
Abstract
Hypercholesterolemia is a major risk factor in atherosclerosis development and lipid-lowering drugs (i.e., statins) remain the treatment of choice. Despite effective reduction of LDL cholesterol in patients, a residual cardiovascular risk persists in some individuals, highlighting the need for further therapeutic intervention. Recently, the CANTOS trial paved the way toward the development of specific therapies targeting inflammation, a key feature in atherosclerosis progression. The pre-existence of multiple drugs modulating both innate and adaptive immune responses has significantly accelerated the number of translational studies applying these drugs to atherosclerosis. Additional preclinical research has led to the discovery of new therapeutic targets, offering promising perspectives for the treatment and prevention of atherosclerosis. Currently, both drugs with selective targeting and broad unspecific anti-inflammatory effects have been tested. In this chapter, we aim to give an overview of current advances in immunomodulatory treatment approaches for atherosclerotic cardiovascular diseases.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Adaptive immunity
- Anti-inflammatory therapy
- Atherosclerosis
- Cardiovascular diseases
- Immunotherapy
- Inflammation
- Innate immunity
1 Introduction
Atherosclerosis is a lipid-driven disease that is characterized by the formation of plaques in the subendothelial space of arteries. Therefore, lipid-lowering drugs such as statins are considered the treatment of choice as their reduction of circulating low-density lipoprotein (LDL) can prevent plaque formation and progression in patients (Ference et al. 2017; Borén et al. 2020). A crucial step in disease initiation involves the retention of LDL and other ApoB-carrying lipoproteins within the vascular wall due to their ability to bind intimal proteoglycans. Subsequent enzymatic and non-enzymatic modifications of these lipoprotein particles result in the formation of damage-associated molecular patterns (DAMPs) that trigger an inflammatory response. This includes the activation of endothelial cells by oxidized phospholipids and other lipid species. In response, the endothelium secretes chemoattractant cytokines like monocyte chemoattractant protein-1 (MCP-1), growth factors (macrophage colony-stimulating factor (MCSF), granulocyte-macrophage colony-stimulating factor (GMCSF)) and upregulates the expression of genes coding for adhesion molecules (VCAM-1 and ICAM-1), allowing the recruitment and differentiation of inflammatory monocytes. Monocytes recruited into the intima become lesional macrophages, which take up oxidized LDL and aggregated LDL via scavenger receptors (CD36, SR-A), ultimately leading to foam cell formation – a rate-limiting step that greatly contributes to the development and progression of atherosclerotic plaques (Borén et al. 2020). Excess accumulation of modified lipoproteins, formation of foam cells, subsequent macrophage cell death, and impaired clearance of these apoptotic cells, all contribute to chronic inflammation in the vascular wall (Borén et al. 2020; Libby 2021). Studies over the past 20 years have focused on the chronic inflammatory nature of atherosclerosis and have demonstrated the detrimental role of inflammation in its pathophysiology. This work led to the consideration of markers of inflammation such as IL-6 and high-sensitivity C-reactive protein (hsCRP) as biomarker for risk stratification of cardiovascular patients (Ridker et al. 2000a, b, 2009). According to this concept, patients with signs of inflammation (i.e., elevated levels of hsCRP or IL-6) are considered at higher risk of experiencing a cardiovascular event during their lifetime compared to individuals without residual inflammation. Additionally, both innate and adaptive immunity have been found to play a crucial role in the initiation, progression, and rupture of atherosclerotic plaques (Gisterå and Hansson 2017; Wolf and Ley 2019; Libby 2021). Since the importance of the immune system in atherosclerotic plaque development is becoming increasingly clear, novel treatments that target both innate and adaptive immunity have been developed with the aim to complement already established lipid-lowering therapies (Bäck et al. 2019). This chapter aims to introduce the reader to these novel anti-inflammatory and immunomodulatory treatments, which range from small molecule drugs to antibodies and vaccination strategies.
To optimally target the inflammatory component of atherosclerosis, one needs to understand the chronic inflammatory nature of this specific process. Chronic inflammation is typically the consequence of a non-resolving inflammation due to an impaired clearance and persistent presence of the inciting trigger combined with a hampered removal of freshly recruited inflammatory cells (Tabas and Glass 2013). In atherosclerosis the main triggers of inflammation are sterile DAMPs that result from the pathological accumulation of lipids in the vascular wall. Therefore, optimal strategies targeting chronic inflammation are either directed toward limiting the generation and activity of disease-specific DAMPs or the inflammatory response they incite. Other strategies aim at dampening inflammation or enhancing the resolution processes. Clearly, the more targeted anti-inflammatory treatments are to a specific pathophysiology, the lower the risk of adverse effects such as a reduction of the host’s capacity to fight infections. Thus, careful assessment of the risks and benefits of each anti-inflammatory strategy is needed during the development of these treatments. Modulating both the innate and adaptive responses offers promising possibilities to limit chronic inflammation in the vascular wall.
2 Targeting Innate Immune Responses
2.1 Targeting the NLRP3 Inflammasome
Accumulating evidence shows that the nucleotide-binding oligomerization domain, leucine-rich repeat-containing receptor (NLR) family pyrin domain-containing 3 (NLRP3) inflammasome plays a central role in plaque formation, growth, and stability. Its activation is typically necessary for the secretion of proinflammatory cytokines, such as IL-1β and IL-18, which in turn activate the innate immune system. Ligation of both pattern recognition receptors (PRRs) and tumor necrosis factor receptor (TNFR) can activate the NF-κB pathway, which promotes the transcription of proinflammatory cytokines like tumor necrosis factor-α (TNF-α) and interleukin IL-6 while also initiating the priming of the NLRP3 inflammasome. The latter involves the production of cytosolic NLRP3 protein as well as the inactive form of IL-1 family cytokines: pro-IL-1β and pro-IL-18 (Kelley et al. 2019). Although several PRRs can trigger the formation of inflammasomes, such as NACHT, LRR, and PYD domains-containing protein 1 (NLRP1), NLR family CARD domain-containing protein 4 (NLRC4), and absent in melanoma 2 (AIM2), the NLRP3 inflammasome displays unique activation properties. A large variety of pathogen-associated molecular patterns (PAMPs) (microbial, viral, fungal molecules) and DAMPs (urate crystals) can activate NLRP3. In the context of atherosclerosis, cholesterol crystals (Duewell et al. 2010) have been suggested as key DAMPs driving NLRP3 activation. Importantly, NLRP3 also senses perturbations of cellular homeostasis such as lysosome rupture, ion channel disturbances (K+ efflux), and mitochondrial stress resulting from various stimuli (Kelley et al. 2019). In its activated form, NLRP3 can recruit the apoptosis-associated speck-like protein which contains a caspase recruitment domain (ASC) and the inactivated form of caspase-1 (pro-caspase-1). The proximity of pro-caspase 1 proteins within the complex leads to caspase-1 activation, which proceeds to the proteolytic activation of pro-IL-1β, pro-IL-18, and the propyroptotic factor gasdermin D (GSDMD) involved in pyroptosis, a lytic cell death necessary for cytokine release.
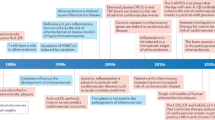
With respect to atherogenesis, important insights came from studies by Duewell and colleagues, who showed that lipid accumulation in the vessel wall leads to the formation of cholesterol crystals which are taken up by macrophages where they activate the NLRP3 inflammasome (Rajamaki et al. 2010; Duewell et al. 2010). In parallel, many studies have investigated the role of NLRP3-derived cytokines (i.e., the IL-1 cytokine family: mainly IL-1β, IL-1α, IL-18, and IL-1R antagonist (IL-1Ra)), as well as proteins involved in NLRP3 activation (ASC, caspase-1) in murine atherosclerosis using transgenic mice. Targeting IL-1 signaling using recombinant IL-1Ra, which competes with IL-1α and IL-1β for binding of IL-1 receptors, in Apoe−/− mice prone to atherosclerosis, resulted in reduced fatty streak formation. This indicates an important role of IL-1Ra in the suppression of lesion development during the early phase of the disease (Elhage et al. 1998; Isoda et al. 2004; Merhi-Soussi et al. 2005). Similarly, overexpression of IL-1Ra in Ldlr−/− mice, fed a high-cholesterol/high-fat diet (containing cholate) for 10 weeks led to a reduction of atherosclerotic lesion area (Devlin et al. 2002). Taken together, these studies suggest that targeting the NLRP3 inflammasome using small molecule inhibitors or targeting NLRP3 products represents promising therapeutic targets for cardiovascular diseases (Fig. 1).

Therapies targeting innate immune responses in atherosclerosis. Proinflammatory cytokines such as IL-6, TNF, IL-1, and IL-18 actively modulate atherosclerosis development. Therefore, several strategies aim at reducing their levels in circulation. The NLRP3 inflammasome greatly contributes to the secretion of such cytokines and several therapeutic approaches have been developed to prevent its deleterious effects. Drugs targeting NLRP3 are of three types: monoclonal antibodies targeting NLRP3 products (i.e., IL1-α-Xilonix, IL1-β-Canakinumab, IL-18-GSK1070806), recombinant proteins (i.e., IL-1-receptor antagonist-Anakinra), and NLRP3 direct inhibitors (i.e., MCC950). While some of these therapies were assessed in large clinical trials (i.e., Canakinumab), others remained inconclusive due to the low number of patients involved in the study (i.e., Xilonix), or were not investigated in humans yet. The potential benefit of such therapies still requires to be evaluated in robust clinical trials
Regarding NLRP3 products, the two IL-1 isoforms (IL-1α and IL-1β) share molecular similarities but are produced by different cellular sources and seem to display different release mechanisms. The release of IL-1α can be inflammasome-dependent or independent, according to the nature of the activating stimulus (Groß et al. 2012). In contrast to IL-1β, the precursor form of IL-1α is active and acts as an alarmin. IL-1α is constitutively expressed as a membrane-bound protein on the surface of various cell types including non-immune cells like epithelial cells. When cells expressing IL-1α undergo cell death, IL-1α is released and acts as an alarmin, inducing IL-1β production and contributing to local inflammation. Additionally, IL-1α can be produced by macrophages, in an inflammasome-independent manner (Freigang et al. 2013). IL-1β on the other hand is mainly produced by hematopoietic cells like dendritic cells, monocytes, and macrophages with its activation requiring (in the majority of cases) the NLRP3 inflammasome. Importantly, IL-1β has been detected in human atherosclerotic plaques and its circulating levels correlate with the severity of coronary atherosclerosis (Galea et al. 1996; Dewberry et al. 2000). Despite these discrepancies, the two IL-1 isoforms share similar downstream effector functions as they both signal via the IL-1 receptor family (IL-1R). Mechanistically, IL-1 signaling impacts various cell types within the atherosclerotic lesion, affecting plaque size and stability. It also contributes to the activation status of endothelial cells, which express adhesion molecules (VCAM-1 and ICAM-1) for the recruitment of leukocytes (Wang et al. 1995). Furthermore, IL-1β induces MCP-1 secretion, which promotes monocyte recruitment and is strongly associated with cardiovascular risk (Jun et al. 2009).
