almost none of them can be simulated quantitatively.
5.3.1 Glyoxal in Superfluid Helium Droplets
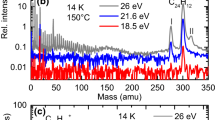
The electronic absorption spectrum of glyoxal recorded by means of depletion spectroscopy is a textbook example for spectral features as expected for solvation in superfluid helium droplets. At appropriate spectral resolution the ZPL at the electronic origin appears as a line resolved rotational fine structure (cf. Fig. 5.2) [13]. While the asymmetric top character expressed by Ray’s asymmetry parameter remained almost unaffected in helium droplets—an increase by only 1% for the ground state and 0.3% for the excited state—the moments of inertia of the S0 state were found increased by factors of 2.87 (A), 2.22 (B), and 2.09 (C). Increased moments of inertia are well known mainly from rotationally resolved IR-spectra of molecules in helium droplets (cf. Chap. 3). The increase of the moments of inertia indicates increasing rotating mass. A countable number of helium atoms rigidly attached to the rotating molecule called solvation layer accounts for the increase quantitatively. Besides this increase of the mass, the observation of free rotation fulfills what is expected from a superfluid solvent with vanishing viscosity. The cryogenic property of helium droplets is expressed by the intensity profile of the rotational fine structure which fits to a temperature of only 0.38(1) K. The ZPL at the electronic band origin is accompanied to the blue by a PW. For glyoxal the spectral shape of the PW fits perfectly to the spectrum of elementary excitations of superfluid helium (cf. Fig. 5.3) [14, 15]. In addition to the free rotation observed for the ZPL the spectral shape of the accompanying PW was a weighty argument for superfluidity and, thus, a milestone in molecular spectroscopy in helium droplets.

Rotationally resolved ZPL at the electronic band origin of glyoxal in superfluid helium droplets having on average 2600 atoms. The red line is a best fit calculated for a free asymmetric top, convoluted with a Lorentzian having a linewidth of dv = 0.035 cm−1 (FWHM). The {j’ ← j”} assignments of the transitions are given for the most prominent lines. (adapted from [13])

a The dispersion curve of elementary excitations in bulk superfluid helium, b the corresponding density of states and c the density of states adopted to a small droplet (red) fitting almost perfectly to the experimental PW in the electronic spectrum of glyoxal in superfluid helium droplets (black) (\(\overline{N }\) = 5500). Adapted from [14]
Remarkably, however, was the change of the moments of inertia upon electronic excitation of glyoxal in helium droplets [13]. This observation was readily explained by the helium attached to the dopant. As mentioned above, electronic excitation is accompanied by the change of the electron density distribution which by itself is of negligible effect on the moments of inertia for the bare molecule. However, in helium droplets the changing electron density has an impact on the attached helium atoms which follow the electron density distribution. Thus, the impact of electronic excitation on the moments of inertia is significantly amplified by the helium solvation layer of the dopant molecule. So far, the helium induced spectral features in the electronic spectrum of glyoxal in helium droplets reflect expectations based on empirical understanding.
However, another observable which did not fit into the empirical modeling of solvation in superfluid helium was the dependence of the line shape within the rotational fine structure on the droplet size [13, 16]. For increasing droplet sizes beyond an average of 3000 helium atoms the line width increased significantly, certainly an indication for inhomogeneous line broadening. There are various line shape determining effects that scale with the average droplet size, namely, the width of the size distribution, the integral strength of dispersion forces of a polarizable environment, the density of states of the dopant inside the droplets—approximated by a particle in a spherical box, the spectral density of surface modes of the droplet, and last but not least vortices as the way a quantum fluid carries angular momentum, to name only the most evident ones. None of these effects succeeded in simulating the experimental observations on the line shapes of rotationally resolved electronic spectra of glyoxal. So far, the droplet size dependence of the line shape of glyoxal in helium droplets remains an open question. None of the helium induced spectral features were explained by specific properties of glyoxal as dopant species. Therefore, these features might be observable for other dopant species as well.
5.3.2 Tetracene in Superfluid Helium Droplets
Tetracene is among the first of organic molecules that have been studied in helium droplets by means of electronic spectroscopy [17]. Its electronic spectrum shown in Fig. 5.4 has undergone a very detailed examination. Neither does the ZPL reveal rotational fine structure nor does the PW reflect the spectral shape expected from superfluid helium [18]. Nevertheless, numerous unexpected spectral features provide information which is relevant for a deeper understanding of microsolvation in helium droplets.

Fluorescence excitation spectrum of the electronic band origin of tetracene in helium droplets (\(\overline{N }\) = 16,000) with v0 = 22,293.4(5) cm−1. The splitting of the ZPL is Δv = 1.1(1) cm−1 whereby the peak at 0 cm−1 is the α-peak and the second and most intense is the β-peak. The phonon wing is enhanced by increasing the laser power from 0.05 to 1.5 mJ/pulse. Both spectra are saturation broadened. (adapted from [19])
A surprising feature in the electronic spectrum of tetracene was a ZPL that is split into a doublet without relation to a rotational fine structure (cf. Fig. 5.4). Pump probe experiments have proven for two independent systems represented by each peak of the doublet [18]. The observation of different dispersed emission spectra upon excitation at each of the two peaks confirmed the presence of two independent solvated systems [20, 21]. In most of the papers on tetracene in helium droplets the two peaks of the doublet are addressed as alpha (low frequency) and beta (high frequency) line which are separated by a gap of about 1 cm−1 whereby the alpha line has roughly 1/3 of the peak intensity of the beta line. Empirical explanation for the doublet splitting by tetracene exhibiting two different configurations of a helium solvation complex was readily on hand and substantiated by quantum chemical modeling [17, 18, 20, 22]. The empirical modeling of helium solvation complexes with a countable number of helium atoms localized on the surface of tetracene in different configurations corresponds to the phenomenon of different sites of molecules in solid matrices. A first purely empirical discussion on possible configurations of solvation complexes [18] was followed by path integral Monte Carlo (PIMC) simulations [22] of a helium solvation layer attached to tetracene embedded into up to 150 helium atoms. Finally, modeling of quantum coherent, but strongly correlated, set of helium atoms adsorbed in a linear arrangement on the quasi-planar molecular surface was capable of reproducing a doublet for the ZPL of tetracene [23]. The experimental spectrum chosen for fitting the doublet simulation was unfortunately a spectrum with poor spectral resolution which does not serve as appropriate experimental reference [24]. Furthermore, the model developed in Ref. [23] relates ZPL splitting to a linear arrangement of helium atoms and thereby serves in addition to explaining the missing of such splitting for a two-dimensional helium solvation layer as present for dopant species such as phthalocyanine and porphin. As a matter of fact, and in contrast to the earliest publication [12], the ZPL of porphin does show a triplet splitting as will be discussed below [25]. Moreover, the model developed in Ref. [23] has never been validated for other dopant species and in particular not for other linear PAH molecules shown in Fig. 5.5.

Fluorescence excitation spectra of the electronic band origin of pentacene (a), tetracene (b), anthracene (c), benzene (e), and perylene (f), and depletion spectrum of the 801 vibronic mode of naphthalene (d). (\(\overline{N }\)~20,000) for (a–e) and \(\overline{N }\)= 8000 for (f)). (adapted from a: [19], b: [19], c: [26], d: [27], e: [28], f: [21])
Further experimental information for the interpretation of helium induced spectral features and in particular for the doublet splitting at the ZPL of tetracene can be collected from comparison with related dopants. Among linear PAH species the electronic spectra of pentacene [11, 19], anthracene [26], naphthalene [27], and benzene [29, 28] have been reported. In contrast to the doublet of tetracene shown in Fig. 5.5b, a singly peaked ZPL was recorded for pentacene at the electronic band origin (cf. Fig. 5.5a) [19]. A different situation was found for anthracene. The ZPL at the electronic band origin in helium droplets was found split into a quartet with almost regular gaps of 1 cm−1 (cf. Fig. 5.5c) which at the blue side merged into a broad and smoothly decreasing signal extending over tens of cm−1. The latter part was assigned to the PW [26]. The characteristic phonon gap of superfluid helium was missing as were two maxima resembling the maxon and roton excitation of a superfluid. Dispersed emission spectra recorded upon excitation at each of the four peaks provided further details. Within the quartet the 1st and the 2nd peak exhibit identical emission spectra which differ from a second emission spectrum recorded upon excitation at the 3rd and 4th peak. Thus, within the quartet in the excitation the 1st and the 3rd line represent individual systems similar as the doublet of tetracene and are therefore assigned accordingly to α and β line. The 2nd and 4th line in the quartet correlate with the 1st and 3rd, respectively, and are therefore assigned as α′ and β′. In comparison to tetracene the spectral gap between the α and β line has doubled and each of the two solvated systems comes with an additional line shifted by 1 cm−1 to the blue. There are two empirical models explaining the α′ and β′ line. Either, the electronic origin of each system is accompanied by a 1 cm−1 van der Waals mode of the solvation complex whose energy dissipates prior to radiative decay. Alternatively, the entire quartet represents four different configurations of an anthracene helium solvation complex whereby the 2nd and the 4th peak represent configurations which are highly metastable in the electronically excited state, and, therefore, relax prior to radiative decay. Even though the series from pentacene to anthracene shows increasing multiplet splitting for decreasing size of the PAH species. A continuation for naphthalene and finally benzene is intuitively unlikely and was refuted by corresponding experiments [27, 29, 28] (cf. Fig. 5.5d, e). The electronic spectrum of naphthalene [27] in helium droplet was found singly peaked which is shown for a vibronic resonance recorded via depletion in Fig. 5.5d. Finally, the ZPL at the electronic band origin of benzene was also singly peaked [29, 28] (c.f. Fig. 5.5d).
Looking finally at perylene consisting of 5 benzene units similar to pentacene, however, in a two-dimensional arrangement, the ZPL at the electronic origin exhibits a rich fine structure with as much as 10 peak maxima within a spectral section of 6 cm−1 [30] a part of which is shown in panel (f) of Fig. 5.5. This multiplet merges into a rather smooth and monotonously decreasing signal which extends over tens of cm−1 assigned to the PW. The multiplet is dominated by a triplet of intense sharp peaks about 0.1 cm−1 in width and with internal gaps of 1.65 cm−1 and 1.50 cm−1, respectively. The leading peak at the low frequency side of the mutiplet is not part of the dominating triplet and is quite low in intensity. Dispersed emission spectra recorded upon excitation at each of the peaks within the ZPL coincide perfectly among each other. Moreover, the leading peak in the dispersed emission coincides with the leading tiny peak of the multiplet in the excitation spectrum. Thus, the first tiny peak resembles the electronic band origin of perylene in helium droplets which contrasts to the assignment reported in the first paper on perylene in helium droplets [30] where the signal to noise limit did not allow for detecting the leading tiny peak.
Further important experimental details have surfaced within the ZPL doublet of tetracene [19]. Upon increased spectral resolution a substructure could be resolved for the more intense β line. In contrast, the spectral shape of the α line remained smooth, however, clearly asymmetric in shape. Within the substructure of the β line, 7 sharp peaks were resolved exhibiting irregular spectral separation among each other as shown in Fig. 5.6 as black line. An attempt to fitting this substructure by an asymmetric rotor with anisotropic angular momentum caused by the pick-up process did not provide convincing results [19, 31]. In case the model of an anisotropic rotor should explain the fine structure, an explanation for the missing of a corresponding fine structure for the alpha line would be needed. Very important in this context was the singly peaked ZPL of pentacene—shifted to the red of the tetracene resonance by roughly 3750 cm−1 as to be expected for an additional carbon ring unit—which shows exactly the same fine structure as recorded for the β line of tetracene [19]. Consequently, this line of pentacene was assigned as β line. It is added in red to the spectrum of tetracene in Fig. 5.6. The spectral identity of the beta line among tetracene and pentacene is another argument against an assignment to rotational fine structure as proposed in Refs. [19] and [31]. It is certainly helium induced and reveals properties of microsolvation which need to be common to tetracene and pentacene. Unfortunately, for anthracene, naphthalene, and benzene the spectral resolution did not suffice to check for a fine structure as resolved for the β line of tetracene and for pentacene.

Adapted from Ref. [19]
High resolution electronic spectrum of the ZPL at the electronic band origin of tetracene (black) and pentacene (red) in superfluid helium droplets (\(\overline{N }\) = 16,000).
An additional important detail came from the investigation of the droplet size dependence of the alpha and beta line of tetracene and in particular of the fine structure of the beta line [19]. As was discussed above already for glyoxal, even the limited range for the average droplet size accessible under subcritical expansion conditions can become effective on the line shape. For tetracene, the droplet size dependence has been investigated for both, the subcritical and the supercritical regime of the droplet source which was accomplished by recording fluorescence instead of depletion as depicted in Fig. 5.7 [19]. The spectral response upon variation of the average droplet size within the subcritical regime was a maximum solvent shift of about 100 cm−1 to the red. Upon transition from sub- to supercritical expansion conditions this solvent shift took a mild turn around to the blue while maintaining the internal gap of 1 cm−1 between the α and the β line (cf. Fig. 5.7 panel (d)). A closer look at the response of the fine structure of the β line upon changing the average droplet size reveals the following details. The fine structure does not shift upon variation of the droplet size as emphasized by dashed vertical lines in Fig. 5.7. Instead, for increasing droplets size the fine structure of the β line passes a maximum intensity and fades away for supercritical expansion conditions at the droplet source. From there on the spectral shape of the β line becomes smooth similar as found for the α line. Its spectral shape is still asymmetric and the peak shifts gradually to the blue with further increasing droplet size as does in parallel also the α line. It should be noted that this blue shift is in the order of the spectral width of the β line and, thus, tiny compared to the overall helium induced red shift of roughly 100 cm−1. Not only the turnaround but in addition the curious disappearance of the fine structure in the β line reveals a fundamental difference for helium droplets generated under subcritical or supercritical expansion conditions. Thereby, one needs to consider, that in the vicinity of the transition from subcritical to supercritical expansion conditions a bimodal droplet size distribution is obtained which may be an expression of instabilities in the droplet source.

source temperatures T0 (from top to bottom). The dashed vertical lines mark the α line and the two most pronounced peaks in the fine structure of the β line in the smallest droplets. Adapted from Ref. [19]
Fluorescence excitation spectra with the α and β line recorded for increasing average droplet size \(\overline{\mathrm{N} }\) obtained by decreasing
Finally, it should be noted that none of the PWs of all of these PAH compounds discussed above fits to the spectral shape of elementary excitations of superfluid helium as reported for glyoxal. Even though it reveals what is expected, the PW recorded for glyoxal is an exception in reflecting the spectral pattern of elementary excitations of superfluid helium as suggested in Fig. 5.3. Under the assumption of a non-superfluid helium solvation layer the PW may be dominated by excitations of the non-superfluid helium solvation layer which is evidenced by rather sharp spectral features already within the spectral section of the phonon gap of superfluid helium. It is rather surprising that the solvation layer of glyoxal evidenced by increased moments of inertia remains consealed within the PW.
In view of all the helium induced spectral features reported for the ZPL at the electronic band origin of tetracene in helium droplets an empirical understanding of microsolvation does not suffice for an explanation. The droplet size dependence exhibiting a turn-around in the solvent shift, the fine structure in the beta line which was identically resolved for pentacene, and, last but not least, the changing line shape upon switching from subcritical to supercritical expansion conditions, do not fit to empirical explanations of helium induced spectral features in electronic spectra.
5.3.3 Phthalocyanine in Superfluid Helium Droplets
Phthalocyanine belongs also to the first samples of organic molecules which were investigated by means of fluorescence excitation spectroscopy in superfluid helium nanodroplets [32]. At the electronic band origin which undergoes a helium induced shift of about 42 cm−1 to the red, a sensitivity of the singly peaked ZPL on the droplet size distribution was immediately recognized. The accompanying PW peaks about 3.8 cm−1 to the blue from the ZPL which is within the range of the phonon gap of superfluid helium. Within a spectral section of roughly 3 cm−1, numerous side maxima are grouped around this most intense center peak of the PW. This spectral substructure indicates a quantized energy level structure as expected for a rather rigid helium solvation layer. Moreover, an investigation of the line shape of the PW at the electronic band origin, its missing response on variation of the droplet size distribution, and corresponding dispersed emission spectra provide strong evidence for the presence of a helium solvation layer and, thus, provide further insight to microsolvation in superfluid helium. Much of the information to be discussed could only be obtained under appropriate spectral resolution, whereby saturation broadening had to be avoided.
The spectral shape of the ZPL at the electronic band origin is asymmetric with a steep rise at the red edge and a tail extending to the blue towards the gas phase resonance frequency [32–34]. Under subcritical expansion conditions in the droplet source, the asymmetry of the ZPL expressed mainly by the spectral width of the tail to the blue decreases with increasing average droplet size and the peak position of the ZPL shifts to the red edge as shown in Fig. 5.8 for temperatures from 15 to 10 K. This behavior reminds of the investigation of electronic transition frequencies under the influence of a finite sized polarizable environment which could be simulated by the so-called excluded volume model [35]. Adapted to the effective size distribution of singly doped helium droplets, the asymmetric line shape of phthalocyanine in helium droplets and its development under variation of the droplet size distribution could be simulated quantitatively [34]. Moreover, the simulation procedure could be applied upside down to deduce the droplet size distribution from the line shape of the ZPL. In this model, the solvent induced shift of electronic transitions of a dopant species is the result of the solvent to solute dispersion interaction whose influence on the dopants energetics is of finite reach. Beyond a certain droplet size, the dopant species does not sense anymore the finite dimension of the droplet. For all droplets exceeding this limit, the dopant experiences bulk conditions. Beyond this limit inhomogeneous line broadening is expected to vanish even though there are still droplets with a broad size distribution.

