
Abstract
Manufacturing immune effector cells (T or NK cells) with CAR-encoding DNA sequences requires efficient and safe genetic engineering procedures. For this purpose, an appropriate genetic vector is chosen according to numerous factors, including the vector genome packaging capacity, cellular tropism, genomic integration, immune toxicity, and other factors. In clinical trials, genomes integrating viral vectors, in particular vectors based on members of the Retroviridae family, such as retroviruses and lentiviruses, have been successfully used for more than 20 years. These vectors contain an RNA genome that when transcribed into double-stranded DNA by reverse transcriptase integrates into the genome of the transduced cell.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


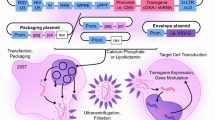
Manufacturing immune effector cells (T or NK cells) with CAR-encoding DNA sequences requires efficient and safe genetic engineering procedures. For this purpose, an appropriate genetic vector is chosen according to numerous factors, including the vector genome packaging capacity, cellular tropism, genomic integration, immune toxicity, and other factors. In clinical trials, genomes integrating viral vectors, in particular vectors based on members of the Retroviridae family, such as retroviruses and lentiviruses, have been successfully used for more than 20 years. These vectors contain an RNA genome that when transcribed into double-stranded DNA by reverse transcriptase integrates into the genome of the transduced cell.
Several precautions are taken to ensure the safe use of such integrating vectors. First, the viral genome is split into three different expression constructs to reduce the risk of recombination events re-establishing replication-competent viruses. Second, long terminal repeats (LTRs) with their enhancer/promoter sequences are deleted, resulting in self-inactivated (SIN) vectors to avoid transactivation of cellular genes in the vicinity of the viral integration site. Third, the viral envelope is pseudotyped with heterologous glycoproteins, such as gibbon ape leukaemia virus (GALV) or vesicular stomatitis virus (VSV)-G protein, to restrict the cell tropism for transduction. The viral vectors have undergone generations of modifications and are classified according to their packaging plasmid. During manufacturing, the use of transduction enhancers, including cationic polymers, lipids, and peptides, such as Retronectin or Vectofusin-1, which is a histidine-rich cationic amphipathic short peptide (Jamali et al. 2019), improves the transduction efficiencies.
Retroviral vectors modified with the LTRs of the myeloproliferative sarcoma virus and an improved 5′ untranslated region, named MP71 retroviral vectors, can achieve high transduction efficiencies in human T cells. While retroviral vectors require actively dividing cells for integration, lentiviral vectors have the capacity to transduce nondividing or slowly proliferating cells and are currently increasingly used for genetic modification of T cells in clinical trials. Cycling T cells can efficiently complete the reverse transcription process of the viral vector, facilitate nuclear import, and enhance the expression of the transgene. Obtaining high virus titres and ultimately sufficient transduction frequencies for production of CAR-T cells on a clinical scale and preserving the T cell phenotype and functional properties after transduction remain a challenge. Despite vector integration into the host genome, T cells have a negligible risk of transformation; thus far, no leukaemia has been observed in T cell-based therapy.
Alternatively, artificial virus-like particles (VLPs) pseudotyped with VSV-G can be used for transfer into haematopoietic cells (Mangeot et al. 2019). DNA packed into transposon vectors, such as sleeping beauty and piggyBac, are transferred to T cells via electroporation (Kebriaei et al. 2016). Transposon-based genetic engineering does not require time-consuming and cost-intensive virus production and is increasingly considered for clinical manufacture of CAR-T cells.
In contrast to integrating DNA transfer technologies, mRNA transfer via electroporation or cationic lipid-mediated transfection produces T cells with transient CAR expression for a few days (Miliotou and Papadopoulou 2020). Such transient CAR-T cell approaches have been investigated and found to produce antitumour reactivity for a limited time to avoid any undesirable effects in patients; however, very few clinical trials using RNA-modified CAR-T cells have been registered.
Genome editing is an upcoming tool to engineer CAR-T cells using specific endonucleases, including meganucleases (MGNs), transcription activator-like effector nucleases (TALENs), megaTAL nucleases, zinc-finger nucleases (ZFNs) and, more recently, clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9-associated nucleases (Pavlovic et al. 2020). These technologies allow insertion of a specific DNA sequence at a predefined emplacement, such as endogenous genetic locus. While efficiently applied in haematopoietic or mesenchymal stem cell modification for years, genome editing in primary T cells has only recently been successfully applied towards efficient CAR-T cell engineering. Examples for potential clinical application are targeting the respective genes for programmed cell death-1 (PD1, CD279), T cell receptor (TCR) α and β chains, CD52, human leukocyte antigens (HLAs), and β2-microglobulin (β2M).