The importance of IL-1α has only recently been appreciated. A small study carried out in coronary artery disease (CAD) patients and healthy control subjects showed higher expression of IL-1α in peripheral blood mononuclear cells (PBMC) of CAD patients (Wæhre et al. 2004). Additionally, IL-1α has been described as the key driver of the senescence-associated secretory phenotype (SASP) of vascular SMCs that can promote vascular inflammation (Clarke et al. 2010; Gardner et al. 2015). Another study carried out by S. Freigang and colleagues also suggests that IL-1α greatly contributes to vascular inflammation in atherosclerosis. Reconstitution of cholesterol fed Ldlr−/− mice with bone marrow from IL-1b−/−, IL-1a−/− and wild-type mice resulted in dramatically reduced atherosclerosis in IL-1α-deficient chimeras but not IL-1β-deficient or wild-type chimeras (Freigang et al. 2013). Similarly, D. Gomez et al showed that antibody-mediated inhibition of IL-1β in Apoe−/− mice with established atherosclerosis failed to decrease lesion size but resulted in adverse plaque remodeling (Gomez et al. 2018). In contrast, other studies suggest beneficial effects of IL-1β targeting in murine atherosclerosis (Kirii et al. 2003; Bhaskar et al. 2011).
Thus, IL-1α and IL-1β may affect plaque formation at different time points in lesion development (Kirii et al. 2003; Duewell et al. 2010; Bhaskar et al. 2011; Gomez et al. 2018; Vromman et al. 2019). While IL-1α appears to be important for the early phase of atherosclerosis, IL-1β seems to be more involved in the progression of established plaques (Vromman et al. 2019). Additionally, Vromman et al. showed that the anatomical location of plaques matters in the response to therapies targeting IL-1 signaling. For example, neutralization of IL-1α or both isoforms using either an anti-IL1α and/or an anti-IL-1β monoclonal antibody reduces the lesion area in aortic roots but not in the brachiocephalic arteries in Apoe −/− mice. Moreover, compensatory outward remodeling was more impaired in the brachiocephalic artery compared to the aortic roots during early atherosclerosis.
In summary, the NLRP3 inflammasome and inflammasome-derived cytokines represent attractive points for therapeutic intervention in cardiovascular diseases. Careful design of preclinical studies will allow better dissection of the mechanistic and physiological aspects of IL-1 inhibition and improvement of the development of new therapies.
2.1.1 Targeting the IL-1 Signaling Pathway
2.1.1.1 Anti-IL-1 β (Canakinumab)
The recent CANTOS (Canakinumab. Anti-Inflammatory Thrombosis Outcome Study) trial has made a profound impact on the field of atherosclerotic cardiovascular disease. It showed for the first time in a large phase 3 clinical trial that therapeutic targeting of inflammation provides a clinical benefit to patients with stable atherosclerosis. The goal of the study was to evaluate the effect of canakinumab, a monoclonal antibody targeting IL-1β, on adverse cardiac events in patients with a history of MI and elevated hsCRP (hsCRP ≥2 mg/L). 10,061 patients, most of whom were on statin therapy, received either placebo or canakinumab at three different doses: 50 mg, 150 mg, or 300 mg, every 3 months. The best results were obtained with the intermediate dose (150 mg) which led to a 15% reduction in the primary endpoint of nonfatal MI, nonfatal stroke, or cardiovascular death. However, as IL-1β is also a crucial cytokine in the host immune response against bacterial infection, its inhibition also significantly increased the number of fatal infections (Ridker et al. 2017). Despite these non-negligible side effects, this study clearly demonstrates a beneficial effect of inhibiting IL-1β signaling.
To better predict positive outcomes of canakinumab therapy in patients, an additional analysis of the CANTOS trial was conducted (Ridker et al. 2018b). Within the canakinumab treated group, patients with hsCRP levels <2 mg/L following treatment had a 25% reduction in major adverse cardiovascular events while no significant benefits were observed in patients with levels ≥2 mg/L. Importantly, cardiovascular mortality and all-cause mortality were both reduced by 31% in patients with hsCRP levels <2 mg/L. This was not the case in patients with levels ≥2 mg/L where no benefits were shown. This analysis clearly documented the need to assess the successful impact on inflammation to optimally appreciate the protective effect of this intervention.
Another analysis published by the CANTOS group indicates that targeting IL-1β alone may not sufficiently alleviate CVD risk, as both IL-6 and IL-18 levels remain predictors of future cardiovascular events in patients receiving anti-IL-1β treatment. Briefly, in patients treated with canakinumab, the risk of major adverse cardiac events (MACE) increased by 15% and 42% for each tertile increase in IL-18 and IL-6 levels, respectively. This suggests a residual inflammatory component that needs to be considered for further cardiovascular risk reduction. Drugs targeting the IL-6 cytokine are currently tested in clinical trials and might benefit patients with sustained elevated IL-6 levels by preventing recurrent CVD events (Biasucci et al. 2020; Ridker et al. 2020).
2.1.1.2 IL-1 Receptor Antagonist (Anakinra)
Anakinra is an antagonist of the IL-1 receptor, blocking the action of both IL-1α and IL-1β isoforms. Since 2001, it has been used as a treatment in patients with rheumatoid arthritis (RA), who are also known to be at high risk of cardiovascular disease (Nurmohamed 2009; Avina-Zubieta et al. 2012; Primdahl et al. 2013). Anakinra was assessed in small clinical trials in patients with myocardial infarction: The “Virginia Commonwealth University-Anakinra Remodeling Trial 1-2-3” (VCU ART1-2-3) and the “Markers of inflammation in non-ST elevation acute coronary syndromes“(MRC ILA-HEART) study (Abbate et al. 2010, 2013, 2020; Morton et al. 2015). In VCU ART, this type of interleukin-1 blockade significantly reduced the systemic inflammatory response with a strong reduction of hsCRP levels in patients with ST-Elevation Myocardial Infarction (STEMI) as well as decreasing the incidence of heart failure. In the MRC ILA-Heart trial, a reduction of IL-6 and hsCRP levels was also observed in non-STEMI (NSTEMI) patients who received Anakinra daily for 2 weeks. However, by day 30, the hsCRP levels in patients treated with Anakinra were significantly higher than in the placebo group, and an unexpected increase of late recurrent ischemic events was observed. Interestingly, a recent study carried out by the Interleukin-1 Genetics Consortium supports the hypothesis that long-term IL-1 inhibition may be associated with an increase in cardiovascular events. In this study, individuals who carried four IL-1Ra raising alleles were found to have an increased odds ratio for coronary heart disease (1.15) compared to those carrying no IL-1Ra raising alleles. Additionally, the concentration of proatherogenic lipids, including LDL cholesterol, increased with each extra allele present (Freitag et al. 2015). However, the VCU-ART study showed no effect on recurrent ischemic events, while there was an increase of MACE upon Anakinra treatment after 1 year in the MRC ILA-Heart trial.
In summary, the relationship between IL-1 targeting and clinical outcomes seems more complex than originally thought. Currently attempted therapeutic strategies include targeting IL-1β (Canakinumab) or both IL-1α and IL-1β with the human recombinant IL-1RA (Anakinra). Further understanding of the distinct roles of the different IL-1 isoforms would enhance our understanding of clinical trial data (CANTOS, VCU ART-1/2/3, MRC ILA-HEART) and improve future trial design.
2.1.2 NLRP3 Inhibitors
Despite the importance of the NLRP3 inflammasome in many inflammatory diseases, there is currently no approved inflammasome inhibitor for the treatment or prevention of atherosclerosis. Nevertheless, many small molecule compounds targeting the NLRP3 inflammasome have been developed, with promising results. Importantly, both canonical and non-canonical NLRP3 activation pathways can be targeted.
2.1.2.1 MCC950
MCC950 is a small molecule inhibitor of the NLRP3 pathway, with a mechanism which is yet to be fully understood (Coll et al. 2015). Nevertheless, it was found to reduce atherosclerotic lesion formation in Apoe−/− mice. This was associated with decreased expression of the adhesion molecules ICAM-1 and VCAM-1, as well as reduced macrophage infiltration within plaques and no change in necrotic core size (Van Der Heijden et al. 2017). This early data suggests that MCC950 represents an interesting candidate to target NLRP3 induced inflammation in atherosclerosis.
2.1.2.2 Tranilast
Tranilast, which was originally used to treat allergy, was shown to inhibit NLRP3 by facilitating its ubiquitination and impairing ASC oligomerization (Huang et al. 2018; Chen et al. 2020). The effect of Tranilast on atherosclerosis was assessed in Watanabe heritable hyperlipidemic rabbits and resulted in decreased atherosclerosis upon treatment (Matsumura et al. 1999). Recently, another study carried out in mice showed that Tranilast decreased the initiation and progression of atherosclerosis in both Ldlr−/− and Apoe−/− prone atherosclerotic mice (Chen et al. 2020). Thus, pharmacologic manipulation of NLRP3 ubiquitination offers a new strategy for protection against atherosclerosis.
Despite encouraging results from animal models, many questions remain to be answered concerning the safety of NLRP3 inhibitors. It will be critical to further examine how these compounds affect the activity of other inflammasomes and how the host immune response is affected in the long run. The initiation of new clinical trials evaluating the benefit of NLRP3 inhibitors in the context of cardiovascular diseases will also depend on these factors.