source temperature indicated in each panel. Helium stagnation pressure was 20 bar. For further details see text. Adapted from [36]
Fluorescence excitation spectra of the ZPL at the electronic band origin of phthalocyanine in helium droplets recorded for increasing average droplet size (from bottom to top) as determined by the droplet
Beyond the bulk limit, the line shape of the ZPL is expected to be dominated by the rotational fine structure of the solvated dopant species. In the case of phthalocyanine accessing the bulk-limit requires droplet sizes of at least 106 helium atoms generated upon supercritical expansion conditions. Experimental results for the ZPL line shape reveal vanishing of the asymmetry upon approaching the transition from subcritical to supercritical expansion conditions in the helium droplet source [33, 34, 36]. Far beyond this limit a very sharp double peak structure is resolved (cf. Fig. 5.8 top panel) which was fitted by the envelope of the rotational band structure calculated for an oblate symmetric top rotor which is an approximative guess for phthalocyanine [37]. As obtained for numerous molecules in helium droplets, also for phthalocyanine the moments of inertia had to be increased by roughly a factor of three compared to the gas phase values. Unfortunately, the rotational fine structure resolved for phthalocyanine in helium droplets beyond the bulk limit is not line resolved [37]. Since the P, Q, and R-branch are merged, it is impossible to deduce the moments of inertia with high precision. Thus, the constants deduced in Ref. [37] represent a perfectly planar symmetric top rotor which is in contradiction not only to the asymmetric top rotor type of phthalocyanine but in addition to the non-planarity of a phthalocyanine helium solvation complex. Nevertheless, the consistency of the rotational band simulation with the experiment could be reached [37].
An important detail in the development of the line shape with the droplet size distribution is counterintuitive to the alleged consistency of helium droplets with the simple model of a polarizable environment. Besides the vanishing inhomogeneity observed for droplets generated under supercritical expansion conditions the ZPL experiences a tiny but clearly measurable shift to the blue [38] (cf. top panel Fig. 5.8) similar as discussed above for tetracene. Such a turn-around of the solvent shift is not expected from the excluded volume model [35]. In the case of tetracene the turnaround was also observed upon passing the limit from subcritical to supercritical expansion conditions in the droplet source so that the turn-around of phthalocyanine confirms fundamental differences in the properties of droplets generated under the two different expansion conditions. Instead, for glyoxal a blue shift was observed already within the subcritical regime of the droplet source. Nevertheless, the success of the excluded volume model applied to simulate the line shape of the ZPL of phthalocyanine in helium droplets generated under subcritical expansion conditions is quite convincing. So far, none of the spectroscopic signatures of phthalocyanine—neither in the ZPL nor in the PW – except of the turn-around of the solvent shift require an explanation based on features characteristic for a quantum fluid. This, however, should not be misunderstood as an indication against superfluidity of helium droplets. The effect of inhomogeneous line broadening as an expression of the droplet size distribution is expected to be ubiquitous throughout an electronic spectrum. Certainly, its strength depends on the van der Waals interaction which is not only dependent on the dopant species but may even be state-specific.
In contrast to the electronic band origin, the line shape at the ZPL of vibronic transitions of phthalocyanine in helium droplets was perfectly Lorentzian as shown in Fig. 5.9 [39]. For those transitions recorded in droplets with average size of 20,000 helium atoms, nothing reminds of the asymmetry due to inhomogeneous line broadening. The corresponding line width varies from peak to peak and can be transformed into the life time of the excited state. Obviously, for the ZPL of vibronic transitions of phthalocyanine the life time of the excited vibronic state is line shape determining and dominates over inhomogeneous line broadening. The average life time of vibronic levels of phthalocyanine in helium droplets was determined to about 15 ± 8 ps for vibrational modes up to 1000 cm−1. No correlation was found between life time and vibrational energy [39]. Beyond 1000 cm−1 the vibrational fine structure of phthalocyanine is obscured by contributions from a second electronically excited state which perturbs the line shapes. The radiative decay time of the first electronically excited state of phthalocyanine is in the order of 10 ns which is three orders of magnitude larger than the life times revealed by Lorentzian line shapes of the vibronically excited states in helium droplets as shown in Fig. 5.9. The missing correlation of the line widths or corresponding life times with vibrational energies speaks against direct dissipation of the vibrational energy into the helium droplet as the life time limiting process. More likely, the life time of the excited states is limited by internal vibrational redistribution (IVR) prior to energy dissipation into the helium droplet followed by radiative decay. Thus, the life times of vibrational states as deduced from the Lorentzian line widths reflect the mode specific IVR probabilities potentially modified by the helium environment. A convincing prove for bad coupling of high energy modes to the helium bath and the promotion of dissipation via IVR came from vibrationally excited hydrogen fluoride (HF) inside helium droplets [40]. Instead of recording a depletion as a result of evaporative cooling after dissipation of the almost 4000 cm−1 of rovibrational excitation the bolometer recorded an accretion from a rovibrationally excited HF molecule.

Fluorescence excitation spectra of ZPL at vibronic resonances of phthalocyanine in helium droplets (\(\overline{N }\)=20,000) (black line) fitted by Lorentzian line shapes (red line). Vibrational frequencies and Lorentzian line widths are added in cm−1. Adapted from [39]
Valuable information on microsolvation of phthalocyanine in helium droplets and in particular on the presence and nature of the solvation layer came from dispersed emission spectra. As known already from matrix isolated molecules, radiative decay of electronically excited molecules in helium droplets originates only and exclusively from the ground level of the first electronically excited state independent of the initially excited level. Thus, radiative decay of vibronically excited dopant molecules is preceded by the dissipation of excitation energy in excess to the electronic band origin. Energy dissipation from the dopant species into the helium droplet reactivates evaporative cooling of the droplet which is the process depletion spectroscopy is based on. Coming back to the valuable information, for phthalocyanine in helium droplets dual emission was found. In addition to the expected emission spectrum coincident in its band origin with the excitation spectrum a second spectrum appeared [41]. The second emission spectrum was identical with respect to Franck–Condon factors (FCF) and vibrational frequencies to the first/expected whereas its helium induced red shift was increased by additional 10.8 cm−1. Its contribution to the integral emission intensity started with about 1.4% upon excitation at the electronic band origin, and increased monotonously with increasing excess excitation energy (cf. Fig. 5.10 from top to bottom). At about 1000 cm−1 of vibrational excess energy its contribution reached roughly 70% and for an excess energy of 15,000 cm−1 it contributed with 98% to the integral emission [39, 41]. Identical vibrational frequencies and FCF in the second spectrum reveals identical dopant species. The only difference is the solvent induced red shift of the electronic transition energy which has increased by roughly 25%. This type of dual emission is a strong indication for two configurations of a rigid helium solvation layer [41]. Prior to emission the decay path including dissipation of vibrational excess excitation energy bifurcates, one of which proceeds without and another with relaxation of the helium solvation layer of the electronically excited dopant species. Thereby, the amount of excess excitation energy drives the relaxation probability of the solvation layer. A four-level scheme summarizes these experimental findings which is shown in Fig. 5.11e. For both electronic states of phthalocyanine involved in the observed electronic transition, the helium solvation layer appears in two configurations. However, the global minimum in the ground state becomes the local minimum in the electronically excited state and vice versa. In both electronic states, relaxation into the corresponding global minimum configuration of the helium solvation layer is possible. The relative intensity of the second emission spectrum images the relaxation probability into the global minimum configuration of the helium solvation layer for the electronically excited dopant. In contrast to the life time read from Lorentzian line widths discussed above, the relaxation probability correlates with the excess excitation energy. Moreover, the correlation between relaxation probability and excess excitation energy is indicative for a barrier between the two helium solvation layer configurations in S1.

Dispersed emission spectra of phthalocyanine in helium droplets recorded upon electronic excitation at the electronic band origin (panel (a)) and at vibronic transitions as indicated in panels (b) to (d). All spectra are plotted twice in order to emphasize the low intensity vibronic transitions. Adapted from [38] and [41]

Left side: Fluorescence excitation spectrum (a), dispersed emission (b), and pump-probe spectrum (c) in the vicinity of the electronic band origin of phthalocyanine in helium droplets. Right side: pump-probe signal intensity (black dots) as function of the time delay between pump and probe laser and fit of exponential decay convoluted with Gaussian beam shape function (d). Four-level scheme explaining dual emission of phthalocyanine in helium droplets (e). For details see text. Adapted from [39]
A pump-probe spectrum shown in panel (c) of Fig. 5.11 reveals a similarly sharp and asymmetric line shape for the excitation of the metastable solvation complex (transition |4 > → |3 > in Fig. 5.11e) as for the stable configuration (transition |1 > → |2 > in Fig. 5.11e) shown in panel (a) of Fig. 5.11. Panel (b) of Fig. 5.11 shows dispersed emission upon excitation with roughly 15,000 cm−1 excess excitation energy that has been the pump process for efficient populating of level |3 >. The integral intensity of the positive peak in panel (c) recorded under variation of the delay time between pump and probe laser is shown in panel (d) of Fig. 5.11. Since pump and probe laser operated in continuous wave mode a time delay was accomplished by shifting the pump laser towards the droplet nozzle. After deconvolution of the gaussian overlap profile of pump and probe beam, a life time of 5.2 μs could be deduced [39]. Since the radiative life time of the electronically excited state in the order of 10 ns is negligible, the 5.2 μs reveal the life time of the metastable solvation complex of phthalocyanine in the electronic ground state. Compared to the pico-second time regime for thermalization of hot dopant species [2], the 5.2 μs life time is quite long and provides evidence for the barrier to the global minimum |1>. On the other hand, it is short enough to complete relaxation in the time between the pick-up and a spectroscopic investigation which explains the missing of corresponding signals in the fluorescence excitation spectrum. As mentioned above, the increase of the helium induced red shift of the electronic transition is surprisingly large. This difference relates to the difference in the electron density distribution of both electronic states of phthalocyanine as depicted schematically in Fig. 5.11e. The spectral response of dual emission is an experimental detail that reflects the charge density distributions and its change upon electronic excitation as sensitized by the helium environment. It serves as a bench mark for theoretical modeling of helium solvation. The observation of dual emission and the sharp peak signal of the metastable solvation complex as recorded in the pump probe experiment speaks for a rather rigid solvation complex most probably sandwich-like and, therefore, speaks also for a non-planar solvation complex. This is an important detail for the simulation of alleged rotational bands discussed in [37].
Additional evidence for the existence of two sandwich-like configurations of a solvation complexes for phthalocyanine in helium droplets came from path-integral-Monte-Carlo simulations (PIMC) [42]. Besides the global minimum configuration with a layer consisting of 24 helium atoms on each side of the planar dopant and both with almost perfect hexagonal structure, a metastable configuration was found with one of the two layers changed to a configuration rather commensurate to the corrugation of the phthalocyanine surface.
Similar multiplet splittings were reported for dispersed emission spectra of Mg-phthalocyanine, and a 1:1 cluster of phthalocyanine and argon in helium droplets [38]. Thereby, for Mg-phthalocyanine the second emission spectrum was shifted by 12.5 cm−1 to the red. Its relative intensity was 90% already without excess excitation energy and approached a value of 99% for an excess excitation energy of only 250 cm−1. According to a triplet splitting in the dispersed emission spectrum the Ar cluster of phthalocyanine exhibits even two additional metastable configurations. Upon increasing the excitation energy, the variation of the intensity profile within the triplet revealed a cascade of two consecutive relaxation steps. In the case of AlCl-phthalocyanine, both the excitation spectrum and the dispersed emission showed doublet features which, however, did not exhibit relaxation among each other [43, 44]. It speaks for an insurmountable barrier between the two systems. As the icing on the cake of the experimental evidence for the relaxation model of a solvation complex, a peak probability for relaxation was found upon excitation with less than 10 cm−1 excess excitation energy, but now injected directly into the mutually relaxing object via excitation of the PW [21]. In purely classical terms, one can conclude that shaking directly on the solvation layer drives the relaxation particularly efficiently even though the excess excitation energy is about a factor of 40 smaller than the smallest vibrational excess energy pumped into the dopant.
According to the empirical interpretation, the doublet in the emission of phthalocyanine is related to the doublet observed in the excitation of tetracene discussed above. Both are explained by the presence of different configurations of a helium solvation layer with the difference that tetracene does not allow for relaxation among the configuration. As seen for tetracene for the first time, multiplet splitting at a ZPL was observed for numerous other dopant species. These multiplet structures are generally understood as the presence of configurational variants of a helium solvation complex. The formation of a solvation complex accompanies helium solvation, however, with dopant specific expression concerning the number of configurational variants and the relaxation dynamics among them. Thereby, relaxation is certainly a response on the change of the electron density distribution upon electronic excitation.
5.3.4 Porphin in Superfluid Helium Droplets
Structurally related to phthalocyanines are porphyrin derivatives. Porphin was also among the first dopant species investigated by electronic spectroscopy in helium droplets [11, 32]. The ZPL at the electronic band origin of porphin in helium droplets exhibits a triplet consisting of a leading intense peak followed to the blue by two tiny peaks at an excess energy of 0.4 cm−1 and 0.7 cm−1, respectively [25]. The intensity profile within the triplet from red to blue was about 10:2:1. The PW is right in the middle of the phonon gap of superfluid helium and exhibits a fine structure qualitatively similar to that of phthalocyanine [11, 25]. Most probably, the ZPL triplet reveals three different configurations of a porphin helium solvation complex. The low oscillator strength and low fluorescence quantum yield of porphin prohibited an investigation of relaxation dynamics by means of dispersed emission spectra.
Similar as discussed for phthalocyanine, the line shape of the ZPL at the electronic origin of porphin is asymmetric, varies with the droplet size distribution, and, therefore, suffers inhomogeneous broadening [45]. However, the steep edge is at the blue side and a tail expands to the red which is inverted compared to the asymmetry found for phthalocyanine. The excluded volume model applied to log-normal droplet size distributions reveals a sharp edge marking the bulk limit of a solvent shift and a tail pointing towards the resonance of the isolated molecule. Accordingly, the inverted asymmetry is indicative for a helium induced solvent shift to the blue. And in fact, applying the excluded volume model with a blue shift the line shape and its dependence on the droplet size distribution can be simulated [45]. As closed shell organic molecule porphin is heliophilic in both electronic states. Whether the solvent shift is to the blue or to the red depends on the difference of the stabilization energy of the two electronic states involved in the transition. Thus, for heliophilic dopant species both a red or a blue shift is possible. What concerns porphin, the option of a helium induced blue shift as suggested by the inverted asymmetry in the line shape is refuted by the gas phase spectra from two independent sources revealing consistently a helium induced red shift of about 8 cm−1 [46, 47].
Another remarkable feature in the droplet size dependence of the line shape was a sharp peak about 0.04 cm−1 in width right on top of the inhomogeneous broadened ZPL [45]. It was observed for average droplet sizes below 10,000 helium atoms. Upon increasing of the droplets size, this peak remained fixed in frequency while the broad and asymmetric part of the ZPL shifted to the blue. Only the peak intensity did vary with the droplet size. This behavior reminds of the response of the fine structure resolved at the beta line of tetracene and on its response on the droplet size (cf. Sect. 5.3.2).
Porphin exhibits a helium induced solvation shift to the red without any doubt. Even though, the asymmetry in the line shape of the ZPL at the electronic band origin and its response to the variation of the droplet size distribution fits to the excluded volume model for a dopant system with a helium induced blue shift. A mistaken assignment of the spectrum to porphin instead to chlorin which is a side-product of the synthesis of porphyrins can safely be excluded. A turnaround of the solvent shift to the blue as discussed above for tetracene and phthalocyanine might be considered as the reason for an inverted asymmetric line shape. However, in this case the turn-around effect of porphin needs to surpass that for phthalocyanine or tetracene by orders of magnitude. Moreover, in contrast to tetracene and phthalocyanine the turnaround of the solvent shift should occur already within the subcritical expansion conditions and far from the transition to supercritical conditions in the droplet source. Thus, the blue shift observed for porphin is qualitatively rather similar to the blue shift reported for the rotationally resolved ZPL at the electronic band origin of glyoxal in helium droplets. Both are recorded within the subcritical expansion regime far from the transition to supercritical conditions at the helium droplet source.
5.3.5 Summary
Electronic spectra of glyoxal, tetracene and related PAH-compounds as well as phthalocyanine and porphin provide strong evidence for the presence of a helium solvation layer rigidly bound to the dopant species. Hence, spectroscopy of molecules doped into helium droplets deals with solvation complexes rather than the bare molecule. Solvation complexes differ from the bare molecule, most evident, in the moments of inertia. Depending on the size and shape of the molecules, several configurations of the solvation complex are to be expected which distinguish in the electronic transition energy as expressed by multiplet splitting at the ZPL and, case dependent, by a relaxation dynamic initiated via electronic excitation. In contrast, vibrational degrees of freedom remain almost unchanged by helium solvation and are insensitive to configurational variants of the helium solvation layer. In addition to the helium solvation layer, the entire droplet body has an influence on the electronic transition frequency of the dopant. In contrast to the solvation layer, which needs to be treated as a quantized multi-particle system of helium atoms attached to the dopant, the droplet body can be treated as a quasi-continuous and polarizable environment. The shift of electronic transition of a dopant depends on the thickness of the polarizable environment. Thus, a distribution of droplet sizes becomes effective in inhomogeneous line broadening. Inhomogeneous line broadening as an explanation for the asymmetry in the line shape of the ZPL works perfectly for phthalocyanine. However, it is put into question by the inverted asymmetry observed for porphin. While the droplet size dependence of the line shape is unquestionable, the excluded volume model does not suffice to explain line shapes in general. Most evident, the phenomenon of a turn-around in the solvent shift does not fit into the excluded volume model. While tetracene an phthalocyanine reveal a correlation between turn around and transition from subcritical to supercritical expansion conditions glyoxal and porphin exhibit turn around within the subcritical regime. Thus, the presence of a dopant helium solvation complex appears to be reasonable. However, several of the helium induced spectroscopic features are not understood.
5.3.6 Low Energy Torsional and Bending Modes in Electronic Spectra of Molecules in Helium Droplets
Dissipation of energy is a key feature accompanying solvation of molecules in helium droplets. Besides the practical benefits of depletion spectroscopy, it is the mechanism that cools molecules in helium droplets to a temperature of 0.38 K within picoseconds. As discussed above, the efficiency of energy dissipation is based on the coupling of the dopant’s vibration to the helium droplet which decreases with increasing vibrational energy as exemplified by vibrationally excited HF molecules inside helium droplet [40]. As outlined above for phthalocyanine (cf. Sect. 5.3.3), the efficiency of energy dissipation may be mediated by IVR into low energy modes prior to energy transfer to the helium droplet. Ligands such as methyl-, buthyl-, ethyl-, phenyl-, cyano-moieties providing low energy torsional or bending modes or simply heavier atoms reducing vibrational frequencies may serve as acceptor modes for IVR and, thereby, promote dissipation of energy into the helium droplet. On the other hand, low temperature conditions in helium droplets are expected to be favorable for resolving vibrational progressions of such low energy modes. At 0.38 K these progressions are free of hot bands and reveal configurational changes accompanying electronic excitation. Electronic spectra of substituted derivatives of phthalocyanine and porphyrin, pyrromethene dyes, and anthracene have been recorded in helium droplets in order to study the influence of the substituents on the dopant to helium interaction.
As explained in Sect. 5.2. electronic spectra recorded as fluorescence excitation or dispersed emission of molecules in helium droplets reveal vibrational frequencies of electronically excited states and of the electronic ground state, respectively. The helium induced shift of vibrational frequencies is on the order of ±1%. A more remarkable effect is line broadening as reported for low energy modes roughly below 300 cm−1 [32]. Low vibrational frequency correlates with large vibrational amplitude which may suffer damping by a solvent. Moreover, the presence of low energy modes might promote IVR and, thus, promotes energy dissipation from high energy modes into the helium droplet. In rather classical terms, the substituents can be seen as kind of antenna supporting the communication between dopant and helium droplet. This was investigated for several substituted derivatives by means of electronic spectroscopy.
5.3.6.1 Low Energy Torsional and Bending Modes of Phthalocyanine and Porphin Derivatives in Helium Droplets
Among the phthalocyanine derivatives the electronic spectrum of 2,9,16,23-tetra-tert-butylphthalocyanine (TTBPc) has been examined [48]. At each of the four six membered rings a tert-buthyl-group substitutes one of the hydrogen atoms in the named position. Besides a vibrational fine structure attributable to the phthalocyanine core unit no additional low frequency progressions of torsional or bending modes of the tert-butyl-moieties appeared in the fluorescence excitation spectrum. However, the ZPL at the electronic band origin appeared split into roughly 20 peaks within the first 3 cm−1 each as narrow as 0.1 cm−1. This large number reveals configurational variants not only with respect to the helium solvation layer. More likely, this compound by itself exhibits stereo isomers which at a temperature of only 0.38 K are spectrally well separated. However, no low energy progressions due to the substituents are observed in the fluorescence excitation spectrum. If at all, the influence of low energy modes of the substituents is a secondary effect observable in vibronic transitions involving normal modes of the phthalocyanine core unit. Below 400 cm−1 the low energy modes of the core unit are significantly attenuated compared to the unsubstituted molecule. Moreover, the line widths of all vibronic resonances exceed that of the electronic band origin by at least an order of magnitude which was not the case for unsubstituted phthalocyanine. Both effects can be rationalized by IVR promoted by the presence of low energy modes of the substituents which is followed by energy dissipation into the helium droplet. The missing of progression of these low energy modes reveals almost identical configuration of the substituents in both electronic states. To our best knowledge, corresponding spectra from the gas phase are not available for this phthalocyanine derivative.