One major application of genome editing is creating “off-the-shelf” allogeneic CAR-T cells to avoid certain limitations associated with autologous T cells, such as the personalized production process, the several weeks of time required for manufacturing, and the risk of manufacturing failure. Such allogeneic CAR-T cells were engineered by genetically eliminating the TCRα constant (TRAC) locus and/or HLA from the T cell surface, reducing the risk of graft versus host disease (GvHD) and allograft rejection. In particular, Torikai et al. combined sleeping beauty transposon-based gene transfer with ZFN-mediated deletion of TCR α and β chains (Torikai et al. 2012); subsequent approaches also eliminated the endogenous TCR (Roth et al. 2018; Legut et al. 2018; Osborn et al. 2016). TALEN-mediated TRAC/CD52 knockout of CD19-specific CAR-T cells (UCART19) was administered to two patients with relapsed ALL in a proof-of-concept study, and no GvHD was reported (Qasim et al. 2017). Several approaches using ZFNs and CRISPR/Cas9, including base editing variants, were used to eliminate HLA class I expression by targeting β2M (Webber et al. 2019) and eliminating the HLA class II transactivator CIITA (Kagoya et al. 2020), all reducing the risk of allogeneic CAR-T cell rejection. To reduce GvHD and fratricide, the CD7 locus was disrupted along with TCRα editing (Gomes-Silva et al. 2017). Eliminating the gene for the TGF-β receptor or PD-1 enhanced CAR-T cell antitumour potency by reducing repression by the tumour stroma (Tang et al. 2020).
Genome editing has also been used to insert the CAR-encoding DNA sequence into the TCR α locus (Eyquem et al. 2017), thereby utilizing the TCR expression machinery for properly regulated CAR expression. Similarly, CAR-encoding DNA was inserted into the TCR α locus, and IL-12-encoding DNA was inserted into the IL2Rα or PDCD1 locus, resulting in CAR-redirected T cell activation along with IL-12 secretion (Sachdeva et al. 2019). Such genome editing approaches can be applied to target other signalling pathways to engineer CAR-T cells with therapeutic outputs in a highly regulated manner. Currently, most of these editing technologies are being explored in mouse models or in a very limited number of patients, making it difficult to draw a definitive conclusion concerning safety and efficacy in the long term.
-
Lentiviral gene transfer is the most frequently applied procedure to engineer CAR-T cells for clinical use.
-
Nonviral transposon-mediated DNA transfer is an upcoming technology to obtain CAR-T cells.
-
Allogeneic “off-the-shelf” CAR-T cells are engineered by genetically eliminating the TCRα constant (TRAC) locus and/or HLA from the T cell surface, reducing the risk of graft versus host disease (GvHD) and allograft rejection.
References
Eyquem J, Mansilla-Soto J, Giavridis T, et al. Targeting a CAR-T o the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–7.
Gomes-Silva D, Srinivasan M, Sharma S, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130:285–96.
Jamali A, Kapitza L, Schaser T, et al. Highly efficient and selective CAR-gene transfer using CD4- and CD8-targeted lentiviral vectors. Mol Ther Methods Clin Dev. 2019;13:371–9.
Kagoya Y, Guo T, Yeung B, et al. Genetic ablation of HLA class I, class II, and the T-cell receptor enables allogeneic T cells to be used for adoptive T-cell therapy. Cancer Immunol Res. 2020;8:926–36.
Kebriaei P, Singh H, Huls MH, et al. Phase I trials using sleeping beauty to generate CD19-specific CAR-T cells. J Clin Invest. 2016;126:3363–76.
Legut M, Dolton G, Mian AA, et al. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood. 2018;131:311–22.
Mangeot PE, Risson V, Fusil F, et al. Genome editing in primary cells and in vivo using viral-derived nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat Commun. 2019;10:45.
Miliotou AN, Papadopoulou LC. In vitro-transcribed (IVT)-mRNA CAR-T therapy development. Methods Mol Biol. 2020;2086:87–117.
Osborn MJ, Webber BR, Knipping F, et al. Evaluation of TCR gene editing achieved by TALENs, CRISPR/Cas9, and megaTAL nucleases. Mol Ther. 2016;24:570–81.
Pavlovic K, Tristán-Manzano M, Maldonado-Pérez N, et al. Using gene editing approaches to fine-tune the immune system. Front Immunol. 2020;11:570672.
Qasim W, Zhan H, Samarasinghe S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR-T cells. Sci Transl Med. 2017;9:eaaj2013.
Roth TL, Puig-Saus C, Yu R, et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. 2018;559:405–9.
Sachdeva M, Busser BW, Temburni S, et al. Repurposing endogenous immune pathways to tailor and control chimeric antigen receptor T cell functionality. Nat Commun. 2019;10:5100.
Tang N, Cheng C, Zhang X, et al. TGF-beta inhibition via CRISPR promotes the long-term efficacy of CAR-T cells against solid tumors. JCI Insight. 2020;5:e133977.
Torikai H, Reik A, Liu PQ, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119:5697–705.
Webber BR, Lonetree CL, Kluesner MG, et al. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat Commun. 2019;10:5222.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this chapter

Cite this chapter
Benabdellah, K., Thomas, S., Abken, H. (2022). Genetic Engineering of Autologous or Allogeneic Immune Effector Cells. In: Kröger, N., Gribben, J., Chabannon, C., Yakoub-Agha, I., Einsele, H. (eds) The EBMT/EHA CAR-T Cell Handbook. Springer, Cham. https://doi.org/10.1007/978-3-030-94353-0_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-94353-0_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-94352-3
Online ISBN: 978-3-030-94353-0
eBook Packages: MedicineMedicine (R0)