2.2 Targeting TNF and Interleukins
2.2.1 Tumor Necrosis Factor (TNF)
Tumor necrosis factor (TNF) is a proinflammatory cytokine with a broad spectrum of functions and targets. It controls inflammatory responses by regulating leukocyte activation, maturation, and cytokine/chemokine release. TNF secretion can be triggered by the recognition of PAMPs and DAMPs, but also by cytokines and interferon such as IL-1, GM-CSF, TGF-β, TNF-α itself (autocrine loop), and IFN-γ (Zelová and Hošek 2013). It is mainly produced by macrophages and T cells, but many other cell types, including non-immune cells such as endothelial cells and smooth muscle cells (SMCs), can also secrete TNF. Importantly, TNF seems to be highly expressed in human atherosclerotic plaques (Rus et al. 1991; Canault et al. 2006). Ohta et al showed that TNF enhances the expression of ICAM-1, VCAM-1, and MCP-1 in endothelial cells, favoring monocyte recruitment into the vascular wall. Additionally, an increase of scavenger receptor A dependent uptake of oxidized LDL by macrophages has been described, as well as decreased IL-1β and INF-γ mRNA expression, which together strongly support a deleterious role of TNF in murine atherosclerosis (Ohta et al. 2005; Xiao et al. 2009). Interestingly, recent studies suggest that TNF might also be involved in the regulation of the NLRP3 inflammasome (Sode et al. 2014; Bauernfeind et al. 2016; McGeough et al. 2017).
TNF is a central mediator of the inflammatory response and its signaling mechanism is well described. Many biological agents interfering with TNF signaling have been developed and tested in clinical trials in the context of inflammatory diseases associated with high cardiovascular risk, such as rheumatoid arthritis (RA), psoriatic arthritis, and Crohn’s disease. For example, treatment with TNF blockers (infliximab or etanercept) in RA is associated with a lower incidence of cardiovascular events (Jacobsson et al. 2005). Two meta-analyses also confirmed that RA patients treated with anti-TNF therapy have a lower risk of cardiovascular events in comparison with patients treated with disease-modifying antirheumatic drugs (DMARDs) (Barnabe et al. 2011; Roubille et al. 2015). Currently, five different anti-TNF drugs are approved for clinical use, which include soluble TNF-receptor (etanercept) and TNF monoclonal antibodies (adalimumab, infliximab, golimumab, and certolizumab pegol). The evidence for their use in atherosclerotic cardiovascular disease will be discussed below.
2.2.1.1 Adalimumab
While adalimumab has been used as a safe therapy in a myriad of inflammatory diseases (Burmester et al. 2020), no large-scale trial regarding its effect on atherosclerotic cardiovascular disease exists. A small study in RA patients showed that treatment with adalimumab improves endothelial-dependent vasodilatation (Gonzalez-Juanatey et al. 2006), findings which were confirmed in a second study by the same authors. In this latter study, a comparison of baseline and post-treatment (12 months) carotid artery wall thickness (intima-media) measurement in RA patients showed no statistical difference (Gonzalez-Juanatey et al. 2012). Nevertheless, these observations suggest that adalimumab may improve the state of subclinical atherosclerosis in RA patients. Improvement of endothelial cell function was also observed in a study involving 14 psoriatic patients (Avgerinou et al. 2011). After 12 weeks of adalimumab treatment, no significant change on hsCRP level compared to baseline was observed, but ICAM-1 expression was decreased and the flow-mediated dilation (FMD), used as a marker of endothelial dysfunction, was improved. A larger study (NCT01722214) included 107 psoriatic patients randomized to receive either adalimumab for 52 weeks or placebo for 16 weeks followed by adalimumab for 36 weeks (per-protocol setting). At 16 weeks, the adalimumab treatment group had no difference in vascular inflammation compared to the placebo control group. At the 52-week time point, there was no difference observed in the ascending aorta with adalimumab, but a modest increase in vascular inflammation in carotids was described (Bissonnette et al. 2017). Thus, in lieu of a large clinical trial, the effect of adalimumab in ASCVD remains unclear.
2.2.1.2 Golimumab
Golimumab is a fully-humanized monoclonal anti-TNF antibody approved for the treatment of RA, ankylosing spondylitis (AS), psoriatic arthritis, and ulcerative colitis. Golimumab has a greater affinity for the soluble (sTNF) vs. the transmembrane form (mTNF). While no data exist regarding the therapeutic benefit of golimumab in atherosclerosis, a pilot study in patients with ASCVD provided encouraging results (Tam et al. 2014). This randomized, double-blind, placebo-controlled trial aimed to assess the efficacy of Golimumab in preventing atherosclerosis progression and arterial stiffness in AS patients. A total of 20 patients received 50 mg of Golimumab monthly, with 21 receiving placebo treatment, for 1 year. After 6 months, no significant change in vascular parameters (i.e. aortic stiffness, carotid intima/media thickness) was observed between the two groups. However, a significant progression of the mean intima-media thickness (IMT) was only seen in the placebo group and not in the golimumab group. Maximum IMT, pulse wave velocity (PWV), and augmentation index (Aix) remained unchanged (paired t-test analysis). Nevertheless, no significant difference concerning vascular parameters was demonstrated between the two groups after 1 year of treatment. Further large-scale studies are needed to fully validate the potential effects seen in this study.
In addition to these findings in AS patients, TNF inhibitors were found to improve the overall pathologic profile of RA and psoriasis patients at high cardiovascular risk. Therapeutic targeting of TNF has shown benefit in the prevention of atherosclerosis in RA (Del Porto et al. 2007), which might also be the case in psoriasis (Sattar et al. 2007). Interestingly, a recent meta-analysis that includes 7,697 coronary artery disease (CAD) patients and 9,655 control patients has investigated the association of TNF gene polymorphisms and CAD susceptibility (Huang et al. 2020). The authors concluded that there was no association of TNF 308G/A, 857C/T, 863C/A, and 1031 T polymorphisms with CAD susceptibility. However, the TNF 238G/A genotype showed a significant association with higher CAD susceptibility in the subgroup of Europeans and North Asians, which suggests a direct role of TNF in CAD. Additionally, several studies described an adverse effect of TNF inhibitors on lipid profiles and described an increase of total cholesterol and triglycerides levels (Curtis et al. 2012; Hassan et al. 2016). In summary, further large-scale studies are required to evaluate the potential benefits of anti-TNF agents.
2.2.2 IL-6
Interleukin-6 (IL-6) is a pleiotropic cytokine involved in different arms of the immune system. It can result in both pro- and anti-inflammatory outcomes which can be explained by the different signaling pathways engaged: the classical pathway, the trans-signaling pathway, and the blockade pathway. The classical pathway occurs in a few cell types such as T cells, hepatocytes, and monocytes. It involves the IL-6 receptor (IL-6R) and glycoprotein 130 (gp130), a signal transducer sub-unit of the IL-6 receptor. In the trans-signaling pathway, the mechanism involves the signal transducer gp130 as well as soluble IL-6 receptor (sIL-6R). In contrast to the classical pathway, trans-signaling can occur in any cell type expressing a membrane-bound gp130 protein. In this scenario, sIL-6 binds to sIL-6R and forms a complex that can activate signaling through gp130 resulting in a greater magnitude of proinflammatory state. IL-6 signaling can also be blocked via the soluble form of gp130 which acts as a natural inhibitor of trans-signaling by binding sIL-6/IL-6R complex (Jostock et al. 2001).
Despite inconclusive data from experimental models (Elhage et al. 2001; Song and Schindler 2004; Schieffer et al. 2004; Schuett et al. 2012), shreds of evidence from translational studies identify IL-6 as a key regulator of atherogenesis. Expression of IL-6 was detected in human atherosclerotic plaques (Seino et al. 1994; Rus et al. 1996; Schieffer et al. 2000). Several studies found that high levels of IL-6 are associated with increased cardiovascular risk in human (Mendall et al. 1997; Ridker et al. 2000b; Fisman et al. 2006; Zakai et al. 2007; Danesh et al. 2008; Lefkou et al. 2010). Among them, a study from J. Danesh and colleagues, who compared serial measurements of serum IL-6 between patients with a history of coronary heart disease and controls. Results showed that long-term exposure to circulating IL-6 was associated with an increased risk of coronary artery disease (Danesh et al. 2008). However, follow-up studies of the CANTOS trial showed that patients with elevated IL-6 levels after canakinumab treatment have no significant benefit as opposed to patients with lower IL-6 levels (Ridker et al. 2018a, 2020).
Collectively, these studies and the studies based on the CANTOS trial suggest an important role of IL-6 in cardiovascular diseases. Targeting the IL-6 axis may be crucial to reduce the residual inflammation responsible for the increased risk of cardiovascular events.
Several clinical trials tested the therapeutic benefit of targeting the IL-6 axis using Tocilizumab (Table 1). However, higher-powered studies are required to better evaluate the safety and efficiency of current IL-6 targeting agents, and appreciate their benefit in preventing atherosclerosis.
2.3 Targeting Chronic Inflammation Using Broad Anti-inflammatory Drugs
Given the importance of inflammation in atherosclerosis, the usage of broad anti-inflammatory drugs could potentially reduce the risk of atherosclerotic events. Well known for their anti-inflammatory properties, methotrexate and colchicine are widely used as main treatments in inflammatory diseases with a high risk of CVD, notably RA, psoriasis, and other rheumatologic disorders (Saag et al. 2008; Singh et al. 2012). The repurposing of these two drugs as a therapeutic approach against atherosclerosis was evaluated in several clinical trials discussed below.
2.3.1 Methotrexate
Treatment with low-dose methotrexate (LD-MTX) reduces the levels of circulating CRP, IL-6, TNF-alpha and cardiovascular events in RA patients (Choi et al. 2002; Wessels et al. 2008; Westlake et al. 2010; Roubille et al. 2015). An important feature of LD-MTX is its ability to increase adenosine production and stimulate the adenosine A2A receptor, which has been shown to promote the expression of several proteins involved in reverse cholesterol transport, thus potentially reducing foam cell formation (Reiss et al. 2008). The potential benefit of LD-MTX in the treatment of atherosclerosis was recently assessed in the cardiovascular inflammation reduction trial (CIRT) which is discussed below.