A larger variety of substituted derivatives has been investigated for porphin [25, 48]. These derivatives are specified by antenna like substituents of alkyl type such as methyl, ethyl, buthyl, or combinations of these moieties situated either at methine or pyrrole sites in the periphery of the planar porphin body. Among them are 5,15-diphenylporphin, 5,10,15,20-tetraphenylporphin, 5,10,15,20-tetramethylporphin, 5,10,15,20-tetrapropylporphin, and 2,7,12,17-tetraethyl-3,8,13,18-tetramethylporphin. All of these derivatives carry two or four substituents situated in such a way that inversion symmetry is maintained. To cut the story short, none of these derivatives show progressions of low energy modes as expected for torsional or bending modes of the corresponding substituents. Therefore, electronic excitation apparently maintains the steric arrangement of the substituents. Line broadening of high frequency vibronic transitions as a signature of promotion of IVR was rather insignificant [25]. In all cases, saturation broadening easily obtained by peak intensities of pulsed dye lasers was a serious side effect and gave an impression of broadened spectra as if they were dominated by the envelop of unresolved low energy progressions [25]. However, by using the moderate photon flux of a cw-dye laser the line widths shrank to an order of 0.1 cm−1 and all the spectrally broad signals vanished entirely. Thus, the broad spectral features can safely be assigned to PW [25]. Under these conditions, rich multiplet splitting of the ZPL at the electronic band origin was resolved. As in the case of the phthalocyanine derivative, the multiplet reveals not only variants of the solvation complex but in addition stereoisomers of the porphin derivative.
One of the porphin derivatives, namely tetraphenylporphin is an interesting example to demonstrate how saturation broadening misleads the interpretation of electronic spectra. Upon saturation broadening the electronic spectrum obtained after doping with a commercial sample of this molecules was dominated by PW which within the first 100 cm−1 exhibits a spectral substructure consisting of a kind of triple-peak feature which then merges into a broad and constantly decreasing signal to the blue [49] as depicted by the upper trace in Fig. 5.12. This feature repeats for two vibronic transitions within the spectral range shown in Fig. 5.12. Upon reducing the photon flux, most of the spectrally broad signal vanishes (cf. lower trace in Fig. 5.12). Now the signal peaks at the electronic band origin and at the two vibronic transitions. At a closer look (cf. inlay in Fig. 5.12), the electronic band origin reveals a ZPL with a single intense peak only 0.05 cm−1 in width [25, 48]. Furthermore, after a gap of about 1 cm−1, a series of similarly sharp peaks follows. Upon increasing the laser intensity, the unstructured PW grows in starting with the same 1 cm−1 gap to the leading peak. As discussed in Refs. [25, 48], the spectroscopic details speak for an assignment to a PW except of the leading sharp and intense peak. Furthermore, there are no contributions of low energy progressions of the four phenyl moieties which speaks for a rigid derivative with almost identical structure in both electronic states. This contrasts to the interpretation of the saturated spectrum as the signature of a floppy molecule [49]. Above all the spectra shown in Fig. 5.12 do not originate from tetraphenylporphin but instead from the corresponding chlorin derivative [25, 48] wel known as side product of the synthesis of porphin derivatives.

5.3.6.2 Low Energy Torsional and Bending Modes of Anthracene Derivatives in Helium Droplets
Various derivatives of anthracene have been investigated by means of electronic spectroscopy in helium droplets. Among them are 9-phenylanthracene, 9-cyanoanthracene, 9-chloroanthracene, 9,10-dichloroanthracene, and three methylated derivatives, namely, 9-methylanthracene, 1-methylanthracene, and 2-methylanthracene [50, 51, 52, 53]. For some of these derivatives corresponding gas phase data have been published [54–63]. Within this series there are four singly substituted derivatives carrying substituents such as a chloro-, cyano-, methyl-, or phenyl-moiety in all cases at the 9-position. Moreover, there are tree derivatives singly substituted with a methyl group at position 9, 1, or 2. Among the singly methylated derivatives the electronic spectra of 1-methylanthracene and 9-methylanthracene do not show low energy torsional or bending progressions neither in the gas phase nor in helium droplets [50] (cf. Fig. 5.13b, c). Obviously, the steric configuration of the methyl substituent is identical in both electronic states. These two anthracene derivatives show rather similar vibronic structure as recorded for anthracene shown in panel (a) of Fig. 5.13. Similar observations were made for 9-cyanoanthracene, 9-chloroanthracene, and 9,10-dichloroanthracene.

Fluorescence excitation spectra of anthracene derivatives in helium droplets (\(\overline{N }\) = 20,000): a anthracene, b 1-methylanthracene, c 9-methylanthracene, d 2-methylanthracene, and e 9-phenylanthracene. The wavenumber scale is related to the corresponding origin given in each panel. In d and e, molecular beam spectra are added in red whose electronic band origin was shifted to coincide with the helium droplet experiment. Vertical lines mark the origin and two prominent vibronic transitions of non-substituted anthracene. Adapted from [50–52]
In contrast, extended low energy progressions were observed for 2-methylanthracene in the gas phase and in helium droplets [50] (cf. Fig. 5.13d). At low temperatures these progressions reveal a twist of the methyl-substituent upon electronic excitation. Remarkably, however, in helium droplets significant line broadening throughout the low energy progressions for 2-methylanthracene was observed. A convolution of the gas phase spectrum with a line broadening function revealed almost perfect coincidence with the helium droplet spectrum. Such a coincidence proves for identical torsional frequencies in both experiments. Identical frequencies reveal identical torsional mass and, thus, no helium atoms attached to the methyl substituent. Moreover, the best fit was obtained for Lorentzian type line broadening an indication for helium induced reduction of the life time of the torsional mode. The low frequency torsion exhibits rather large amplitude motion which accomplishes efficient coupling to the helium environment. At this point it needs to be mentioned that line broadening was observed throughout the entire progression. We will come back to this issue below in the discussion of pyrromethene dye molecules. Similar line broadening was recorded for 9-phenylanthracene shown in panel (e) of Fig. 5.13 which was explained accordingly.
Slight modifications of the intensity pattern were observed in the helium droplet spectra. The process of dissipation of excess excitation energy into the helium droplet may become influential on the decay path of the excited dopant. As a consequence, the fluorescence quantum yield and, thus, the intensity recorded by means of fluorescence excitation might differ in helium droplets compared to gas phase [50, 53]. In the best case, dark states exhibiting a non-radiating relaxation for the isolated molecule can be bypassed to a fluorescent decay path. In the worst case, the bypassing may extinguish radiative decay.
5.3.6.3 Low Energy Torsional and Bending Modes of Pyrromethene Dyes in Helium Droplets
Dominant torsional and bending modes of aryl-, alkyl-, phenyl- and cyano-substituents in electronic spectra are known for borondipyrromethene dye molecules. Supersonic jet spectra revealed spectrally well resolved and extended low frequency progressions [64, 65]. Even the non-substituted 4-boro-3a,4a-diaza-s-indacene or borondipyrromethene (BDP) shows a progression of a flopping mode of the BF2 unit. According to extended progressions of low energy modes recorded from cold samples, electronic excitation of these derivatives is accompanied by significant rearrangement of the steric configuration of the substituents. In addition to BDP the following derivatives have been investigated in the gas phase as well as in helium droplets by means of electronic spectroscopy: 8-phenylpyrromethene-difluoroborat (8-PhPM), 1,3,5,7,8-pentamethylpyrromethene-difluoroborat (PM546), 1,3,5,7,8-pentamethyl-2,6-diethylpyrromethene-difluoroborat (PM567), 1,2,3,5,6,7-hexamethyl-8-cyanopyrromethene-difluoroborat (PM650) [53, 64, 65]. Gas phase spectra were recorded from a supersonic molecular jet which allowed to even resolve the rotational band contour at the electronic band origin. Thus, the rotational temperature could be determined to about 10 K. Upon doping into superfluid helium droplets, molecules are cooled down to 0.38 K for all internal degrees of freedom. Thus, hot bands are eliminated entirely and electronic spectra should exhibit reduced spectral density compared to the gas phase. This, however, was only the case for BDP where the band of the BF2 flopping mode was missing. All the other derivatives undergo a counterintuitive development from spectrally resolved progressions of low energy modes in the gas phase to a kind of envelope of these progressions recorded in helium droplets as shown in the left column of Fig. 5.14 for three of the substituted pyrromethene derivatives. Similar as discussed above for 9-phenylanthracene and 2-methylanthracene, the helium droplet spectra resemble a convolution of the gas phase spectra with a line broadening function. As in the case of the anthracene derivatives, the torsional and bending modes show identical frequencies in both experiments. Thus, helium solvation maintains the mass involved in torsional or bending motion.

