
Abstract
A Recurrent Neural Network (RNN) trained with a set of molecules represented as SMILES strings can generate millions of different valid and meaningful chemical structures. In most of the reported architectures the models have been trained using a canonical (unique for each molecule) representation of SMILES. Instead, this research shows that when using randomized SMILES as a data amplification technique, a model can generate more molecules and those are going to accurately represent the training set properties. To show that, an extensive benchmark study has been conducted using research from a recently published article which shows that models trained with molecules from the GDB-13 database (975 million molecules) achieve better overall chemical space coverage when the posterior probability distribution is as uniform as possible. Specifically, we created models that generate nearly all the GDB-13 chemical space using only 1 million molecules as training set. Lastly, models were also trained with smaller training set sizes and show substantial improvement when using randomized SMILES compared to canonical.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others

Keywords
- Cheminformatics
- Molecular generative models
- Randomized SMILES
- Molecular databases
- Recurrent Neural Networks
- Benchmarking
1 Introduction
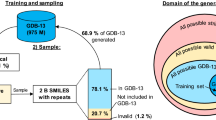
Molecular deep generative models have emerged as a powerful tool to generate chemical space [6] and obtain optimised compounds [2, 5]. Models trained with a set of drug-like molecules can generate molecules that are similar but not equal to those in the training set, thus spanning a bigger chemical space than that of training data. The most popular architecture uses Recurrent Neural Networks (RNNs) and the SMILES syntax [7] to represent molecules. Nevertheless, a recent publication [1] shows that this architecture introduces bias to the generated chemical space. To be able to prove that, models were created with a subset of GDB-13 [4], a database that holds most drug-like molecules up to 13 heavy atoms, and sampled with replacement 2 billion times. At most, only 68% of GDB-13 could be obtained from a theoretical maximum of 87%, which would be from a sample of the same size from an ideal model that has a uniform probability of obtaining each molecule from GDB-13.
This study uses the previous research as a starting point and focuses on benchmarking RNN with SMILES trained with subsets of GDB-13 of different sizes (1 million and 1000 molecules) and with different variants of the SMILES notation. One of those variants, randomized SMILES, can be used as a data amplification technique and is shown to generate more diversity [3]. When the right data representations and hyperparameter combinations are chosen, models are able to generate more diversity and learn to better generalise the training set information.
2 Methods
The model architecture used is similar to the one used in [1, 5]. The training set sequences are pre-processed, and for each training epoch the entire training set is shuffled and subdivided in batches. The encoded SMILES strings of each batch are input token by token to an embedding layer, followed by several layers of RNN cells. Between the inner RNN layers there can be dropout layers. Then, the output from the cells is squeezed to the vocabulary size by a linear layer and a softmax is performed to obtain the probabilities of sampling each token in the next position. This is repeated for each token in the entire sequence.
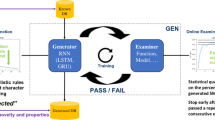
The models were optimised for the hyperparameter combinations shown in Table 1. Also, training sets were set up with canonical SMILES and randomized SMILES. In the case of the randomized SMILES, each training epoch had a different permutation. For each combination of hyperparameters a model was trained and a sample with replacement of 2 billion SMILES strings was performed (Fig. 1). Then, three ratios were calculated from the percentages obtained that characterise the three main properties that the output domain should have: uniformity (even posterior probability for each molecule), completeness (all molecules from GDB-13) and closeness (no molecules outside of GDB-13 should be generated). Lastly, the UCC, a ratio obtained from the other three was used as a sorting criteria for all the models.

Training and sampling process used for each model in the benchmark and the formulas for the ratios calculated from the sample.
3 Results
Table 2 shows the results for the models with highest UCC score of each training set size with each SMILES variant. 1M models trained with randomized SMILES are overall better than those trained with canonical SMILES. This might be due to the additional information the model has from molecules in the training set when they are input as different randomized SMILES each epoch. Notice especially that the completeness is at 0.95, which indicates that the model is theoretically able to reproduce mostly all of GDB-13 given enough sampling. On the other hand, models trained with 1000 SMILES have much lower performance, as there is not enough information in the training sets to be able to generalise the entire database. Nevertheless, the randomized SMILES model has an even better performance compared to the canonical SMILES one. Namely, a model trained with canonical SMILES can only reach 52% valid molecules, whereas the randomized SMILES model learns much better (82%). This shows that randomized SMILES add more information to the model and effectively increase its learning capability without having to add additional data to the training set.
References
Arús-Pous, J., Blaschke, T., Ulander, S., Reymond, J.L., Chen, H., Engkvist, O.: Exploring the GDB-13 chemical space using deep generative models. J. Cheminform. 11(1), 20 (2019). https://doi.org/10.1186/s13321-019-0341-z
Awale, M., Sirockin, F., Stiefl, N., Reymond, J.l.: Drug analogs from fragment based long short-term memory generative neural networks (2018). https://doi.org/10.26434/chemrxiv.7277354.v1, https://chemrxiv.org/articles/Drug_Analogs_from_Fragment_Based_Long_Short-Term_Memory_Generative_Neural_Networks/7277354
Bjerrum, E.J.: SMILES Enumeration as Data Augmentation for Neural Network Modeling of Molecules. arXiv March 2017. http://arxiv.org/abs/1703.07076
Blum, L.C., Reymond, J.L.: 970 million druglike small molecules for virtual screening in the chemical universe database GDB-13. J. Am. Chem. Soc. 131(25), 8732–8733 (2009). https://doi.org/10.1021/ja902302h
Olivecrona, M., Blaschke, T., Engkvist, O., Chen, H.: Molecular de novo design through deep reinforcement learning. J. Cheminform. 9(1) (2017). https://doi.org/10.1186/s13321-017-0235-x, http://arxiv.org/abs/1704.07555
Segler, M.H.S., Kogej, T., Tyrchan, C., Waller, M.P.: Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 4(1), 1–17 (2018). https://doi.org/10.1021/acscentsci.7b00512, http://arxiv.org/abs/1701.01329
Weininger, D.: SMILES, a chemical language and information system: 1: introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28(1), 31–36 (1988). https://doi.org/10.1021/ci00057a005
Acknowledgements
This project is supported financially by the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 676434, “Big Data in Chemistry” (“BIGCHEM” http://bigchem.eu).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2019 The Author(s)
About this paper

Cite this paper
Arús-Pous, J. et al. (2019). Improving Deep Generative Models with Randomized SMILES. In: Tetko, I., Kůrková, V., Karpov, P., Theis, F. (eds) Artificial Neural Networks and Machine Learning – ICANN 2019: Workshop and Special Sessions. ICANN 2019. Lecture Notes in Computer Science(), vol 11731. Springer, Cham. https://doi.org/10.1007/978-3-030-30493-5_68
Download citation
DOI: https://doi.org/10.1007/978-3-030-30493-5_68
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-30492-8
Online ISBN: 978-3-030-30493-5
eBook Packages: Computer ScienceComputer Science (R0)