2.3.1.1 Cardiovascular Inflammation Reduction Trial (CIRT)
The CIRT trial was performed in parallel with the CANTOS trial and aimed to determine if LD-MTX could reduce cardiovascular events in patients with stable atherosclerosis and high cardiovascular risk (Everett et al. 2013). The double-blind trial included 4,786 patients with a history of myocardial infarction or multivessel coronary disease, as well as either type 2 diabetes or metabolic syndrome. Patients were randomized and treated with either LD-MTX (2,391) or placebo (2,395). The final primary endpoint was a composite of nonfatal myocardial infarction, nonfatal stroke, cardiovascular death, and hospitalization for unstable angina that led to urgent revascularization. After a median follow-up of 2.3 years, the primary endpoint occurred in 201 patients in the methotrexate group and 207 patients in the placebo group (HR 0.96, 95% CI 0.79–1.16). Also, treatment with LD-MTX did not reduce plasma levels of CRP, IL-1β, IL-6, TNF-α. No significant difference between the two groups for any component of the primary endpoint was found.
Considering the success of the CANTOS trial and the beneficial effect seen with LD-MTX in RA patients, the CIRT trial results are disappointing. While CANTOS targeted a specific and well-defined pathway, the CIRT trial used a drug with a large spectrum of anti-inflammatory effects with molecular consequences which are not yet understood. Besides the different mechanisms of action, an additional explanation for the discrepant results between CIRT and CANTOS may be differences in the study design of the two studies. Patients recruited to the CIRT trial had a lower residual inflammatory profile (median baseline CRP: 1.6 mg/L) compared to patients from the CANTOS trial, where patient recruitment was based on hsCRP levels ≥2 mg/L as a marker of high residual inflammation (median baseline CRP: 4.2 mg/L). In support of this explanation, treatment of RA patients with LD-MTX led to a reduction of hsCRP (Westlake et al. 2010) and decreased cardiovascular risk.
These data demonstrate that the design of future clinical trials needs to consider inclusion criteria that target individuals with a high inflammatory profile.
2.3.2 Low-Dose Colchicine
Colchicine is an inexpensive anti-inflammatory drug used in patients with gout, familial Mediterranean fever, and pericarditis. By preventing microtubule assembly, colchicine can disrupt inflammasome activation, microtubule-based inflammatory cell chemotaxis, phagocytosis, and other host immune mechanisms. The first benefit of colchicine in ischemic heart disease (IHD) has been observed in familial Mediterranean fever patients (Langevitz et al. 2001). Patients diagnosed with FMF show a sustained inflammatory response which puts them at a higher risk of suffering a heart attack in comparison with the general population. Langevitz and colleagues showed that treatment with colchicine lowered the risk of IHD in FMF patients compared to untreated patients with other inflammatory conditions and similar rates of risk factors. Also, patients taking colchicine achieved a frequency of IHD comparable to the general population. To assess the potential benefit of colchicine in atherosclerosis, several clinical trials such as the low-dose colchicine trial (LoDoCO), or more recently the colchicine cardiovascular outcomes trial (COLCOT), have been carried out and will be discussed below.
2.3.2.1 LoDoCo-MI and LoDoCo-1/2 Trials
In 2007, a small pilot study had demonstrated the potential of low-dose colchicine to reduce hsCRP levels, independently of lowering cholesterol (Atorvastatin), in patients with stable coronary artery disease (Nidorf and Thompson 2007). However, in the randomized and placebo-controlled LoDoCo (Low-Dose Colchicine)-MI study (237 patients), colchicine at 0.5 mg/day for 30 days failed to reduce hsCRP levels (≤ 2 mg/L) in patients with recent MI (Hennessy et al. 2019).
Subsequently, the LoDoCo (Low-Dose Colchicine) trial was designed as a prospective study to assess the efficiency and safety of long-term low-dose colchicine usage in patients with stable coronary disease. 532 patients on lipid-lowering medication and antithrombotic therapy were randomized and treated with a daily dose of colchicine (0.5 mg/mL) or no colchicine. After a median follow-up of 3 years, the primary outcome (acute coronary syndrome, out-of-hospital cardiac arrest, or non-cardioembolic ischemic stroke) had occurred in 5.3% of patients in the low-dose colchicine group, as opposed to 16% in the non-treated group (Nidorf et al. 2013). To validate the observations from the LoDoCo study, a large-scale study (LoDoCo 2) involving 5,522 patients was conducted. LoDoCo 2 confirmed the significant treatment benefit of low-dose colchicine in reducing CVD risk in patients with stable coronary disease. The primary endpoint was a composite of cardiovascular death, spontaneous (non-procedural) myocardial infarction, ischemic stroke, or ischemia-driven coronary revascularization. The secondary endpoint was a composite of cardiovascular death, spontaneous MI, or ischemic stroke. Primary endpoints occurred in 6.8% of patients in the low-dose colchicine group as opposed to 9.6% in the placebo-treated group, and secondary endpoints occurred in 4.2% in the colchicine group and 5.7% in the placebo group. However, an increase in death from non-cardiovascular diseases was observed in the colchicine treated group, which further justifies the need for more targeted therapies that limit undesirable side effects (Nidorf et al. 2019, 2020; Pradhan 2021).
2.3.2.2 COLCOT Trial
Another recent double-blind randomized placebo-controlled colchicine intervention study, named COLCOT (colchicine cardiovascular outcomes trial), aimed to assess the benefit of colchicine in patients with a recent acute MI (Tardif et al. 2019). In total, 4,745 patients were divided into two groups that were given oral colchicine at 0.5 mg/day or a placebo. The primary efficacy endpoint was a composite of death from cardiovascular causes, myocardial infarction, stroke, urgent hospitalization for angina leading to coronary revascularization, or resuscitated cardiac arrest. At a median of 23 months, the primary composite endpoint had occurred in 5.5% of the treatment group and 7.1% in the placebo group (hazard ratio, 0.77). Remarkably, the hazard ratio for stroke (HR = 0.26) and hospitalization for angina (HR = 0.50) indicate an important benefit in the colchicine treated group for these two particular primary endpoints. Overall, colchicine showed a good safety and tolerability profile despite a slight but significant increase in pneumonia occurrence (0.9%) in the colchicine group compared to the placebo group (0.4%). There were no differences in peripheral blood counts and CRP levels among the treated group compared to the placebo. Recent follow-up analysis suggests that the main benefit of colchicine therapy occurs when treatment is initiated in the first 3 days post-MI (Bouabdallaoui et al. 2020).
Additionally, the repurposed usage of low-dose colchicine has shown multiple benefits for the treatment of stable coronary artery diseases. A recent pilot study carried out in 80 patients with recent acute coronary syndrome suggests that low-dose colchicine uptake on a regular basis can efficiently stabilize plaques on top of optimal medical therapy (Vaidya et al. 2018). Further upcoming data from large-scale trials such as CLEAR-Synergy (4,000 patients with ST-elevation myocardial infarction undergoing percutaneous coronary intervention (NCT03048825)) will help to validate its efficiency and tolerability in coronary diseases.
2.4 Targeting Chemokine Signaling
Considerable work has been carried out on the identification of inflammatory mediators that actively participate in the progression of atherosclerosis which has increased the number of potential new therapeutic targets. Among them, chemokines were identified as key players in leukocyte recruitment/adhesion and activation as well as promotion of local inflammation in arteries. The contribution of chemokines to plaque formation/destabilization and thrombus formation has been extensively reviewed elsewhere (Zernecke and Weber 2014).
Despite the proven relevance of specific chemokines in atherogenesis, the clinical evaluation of drugs targeting specific chemokines or chemokine pairings has not progressed much. This is mainly due to the complexity of the “working network” within which chemokines operate and their critical role in fundamental host defense.
Several chemokines and their receptors such as CCL5/CCR5 (including heterodimeric interactions), CCL2/CCR2, CX3CL1/CX3CR1 are an active part of disease progression (Combadière et al. 2008). Drugs targeting those axes were developed in the context of other diseases like human immunodeficiency virus infections and facilitate the establishment of new clinical studies involving patients with cardiovascular diseases. Currently, drugs used to disrupt chemokine–receptor interactions rely on four main strategies: the modification of the chemokine N-terminal domain, the synthesis of small molecules used as a receptor antagonist, the usage of specific antibodies targeting chemokines, and other drugs interfering with chemokines’ heterodimeric interactions. Here, we will focus on promising chemokine targeting therapies with available clinical data in the context of atherosclerosis.
2.4.1 Targeting the CCR5 Axis
Expression of CCL5 and CCR5 has been identified in atherosclerotic plaques more than 15 years ago (Wilcox et al. 1994; Pattison et al. 1996; Veillard et al. 2004). Atheroprotective effects resulting from the perturbation of CCR5/RANTES (Regulated on Activation, Normal T cell Expressed and Secreted) interactions were described in several mouse studies. The genetic inactivation of CCR5 in chow or high-fat diet-fed Apoe−/− mice was shown to be protective in advanced atherosclerosis, but not in the early phase of the disease. The atheroprotective effect was associated with a decrease of macrophage numbers in plaques and reduced levels of circulating IL-6. The plaque quality was also affected by an increased content of SMCs, promoting plaque stabilization (Kuziel et al. 2003; Braunersreuther et al. 2007; Quinones et al. 2007). Moreover, a natural variant of CCR5 (CCR5Δ32 polymorphism) which reduces the cell surface expression of CCR5, is associated with a reduced risk of coronary artery diseases and MI (Szalai et al. 2001; González et al. 2001). Thus, the CCR5 pathway appears to be an interesting potential target for antiatherosclerotic therapy.
2.4.1.1 Maraviroc
The CCR5 inhibitor maraviroc is one of the few small molecules targeting a chemokine/receptor interaction approved by the US Food and Drug Administration and the European Medicines Agency as a preventive treatment for HIV (2007). HIV-associated inflammation increases HIV patients’ risk of cardiovascular disease (Shah et al. 2018), which suggests a potential role of maraviroc in modulating the atherogenic risk of these patients. This was confirmed preclinically as the treatment of atherosclerosis-prone Apoe−/− mice with maraviroc has been found to reduce atherosclerotic lesion development (Cipriani et al. 2013).