Fluorescence excitation spectra of three pyrromethene dye molecules (PM650 (a) (d), 8-PhPM (b) (e), and PM567 (c) (f)) recorded in helium droplets (black line) compared with corresponding spectra recorded in a molecular beam (grey line). Right column zooms into the electronic band origin for which a fine structure was resolved in helium droplets (red section). Adapted from [44]
However, at a closer look depicted in the right column of panels in Fig. 5.14, one can recognize that for all of the pyrromethene derivatives the ZPL at the electronic band origin shows a spectrally well resolved multiplet with line widths in the order of 0.1 cm−1. Only this particular ZPL at the electronic band origin is excluded from line broadening. In contrast to vibronic excitations the electronic band origin does not carry excess excitation energy and, therefore, no energy dissipation takes place. This observation is a strong indication for line broadening due to reduced excited state life time accomplished by highly efficient energy dissipation.
Coming back to 9-phenylanthracene and 2-methylanthracene, line broadening was also observed at the electronic band origin. Therefore, energy dissipation cannot be responsible for line broadening in the spectra of anthracene derivatives. Either intramolecular processes such as internal conversion (IC) or intersystem crossing (ISC) are involved and significantly promoted by the helium environment or the line broadening is an expression of severe perturbation of the closer helium environment induced by electronic excitation of the anthracene derivatives. For the two anthracene derivatives, the concomitant change of the electron density distribution enforces the substituent, either a methyl or a phenyl moiety, to undergo a twist of 60° and 30°, respectively [50, 66] as deduced from the Franck–Condon-pattern of the corresponding torsional progression. It is rather unlikely that such a change of the electron density distribution will not be sensitized in addition by the helium environment.
5.3.6.4 Summary
In summary, the substituted derivatives of phthalocyanine and porphin, anthracene, and pyrromethene have shown that helium solvation is of minor influence on the substituents and their intramolecular dynamics. Torsional and bending frequencies are almost identical as in the gas phase. Moreover, the spectral width of corresponding progressions which reflect the change of the equilibrium configuration of the steric arrangement of the substituents upon electronic excitation was also found unaltered in helium droplets. The major impact of the helium environment was line broadening. Life time broadening is certainly involved and induced by highly efficient energy dissipation particularly from low energy and large amplitude torsional and bending modes into the helium droplet. However, a perturbation of the entire solvation complex, namely, the helium solvation layer covering the dopant cannot be ruled out. To our best knowledge, there is no example where spectral resolution of low energy and large amplitude vibrational, torsional or bending modes has made profit from low temperatures in helium droplets with respect to spectral resolution.
5.4 Van Der Waals Clusters Generated in Helium Droplets
One of the exciting opportunities offered by helium droplets is the formation of clusters with well-defined stoichiometry. Although the droplet size as well as the doping process are subject to statistical distributions, individual spectroscopic signals can be assigned to clusters well defined with respect to the stoichiometry. A safe assignment of the cluster stoichiometry is particularly warranted for small clusters of less than say ten individual units which is the relevant range of sizes discussed in the following. For this range of size the number of configurational variants is an issue. The doping is accomplished via pick-up on the flight of the droplet beam through a compartment called pick-up unit providing the dopant species at a tunable particle density. Pick up via consecutive collisions of a droplet with a number of k dopant particles obeys Poisson statistics and, thus, depends on the droplet size, the dopant particle density, and the length of the path of the droplets through the pick-up unit. The underlying cluster stoichiometry can be deduced for each individual cluster signal by recording its intensity under variation of the particle density in the pick-up unit [67]. The number k of dopant particles is the fitted parameter to obtain an overlap of the experimental intensity profile with a Poisson distribution. In contrast to a standard mass spectrometer that measures the mass to charge ratio after ionizing the clusters, the procedure in helium droplets is entirely non-destructive. Thus, an unequivocal assignment of the nascent cluster size is obtained. Finally, the clusters generated in helium droplets are cooled down to the droplet temperature of 0.38(1) K for all internal degrees of freedom whereby from other sources providing clusters in the gas phase internal temperatures up to the boiling point of the clusters are obtained. The sub 1 K temperature eliminates hot bands almost entirely. Moreover, metastable cluster configurations may be stabilized. From the very beginning, it was not surprising that electronic spectra of clusters generated in helium droplets show large numbers of cluster signals arising from cluster configurations that are stabilized only in helium droplets [17] as will be discussed in the following. Thereby, electronic spectroscopy is selective not only for different stoichiometry but in addition for configurational variants within a single cluster stoichiometry.
5.4.1 Van Der Waals Clusters of Tetracene with Argon Atoms
The formation of clusters inside a helium droplet by multiple doping via consecutive pick-up has first been demonstrated by the group of J. Peter Toennies via mass spectrometric detection [67]. Not much later, hetero clusters of tetracene and argon have been investigated by means of electronic spectroscopy [17]. In contrast to previous investigations of tetracene-argon clusters generated in a seeded beam expansion [68–72], the electronic spectra from clusters in helium droplets reveal spectrally much sharper lines. Vibrational and rotational hot bands as present in a supersonic jet experiment are eliminated almost entirely in helium droplets. For clusters in helium droplets consisting of tetracene and a single argon atom the most intense signal exhibits the largest argon induced red shift. Two additional signals were identified as configurational variants of the same cluster size exhibiting reduced red shift and intensity. The stoichiometry was determined unequivocally as described above. The assignment of the signals to different conformers was substantiated by pump-probe experiments. Further insight into the individual structure of these three clusters was not revealed by the experimental data.
In Ref. [17] a number of 21 signals of clusters consisting of a single tetracene molecule and up to almost five argon atoms have been recorded within a spectral range of 150 cm−1 to the red of the electronic band origin of tetracene. The assignment of stoichiometric and of isomeric variants for clusters with more than one argon atom was based on empirical rules of additivity of argon induced red shifts as deduced from extensive gas phase studies of van der Waals clusters consisting of a single chromophore and a well-defined number of rare gas atoms [71, 72]. For these clusters, helium droplets provide a unique option for further analyzing the cluster structure which allows to validate these empirical rules. The option for cluster analysis goes beyond an assignment of the stoichiometry and is based on variation of the pick-up sequence of the chromophore and the rare gas atoms. Upon initial doping with rare gas atoms, the chromophore approaches a rigid rare gas cluster inside the helium droplet and most probably attaches to the cluster surface. The favored cluster configuration is single-sided in the attachment of rare gas with respect to the chromophore. Upon changing the pick-up sequence, the chromophore inside the helium droplet attracts one rare gas atom after the other. In case of a planar chromophore, the cluster configuration will be a double-sided attachment of the rare gas and single-sided configurations are less likely. Comparison of peak intensities for both pick-up sequences at identical conditions in the doping cell allows to identify single-sided and double-sided rare gas attachment to the chromophore.
Applying those methods in order to analyze the configuration of the tetracene-argon clusters in helium droplets, the limitations of the empirical additivity rule became obvious [73]. The advantage in terms of structural analysis as accomplished by the alternation of the pickup sequence is slightly relativized by the authors for good reason. In helium droplets the process of cluster formation proceeds under permanent dissipation of energy into the helium environment. Therefore, the path from separate components to the cluster is different from the path of cluster growth in a seeded beam expansion and so are the resulting cluster configurations.
The spectral shift of electronic resonances among configurational variants of a certain cluster size is on the order of frequencies expected for van der Waals modes. Therefore, the number of isomeric configurations does not necessarily correlate with the number of signals assigned to a particular cluster stoichiometry. For the 1:1 cluster of tetracene-argon the identification of band origins was accomplished by pump-probe experiments [17]. An alternative rather simple method to identify electronic band origins is provided by dispersed emission spectra. It is another uniqueness offered by helium droplets as host. In contrast to electronic band origins low energy modes carry excess excitation energy that dissipates prior to radiative decay which than occurs red shifted. Thus, a coincidence of the band origin in the dispersed emission spectrum with the excitation frequency identifies the corresponding resonance in excitation as electronic band origin which can be counted as one of the isomeric variants of the corresponding cluster size. Instead, a red shift in the electronic band origin of the dispersed emission with respect to the excitation frequency speaks against an assignment to a band origin. As a kind of drawback, one needs to keep in mind that besides dissipation of excess excitation energy a red shifted emission may reveal the relaxation of a particular helium solvation complex as observed for phthalocyanine in helium droplets (cf. Fig. 5.10). While emission coincident with the excitation proves for an electronic band origin of a particular cluster configuration a red shifted emission does not necessarily speak against it.
5.4.2 Van Der Waals Clusters of Anthracene with Argon Atoms
Among numerous other examples of clusters, those of a single anthracene molecule and a variable number of argon atoms have been investigated in helium droplets as well as in the gas phase. The initial motivation for the investigation of clusters consisting of a single anthracene molecule and increasing numbers of argon atoms was the interest in the transition from the isolated anthracene molecule to the fully solvated chromophore. Thereby, shell structures and specific interactions between chromophore and environment were expected [74, 75]. Stoichiometric analysis of such clusters generated in a seeded beam expansion proposed the presence of isomeric variants for all cluster sizes [57, 76–79]. A major breakthrough in the analysis of cluster configurations could be accomplished by rotationally resolved spectroscopy. Therefore, a smart approach was chosen by means of rotational coherence spectroscopy which allows to observe molecular rotation in the time domain. Thus, the limitations set by the spectral resolution in the frequency domain are eliminated by the time domain. As a result, for the clusters of anthracene with up to three argon atoms only a single configuration could be identified [80]. A final statement including clusters with up to 6 argon atoms came from an extensive study where clusters were interrogated by a broad set of experimental diagnostics based on mass-selective, fragmentation-free, two-color resonant two-photon ionization and laser induced fluorescence as well as a theoretical modeling of the ionization energy [81]. According to this study which refers also to almost all of the previous publications the clusters of anthracene with up to at least 6 argon atoms generated in a seeded supersonic beam expansion exhibit only a single configuration. The corresponding electronic band origins extrapolated to up to 8 argon atoms are depicted in Fig. 5.15 as black squares.
The investigation of anthracene argon clusters in superfluid helium droplets contrasts to the gas phase experiment [82]. By means of dispersed emission spectra, within the spectral range of 160 cm−1 to the red of the electronic band origin of bare anthracene a number of 13 electronic band origins have been identified. By means of Poisson intensity profiles the band origins were assigned stoichiometrically. In Fig. 5.15, all red symbols mark electronic band origins sorted according to the cluster size given by the number of argon atoms. In helium droplets, a single argon atom finds two different sites whereas a maximum of five configurations were identified for two argon atoms attached to anthracene. Moreover, clusters with five and more argon atoms did not show spectrally resolved lines. Instead, spectrally broad signal was recorded. Therefore, these larger clusters could not be recorded individually and, thus, further stoichiometric assignment was not possible. By alternation of the pick-up sequence single-sided and double-sided cluster configurations could be assigned [82]. Clusters with two or four argon atoms exhibited single-sided and double-sided configurations while the cluster with three argon atoms appeared only double sided.

Frequency position of electronic band origin of anthracene and its clusters with 1 up to 8 argon atoms as revealed from gas phase experiments (black squares) and from a helium droplet experiment (red). Highlighted are two series from helium droplets as full red squares and full red triangles which develop with similar gradient as gas phase clusters. Adapted from Ref. [82]

3-dimensional plot of the minimum surface of the interaction potential of phthalocyanine in the electronic ground state and argon with phthalocyanine in the x–y plane. Axes are scaled in Å. The potential energy scales from pink/weak to red/strong. It is calculated from pair potentials parameters as given in Ref. [42]
It is striking that the stoichiometric gradient of the electronic band origin for clusters exhibiting the larges red shift in helium droplets—marked by red squares and labeled as SR1 in Fig. 5.16.—is very similar to that reported for gas phase conditions—marked by black squares. The gradients of linear extrapolations amount to −39(2) cm (black dashed line) and −40(1) cm (red dashed line), respectively. Moreover, the assignment to single-sided and double-sided configurations within SR1 confirmed the results deduced from the gas phase studies. In the latter case, the structural assignment required theoretical input in addition to the experimental data. The configurations identified from rotational coherence spectroscopy for clusters with up to three argon atoms [80] were also consistent with the configurations reported in Ref. [81] and those from helium droplets [82]. Among additional isomeric variants identified in helium droplets a second series—highlighted in Fig. 5.15 by red full triangles (labeled as SR2)—exhibits a similar stoichiometric gradient of −43(1) cm (red full line) as in the gas phase. According to the analysis performed in Ref. [82] these clusters of SR2 exhibit identical configuration with respect to single or double-sided occupation as those labeled SR1. The argon induced red shift of the cluster with a single argon atom from SR1 is similar to that in the gas phase which speaks for identical cluster configurations. The red shift of the corresponding cluster from SR2 in helium droplets reveals significant shielding of the argon induced solvent shift which continues similarly for the larger clusters of SR2. The reduction of the red shift in SR2 compared to SR1 is similar for each cluster size. The reduced argon induced red shift in combination with a similar stoichiometric gradient might be indicative for cluster configurations similar to those of SR1, whereby one argon atom is located in a more distant position most probably shielded by the helium solvation layer. It is evident that the effect of shielding of a single argon atom on the argon induced red shift decreases with increasing cluster size and vice versa as can be recognized from the linear extrapolation for SR2.
The number of theoretically proposed isomeric variants of the clusters of anthracene with up to three argon atoms listed for gas phase conditions in Ref. [80] contrasts to the single isomer identified experimentally. In contrast to both, the large number of isomeric variants identified in helium droplets which exceeds what is proposed in Ref. [80] speaks for an involvement of helium atoms as part of the solvated clusters. A deeper insight into the internal structure of those clusters requires detailed information on the mass distribution as revealed by a rotationally resolved spectroscopy similar as presented in Chaps. 3 or 8.
Additional peculiarities of cluster formation in helium droplets are revealed by electronic spectra for clusters of anthracene with five and more argon atoms. In contrast to corresponding gas phase spectra with spectrally isolated peaks the signals of those larger clusters in helium droplets merge into a broad and unstructured feature which does not allow to address individual clusters. Without helium droplets a broad signal might be an indication for intrinsic fluctionality of the clusters. However, inside helium droplets spectral broadening might also reveal an increased number of isomeric configurations possibly involving in addition a variable number of helium atoms. A recent theoretical investigation of those clusters embedded into 1000 helium atoms reported rigid cluster configurations for up to 9 argon atoms and, thus, excluded fluctionality [83]. Moreover, asymmetric cluster configurations with single-sided attachment of all argon atoms to the anthracene molecule were found favored. The latter disagrees with the experiment [81, 82], and it is not clear in how far the theoretical treatment allows to exclude cluster configurations involving helium atoms other than simply a solvation layer. A continuation of theoretical investigations for those clusters generated in helium droplets might start with confirming cluster configurations known from gas phase experiments and continue with investigation of their response to embedding into a helium bath. It should be continued by cluster formation inside helium droplets which then accounts for the possible involvement of helium atoms as part of the cluster.
5.4.3 Van Der Waals Clusters of Phthalocyanine with Argon Atoms
A cluster consisting of a single phthalocyanine molecule with a single argon atom was already addressed in Sect. 5.3.3 in the context of relaxation dynamics of helium solvated compounds induced by electronic excitation. The dispersed emission spectrum revealed two additional configurations accessible via relaxation of the electronically excited cluster [21].
Besides the cluster whose emission revealed additional metastable configurations which exhibit increased red shifts, the fluorescence excitation spectrum revealed additional configurations of this cluster with reduced red shift. While the first cluster exhibited an argon induced red shift of 15 cm−1 identical to that reported from the gas phase [84], the argon induced red shift of a second cluster signal was only 1.3 cm−1. Probably, a third isomer of this cluster size exists with an argon induced red shift of 3.6 cm−1. Looking at the phthalocyanine to argon potential hypersurface deduced from pair potentials shown in Fig. 5.15 (such a potential model was also used for PIMC simulations on the solvation of phthalocyanine in helium droplets [42]) only a single global minimum about 3 Å above the center of mass of phthalocyanine was found. No additional local minima are present that could trap an argon atom even at low temperatures. Figure 5.16 shows a 3-dimensional plot representing the surface of the potential minimum for the electronic ground state. The color expresses the binding energy rising from pink to red. The red spot constitutes the global minimum with a binding energy of about 670 cm−1. Besides the global minimum, additional cluster configurations require stabilization from the helium environment. The tiny red shift of two additional phthalocyanine-argon clusters is most probably induced by helium solvation. Moreover, instead of helium induced stabilization possibly in the periphery of the chromophore the argon atom might be shielded from phthalocyanine by the helium solvation layer.
According to a semiempirical model deduced from experimental studies of hetero clusters consisting of a single aromatic chromophore and variable numbers of rare gas atoms [72], the attachment of a rare gas atom, say argon atom to a chromophore inside helium droplets means replacing helium atoms by an argon atom [17]. According to Refs. [72], the argon induced red shift in helium droplets is reduced compared to argon attachment in the gas phase. In the case of the phthalocyanine-argon cluster the experimentally observed argon induced red shift amounts to 15 cm−1 in the gas phase and in helium droplets. Obviously, sometimes empirical rules fail and the experiment asks for a solid theoretical model.
Besides relaxation dynamics of isomeric configurations among each other induced by electronic excitation as discussed in Sect. 5.3.3 for the most abundant phthalocyanine argon cluster, dispersed emission spectra revealed perceived dissociation [85]. Upon vibronic excitation of the most prominent cluster with only one argon atom not only the triple emission was observed that reveals a cascade of configurational relaxations. In addition, the emission spectrum showed a weak contribution of bare phthalocyanine [85]. According to the argon induced red shift of only 15 cm−1, the first vibronic transition of the cluster suffices to exceed the electronic band origin of bare phthalocyanine. Thus, the balance of energy allows for dissociative relaxation of the vibronically excited cluster. However, according to the binding energy of argon at the global minimum, dissociation needs to pass a barrier of about 676 cm−1 [83, 84]. In case the signal contribution from bare phthalocyanine should be due to dissociation of the cluster, the barrier needed a helium induced reduction to only 16% which is unlikely. In the meantime, we have repeated the experiment with the simple modification that doping with argon was eliminated. Upon excitation still at the frequencies of vibronic transitions of the argon cluster which means about 15 cm−1 to the red of vibronic excitations of bare phthalocyanine, dispersed emission could be detected exclusively from bare phthalocyanine while the cluster signal had vanished. Even upon detuning the laser from the resonances of the argon cluster but still far from any resonance of bare phthalocyanine, the emission of phthalocyanine could be detected. Thus, the additional emission from bare phthalocyanine had nothing to do with the phthalocyanine-argon cluster and its dissociation. Instead, it came from excitation of bare phthalocyanine as accomplished by excitation at the high frequency tail of the PW. In addition to what was discussed for TPP in Fig. 5.12, this is another example for how easy one can be misled by helium induced spectral features.
Another curious dynamic process was observed in the dispersed emission of a cluster with two argon atoms in a single sided configuration. Upon vibronic excitation of this cluster, dual emission was observed. Besides the emission of the single-sided cluster that of a double-sided cluster of identical stoichiometry was detected. Without vibrational excess excitation this signal contribution was absent [85]. Such a vibronically induced isomerization from single-sided to double-sided cluster configuration suggests tunneling through the center of mass of phthalocyanine as isomerization coordinate. However, this is rather speculative and requires further experimental investigations.
5.4.4 Summary
In summary, the pick-up process is an ideal experimental method for multiple doping of helium droplets and thus, for designing clusters of well defined stoichiometry. In particular the generation of heterogeneous clusters by consecutive pick-up of the various components is a very favorable experimental technique. The identification of the number of isomeric variants is readily accomplished by means of dispersed emission spectra. Further structural information can be obtained from alternating the pick-up sequence of different cluster components. The low temperature conditions allow for elimination of hot bands and, thus, for unprecedented spectral clarity of cluster spectra. Thus, even tiny local minima in the configuration potential might be stabilized and detected. Besides stabilization of local minima in the configuration space of the cluster, additional configurations involving helium atoms cannot be excluded. An extreme scenario as proposed for anthracene-argon clusters is the attachment of cluster components to the helium solvation layer of another subunit. In this respect, the investigation of clusters generated inside helium droplets opens new perspectives beyond what is found under gas phase conditions.
5.5 Elementary Chemical Reactions in Helium Droplets
For the investigation of elementary chemical processes, helium droplets are known to serve as a cryogenic reactor which provides insight to low energy reaction paths otherwise not accessible. This is mainly due to highly efficient dissipation of excitation energy from the dopant system to the helium droplet prior to reactive encounter and throughout the reaction process. Chemistry induced by electronic excitation is certainly the key aspect of this section. Thereby, intramolecular dynamics is readily investigated by means of dispersed emission spectroscopy as discussed above in the context of heterogeneous clusters and solvation complexes in helium droplets. In the following three benchmark experiments will be discussed which address first a bimolecular elementary exchange reaction, namely, N2O + Ba → BaO* + N2, secondly, a photodissociation process, namely cleavage of iodine from CH3I and CF3I, and, finally, excited state intramolecular proton transfer (ESIPT) for 3-hydroxyflavone. For all three reactions, the helium droplet experiment is contrasted with corresponding experiments in the gas phase.
5.5.1 Bimolecular Reaction of Barium with Nitrous Oxide
The first bimolecular chemical reaction investigated in helium droplets was the formation of barium oxide from the reaction of barium atoms with nitrous oxide, Ba + N2O → BaO* + N2 [86]. The interesting aspect of this reaction is the formation of an electronically excited product molecule which decays radiatively. The emitted chemiluminecence is readily detected by means of a grating spectrograph. The investigation of elementary chemical reactions under single collision condition allows to study this exchange reaction in its very details. The dispersed spectrum of the chemiluminescence provides insight into the internal energy distribution of the BaO* product molecules whereas tuning the collision energy provides insight into the entrance channel. The reaction of Ba with N2O has been investigated long before in the gas phase [87–91] and in addition on argon clusters [92, 93].
In the gas phase, crossed beam experiments have been performed in order to accomplish single collision conditions. The chemiluminescence of the BaO* upon reaction with nitrous oxide was found to cover almost the entire range of the visible spectrum and is shown in panel (a) of Fig. 5.17 for two different collision energies. Not much of a fine structure could be resolved and the maximum as well as the width of the emission was found to depend on the collision energy [90]. In contrast, upon replacing nitrous oxide by nitrogen dioxide the chemiluminescence spectrum was shifted slightly to the red and showed a clear fine structure which could be assigned to vibronic bands of the barium oxide product molecule [87]. The difference in the spectra reveal different energy distribution in the exit channel of both reaction systems. Besides the collision energy, the internal energy of the Ba educt has been varied. In addition to the ground state, electronically excited states of the barium atom [88] were studied for the reaction with nitrous oxide and in addition with nitrogen dioxide and also with ozone. In all cases electronic excitation of Ba became effective on the internal state distribution of the BaO* product molecule.

Chemiluminescence spectra of BaO* generated from the bimolecular reaction of Ba + N2O in the gas phase for two collision energies (a) (Ref. [90]), on argon clusters (b) (Refs. [92, 93]), on helium droplets (\(\overline{N }\)~20,000) (c) (Ref. [86]) and on Xe15 clusters inside helium droplets (\(\overline{N }\)~20,000) (d) (Ref. [86]). b and c reveal signal from hot BaO* having left the cluster/droplet and from cold BaO* residing on or in the cluster/droplet as indicated by two cartoons in panel (b). In d BaO* escaped from the xenon cluster, however, radiates still inside the helium droplet
In a second experiment the same reaction system was studied on the surface of argon clusters [92, 93]. The purpose of these studies was to investigate the influence of solvents on elementary chemical processes. As a result, the chemiluminescence spectrum revealed two contributions shown in panel (b) of Fig. 5.17. One part of the spectrum was almost identical to the broad and unstructured spectrum recorded in the gas phase except of a slight spectral shift and much higher signal intensity. Superimposed to this a series of well separated peaks were recorded. Two reaction processes were proposed in order to explain the two signal contributions. In one case the reaction proceeds on the surface of the argon cluster so that the hot BaO* product molecule can leave the argon cluster prior to emission. In the second case the reaction happens inside the argon cluster. Thus, the internally hot BaO* product molecule cools down via energy dissipation into the argon cluster prior to radiative decay. In this case, hot bands are eliminated. The authors identify two major solvent effects. First, the effective reaction cross section was increased to the capture cross section of the Ba atom by the N2O-doped argon cluster, which to a reasonable approximation corresponds to the geometric cross section of the argon cluster. Secondly, the collision energy in the entrance channel of the reaction was given by the mobility of the educt moieties on or in the argon cluster and, therefore, not a variable parameter as in the molecular beam experiment. Besides the latter issue providing rather indefinite energetic conditions, the approach on or in an argon cluster does not warrant for single collision conditions as in the molecular beam experiment. Finally, chemiluminescence from the reaction of barium in a single collision process with a cluster of the N2O instead of a single N2O molecule on an argon cluster was found to be rather quenched [91].
This bimolecular reactive experiment has been repeated in helium droplets [71, 86]. Thereby, helium droplets had first been doped with barium atoms and secondly with N2O. Chemiluminescence has been collected by an optical fiber bundle and guided to a spectrograph. The corresponding spectrum shown in panel (c) of Fig. 5.17 was similar to that recorded for the reaction on argon clusters. Again, two contributions of chemiluminescence could be identified, one of which shows a broad signal across the entire visible spectral range and a second consisting of sharp peaks superimposed to the first. The former originates from hot product molecules and the latter from internally cold BaO*. The contribution from hot barium oxide reveals high escape probability from the helium droplet as accomplished by the heliophobic barium atoms shifting the reaction towards the surface of the droplet. Cold BaO* shows that product molecules remaining in contact with the helium droplet. One of the remarkable differences to the argon solvated reaction was a smaller solvent shift for the cold BaO* as to be expected for less polarizable helium as host. Furthermore, the vibronic lines of cold chemiluminescence were much narrower because of the lower temperature in helium droplets and possibly reduced inhomogeneous line broadening.
In a next step the experiment in helium droplets was modified in order to promote reaction inside the droplet and suppress reaction on the droplet surface. By additional doping of the helium droplets with on average 15 xenon atoms prior to doping of the reactants, the Ba atom was attracted by the xenon cluster into the helium droplet. Thus, the reactive encounter proceeds on the surface of the xenon cluster inside the helium droplet so that the escape probability of hot BaO* from the helium droplet prior to radiative decay became negligible. Under these conditions the signal from hot BaO* vanished almost entirely (cf. Fig. 5.17d). Despite additional doping with xenon the cold chemiluminescence spectrum was not shifted with respect to the corresponding signal without xenon doping. The missing spectral shift as expected for an attachment to the xenon cluster is indicative for detachment of the product from the xenon cluster inside the helium droplet. Similar as for argon clusters as host, helium droplets act as a catalyst by increasing the effective reactive cross section by orders of magnitude [86].
Along the path from the gas phase to the argon solvated and finally to the helium solvated version of this bimolecular reaction the focus has changed. While the gas phase experiment provides insight into an elementary chemical reaction process including energetic conditions in the entrance and exit channel under single collision conditions, argon or helium solvation reveals the influence of the solvent on the products in the exit channel. In fact, the effective reactive cross section is blown up to the pick-up cross section of the educts by the host cluster. However, single collision conditions are not warranted in helium droplets or in and on argon clusters. Any details of the reaction process other than the identification of BaO* as reaction product got lost and the information revealed from the chemiluminescence spectrum recorded from xenon doped helium droplets does not go far beyond mass spectrometric detection.
5.5.2 Photolysis of Iodomethane and Perfluorated Iodomethane in Helium Droplets
One of the most detailed investigations of chemistry inside superfluid helium droplets reports on the photolysis of iodomethane and its perfluorated isomer by means of velocity map ion imaging (VMII) [94–97]. One key issue of this investigation was the influence of helium droplets as a solvent on the energetics of a photoinduced unimolecular dissociation. In particular, possible expression of vanishing viscosity—a characteristic property of superfluidity—in the velocity distribution of photo fragments generated inside helium droplets was of interest. As in the case of the bimolecular chemiluminescent reaction discussed above, such an investigation profits from comparison with corresponding experimental data under gas phase conditions which are available for the chosen system [98]. Both, the gas phase experiment as well as the corresponding helium droplet experiment are benchmark studies which are making use of the capability of VMII in full depth. The authors of the gas phase experiment [98] are the pioneers in the development of VMII. The ideal photolysis process demonstrating the capability of VMII is photolysis with an abstraction of only a single atom. The atomic fragment may access only few energetically well separated electronic states, in the case of iodine two spin states. The other fragment, a molecular radical, appears with internal energy distributed over a variety of rovibrational states, if energetically accessible also in different electronic states. The elegance of VMII as experimental technique [99] lies in the wealth of information that can be obtained from the imaging of the three-dimensional velocity distribution of only the atomic photolysis product. According to conservation laws for energy and linear momentum the velocity map of the atomic fragment reveals kinetic energies of both photofragments as well as the internal energy distribution of the non-detected molecular fragment. The internal energy of the detected fragment is revealed by the VMII detection process. Finally, the spatial distribution of the fragment velocity reveals details on the intramolecular dissociation coordinate as well as on the timing of the dissociation process. Further details can be deduced from correlation of vector quantities obtained by polarization sensitive photolysis and detection schemes which, however, will not be subject of the experiments discussed in the following sections.
The gas phase experiment reported in Ref. [98] headed for the investigation of the dynamics of the photolysis of iodomethane. CH3I was prepared at low temperature accomplished by a seeded beam expansion. Thus, the energetic conditions are under control by the frequency of the photolysis laser. The entrance channel for the dissociation of CH3I → CH3 + I was accessed by electronic excitation into the so-called A-band of the CH3I molecule [98]. With the variation of the photon energy across the A-band the starting point for photolysis varies among three different electronically excited states which are assigned as 1Q1, 3Q0, and 3Q1. At the Franck–Condon point the three levels scale with decreasing energy as listed. All three are repulsive with respect to the I-C coordinate. Among the three,1Q1 and 3Q1 converge to CH3 and I in the electronic ground state, whereas 1Q0 converges to I*(2P1/2). This latter state exhibits a conical intersection with the 1Q1 state.
Velocity map ion images of the methyl radical shown in black and white in the left panel of Fig. 5.18 distinguish two exit channels for which the different velocities fit perfectly to the energy difference of the two spin states of the atomic iodine fragment. Besides other important details on the photolysis of CH3I such as energy disposal into kinetic and internal degrees of freedom as function of the photolysis energy, the anisotropy in the spatial distribution of the fragments revealed information on the probability of curve crossing at the conical intersection. Corresponding data for the perfluorated analog of iodomethane allowed for comparison of the heavy-light product combination for CH3I with a rather equally weighted version for CF3I. So far, from the gas phase experiment [96] only those results are reviewed which are relevant for comparison with the helium droplet experiment.

Left panel: Velocity map ion image of CH3 radicals generated by photolysis of CH3I. Map in black and white recorded from gas phase adapted from [98]. Center colored map recorded from superfluid helium droplets adapted from [95]. Right panel: Velocity distributions of CH3 radicals from the gas phase resolves two channels in correlation with I* (2P1/2) and I (2P3/2). Inlay right panel: velocity distribution from the helium droplet experiment
About a decade later, the photolysis of CH3I and CF3I was performed in superfluid helium droplets. In general, the influence of the solvent on the photolysis dynamics was of interest. Three consecutive papers [95–97] under the common title “Photodissociation of alkyl iodides in helium droplets” have been published each with an individual subtitle, namely, “Kinetic energy transfer”, “Solvation dynamics”, and “Recombination”. In the first paper, the VMII of the fragments revealed substantial dissipation of kinetic energy from the photolysis products into the helium environment. The VMII of the CH3 radical added in color to VMII from the gas phase experiment in Fig. 5.18 revealed a singly peaked velocity distribution (inset in right panel of Fig. 5.18) at on average less than 10% of the doubly peaked gas phase velocities plotted in the left panel of Fig. 5.18. For the heavier CF3 radical the kinetic energy loss amounts to about 60% also without any signature of two channels as observed for the two electronic states of the iodine fragment in the gas phase. The heavier the product the less the energy loss and the larger the droplets the larger the energy loss of the escaping fragments. The angular distribution of the velocity map revealed anisotropy, however, substantially reduced for the light methyl radical fragment. Beyond an escape velocity of 600 m/s the anisotropy reached a constant average of about 70% of the gas phase value. Below 600 m/s the remaining anisotropy decreased with decreasing fragment velocity. Similar observations were reported for the heavier CF3 radical whereby fragments with an escape velocity of more than 400 m/s reached an anisotropy almost identical to the gas phase experiment. Again, the heavier fragment is less perturbed by the helium environment. Due to numerous other experimental details in combination with a very careful analysis of the influence of the droplet size, the effect of helium solvation on the dynamics of the photolysis could be explained by a classical model of binary collisions of the fragments with individual helium atoms on their way out of the droplet. By means of quasi classical trajectory calculations the experimental velocity distributions could be reproduced quantitatively for both fragments of the photolysis of iodomethane and the perfluorated variant. Instead of characteristic features due to vanishing viscosity of superfluid helium, the influence of the helium environment on the photofragment dynamics revealed the image of a classical solvent.
The second article under the subtitle “Solvation dynamics” [96] addresses the solvation of the photolysis fragments along their path leaving the helium droplet. Solvation means the formation of clusters consisting of a photofragment and variable numbers of rigidly attached helium atoms. The corresponding cluster size distribution as deduced from mass selective VMII was recorded for clusters having left the helium droplet. Thereby it was found that the formation of such clusters took place along the escape path of the fragments. The dynamics of attachment and detachment by consecutive collisions along the escape path is responsible for the cluster size distribution. Since the fragment velocity ratio is inverse proportional to the mass ratio, the iodine fragment from CH3I is much slower than that from CF3I. Slow fragment velocity was found to favor cluster formation. The dependence of the cluster size distribution on the escape velocity revealed a cluster growth along the escape path of the fragments through the droplet. Moreover, clusters generated as a result of photolysis could be excluded. In the case of the methyl radical, the protonated isomer did form clusters whereas the perfluorated isomer did not. The higher internal energy of the perfluorated fragment was made responsible for the missing of cluster formation.
The comparison of helium clusters of the iodine fragment generated either from the protonated or the perfluorated compound revealed further insight into the formation/solvation dynamics. For the slow iodine fragment the cluster size distribution was almost insensitive to the droplet size distribution. Instead, for the fast iodine fragment the cluster size distribution shifted to larger clusters with increasing droplet radius. For slow iodine fragments a dynamic equilibrium between attachment and detachment is reached whereas fast iodine fragments escape before reaching such an equilibrium.
Finally, the observation of drastically reduced fragment velocities raises the question on the possibility of recombination as discussed in Ref. [97]. Indeed, the authors could positively prove that one out of several overlapping signal contributions within the VMI images recorded for the nascent mass of CH3I shown in Fig. 5.19 was definitely the result of recombination inside the droplet. Besides the recombination signal marked in panel d) in Fig. 5.19 additional signals from background gas in the detection chamber (panel a)), an effusive beam emerging from the pick-up chamber (panel b)), and a contribution from escaped IHe4 fragment clusters (panel c)) due to imperfect mass selection were detected. With increasing helium droplet size the recombination signal increased. Moreover, evidence was provided that the cut off in the helium cluster size distribution of solvated fragments was due to a vanishing escape probability beyond a certain cluster size. For the perfluorated variant no recombination could be observed. The velocities of the photo fragments from the perfluorated variant did not allow for sufficient deceleration to accomplish recombination. Therefore, corresponding VMII did not show a signal in correspondence to signal (d) (cf. Ref. [97]). In addition, signal (c) is missing since there were no fragment clusters close to the mass of the perfluorated iodomethane.