A recent pilot study in HIV patients showed that maraviroc treatment decreases carotid intima-media thickness, reflecting potential anti-atherogenic effects (Francisci et al. 2019). Additionally, an anti-human CCR5 monoclonal antibody (HGS004-Human Genome Sciences), tested in a clinical trial in HIV patients, showed a good safety profile and could be an interesting candidate for atherosclerosis therapy (Lalezari et al. 2008). Finally, a phase 4 clinical trial evaluating the efficacy of maraviroc in modulating atherosclerosis in HIV patients has been completed (NCT03402815) and results are forthcoming. This data will provide crucial information regarding the future of CCR5 axis targeting therapy in atherosclerosis.
2.4.2 Targeting the CCR2 Axis
Many pieces of evidence from experimental models demonstrate the proatherogenic role of CCR2 and its ligand CCL2/MCP-1 (Monocyte chemoattractant protein 1) (França et al. 2017). Notably, CCR2 signaling can promote monocyte recruitment to atherosclerotic lesions and foam cell formation (Boring et al. 1998; Bobryshev 2006). A recent discovery by Winter et al showed that the recruitment of myeloid cells within atherosclerotic lesions is subject to circadian regulation (Winter et al. 2018). This establishes a new concept of chrono-pharmacology-based therapy for the treatment of atherosclerosis, which is particularly relevant for the usage of CCR2 antagonists given their role in the promotion of monocyte recruitment (Winter et al. 2018).
2.4.2.1 MNL1202
In an effort to develop a specific antagonist of CCR2, a humanized antibody MNL1202 has been generated and tested in a phase 2 clinical trial in patients at risk for CVD (with at least 2 risk factors for atherosclerotic CVD, hsCRP level >3 mg/L, and receiving or not receiving lipid-lowering drugs). The direct blocking of CCR2 with MLN1202 was well tolerated and significantly reduced hsCRP levels in the antibody-treated group compared to the placebo-treated group (Gilbert et al. 2011). Thus MLN1202 appears to be a promising candidate to target atherosclerosis-associated inflammation.
Despite encouraging preclinical results, few molecules targeting chemokines and their receptors have been approved for clinical use yet. The fundamental role of chemokine signaling during both innate and adaptive immune response suggests that its targeting likely critically impairs the host immune response. For example, treatment with the CCL5 variant Met-RANTES or deficiency in CCL5 delays viral clearance by macrophages and impairs T-cell function (Makino et al. 2002). In this regard, the specific targeting of chemokine heteromerization represents a good alternative as it poses fewer risks of immunological side effects than chemokine antagonists. Up to now, 11 heteromeric chemokine pairs have been reported (Koenen and Weber 2010), expanding the therapeutic possibilities. Importantly, preclinical studies are carried out in a pathogen-free context, which is not representative of human pathogen exposure. Thus, future studies must assess the risk of long-term exposure to such chemokine targeting therapies. The complexity of the chemokine network, the lack of data concerning the toxicity of their targeting agents, and the availability of other, safer therapeutic approaches have drastically slowed down the translation of chemokine targeted therapies to human cardiovascular disease. Future studies should include a parallel assessment of risks and benefits provided by molecules targeting chemokine interactions to better appreciate the net effect on patients with cardiovascular diseases.
3 Modulation of the Adaptive Immune Response
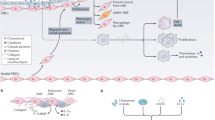
Adaptive immunity plays a key role in driving and modulating the pathogenesis of atherosclerosis (Sage et al. 2018; ; Wolf and Ley 2019; Libby 2021). The adaptive branch of immunity consists primarily of T and B cells, both of which have the capacity to induce immune responses that are specifically targeted to disease-associated antigens. Adaptive immunity therefore represents an attractive therapeutic target as its modulation could allow for a more specific response, potentially reducing the risks of weakened host defenses associated with broad anti-inflammatory therapies (Fig. 2).

Targeting adaptive immune responses in atherosclerosis. Regulatory T cells (Tregs) play a protective role in atherosclerosis via their immunosuppressive capacities such as the secretion of IL10 and TGFβ. They can suppress the activity of proatherogenic effector T cells such as TH1 cells. Treg numbers can be increased in a non-specific manner via the use of low-dose IL2 or in an antigen-specific manner by immunization with ApoB100-derived peptides. Furthermore, blocking effector T cell activation by inhibiting the interaction between co-stimulatory CD80/CD86 on antigen-presenting cells such as B cells and CD28 on T cells has been shown to reduce atherosclerosis in mice. B2 cells and many of their effector functions including some proatherogenic antibody isotypes such as IgE have been shown to promote atherogenesis. Thus, B cell depleting antibodies such as anti-CD20 or anti-BAFF antibodies have been shown to protect mice from atherosclerosis. Additionally, the depletion of proatherogenic antibodies such as IgE may have beneficial effects. On the other hand, immunization with modified LDL has been shown to induce protective antibody responses (both IgG and IgM) against oxidation-specific epitopes (OSE) that ameliorate atherosclerosis in preclinical studies
3.1 Strategies Targeting the Homeostatic Balance Between Regulatory and Effector T Cells
Although no therapies targeting T cells are currently in clinical use for the treatment and prevention of cardiovascular disease, some therapeutic approaches have shown promise in preclinical models and are being evaluated in clinical trials. In atherosclerosis-prone mice, regulatory T cells (Tregs) have been shown to be beneficial both in the progression and regression of atherosclerosis (Ait-Oufella et al. 2006; Mor et al. 2007; Klingenberg et al. 2013; Sharma et al. 2020). In contrast, many effector T cells are thought to be proatherogenic, particularly IFNγ-secreting T helper 1 (Th1) (Saigusa et al. 2020; Buono et al. 2005). In humans, cardiovascular disease is associated with an imbalance in Treg and effector T cells, where Tregs are decreased in patients with acute coronary syndrome and low numbers are associated with increased rates of acute coronary events (Mor et al. 2006; Han et al. 2007; Cheng et al. 2008; Ammirati et al. 2010; Wigren et al. 2012).
Therefore, the aim of T cell-targeted therapeutic approaches in atherosclerosis is to change the homeostatic balance of different T cell subsets by expanding atheroprotective, immunosuppressive Tregs while suppressing putatively proatherogenic effector T cell populations (Foks et al. 2015). This could be achieved on the one hand by the stimulation of non-specific Tregs by targeting pathways such as interleukin-2 (IL-2). On the other hand, specific Tregs can be expanded with the use of tolerogenic vaccines against atherosclerosis-relevant antigens.
3.1.1 Low-Dose IL-2 Therapy
Interleukin-2 is a cytokine that plays a key role in the regulation of T cell activity and survival. Although IL-2 has the ability to regulate all T cells, it appears to be particularly important in the development and survival of Tregs (Boyman and Sprent 2012). The activation threshold for Tregs is lower than for other effector T cells (Yu et al. 2009), which may explain the somewhat paradoxical effect of IL-2 therapy. While high-dose IL-2 therapy is thought to promote effector immunity, low-dose IL-2 stimulates immunosuppressive Tregs (Klatzmann and Abbas 2015). A potential added benefit of this therapy could be the IL-2 mediated expansion of innate lymphoid cells 2 (ILC2) (Van Gool et al. 2014). ILC2 cells represent a rare subset of immune cells that likely exert atheroprotective functions via the secretion of type-2 cytokines such as IL-5 and IL-13 (Binder et al. 2004; Cardilo-Reis et al. 2012; Newland et al. 2017). Preclinical studies have shown that administration of recombinant IL-2 complexed with monoclonal antibodies against IL-2 increases the levels of Tregs and reduces atherosclerotic plaque burden in mice (Foks et al. 2011; Dinh et al. 2012; Proto et al. 2018).
The IL-2 analogue aldesleukin is currently approved for the treatment of advanced renal cell carcinoma and metastatic melanoma. In cardiovascular disease, low-dose IL-2 therapy is being evaluated in clinical trials with the aim to increase Treg numbers, thereby limiting post-ischemic inflammatory responses and promoting myocardial healing in the setting of ischemic heart disease and acute coronary syndrome. The low-dose Interleukin-2 in patients with stable ischemic heart disease and acute coronary syndromes (LILACS) study is a randomized, placebo-controlled, double-blind phase I/IIa clinical trial that aims to assess the safety and tolerability of escalating doses of recombinant IL2, as well as its ability to alter Treg, effector T cell and other immune cell populations (NCT03113773; expected completion date 06/2021; Zhao et al. 2018). Preliminary results indicate the treatment is well tolerated and induces robust increases in Tregs without affecting effector T cells. Interestingly, a dose-dependent reduction in B cells was also seen, which may have further beneficial effects on atherosclerosis (Zhao et al. 2020a). The subsequent low-dose IL-2 for the Reduction of Vascular Inflammation In Acute Coronary Syndromes (IVORY) phase II trial will assess changes in vascular inflammation in patients with acute coronary syndromes (NCT04241601; expected completion date 01/2024; Zhao et al. 2020b).
One limitation of low-dose IL-2 therapy are potential off-target effects, which include mild increases in NK cells and eosinophils (Zhao et al. 2020a). Furthermore, initially protective Tregs may undergo a phenotypic switch toward a proinflammatory TH1-like phenotype with progressing disease in mice (Li et al. 2016; Butcher et al. 2016; Wolf et al. 2020). It remains to be addressed whether this switch can occur in humans.
3.1.2 Alternative Means to Expand Regulatory T Cells
Alternative options to harness the immunosuppressive capacity of Tregs is by ex vivo expansion of Tregs, where autologous naïve Tregs are isolated from the blood and expanded in vivo using a cytokine cocktail or antigenic stimuli. The adoptive transfer of Tregs has been tested in humans in the setting of transplantation (Trzonkowski et al. 2009; Brunstein et al. 2011; Di Ianni et al. 2011; Mathew et al. 2018) and type 1 diabetes (Marek-Trzonkowska et al. 2014), but not cardiovascular disease. Another way to expand Tregs is using tolerogenic non-FcR binding anti-CD3 antibodies such as teplizumab, which have shown promise in clinical trials for the treatment and prevention of type 1 diabetes (Herold et al. 2013, 2019). However, the effect of anti-CD3 on atherosclerosis has so far only been addressed in preclinical murine studies alone (Steffens et al. 2006; Kita et al. 2014) or in combination with IL-2 complex (Kasahara et al. 2014), where it was associated with reduced atherosclerosis progression or increased Treg-dependent plaque regression.