Velocity map image of non-solvated CH3I molecules from the detection volume of the ion imaging setup for average droplet radii of 40 Å. The time delay between UV dissociation and femtosecond ionization pulses was set to 50 ns. Note that the images contain signals from parent molecules present as background gas in the detection chamber (a) and of an effusive beam emerging from the doping chamber (b). The images of CH3I furthermore contain contributions from escaping IHe4 fragments (c) whose mass differs by only 1 amu from the mass of CH3I. The recombination signal (d) is readily observed for CH3I. Adapted from Ref. [97]
The investigation of photolysis of methyl iodide inside helium droplets is one of the most profound investigations of helium droplets acting as a solvent on a dopant system and its chemical dynamics. The wealth of experimental details reported in three consecutive papers [95–97] preceded by a letter [94] speaks for itself and goes far beyond what is reviewed above. However, in comparison with the gas phase experiment revealing the very details of the energetics in the entrance and the exit channel of the photolysis of methyl iodide and its perfluorated variant, not much is left over in the helium droplet experiment. Again, the continuous and highly efficient dissipation of internal and kinetic energy into the helium droplet alters the conditions for molecular dynamics substantially. The helium droplet experiment changes the focus from investigating properties of the dopant system to the influence of helium solvation on this system and its chemical dynamics. In this respect Refs. [94–97] are a benchmark project that provided deep insight into solvation in helium droplets.
5.5.3 Excited State Intramolecular Proton Transfer (ESIPT) in Superfluid Helium Droplets
The third example studying molecular dynamics in helium droplets is ESIPT of 3-hydroxyflavone. ESIPT is a unimolecular process that is initiated by electronic excitation. According to Born–Oppenheimer approximation, the process of electronic excitation starts with a change of the electron density distribution and only afterwards the nuclei follow. In the case of ESIPT, the nuclear response is a proton transfer to another position. As depicted in Fig. 5.20 a hydrogen atom jumps from one oxygen to another oxygen. This rearrangement is enforced by the change in the electron density distribution accompanying electronic excitation which stabilizes the tautomer (T*) compared to the normal form (N*). As a consequence of ESIPT upon excitation at the electronic band origin of the normal form at about 351 nm, emission is recorded only in the green at and beyond 500 nm originating from the tautomer. In the electronic ground state, the energetic conditions between tautomer and normal form are inverted. The corresponding relaxation process is called back proton transfer (BPT). Under room temperature conditions this molecule is found exclusively in the normal form. Thus, electronic excitation at around 351 nm is the initiating step of a photocycle which is continued by ESIPT followed by radiative decay of the keto form and finished by BPT as depicted in Fig. 5.20. Within this photocycle the time constants for ESIPT and BPT are characteristic quantities. Moreover, the influence of solvents on ESIPT and BPT are important details in order to elucidate such chemical processes.

Jablonski diagram depicting ESIPT and BPT of 3-hydroxyflavone. Left side shows the normal form (N / N*) and right side shows the tautomeric form (T/T*)
While ESIPT of 3-hydroxyflavone was known for long the particular dynamics was a matter of long-lasting investigations [100, 101]. The missing of any emission from the normal form (N) revealed that the rate constant for ESIPT exceeds the corresponding quantity for radiative decay. The rise time of tautomeric emission of 3-hydroxyflavone doped into an argon matrix revealed an upper limit in the time constant for ESIPT of 2 ps [102]. In those days, time resolved spectroscopy in the picosecond regime was a challenge. However, upon approaching the limits in the time domain, corresponding information can readily be obtained from the frequency domain. This has been done successfully by means of a spectrally highly resolved fluorescence excitation spectrum recorded from a seeded supersonic jet of 3-hydroxyflavone [103]. The line shape at the electronic band origin was almost perfectly reproduced by a Lorentzian type exhibiting a width of 4.1 cm−1. After deconvolution of minor contributions of rotational bands and of the laser band width a purely Lorentzian contribution with a spectral width of 3.9 cm−1 was determined which corresponds to an excited state life time of 1.4 ps. Since emission occurred exclusively from the tautomer the homogeneous line width reveals the life time of the electronically excited normal form that decays via ESIPT. Thus, the excited state life time corresponds to the rate constant for ESIPT which amounts to 740 GHz. A possible influence of the steric configuration of the phenyl moiety in the electronically excited system was a matter of discussion. A twist could be confirmed by the intensity pattern within a low energy vibronic progression of 45 cm−1 in the excitation spectrum assigned to the phenyl torsion. Upon deuteration the emission was also exclusively of tautomeric origin. However, the line width in the electronic spectrum of the deuterated isomer was significantly smaller as compared to the protonated isomer which is indicative for a reduced ESIPT rate constant. Moreover, the line shape was dominantly of Gaussian type which did not allow for identifying a homogeneous contribution by means of deconvolution [104].
Further insight into the influence of the phenyl moiety on ESIPT has been obtained from corresponding experimental data from 3-hydroxychromone and from 2-(2-naphthyl)-3-hydroxychromone [105]. For all three compounds a Lorentzian line shape recorded at the electronic band origin revealed rate constants for ESIPT of 655 GHz for 3-hydroxyflavone, 145 GHZ for 2-(2-naphthyl)-3-hydroxychromone, and 1770 GHz for 3-hydroxychromone. Obviously, the two PAH substituents impede ESIPT the more the larger the substituent.
It was also known for long that a protic or polar solvent impedes ESIPT as revealed by ultraviolet emission from the solvated 3-hydroxyflavone [101]. The influence of water as a protic solvent was investigated for clusters consisting of a single 3-hydroxyflavone molecule and in addition one or two water molecules as generated in a seeded molecular beam experiment [105]. Dispersed emission revealed that ESIPT was blocked by adding water. Only a single water molecule sufficed to suppress ESIPT entirely. This result needs to be confronted with the observation of dual emission, namely, of normal and tautomeric origin obtained from 3-hydrxychromone derivatives dissolved in neat water [106]. The authors suggest to use the gradually changing emission of 3-hydroxychromone dyes as sensors for protic impurities and in particular of water in solutions. Another supersonic jet experiment on clusters of 3-hydroxyflavone with one or two water molecules revealed an obstacle to proton transfer only for the attachment of two water molecules, whereby reactivation of ESIPT could be accomplished upon sufficient excess excitation energy, indicative for a barrier in the proton transfer path [107, 108]. These results were obtained from mass selective IR/R2PI spectra and interpreted in combination with theoretical vibrational frequencies obtained by means of DFT and TDDFT calculations for the clusters of 3-hydroxyflavone with one or two water molecules. In summary, the experiments discussed above are benchmarks in featuring ESIPT of 3-hydroxyflavone as isolated molecule and under the influence of water on a molecular scale. While results for the isolated molecule are consistent the influence of water on ESIPT revealed inconsistencies among the different experiments.
For the investigation of ESIPT and in addition of further steps in the photocycle depicted in Fig. 5.20, helium droplets implemented as host can provide new details. In particular the influence of solvents such as water on a molecular scale can be realized even selective for isomeric variants of stoichiometrically selected complexes. Moreover, the efficient dissipation of rovibrational energy into the helium droplets allows for cooling of the electronically excited tautomer prior to radiative decay. With the expectation to record vibrationally resolved dispersed emission, the rate constant for BPT might be accessible from a line shape analysis of the electronic band origin similar as reported for ESIPT from the line shape in the excitation [102–105]. Last but not least, the influence of helium solvation on ESIPT is of interest. Thus, fluorescence excitation spectra and dispersed emission spectra of 3-hydroxyflavone and of its clusters with water have been recorded [109]. Dispersed emission spectra of 3-hydroxyflavone in helium droplets upon excitation at about 351 nm showed exclusively emission at and beyond 500 nm. This was a clear prove for unhindered ESIPT inside superfluid helium droplets as in the gas phase. In contrast to gas phase experiments, the dispersed emission of 3-hydroxyflavone in helium droplets revealed a vibrational fine structure as was expected from efficient cooling of the tautomer prior to radiative decay (cf. Fig. 5.21). This is similar to the cooling of electronically excited BaO* reaction products prior to radiative decay as discussed in Sect. 5.5.1. In the present cases the cooling allows to obtain information on the vibrational fine structure of the ground state of the metastable tautomer of 3-hydroxyflavone which otherwise is hardly accessible. Moreover, the dispersed emission of a rovibrationally cold tautomer allows for line shape analysis similar as done in Refs. [102–105] for the excitation spectrum. The line shape analysis at the electronic band origin revealed a Voigt profile with a Gaussian component of 27.2 cm−1 in width and a Lorentzian component of 22.5 cm−1 in width [109]. The latter represents the homogeneous contribution to the electronic transition of the tautomer which is determined by the life time of both electronic states involved in the transition. The contribution of the electronically excited state life time in the order of 10 ns [105] to the Lorentzian line width is negligible. Thus, the Lorentzian contribution can be attributed solely to homogeneous line broadening due to BPT. Accordingly, the rate constant for BPT is about 4.2 THz which corresponds to a time constant of 236 fs. An almost identical time constant was found from corresponding investigations in a Shpol’skii matrix [110]. So far, the influence of helium droplets on ESIPT in 3-hydroxyflavone could be classified as negligible. However, the cryogenic capability of helium droplets allowes to partly resolve the vibrational fine structure of the tautomer’s electronic ground state and, thereby, deduce the rate constant for BPT.

As reported from gas phase experiments, the clusters of 3-hydroxyflavone with one or two water molecules suffer a red shift in the electronic band origin. Thus, 351 nm for excitation should excite both, bare 3-hydroxyflavone and in addition its clusters with water. Upon additional doping of the helium droplets with water post to 3-hydroxyflavone dispersed emission upon excitation at 351 nm showed spectrally well resolved cluster signals about 400 cm−1 further to the blue of bare 3-hydroxyflavone. According to the Poisson intensity profiles individual resonances could be assigned to clusters of 3-hydroxyflavone with one and with two water molecules as depicted in Fig. 5.22 as blue and green section, respectively. The red section is the electronic band origin of bare 3-hydroxyflavone. Only tautomeric emission was recorded for the clusters with one or two water molecules. The corresponding size distribution defined by the number of attached water molecules as revealed by a Poisson distribution is added as circles in the inlay using the same color code as in the spectrum. Upon increasing the pick-up probability to obtain the cluster size distribution plotted in black squares which corresponds to an average cluster size of four water molecules, a minor contribution of blue emission could be detected as a signature for hindered ESIPT. Surprisingly, however, this emission was spectrally broad without any kind of vibronic fine structure which contrasts to the emission of the tautomer whether from bare 3-hydroxyflavone or from the clusters with one or two water molecules. Under the given pick-up conditions only the largest clusters—probably with eight and more water molecules—might be responsible for hindered ESIPT. Instead of clarifying the inconsistency reported on the influence of water on ESIPT in 3-hydroxyflavone [105–108], the helium droplet experiment provides a third result [109] which is inconsistent to the gas phase data.

Dispersed emission spectra of 3-hydroxyflavone and its clusters with water in superfluid helium droplets (\(\overline{N }\) = 5500). The black spectrum was recorded for a cluster size distribution shown as black squares in the inset. Colored spectrum marks signal of bare 3-hydroxyflavone in red, of clusters with one water molecule in blue and with two water molecules in green. The corresponding cluster size distribution is added to the inset. (Adapted from [109])
To avoid speculations about the missing of vibrational fine structure in the blue emission of larger clusters with water, the investigation of the fluorescence excitation spectrum of 3-hydroxyflavone in helium droplets might provide explanations. However, instead of a vibrational fine structure as resolved in the gas phase experiment [102–104] shown as black line in Fig. 5.23, the spectrum from helium droplets showed two spectrally broad electronic bands starting at about 352 nm and 349 nm, respectively, as shown in the same figure in red. The helium induced blue shift is hard to quantify without a peak at the electronic band origin. A scenario responsible for vanishing of the vibrational fine structure is not evident. Upon increasing line widths in the gas phase spectrum, a factor of 10 suffice to hide the fine structure resolved in the gas phase. In this case, the factor of 10 in the line width can be interpreted as the helium induced increase of the ESIPT rate constant. Alternatively, a perturbation of the closer helium environment caused by electronic excitation of 3-hydroxyflavone might be a reason for line broadening. This scenario is similar to the explanation of line broadening for 9-phenylanthracene and 2-methylanthracene in helium droplets.

The investigation of ESIPT for 3-hydroxyflavone in helium droplets and in addition its response on attachment of water molecules revealed further insight into helium solvation. This experiment profited in the first place from highly efficient energy dissipation prior to radiative decay and in addition from stoichiometrically perfectly controlled cluster formation with water. The first advantage provided access to the BPT rate constant from line shape analysis in the cold emission of the tautomer. The latter gave access to the ESIPT under the influence of single water molecules. However, instead of clarifying in particular the effect of water on the ESIPT process, the helium droplet experiment was inconsistent to all of the previous results reported from gas phase experiments. As a matter of fact, the configuration of clusters generated in helium droplets might differ from those in the gas phase. However, to our best knowledge, there have never been reports on clusters in helium droplets that definitely excluded those configurations obtained in the gas phase. The stoichiometry of the clusters in helium droplets is unequivocal and so are all dispersed emission spectra. The problem of vanished vibrational fine structure in the excitation spectrum of the normal form and in addition in the corresponding dispersed emission from larger water clusters is an open issue which deserves further attention. In summary, helium solvation exhibits a significant influence of the helium environment on ESIPT in 3-hydroxyflavone and on its clusters with water.
5.5.4 Summary
The cryogenic capability of helium droplets has a significant influence on chemical processes whether bimolecular or unimolecular. Energy deposited in rovibrational degrees of freedom is instantaneously dissipated into the helium environment and, thus, the reaction path is forced to proceed without rovibrational contributions in the entrance channel as well as for reactive intermediates. Helium droplets as cryogenic reactor provide ideal conditions for the investigation of low temperature chemistry. The influence of solvents on a molecular scale in chemical processes is readily accessible under perfect control of the cluster stoichiometry. Nevertheless, one needs to consider that the attachment of solvent molecules—in the present case of water—to the reactive system—in the present case to 3-hydroxyflavone—allows for configurations that are not accessible without the helium environment.
5.6 Concluding Remarks on Electronic Spectroscopy of Molecules in Superfluid Helium Droplets
This article is far from a review on the title subject. Reviews on the title subject as well as on work related to helium droplets are listed in Appendix A of this book. Instead, this article aimed to highlight peculiar properties of helium droplets as cryogenic superfluid host for studying molecular systems and molecular dynamics by means of electronic spectroscopy. Several of the expectations that initiated and fueled the development of helium droplet sources and its application in molecular spectroscopy also described in Chap. 1 of this book were confirmed by corresponding experiments. Numerous rather surprising experimental observations could be explained by empirical conclusions and evidence-based arguments. However, some of the details of experimental observations were counterintuitive and thus do neither fit to theoretical nor empirical models. The experimental results presented in this article were selected with the idea to report on both, the expected and evident features of helium solvation as observed for glyoxal (Figs. 5.2 and 5.3) and in addition those which are not understood (cf. Figs. 5.5, 5.6, 5.7, 5.8, 5.16, and 5.23). Providing explanations for the latter is certainly a challenge and is vital for making use of the full capacity of helium droplets as cryogenic host. Among unsolved problems, the relation of line shapes and solvent shifts to the droplet size distribution stands out (Figs. 5.7, 5.8, and corresponding data for glyoxal from Ref. [16]). While the adaption of the excluded volume model to finite sized helium droplets is an unquestionable approach to handle the influence of a polarizable environment on the dopant species it does not suffice to describe all of what was reported experimentally on line shapes in helium droplets. As addressed also in Chap. 1, the source conditions such as nozzle temperature, stagnation pressure, and nozzle diameter are influential on the helium droplets. Whether from subcritical or supercritical expansion the change observed in the line shapes does not simply reflect what is expected from the accompanying change in the droplet size. Most curious was a turn-around in the helium solvation shift of electronic transitions as reported for tetracene (Fig. 5.7) and phthalocyanine at the transition from subcritical to supercritical droplet source conditions (Fig. 5.8 top panel). However, a kind of turn-around was also observed for glyoxal and in a different way for porphin, however, in both cases far from a transition in the droplet source conditions.
Besides sophisticated details revealed by the line shape and the overall line shift, the multiplet splitting observed at the ZPL of numerous dopant species deserves further investigations. Numerous experimental results similar as discussed above for a series of PAH species (cf. Fig. 5.5), for a series of derivatives of anthracene (Fig. 5.13), of pyrromethene dye molecules (Fig. 5.14), or of porphin can be taken as a guideline to develop an explanation possibly based on configurational variants of a helium solvation complex which needs to be manifested by theoretical modeling. It is the ultimate challenge for quantum chemical treatment of many particle systems steered by dispersion forces.
The phenomenon of multiplet splitting of the ZPL reveals insight into microsolvation. In addition, it is a key issue of studying van der Waals clusters and, in case of non-polar dopant species, a perfect example for cluster formation driven purely by dispersion interaction. In combination with electronic spectroscopy the investigation of heterogeneous clusters profits immensely from helium droplets as cryogenic host. In particular heterogeneous clusters consisting of a single chromophore molecule and a certain number of atoms, mostly rare gas atoms, or small molecules such as water, oxygen, nitrogen, or hydrogen, reveal information on microsolvation including details such as rigidity or shell structures on a molecular scale. Compared to alternative techniques of cluster generation as accomplished by seeded beam expansion, helium droplets as host provide unique advantages with respect to control of cluster stoichiometry and cluster temperature. However, one needs to keep in mind that the wealth of cluster configurations that are generated under support of the cryogenic environment and which can be addressed selectively by means of electronic spectroscopy might include species which involve helium atoms in addition as proposed for anthracene argon clusters (Fig. 5.16). Further experimental data and a deeper experimental insight into the cluster configuration is needed. Nevertheless, electronic spectra of van der Waals clusters generated in helium droplets are a valuable source of experimental data for the improvement of quantum chemical models dealing with dispersion interaction and with many particle systems that additionally allow to interpret and predict the splitting at the ZPL in electronic spectra.
Chemistry inside helium droplets deserves particular attention. Much of what happens under gas phase conditions is significantly modified by the helium environment as exemplified by the velocity map ion image of CH3 fragment from the gas phase and from helium droplets shown in Fig. 5.18. The efficient dissipation of energy from the reaction system to the helium droplet which is active during the entire reaction process from the entrance channel to the exit channel has an immense impact on the reaction path. Exceeding the Landau velocity transforms the superfluid environment into a normal fluid with significant consequences on among others the kinetic energy distribution (cf. Fig. 5.18). As shown for the BaO* product molecule in Fig. 5.17 and for the tautomer of 3-hydroxyflavone in Fig. 5.21, reaction products as well as reactive intermediates are cooled prior to radiative decay. Thus, emission spectra are cleaned from hot bands and reveal vibrational fine structure of the electronic ground state which in the case of the metastable tautomer of 3-hydroxyflavone is otherwise not accessible. However, the genuine energy distribution as characterizing feature of molecular dynamics is entirely lost due to permanent dissipation of energy into the helium droplets. Low temperature chemistry and within this context the investigation of tunneling processes find unprecedented potential by using superfluid helium droplets as a cryogenic reactor.
Helium droplets and solvation of molecules in helium droplets bears numerous secrets that still need to be revealed. The exceptional sensitivity of electronic spectroscopy plays a key role in this endeavor. Quantitative understanding of helium droplets as nano-scaled quantum fluid and molecular solvation inside them is the ultimate goal. This is mandatory in order to make use of the full capacity of superfluid helium nanodroplets as cryogenic host for studying molecules, their dynamics, and fundamental chemical processes. Continuing work in this field warrants for gain of knowledge for all the various aspects offered by superfluid helium nanodroplets.
Notes
- 1.
Exceptions are the para-hydrogen matrix and methane as dopant [10].
References
J.P. Toennies, A.F. Vilesov, Sectroscopy of atoms and molecules in liquid helium. Annu. Rev. Phys. Chem. 49, 1–41 (1998)
F. Stienkemeier, A.F. Vilesov, Electronic spectroscopy in He droplets. J. Chem. Phys. 115, 10119 (2001). https://doi.org/10.1063/1.1415433
J.P. Toennies, A.F. Vilesov, Suprafluide Heliumtröpfchen: außergewöhnlich kalte Nanomatrices für Moleküle und molekulare Komplexe. Angew. Chem. 116, 2674–2702 (2004). https://doi.org/10.1002/ange.200300611
J.P. Toennies, A.F. Vilesov, Superfluid helium droplets: a uniquely cold nanomatrix for molecules and molecular complexes. Angew. Chem. Int. Ed. 43, 2622–2648 (2004). https://doi.org/10.1002/anie.200300611
C. Callegari, K.K. Lehmann, R. Schmied, G. Scoles, Helium nanodroplet isolation rovibrational spectroscopy: methods and recent results. J. Chem. Phys. 115, 10090 (2001). https://doi.org/10.1063/1.1418746
M. Hartmann, R.E. Miller, J.P. Toennies, Rotaitionally resolved spectroscopy of SF6 in liquid helium clusters: a molecular probe of cluster temperature. Phy. Rev. Lett. 75, 1566–1569 (1995)
M.Y. Chio, G.E. Douberly, T.M. Falconer, W.K. Lewis, C.M. Lindsay, J.M. Merritt, P.L. Stiles, R.E. Miller, Infrared spectroscopy of helium nanodroplets: novel methods for physics and chemistry. Int. Rev. Phys. Chem. 25, 15–75 (2006). https://doi.org/10.1080/01442350600625092
G. Scoles (ed.) Atomic and Molecular Beam Methods, vol. 1 (Oxford University Press Inc, 1988) ISBN: 0195042808 (ISBN13: 9780195042801); vol. 2, (Oxford University Press Inc, 1988) ISBN: 0195042816 (ISBN13: 9780195042818)
I.R. Dunkin, Matrix-Isolation Techniques—A Practical Approach (Oxford University Press, Oxford, 1998). ISBN 0-19-855863-5
S. Tam, M.E. Fajardo, H. Katsuki, H. Hoshina, T. Wakabayashi, T. Momose, High resolution infrared absorption spectra of methane molecules isolated in solid parahydrogen matrices. J. Chem. Phys. 111, 4191–4198 (1999). https://doi.org/10.1063/1.479717
M. Hartmann, A. Lindinger, J.P. Toennies, A.F. Vilesov, The phonon wings in the (S1← S0) spectra of tetracene, pentacene, porphin and phthalocyanine in liquid helium droplets. Phys. Chem. Chem. Phys. 4, 4839–4844 (2002)
A. Lindinger, E. Lugovoj, J.P. Toennies, A.F. Vilesov, Splitting of the zero phonon lines of indole, 3-methyl indole, tryptamine and n-acetyl tryptophan amide in helium droplets Z. Phys. Chem. 215, 401–416 (2001). https://doi.org/10.1524/zpch.2001.215.3.401
N. Pörtner, J.P. Toennies, A.F. Vilesov, The observation of large changes in the rotational constants of glyoxal in superfluid helium droplets upon electronic excitation. J. Chem. Phys. 117, 6054–6060 (2002). https://doi.org/10.1063/1.1502643
M. Hartmann, J.P. Toennies, A.F. Vilesov, G. Benedek, Spectroscopic evidence for superfluidity in liquid He droplets. Czec. J. Phys. 46, 2951–2956 (1996)
M. Hartmann, F. Mielke, J.P. Toennies, A.F. Vilesov, Direct spectroscopic observation od elementary excitations in superfluid he droplets. Phys. Rev. Lett. 76, 4560–4563 (1996)
N. Pörtner, MPI für Strömungsforschung. Göttingen, Germany, Bericht 10 (200x)
M. Hartmann, A. Lindinger, J.P. Toennies, A.F. Vilesov, Laser-induced fluorescence spectroscopy of van der Waals complexes of tetracene–ArN (N≤5) and pentacene–Ar within ultracold liquid He droplets. Chem. Phys. 239, 139–149 (1998). https://doi.org/10.1016/S0301-0104(98)00250-X
M. Hartmann, A. Lindinger, J.P. Toennies, A.F. Vilesov, Hole-burning studies of the splitting in the ground and excited vibronic states of tetracene in helium droplets. J. Phys. Chem. A 105, 6369–6377 (2001). https://doi.org/10.1021/jp003600t
N. Pörtner, J.P. Toennies, A. Vilesov, F. Stienkemeier, Anomalous fine structures of the 000 band of tetracene in large He droplets and their dependence on droplet size. Mol. Phys. 110, 1767–1780 (2012). https://doi.org/10.1080/00268976.2012.679633
D. Pentlehner, R. Riechers, B. Dick, A. Slenczka, U. Even, N. Lavie, R. Brown, K. Luria, Rapidly pulsed helium droplet source. Rev. Sci. Instrum. 80, 043302 (2009). https://doi.org/10.1063/1.3117196
R. Lehnig, A. Slenczka, Spectroscopic investigation of the solvation of organic molecules in superfluid helium droplets. J. Chem. Phys. 122, 244317 (2005). https://doi.org/10.1063/1.1946739
H.D. Whitley, J.L. DuBois, K.B. Whaley, Spectral shifts and helium configurations in 4HeN–tetracene clusters. J. Chem. Phys. 131, 124514 (2009). https://doi.org/10.1063/1.3236386
H.D. Whitley, J.L. DuBois, K.B. Whaley, Theoretical analysis of the anomalous spectral splitting of tetracene in He droplets. J. Phys. Chem. A 115, 7220–7233 (2011). https://doi.org/10.1021/jp2003003
S. Krasnokutski, G. Rouille, F. Huisken, Electronic spectroscopy of anthracene molecules trapped in helium nanodroplets. Chem. Phys. Lett. 406, 386–392 (2005). https://doi.org/10.1016/j.cplett.2005.02.126
R. Riechers, D. Pentlehner, A. Slenczka, Microsolvation in superfluid helium droplets studied by the electronic spectra of six porphyrin derivatives and one chlorine compound. J. Chem. Phys. 138, 244303 (2013). https://doi.org/10.1063/1.4811199
D. Pentlehner, A. Slenczka, Microsolvation of anthracene inside superfluid helium nanodroplets. Mol. Phys. 110, 1933–1940 (2012). https://doi.org/10.1080/00268976.2012.695406
A. Lindinger, Ph. D. thesis, Universität Göttingen, Germany (1999)
R. Schmied, P. Carcabal, A.M. Dokter, V.P.A. Lonij, K.K. Lehmann, G. Scoles, UV spectra of benzene isotopomers and dimers in helium nanodroplets. J. Chem. Phys. 121, 2701–2711 (2004). https://doi.org/10.1063/1.1767515
E. Loginov, A. Braun, M. Drabbels, A new sensitive detection scheme for helium nanodroplet isolation spectroscopy: application to benzene. Phys. Chem. Chem. Phys. 10, 6107–6114 (2008). https://doi.org/10.1039/B807911A
P. Carcabal, R. Schmied, K.K. Lehmann, G. Scoles, Helium nanodroplet isolation spectroscopy of perylene and its complexes with oxygen. J. Chem. Phys. 120, 6792–6793 (2004). https://doi.org/10.1063/1.1667462
N. Pörtner, A.F. Vilesov, M. Havenith, Spontaneous alignment of tetracene molecules in 4He droplets. Chem. Phys. Lett. 368, 458–464 (2003). https://doi.org/10.1016/S0009-2614(02)01903-6
M. Hartmann, MPI für Strömungsforschung, Göttingen, Germany, Bericht 10 (1997)
A. Slenczka, B. Dick, M. Hartmann, J.P. Toennies, Inhomogeneous broadening of the zero phonon line of phthalocyanine in superfluid helium droplets. J. Chem. Phys. 115, 10199 (2001). https://doi.org/10.1063/1.1409353
B. Dick, A. Slenczka, Inhomogeneous line shape theory of electronic transitions for molecules embedded in superfluid helium droplets. J. Chem. Phys. 115, 10206 (2001). https://doi.org/10.1063/1.1409354
J. Jortner, Cluster size effects. Z. Phys. D 24, 247–275 (1992). https://doi.org/10.1007/BF01425749
S. Fuchs, J. Fischer, A. Slenczka, M. Karra, B. Friedrich, Microsolvation of phthalocyanine molecules in superfluid helium nanodroplets as revealed by the optical line shape at electronic origin. J. Chem. Phys. 148, 144301 (2018). https://doi.org/10.1063/1.5022006
R. Lehnig, M. Slipchenko, S. Kuma, T. Momose, B. Sartakov, A. Vilesov, Fine structure of the (S1←S0) band origins of phthalocyanine molecules in helium droplets. J. Chem. Phys. 121, 9396–9405 (2004). https://doi.org/10.1063/1.1804945