3.2 Strategies Targeting B Cells and Humoral Immunity
B cells play a well-established role in the pathogenesis of atherosclerosis, with different B cell subsets having distinct effects on the pathogenesis of atherosclerosis (Sage et al.). B cells exert their function primarily via the generation of humoral immunity in the form of highly specific antibodies, but they also have the capacity to act as antigen-presenting cells to T cells and to secrete cytokines. The most abundant B cell type, follicular B2 cells, as well as B cell-derived germinal center B cells that arise upon interaction with follicular T helper cells, are generally considered proatherogenic (Clement et al. 2015; Tay et al. 2018; Centa et al. 2019; Bagchi-Chakraborty et al. 2019). In contrast, marginal zone B2 cells may protect from atherosclerosis by suppressing T follicular helper cells (Nus et al. 2017) and potentially via the secretion of atheroprotective IgM (Grasset et al. 2015). B1 cells, which can be further subdivided into B1a and B1b cells, represent an innate-like B cell type that can secrete germline-encoded natural antibodies that are typically of the IgM class in a T cell-independent manner. Their atheroprotective function, which is dependent on their ability to secrete IgM, is well-established in mice (Lewis et al. 2009; Kyaw et al. 2012; Rosenfeld et al. 2015; Gruber et al. 2016; Tsiantoulas et al. 2017). Other minor B cell subsets include B1 cell derived innate response activator (IRA) B cells that can promote atherosclerosis via the secretion of GM-CSF and the subsequent induction of Th1-differentiation (Hilgendorf et al. 2014) and IL-10-secreting B regulatory cells (Bregs), whose role in atherosclerosis remains controversial (Strom et al. 2015; Sage et al. 2015; Douna et al. 2019).
Although most evidence on the role of B cells in atherosclerosis stems from preclinical studies, B cells also appear to be key players in human cardiovascular disease. Patients suffering from autoimmune diseases such as systemic lupus erythematosus (SLE) and rheumatoid arthritis have an increased risk of cardiovascular disease independently of other risk factors (Hollan et al. 2013). A genome wide association study (GWAS) and transcriptome analysis of the Framingham Heart Study cohort recently revealed genes associated with B cell activation and differentiation to be the most deregulated genes between patients with and without coronary heart disease, suggesting a key function for B cells in modulating atherosclerosis (Huan et al. 2013). B cells are present in the vascular wall of human atherosclerotic plaques (Hamze et al. 2013; Kortelainen and Porvari 2014), where they may undergo antigen-driven clonal expansion ((Burioni et al. 2009). However, it is currently not clear how findings from murine B cell subsets and their functions in cardiovascular disease translate to human B cells. For example, although B1 cells are a well-described atheroprotective subset in mice, the nature and existence of the human equivalent of murine B1 cells remain controversial (Griffin et al. 2011; Upadhye et al. 2020). McNamara and colleagues have recently found a subset of CXCR4-expressing B1 cells that correlated with the levels of IgM directed against MDA-LDL and that inversely correlated with plaque volume and stenosis (Upadhye et al. 2019). Furthermore, increased levels of CD86-expressing CD19+ B cells were found to associate with an increased risk of stroke, while CD40-expressing CD19+ B cells showed a negative association (Mantani et al. 2014).
Overall, the aim of B cell-targeted therapy is to reduce the proatherogenic effects of follicular B2 cells, ideally while preserving atheroprotective IgM-secreting B1 cells (Porsch and Binder 2019). Currently, B cell depletion therapy is primarily used in the treatment of B cell malignancies and in the treatment of autoimmune diseases. An alternative therapeutic target is humoral immunity. On the one hand, putatively proatherogenic antibody classes such as IgE may be neutralized. On the other hand, passive and active immunization strategies may be harnessed to specifically target atherosclerosis-associated antigens such as oxidation-specific epitopes.
3.2.1 Anti-CD20 Mediated B Cell Depletion (Rituximab)
Given the strong evidence for a proatherogenic role of follicular B2 cells, the major B cell type in the body, some high-risk patients may benefit from their depletion. Indeed, preclinical studies suggest that anti-CD20 mediated B cell depletion could ameliorate the development of several cardiovascular pathologies including atherosclerosis (Ait-Oufella et al. 2010; Kyaw et al. 2010), myocardial infarction (Zouggari et al. 2013), myocardial infarction-accelerated atherosclerosis (Kyaw et al. 2021), abdominal aortic aneurysm (Schaheen et al. 2016), and hypertension (Chan et al. 2015).
In mice, anti-CD20 treatment preferentially depletes B2 cells, while preserving atheroprotective B1 cells (Hamaguchi et al. 2005). In line with that, anti-CD20 treatment of Ldlr−/− and Apoe−/− mice potently decreases B2 cell numbers and IgG levels and induces significant reductions in atherosclerosis (Ait-Oufella et al. 2010; Kyaw et al. 2010). In the context of acute myocardial infarction, Zouggari et al. could show that anti-CD20 mediated B2 cell depletion was associated with reduced inflammation and CCL7-mediated monocyte mobilization while improving cardiac function, reducing infarct size and fibrosis (Zouggari et al. 2013). Furthermore, anti-CD20 mediated B2 cell depletion significantly reduced myocardial infarction-accelerated atherosclerosis (Kyaw et al. 2021).
Rituximab is a CD20-targeted monoclonal antibody that has been approved for the treatment of some B cell malignancies (NHL, CLL) and autoimmune diseases (rheumatoid arthritis, granulomatosis with polyangiitis, pemphigus vulgaris) for more than two decades. Given its long clinical use, there is increasing evidence on its effect on cardiovascular parameters. Although a recent meta-analysis could not show an effect on short-term cardiovascular adverse events in randomized placebo-controlled trials involving rituximab, long-term follow-up data is lacking (Morris-Rosenfeld et al. 2014). Similarly, a study assessing the effects of biologic and conventional disease-modifying antirheumatic drugs on incident cardiovascular disease in rheumatoid arthritis patients with a mean follow-up of 4 years could find no beneficial effects of rituximab on cardiovascular risk, although the number of patients on rituximab in this study was relatively low (Ozen et al. 2020). However, several small human studies could show reductions in carotid intima-media thickness (Kerekes et al. 2009; Benucci et al. 2013; Novikova et al. 2016), improved flow-mediated dilation (Kerekes et al. 2009; Benucci et al. 2013; Hsue et al. 2014) and decreased arterial thickness (Provan et al. 2015) in patients receiving rituximab.
The Rituximab in patients with acute ST-elevation myocardial infarction (RITA-MI) trial was a prospective, open-label, single-arm phase I/IIa trial testing a single intravenous injection of rituximab in patients with STEMI within 48 h of symptom onset with a 6-month follow-up. Preliminary data suggests the treatment is well tolerated at all doses tested and resulted in a rapid, robust reduction of circulating B cells by a mean 96.3% that persisted throughout the follow-up period in a dose-dependent manner. Interestingly, immunoglobulin levels were not affected at that point. Additionally, clinical cardiac parameters including ejection fraction and (cardiac) biomarkers such as C-reactive protein (CRP) and B-type natriuretic peptide (BNP) were improved at follow-up (NCT03072199; Zhao et al. 2021). A subsequent phase IIb trial, RITA-MI 2, will be carried out in the form of a placebo-controlled randomized trial, which will allow further assessment of whether anti-CD20-mediated B cell depletion provides clinical benefits compared to placebo treatment (Zhao et al. 2021). The ICFEr-RITU2 trial is another phase II clinical trial assessing the safety of rituximab and the effect of rituximab on cardiac fibrotic remodeling in patients with chronic heart failure with reduced ejection fraction (HFrEF; NCT03332888; Sánchez-Trujillo et al. 2019). Furthermore, the risk of atherosclerotic cardiovascular disease was reduced in patients receiving rituximab following kidney transplantation (Kim et al. 2019), further supporting the potential of anti-CD20 mediated B cell depletion in the treatment of cardiovascular disease.
3.2.2 Other B Cell Depleting Antibodies
In addition to rituximab, there are emerging targets on B cells such as CD19 (inebilizumab, blinatumomab) and CD22 (inotuzumab ozogamicin) that are being targeted to deplete B cells in cancer and autoimmune disease (Hofmann et al. 2018). However, these receptors are also expressed on B1 cells, and may thus interfere with atheroprotective B cell effects. Additionally, there are therapies targeting receptors expressed on antibody-secreting plasma cells such as CD38 (daratumumab) and SLAMF7 (elotuzumab), which are used in the treatment of multiple myeloma (Radocha et al. 2021). However, the effect of such B cell depleting agents on cardiovascular disease has not been assessed directly.
3.2.3 B Cell Depletion via Targeting of the BAFF/APRIL System
An alternative means to deplete B cells is via the targeting of the B cell activating factor (BAFF)/A Proliferation-Inducing Ligand (APRIL) system, which is made up of soluble mediators that are required for B cell survival. BAFF appears to be particularly important in the survival of B2 cells (Rauch et al. 2009). In preclinical studies, BAFFR deficiency and anti-BAFFR mAb administration have been shown to robustly and selectively deplete B2 cells, thus decreasing atherosclerotic lesion formation (Kyaw et al. 2012; Sage et al. 2012) and improving recovery from myocardial infarction (Zouggari et al. 2013). However, there is also evidence that BAFF may promote Breg differentiation (Yang et al. 2010) and that BAFF overexpression might protect from atherosclerosis (Jackson et al. 2016). BAFF may also exert atheroprotective functions via inhibition of TLR9-IRF7-dependent proinflammatory signaling in macrophages. Indeed, BAFF neutralization promoted lesion formation in atherosclerosis-prone mice and increased lesional CXCL10 content, suggesting potential adverse effects of BAFF-targeted therapies (Tsiantoulas et al. 2018). Belimumab is a BAFF-targeting antibody that is approved for the treatment of SLE, which robustly depletes B cells, plasma cells, and immunoglobulins. So far, the effect of belimumab on cardiovascular disease has not been assessed in human patients.