R. Lehnig, A. Slenczka, Microsolvation of phthalocyanines in superfluid helium droplets. ChemPhysChem 5, 1014–1019 (2004). https://doi.org/10.1002/cphc.200400022
D. Pentlehner, R. Riechers, A. Vdovin, G.M. Pötzl, A. Slenczka, Electronic spectroscopy of molecules in superfluid helium nanodroplets: an excellent sensor for intramolecular charge redistribution. J. Phys. Chem. A 115, 7034–7043 (2011). https://doi.org/10.1021/jp112351u
K. Nauta, R.E. Miller, Metastable vibrationally excited HF (v=1) in helium nanodroplets. J. Chem. Phys. 113, 9466–9469 (2000). https://doi.org/10.1063/1.1319965
R. Lehnig, A. Slenczka, Emission spectra of free base phthalocyanine in superfluid helium droplets. J. Chem. Phys. 118, 8256–8260 (2003). https://doi.org/10.1063/1.1565313
H.D. Whitley, P. Huang, Y. Kwon, K.B. Whaley, Multiple solvation configurations around phthalocyanine in helium droplets. J. Chem. Phys. 123, 054307 (2005). https://doi.org/10.1063/1.1961532
A. Slenczka, Electronic spectroscopy of phthalocyanine and porphyrin derivatives in superfluid helium nanodroplets. Molecules 22, 1244 (2017). https://doi.org/10.3390/molecules22081244
T. Premke, E.-M. Wirths, D. Pentlehner, R. Riechers, R. Lehnig, A. Vdovin, A. Slenczka, Microsolvation of molecules insuperfluid helium nanodroplets revealed by means of electronic spectroscopy. Front. Chem. Phys. Chem. Chem. Phys. 2, 51 (2014). https://doi.org/10.3389/fchem.2014.00051
J. Fischer, S. Fuchs, A. Slenczka, M. Karra, V. Friedrich, Microsolvation of porphine molecules in superfluid helium nanodroplets as revealed by optical line shape at the electronic origin. J. Chem. Phys. 149, 244306 (2018). https://doi.org/10.1063/1.5052615
U. Even, J. Jortner, Z. Berkovitch-Yellin, Electronic excitations of the free-base porphine–Ar van der Waals complex. Can. J. Chem. 63, 2073 (1985). https://doi.org/10.1139/v85-342
F. Schlaghaufer, unpublished results (2020)
R.E.F.E. Riechers, High-resolution spectroscopy in superfluid helium droplets. Investigation of vibrational fine structures in electronic spectra of phthalocyanine and porphyrin derivatives, Dissertation, Universität Regensburg, Regensburg (2011)
C. Callegari, W.E. Ernst, Helium droplets as nanocryostats for molecular spectroscopy—from the vacuum ultraviolet to the microwave regime, 1551–1594, in Handbook of High-resolution Spectroscopy, ed. by M. Quack, F. Merkt (Wiley, Ltd., 2011). ISBN: 978-0-470-74959-3. http://onlinelibrary.wiley.com/book/https://doi.org/10.1002/9780470749593
D. Pentlehner, C. Greil, B. Dick, A. Slenczka, Line broadening in electronic spectra of anthracene derivatives inside superfluid helium nanodroplets. J. Chem. Phys. 133, 1–9 114505 (2010). https://doi.org/10.1063/1.3479583
D. Pentlehner, A. Slenczka, Electronic spectroscopy of 9,10-dichloroanthracene inside helium droplets. J. Chem. Phys. 138, 024313 (2013). https://doi.org/10.1063/1.4773894
D. Pentlehner, A. Slenczka, Helium induced fine structure in the electronic spectra of anthracene derivatives doped into superfluid helium nanodroplets. J. Chem. Phys. 142, 014311 (2015). https://doi.org/10.1063/1.4904899
D. Pentlehner, Perturbations of electronic transitions of organic molecules in helium droplets generated with a new pulsed droplet source, Dissertation, Universität Regensburg, Regensburg (2010)
S. Hirayama, A comparative study of the fluorescence lifetimes of 9-cyanoanthracene in a bulb and supersonic free jet. J. Chem. Phys. 85, 6867–6874 (1986). https://doi.org/10.1063/1.451424
A. Amirav, J. Jortner, S. Okajima, E.C. Lim, Manifestation of intramolecular vibrational energy redistribution on electronic relaxation in large molecules. Chem. Phys. Lett. 126, 487–494 (1986). https://doi.org/10.1016/S0009-2614(86)80162-2
A. Amirav, Rotational and vibrational energy effect on energy-resolved emission of anthracene and 9-cyanoanthracene. Chem. Phys. 124, 163–175 (1988). https://doi.org/10.1016/0301-0104(88)87147-7
A. Amirav, C. Horwitz, J. Jortner, Optical selection studies of electronic relaxation from the S1 state of jet-cooled anthracene derivatives. J. Chem. Phys. 88, 3092–3111 (1988). https://doi.org/10.1063/1.453953
A. Amirav, J. Jortner, Laser-free absorption and fluorescence spectroscopy of large molecules in planar supersonic expansions. Chem. Phys. Lett. 94, 545–548 (1983). https://doi.org/10.1016/0009-2614(83)85052-0
A. Amirav, M. Sonnenschein, J. Jortner, Absolute fluorescence quantum yields of large molecules in supersonic expansions. Chem. Phys. 88, 4214–4218 (1984). https://doi.org/10.1021/j150663a005
F. Tanaka, S. Yamashita, S. Hirayama, A. Adach, K. Shobatake, Fluorescence decays of 9,10-dichloroanthracene and its van der waals complexes with rare gas atoms in supersonic free jets. Chem. Phys. 131, 435–442 (1989). https://doi.org/10.1016/0301-0104(89)80188-0
A. Penner, A. Amirav, V. Jortner, A. Nitzan, J. Gersten, Solvation effects on molecular pure radiative lifetime and absorption oscillator strength in clusters. J. Chem. Phys. 93, 147–159 (1990). https://doi.org/10.1063/1.459613
N. Ben-Horin, D. Barhatt, U. Even, J. Jortner, Spectroscopic interrogation of heterocluster isomerization. II. Spectroscopy of (9,10 dichloroanthracene)⋅(rare gas)n heteroclusters. J. Chem. Phys. 97, 6011–6031 (1992). https://doi.org/10.1063/1.463712
A. Penner, A. Amirav, Vibrational predissociation of 9,10-dichloroanthracene—mixed and homo rare gas atom clusters. J. Chem. Phys. 99, 9616–9629 (1993). https://doi.org/10.1063/1.465495
A. Stromeck-Faderl, D. Pentlehner, U. Kensy, B. Dick, High-resolution electronic spectroscopy of the BODIPY chromophore in supersonic beam and superfluid helium droplets. ChemPhysChem 12, 1969–1980 (2011). https://doi.org/10.1002/cphc.201001076
A. Stromeck-Faderl, Hochauflösende Spektroskopie an isolierten Molekülen im Überschall-Düsenstrahl—Untersuchung von Pyrromethen-Farbstoffen, Dissertation, Universität Regensburg, Regensburg (2009)
D.W. Werst, W.R. Gentry, P.F. Barbara, The S0 and S1 torsional potentials of 9-Phenylanthracene. J. Phys. Chem. 89, 729–732 (1985). https://doi.org/10.1021/j100251a001
M. Lewerenz, B. Schilling, J.P. Toennies, Successive capture and coagulation of atoms and molecules to small clusters in large liquid helium clusters. J. Chem. Phys. 102, 8191–8207 (1995). https://doi.org/10.1063/1.469231
A. Amirav, U. Even, J. Jortner, Excited-state energetics and dynamics of pentacene-rare gas complexes. J. Phys. Chem. 85, 309–312 (1981). https://doi.org/10.1021/j150604a002
S. Leutwyler, J. Jortner, The adsorption of rare-gas atoms on microsurfaces of large aromatic molecules. J. Phys. Chem. 91, 5558–5568 (1987). https://doi.org/10.1021/j100306a014
W.M. Van Herpen, W.L. Meerts, A. Dymanus, Rotationally resolved laser spectroscopy of tetracene and its van der Waals complexes with inert gas atoms. J. Chem. Phys. 87, 182–190 (1987). https://doi.org/10.1063/1.453613
N. Ben-Horin, U. Even, J. Jortner, S. Leutwyler, Spectroscopy and nuclear dynamics of tetracene–rare‐gas heteroclusters. J. Chem. Phys. 97, 5296–5315 (1992). https://doi.org/10.1063/1.463790
E. Shalev, N. Ben-Horin, U. Even, J. Jortner, Electronic spectral shifts of aromatic molecule–rare‐gas heteroclusters. J. Chem. Phys. 95, 3147–3166 (1991). https://doi.org/10.1063/1.460872
N. Pörtner, A.F. Vilesov, M. Havenith, The formation of heterogeneous van der Waals complexes in helium droplets. Chem. Phys. Lett. 343, 281–288 (2001). https://doi.org/10.1016/S0009-2614(01)00648-0
T.R. Hays, V. Henke, V. Selzle, E.W. Schlag, Anthracene-argon complexes in a supersonic jet; spectra and lifetimes. Chem. Phys. Lett. 77, 19–24 (1981). https://doi.org/10.1016/0009-2614(81)85591-1
A. Amirav, U. Even, J. Jortner, Electronic-vibrational excitations of aromatic molecules in large argon clusters. J. Phys. Chem. 86, 3345–3358 (1982). https://doi.org/10.1021/j100214a017
W.F. Henke, W. Yu, H.L. Selzle, E.W. Schlag, D. Wutz, S.H. Lin, Theoretical study of electronic spectral shifts of van der Waals complexes. Chem. Phys. 97, 205–215 (1985). https://doi.org/10.1016/0301-0104(85)87032-4
A. Heikal, L. Bañares, D.H. Semmes, A.H. Zewail, Real-time dynamics of vibrational predissociation in anthracene-Arn (n = 1, 2, 3). Chem. Phys. 156, 231–250 (1991). https://doi.org/10.1016/0301-0104(91)80092-V
T. Chakraborty, E.C. Lim, Study of van der Waals clusters of anthracene by laser-induced fluorescence in a supersonic jet: evidence for two structurally different dimers. J. Phys. Chem. 97, 11151–11153 (1993). https://doi.org/10.1021/j100145a004
M.C.R. Cockett, K. Kimura, A study of anthracene–Arn (n=0–5) in the ground cationic state by laser threshold photoelectron spectroscopy: Selective ionization of complex isomers formed in the free jet expansion. J. Chem. Phys. 100, 3429–3441 (1994). https://doi.org/10.1063/1.466386
S.M. Ohline, J. Romascan, P.M. Felker, Rotational coherence spectroscopy of aromatic-(Ar)n clusters: geometries of anthracene-(Ar)n, 9,10-dichloroanthracene-Ar, and tetracene-Ar. J. Phys. Chem. 99, 7311–7319 (1995). https://doi.org/10.1021/j100019a014
D. Uridat, V. Brenner, I. Dimicoli, J. Le Calvé, P. Millié, M. Mons, F. Piuzzi, Existence of two internal energy distributions in jet-formed van der Waals heteroclusters: example of the anthracene–argonn system. Chem. Phys. 239, 151–175 (1998). https://doi.org/10.1016/S0301-0104(98)00269-9
E.-M. Lottner, A. Slenczka, Anthracene−argon clusters generated in superfluid helium nanodroplets: new aspects on cluster formation and microsolvation. J. Phys. Chem. A 124, 311–321 (2020). https://doi.org/10.1021/acs.jpca.9b04138
F. Calvo, E. Yurtsever, The metastable structures of anthracene‑argon clusters inside helium nanodroplets. Theor. Chem. Acc. 140, 21 (2021). https://doi.org/10.1007/s00214-021-02721-4
S.H. Cho, M. Yoon, S.K. Kim, Spectroscopy and energy disposal dynamics of phthalocyanine–Arn (n=1,2) complexes generated by hyperthermal pulsed nozzle source. Chem. Phys. Lett. 326, 65–72 (2000). https://doi.org/10.1016/S0009-2614(00)00777-6
R. Lehnig, J.A. Sebree, A. Slenczka, Structure and dynamics of phthalocyanine-Argonn (n=1-4) complexes studied in helium nanodroplets. J. Phys. Chem. A 111, 7576–7584 (2007). https://doi.org/10.1021/jp0708493
E. Lugovoj, J.P. Toennies, A. Vilesov, Manipulating and enhancing chemical reactions in helium droplets. J. Chem. Phys. 112, 8217–8220 (2000). https://doi.org/10.1063/1.481426
C. Ottinger, R.N. Zare, Crossed beam chemiluminescence. Chem. Phys. Lett. 5, 243-248 (1970). https://doi.org/10.1016/0009-2614(70)85016-3
J.W. Cox, P.J. Dagdigian, Singlecollision chemiluminescence study of the Ba(1 S,3 D)+NO2, N2O, O3 reactions. J. Chem. Phys. 79, 5351–5259 (1983). https://doi.org/10.1063/1.445698
C.D. Jonah, R.N. Zare, C. Ottinger, Crossed‐beam chemiluminescence studies of some group IIa metal oxides. J. Chem. Phys. 56, 263–274 (1972). https://doi.org/10.1063/1.1676857
C. Alcaraz, P. de Pujo, J. Cuvellier, J.M. Mestdagh, Collision energy dependence of the chemiluminescent reaction: Ba+N2O→BaO+N2. J. Chem. Phys. 89, 1945–1949 (1988). https://doi.org/10.1063/1.455092
J.P. Visticot, J.M. Mestdagh, C. Alcaraz, J. Cuvellier, J. Berlande, Reaction of barium atoms with N2O clusters. J. Chem. Phys. 88, 3081–3085 (1998). https://doi.org/10.1063/1.453951
A. Lallement, J. Cuvellier, J.M. Mestdagh, P. Meynadier, P. de Pujo, O. Sublemontier, J.P. Visticot, J. Berlande, X. Biquard, Reaction between (N2O, Ar) binary clusters and barium atoms. Chem. Phys. Lett. 189, 182–188 (1992). https://doi.org/10.1016/0009-2614(92)85120-Y
J.M. Mestdagh, V. Gaveau, C. Gee, O. Sublemontier, J.P. Visticot, Cluster isolated chemical reactions. Int. Rev. Phys. Chem. 16, 215–247 (1997). https://doi.org/10.1080/014423597230280
A. Braun, V. Drabbels, Imaging the translational dynamics of CF3 in liquid helium droplets. Phys. Rev. Lett. 93, 253401 (2004). https://doi.org/10.1103/PhysRevLett.93.253401
A. Braun, M. Drabbels, Photodissociation of alkyl iodides in helium nanodroplets. I. Kinetic energy transfer. J. Chem. Phys. 127, 114303 (2007). https://doi.org/10.1063/1.2767261
A. Braun, M. Drabbels, Photodissociation of alkyl iodides in helium nanodroplets. II. Solvation dynamics. J. Chem. Phys. 127, 114304 (2007). https://doi.org/10.1063/1.2767262
A. Braun, M. Drabbels, Photodissociation of alkyl iodides in helium nanodroplets. III. Recombination. J. Chem. Phys. 127, 114305 (2007). https://doi.org/10.1063/1.2767263
A.T.J.B. Eppink, D.H. Parker, Methyl iodide A-band decomposition study by photofragment velocity imaging. J. Chem. Phys. 109, 4758–4767 (1998). https://doi.org/10.1063/1.477087
A.T.J.B. Eppink, D.H. Parker, Velocity map imaging of ions and electrons using electrostatic lenses: application in photoelectron and photofragment ion imaging of molecular oxygen. Rev. Sci. Instr. 68, 3477–3485 (1997). https://doi.org/10.1063/1.1148310
P.K. Sengupta, M. Kasha, Excited state proton-transfer spectroscopy of 3-hydroxyflavone and quercetin. Chem. Phys. Lett. 68, 382–385 (1979). https://doi.org/10.1016/0009-2614(79)87221-8
D. McMorrow, M. Kasha, Intramolecular excited-state proton transfer in 3-hydroxyflavone. Hydrogen-bonding solvent perturbations. J. Phys. Chem. 88, 2235–2243 (1984). https://doi.org/10.1021/j150655a012
B. Dick, N.P. Emsting, Excited-state intramolecular proton transfer in 3-hydroxylflavone isolated in solid argon: fluoroescence and fluorescence-excitation spectra and tautomer fluorescence rise time. J. Phys. Chem. 91, 4261–4265 (1987). https://doi.org/10.1021/j100300a012
N.P. Ernsting, B. Dick, Fluorescence excitation of isolated, jet-cooled 3-hydroxyflavone: the rate of excited state intramolecular proton transfer from homogeneous linewidths. Chem. Phys. 136, 181–186 (1989). https://doi.org/10.1016/0301-0104(89)80045-X
A. Mühlpfordt, T. Bultmann, N.P. Ernsting, B. Dick, Excited-state intramolecular proton transfer in jet-cooled 3-hydroxyflavone. Deuteration studies, vibronic double-resonance experiments, and semiempirical (AM1) calculations of potential-energy surfaces. Chem. Phys. 181, 447–460 (1994). https://doi.org/10.1016/0301-0104(93)E0448-5
A. Ito, Y. Fujiwara, M. Itoh, Intramolecular excited-state proton transfer in jet-cooled 2-substituted 3-hydroxychromones and their water clusters. J. Chem. Phys. 96, 7474–7482 (1992). https://doi.org/10.1063/1.462398
A.S. Klymchenko, A.P. Demchenko, 3-Hydroxychromonedyes exhibiting excited-state intramolecular proton transfer in water with efficient two-band fluorescence. New J. Chem. 28, 687–692 (2004). https://doi.org/10.1039/B316149H
K. Bartl, A. Funk, M. Gerhards, IR/UV spectroscopy on jet cooled 3-hydroxyflavone (H2O)n (n=1,2) clusters along proton transfer coordinates in the electronic ground and excited states. J. Chem. Phys. 129, 234306 (2008).https://doi.org/10.1063/1.3037023
K. Bartl, A. Funk, K. Schwing, H. Fricke, G. Kock, H.-D. Martin, M. Gerhards, IR spectroscopy applied subsequent to a proton transfer reaction in the excited state of isolated 3-hydroxyflavone and 2-(2-naphthyl)-3-hydroxychromone. Phys. Chem. Chem. Phys. 11, 1173–1179 (2009). https://doi.org/10.1039/B813425A
R. Lehnig, D. Pentlehner, A. Vdovin, B. Dick, A. Slenczka, Photochemistry of 3-hydroxyflavone inside superfluid helium nanodroplets. J. Chem. Phys. 131, 194307 (2009).https://doi.org/10.1063/1.3262707
A.N. Bader, F. Ariese, C. Gooijer, Proton transfer in 3-hydroxyflavone studied by high-resolution 10 K laser-excited Shpol’skii spectroscopy. J. Phys. Chem. A 106, 2844–2848 (2002). https://doi.org/10.1021/jp013840o
R. Lehnig, A. Slenczka, Quantum solvation of phthalocyanine in superfluid helium droplets. J. Chem. Phys. 120, 5064–5066 (2004). https://doi.org/10.1063/1.1647536
Acknowledgements
We gratefully acknowledge critical comments from J. Peter Toennies. A.S. would like to acknowledge the tremendous contribution of his PhD students who have shared part of their life time to perform excellent experimental work in the Regensburg helium droplet laboratory. Among those are Rudolf Lehnig, Dominik Pentlehner, Ricarda Riechers, Eva-Maria Lottner née Wirths, Tobias Premke. Guest scientists who have joined the laboratory at different times are Alexander Vdovin, Joshua A. Sebree, Lars Christansen, and Jens H. Nielsen. Finally, A.S. acknowledges the continuous support the helium droplet laboratory received from Bernhard Dick and his successor Patrick Nürnberger as chairholders at the Regensburg Institute for Physical and Theoretical Chemistry.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this chapter

Cite this chapter
Schlaghaufer, F., Fischer, J., Slenczka, A. (2022). Electronic Spectroscopy in Superfluid Helium Droplets. In: Slenczka, A., Toennies, J.P. (eds) Molecules in Superfluid Helium Nanodroplets. Topics in Applied Physics, vol 145. Springer, Cham. https://doi.org/10.1007/978-3-030-94896-2_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-94896-2_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-94895-5
Online ISBN: 978-3-030-94896-2
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)