3.2.4 IgE Neutralization (Omalizumab)
The primary effector function of B cells is the generation of humoral immunity in the form of antigen-specific antibodies. There is ample epidemiological and preclinical experimental evidence suggesting an important role for humoral immunity and different antibody classes in atherosclerosis (Khamis et al. 2016; Sage et al. 2017; Tay et al. 2018; Centa et al. 2019; van den Berg et al. 2019). IgE is a class-switched, typically follicular B cell-derived antibody class that is associated with allergic responses. It can trigger proinflammatory responses via interaction with FcεRI receptors on mast cells, basophils, macrophages, and vascular cells (Wu and Zarrin 2014). Plasma IgE levels correlate with coronary heart disease in humans (Kounis and Hahalis 2016). Interestingly, patients suffering from asthma are also at increased risk for cardiovascular disease (Tattersall et al. 2015). Preclinical studies suggest a proatherogenic role for IgE in Apoe−/− and Ldlr−/− mice (Wang et al. 2011; Wezel et al. 2015; Tsiantoulas et al. 2017; Zhang et al. 2020). Antibody-mediated IgE neutralization was found to reduce atherosclerotic lesion formation in Ldlr−/− mice in the context of secreted IgM deficiency (Tsiantoulas et al. 2017).
Omalizumab is an IgE-neutralizing antibody that interferes with the interaction of IgE and its receptors, resulting in the reduction of IgE levels. It is approved for the treatment of severe asthma and chronic idiopathic urticaria. Although a clinical trial suggested potential cardiovascular side effects of omalizumab treatment, this clinical trial was biased by the dramatically increased cardiovascular disease burden at baseline in the patients receiving omalizumab (Iribarren et al. 2017a). A subsequent pooled analysis of randomized, double-blind placebo-controlled trials involving omalizumab did not show an increased cardiovascular risk (Iribarren et al. 2017b) Therefore, the effect of IgE neutralization on cardiovascular disease remains to be evaluated in a clinical trial setting.
3.3 Strategies Targeting the Interaction Between T Cells and Antigen-Presenting Cells
Efficient T cell activation is crucially dependent on the engagement of co-stimulatory pathways. It has been hypothesized that T cells require two signals in order to become fully activated, one being antigen-presentation via MHC-molecules, the other being co-stimulation by receptors expressed on the surface of antigen-presenting cells such as dendritic cells, macrophages, and B cells. Inhibition of such co-stimulatory pathways may therefore interfere with the activation, proliferation, and differentiation of T cells, which may have beneficial effects on atherosclerosis. In contrast, targeting co-inhibitory pathways with the use of immune checkpoint inhibitors in cancer therapy may enhance T cell activity, which could have detrimental effects on cardiovascular disease.
3.3.1 Targeting Co-stimulatory Pathways
3.3.1.1 Abatacept
CD80 (B7-1) and CD86 (B7-2) are expressed on activated antigen-presenting cells, where they can interact with both the co-stimulatory CD28 receptor and the inhibitory CTLA4 receptor on T cells (Sharpe and Freeman 2002).
Abatacept is a recombinant biologic consisting of the Ig-domain of CTLA4 and a human IgG1 Fc region. It binds CD80/CD86 with high affinity, thus blocking the interaction of CD80/CD86 with the co-stimulatory CD28 receptor and activation of T cells. Preclinical studies suggest a primarily proatherogenic role for CD80/CD86. However, CD80/CD86 may also be required for the efficient induction of Tregs (Ait-Oufella et al. 2006). However, abatacept was shown to reduce atherosclerosis in different murine models of atherosclerosis (Ma et al. 2013; Ewing et al. 2013). Abatacept is currently clinically approved for the treatment of rheumatoid, juvenile idiopathic, and psoriatic arthritis, while the highly similar belatacept is used in patients receiving kidney transplants. Some clinical trials suggest slightly reduced cardiovascular disease risk in patients receiving abatacept compared to TNF inhibitors (Zhang et al. 2016; Kang et al. 2018; Jin et al. 2018; Hsieh et al. 2020).
3.3.1.2 CD40-CD40L
Another important co-stimulatory signal is provided by the CD40-CD40L interaction, a member of the tumor necrosis factor receptor (TNFR) family. Targeting this pathway reduced atherosclerosis in mice (Lutgens et al. 2000, 2010; Schönbeck et al. 2000), but not in others (Zirlik et al. 2007). Additionally, CD40-signaling intermediates, the so-called TNF-receptor associated factors (TRAFs), have been targeted successfully by a small molecule inhibitor in mouse models (Lutgens et al. 2010; Chatzigeorgiou et al. 2014). Importantly, no immunosuppressive side effects were observed with this inhibitor (Seijkens et al. 2018) and a newly developed delivery system, linking a TRAF6 inhibitor to a HDL particle, was found to be safe in non-human primates (Lameijer et al. 2018). Based on these initial findings, further assessment of CD40-TRAF inhibition needs to be carried out to verify its role in human atherosclerosis. While CD40-CD40L targeting therapeutics have not been approved for clinical use yet, several antagonistic antibodies are currently being evaluated in clinical trials for the treatment of inflammatory conditions (Karnell et al. 2019).
3.3.1.3 Co-inhibitory Pathways
Recent years have seen the emergence of cancer immunotherapy and the widespread use of immune checkpoint inhibitors. Immune checkpoint inhibitors are used clinically with the aim to enhance anti-tumor immunity by T cells. Many of these drugs target pathways that have been described to play a proatherogenic role. Given the prominent role of effector T cells and these immunomodulatory pathways in cardiovascular disease, such T cell-simulating therapy is expected to have detrimental effects on atherosclerosis (Simons et al. 2019). Importantly, a recent study by Drobni et al. could demonstrate a threefold increased risk for cardiovascular events and more than threefold increased aortic plaque volumes in patients receiving immune checkpoint inhibitor therapy (Drobni et al. 2020). Additionally, atherosclerotic mice which underwent short-term check point inhibition therapy developed a more vulnerable, proinflammatory atherosclerotic phenotype due to an access of effector T cells, endothelial activation, and increased CD8+ T cell recruitment (Poels et al. 2020). This study highlights the importance of cardiovascular risk awareness in cancer patients receiving checkpoint inhibitor immunotherapy.
3.3.1.4 Vaccination Strategies
Vaccines represent an attractive future therapeutic avenue, as they may provide a durable, antigen-specific approach, thus limiting adverse effects on the host immune response. However, vaccination approaches remain challenging, as they require the targeting of specific self-antigens that may trigger unwanted autoimmune responses. Unlike traditional vaccines that aim at enhancing the activity of the adaptive immune system against pathogens, the idea behind immunization strategies against atherosclerosis is either to induce immunological tolerance or to neutralize atherosclerosis-specific antigens without inducing a considerable inflammatory response. Additionally, passive immunization strategies offer the opportunity to directly administer antibodies that may modulate inflammatory and metabolic pathogenic mechanisms in atherosclerosis, although their effect is transient.
3.3.2 Immunization Against LDL-Related Antigens
LDL and particularly its modified forms represent well-established immunogenic epitopes that have previously been shown to play a role in atherosclerosis and to be recognized by various components of the immune system. LDL can undergo oxidative modifications that result in the formation of a variety of lipid peroxidation-derived oxidation-specific epitopes (OSE), including phosphorylcholine (PC)-containing oxidized phospholipids and malondialdehyde (MDA), which are highly immunogenic and trigger inflammation in the vascular wall. Notably, OSE are targeted by specific circulating antibodies that have been shown to neutralize their proinflammatory activities. Thus, several experimental studies have addressed the effect of immunization with modified LDL or parts of its main lipoprotein component, Apolipoprotein B (ApoB).
Palinski and colleagues were able to show that immunization with malondialdehyde (MDA)-modified LDL reduced atherogenesis in LDLR-deficient rabbits (Palinski et al. 1995), which was further confirmed by several groups using modified LDL in rabbits (Ameli et al. 1996; Nilsson et al. 1997; Asgary et al. 2007) and mice (George et al. 1998; Freigang et al. 1998; Zhou et al. 2001; Binder et al. 2004; Van Puijvelde et al. 2006). Specific advanced MDA adducts have been shown to be particularly immunogenic, which may further impact the effect of vaccination on atherogenesis (Gonen et al. 2014).
Although the mechanisms responsible for the atheroprotective effects observed in these studies have not been fully elucidated, several pathways play a role, including the induction of neutralizing antibodies that interfere with the proinflammatory effects of these OSE. However, even though several preclinical studies have shown a prominent induction of humoral immunity upon immunization (George et al. 1998; Freigang et al. 1998; Zhou et al. 2001; Binder et al. 2004) and the inverse association between plaque size and antibody levels against OSEs (Zhou et al. 2001), it is currently not clear how these findings translate to the clinic.
Passive immunization via administration of antibodies against OSE has shown promise in preclinical models (Schiopu et al. 2004, 2007; Poulsen et al. 2016). The phase II GLACIER trial failed to show reduced vascular inflammation following administration of a recombinant IgG1 antibody against MDA-modified ApoB100 (Lehrer-Graiwer et al. 2015). However, the relatively short follow-up period, the use of FDG-PET instead of coronary artery assessments, and the patient selection criteria may have been a limitation of this study. Given the proposed atheroprotective role of IgM against oxidation-specific epitopes, it will be interesting to assess the effect of passive administration of IgM antibodies (or single chain and other class switch variants thereof) on human atherosclerosis in the future. One attractive candidate is the IgM antibody E06, which binds the phosphocholine (PC) headgroup of oxidized phospholipids (oxPLs), thus blocking the proinflammatory properties of oxPL and the uptake of oxLDL by macrophages (Shaw et al. 2000; Que et al. 2018).
3.3.3 Tolerogenic Vaccination
Another proposed atheroprotective mechanism is tolerization, which is associated with the induction of immunosuppressive Tregs that may limit inflammation via IL10, TGFβ and the suppression of T effector functions. Several preclinical studies found the reduction in atherosclerotic lesion formation upon administration of modified LDL or ApoB100-derived peptides to be associated with significant increases in Treg numbers (Fredrikson et al. 2008; Wigren et al. 2011; Herbin et al. 2012).
In addition to lipid-derived antigens, peptide-based vaccination strategies based on ApoB100-derived peptides, many of which are recognized by autoantibodies from sera of patients with coronary heart disease, have also shown promise in preclinical studies (Fredrikson et al. 2003). Administration of several ApoB100 peptides, including P210, P45, P74, (Fredrikson et al. 2003, 2005, 2008; Dunér et al. 2021), P2 (Chyu et al. 2005), P3 (Tse et al. 2013), P18 (Kimura et al. 2018), P265 and P295 (Gisterå et al. 2017), has been shown to reduce lesion formation in murine atherosclerosis
However, several challenges remain before the clinical implementation of inflammation-restraining tolerogenic T cell vaccine is possible. Given that the interaction of TCRs with their cognate peptide is dependent on the efficient presentation on MHC type II molecules, HLA types need to be matched. The necessity for HLA matching has resulted in tetramer-based approaches to allow the identification of antigen-specific T cell populations (Kimura et al. 2017, 2018). The considerable heterogeneity of the human HLA locus and the need to adapt peptides to individual HLA types present a major challenge in the design of T-cell based peptide vaccines. Furthermore, the efficient induction of tolerance critically depends on dose, choice of adjuvant, and the route of administration (Van Puijvelde et al. 2006, 2007; Klingenberg et al. 2010; Czerkinsky and Holmgren 2012).
3.3.4 Immunization Against Pathogen-Derived Antigens
Some studies suggest that vaccination against common diseases such as influenza and pneumonia can reduce cardiovascular risk (Ren et al. 2015; Barnes et al. 2015). A double-blind randomized placebo-controlled clinical trial (Influenza Vaccination After Myocardial Infarction, IAMI; NCT02831608) is currently underway to assess the effect of influenza vaccination on cardiovascular outcomes in patients with preexisting cardiovascular disease (Fröbert et al. 2017). While it cannot be excluded that inflammation associated with infections may affect the plaque, the potential beneficial effect of pneumococcal vaccination may be due to its ability to induce antibodies that react with the oxidation-specific epitope, PC (Binder et al. 2003; Caligiuri et al. 2007). Initial studies have evaluated the consequences of the molecular mimicry between oxidized LDL and the CPS of pneumococci and demonstrated that immunization of Ldlr−/− mice with pneumococcal extracts induced PC-specific IgM and reduced lesion formation (Binder et al. 2003). A similar effect has been observed when Apoe−/− mice were immunized with PC-conjugated KLH (Caligiuri et al. 2007).
These studies suggested the possibility that existing pneumococcal vaccines could be repurposed to trigger anti-atherogenic immunity in humans. However, PC is not a major antigenic component of pneumococcal vaccines due to poor immunogenicity, and several studies indicated that unlike natural infections vaccination with existing vaccines does not trigger robust anti-OxLDL antibody titers. Small-scale clinical trials of the 13-valent conjugated pneumococcal vaccine Prevnar-13 did not find an induction of antibodies against oxLDL or phosphorylcholine in a small cohort of patients with metabolic diseases (Grievink et al. 2020; Shiri-Sverdlov et al. 2021), which may be due to the fact that PC is only a minimal constituent of Prevnar-13. The AUSPICE trial to assess the effect of the pneumococcal polysaccharide vaccine Pneumovax 23 on antibody levels against oxidation-specific epitopes and several cardiovascular disease markers in 4725 patients is currently underway (ACTRN12615000536561; Ren et al. 2016).
Heat shock proteins (HSPs) represent a group of highly conserved stress-associated proteins that can act as autoantigens either due to cross-reactivity against microbial-derived HSPs or following their expression during endothelial injury (Wick et al. 2014). Preclinical studies show that administration of microbial HSPs or HSP-related peptides protects mice from atherosclerosis (Maron et al. 2002; Van Puijvelde et al. 2007; Jing et al. 2011; Klingenberg et al. 2012; Grundtman et al. 2015). The atheroprotective mechanism may involve induction of cross-reactive antibodies against MDA-LDL (Kyrklund et al. 2020) or tolerogenic activation of Tregs (Van Puijvelde et al. 2007).
4 Conclusion
The immune system represents a versatile and essential part of human homeostasis which harbors great potential for the treatment and prevention of atherosclerosis. While the identification of relevant therapeutic targets remains challenging, it has greatly improved over the last decade as various anti-inflammatory and immunomodulatory approaches have been studied. These range from modulation of the innate immune system by interfering with cytokines and their production, chemokines and general inflammation, to the adaptive immune system where B cells, T cells, their interaction and their potential in immunization are utilized. While these approaches have proven promising in multiple preclinical studies, with some translation into human trials, a wider application and breakthrough into the clinic is yet to occur. One factor which slows the translation of preclinical findings into the clinic is the immune system’s essential role in fighting infections. Unspecific modulation or downregulation of the immune system can lead to unwanted side effects such as increased infections. It is thus important to conduct further research in order to harvest the positive potential while minimizing side effects. Research into potential biomarkers such as hsCRP and IL-6 – but with even greater specificity for the atherogenic disease process – represents an important step toward solving this issue, as they allow the identification of patients who are most likely benefiting from immunomodulatory treatment. Moreover, highly specific biomarkers would also be needed to monitor a successful response to anti-inflammatory or immunomodulatory treatments. Additionally, further research into the immune system’s role in human atherosclerosis, combined with large-scale clinical trials will pave the way for immunomodulatory treatments to become a realistic addition to standard atherosclerosis therapies.
References
Abbate A, Kontos MC, Grizzard JD et al (2010) Interleukin-1 blockade with Anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra remodeling trial [VCU-ART] pilot study). AJC 105:1371–1377. https://doi.org/10.1016/j.amjcard.2009.12.059
Abbate A, Van Tassell BW, Biondi-Zoccai G et al (2013) Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia commonwealth university-anakinra remodeling trial (2) (vcu-art 2) pilot study]. Am J Cardiol 111:1394–1400. https://doi.org/10.1016/j.amjcard.2013.01.287
Abbate A, Trankle CR, Buckley LF et al (2020) Interleukin-1 blockade inhibits the acute inflammatory response in patients with ST-segment-elevation myocardial infarction. J Am Heart Assoc 9:e014941. https://doi.org/10.1161/JAHA.119.014941
Ait-Oufella H, Salomon BL, Potteaux S et al (2006) Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med 12:178–180. https://doi.org/10.1038/nm1343
Ait-Oufella H, Herbin O, Bouaziz J-D et al (2010) B cell depletion reduces the development of atherosclerosis in mice. J Exp Med 207:1579–1587. https://doi.org/10.1084/jem.20100155
Ameli S, Hultgårdh-Nilsson A, Regnström J et al (1996) Effect of immunization with homologous LDL and oxidized LDL on early atherosclerosis in hypercholesterolemic rabbits. Arterioscler Thromb Vasc Biol 16:1074–1079. https://doi.org/10.1161/01.ATV.16.8.1074
Ammirati E, Cianflone D, Banfi M et al (2010) Circulating CD4+CD25hiCD127lo regulatory T-cell levels do not reflect the extent or severity of carotid and coronary atherosclerosis. Arterioscler Thromb Vasc Biol 30:1832–1841. https://doi.org/10.1161/ATVBAHA.110.206813
Asgary S, Saberi SA, Azampanah S (2007) Effect of immunization against ox-LDL with two different antigens on formation and development of atherosclerosis. Lipids Health Dis 6:32. https://doi.org/10.1186/1476-511X-6-32
Avgerinou G, Tousoulis D, Siasos G et al (2011) Anti-tumor necrosis factor alpha treatment with adalimumab improves significantly endothelial function and decreases inflammatory process in patients with chronic psoriasis. Int J Cardiol 151:382–383. https://doi.org/10.1016/j.ijcard.2011.06.112
Avina-Zubieta JA, Thomas J, Sadatsafavi M et al (2012) Risk of incident cardiovascular events in patients with rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis 71:1524–1529. https://doi.org/10.1136/annrheumdis-2011-200726
Bäck M, Yurdagul A, Tabas I et al (2019) Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol 16:389–406
Bagchi-Chakraborty J, Francis A, Bray T et al (2019) B cell Fcγ receptor IIb modulates atherosclerosis in male and female mice by controlling adaptive germinal center and innate B1-cell responses. Arterioscler Thromb Vasc Biol 39:1379. https://doi.org/10.1161/ATVBAHA.118.312272
Barnabe C, Martin B-J, Ghali WA (2011) Systematic review and meta-analysis: anti-tumor necrosis factor α therapy and cardiovascular events in rheumatoid arthritis. Arthritis Care Res (Hoboken) 63:522–529. https://doi.org/10.1002/acr.20371
Barnes M, Heywood AE, Mahimbo A et al (2015) Acute myocardial infarction and influenza: a meta-analysis of case-control studies. Heart 101:1738–1747. https://doi.org/10.1136/heartjnl-2015-307691
Bauernfeind F, Niepmann S, Knolle PA, Hornung V (2016) Aging-associated TNF production primes Inflammasome activation and NLRP3-related metabolic disturbances. J Immunol 197:2900–2908. https://doi.org/10.4049/jimmunol.1501336
Benucci M, Saviola G, Manfredi M et al (2013) Factors correlated with improvement of endothelial dysfunction during rituximab therapy in patients with rheumatoid arthritis. Biol Theory 7:69–75. https://doi.org/10.2147/BTT.S39182
Bhaskar V, Yin J, Mirza AM et al (2011) Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in apolipoprotein E-deficient mice. Atherosclerosis 216:313–320. https://doi.org/10.1016/j.atherosclerosis.2011.02.026
Biasucci LM, Pedicino D, Liuzzo G (2020) Promises and challenges of targeting inflammation to treat cardiovascular disease: the post-CANTOS era. Eur Heart J 41:2164–2167. https://doi.org/10.1093/eurheartj/ehz586
Binder CJ, Hörkkö S, Dewan A et al (2003) Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med 9:736–743. https://doi.org/10.1038/nm876
Binder CJ, Hartvigsen K, Chang M-K et al (2004) IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J Clin Invest 114:427–437. https://doi.org/10.1172/JCI20479
Bissonnette R, Harel F, Krueger JG et al (2017) TNF-alpha; antagonist and vascular inflammation in patients with psoriasis vulgaris: a randomized placebo-controlled study. J Invest Dermatol 137:1638. https://doi.org/10.1016/j.jid.2017.02.977
Bobryshev YV (2006) Monocyte recruitment and foam cell formation in atherosclerosis. Micron 37:208–222
Borén J, John Chapman M, Krauss RM et al (2020) Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European atherosclerosis society consensus panel. Eur Heart J 41:2313–2330
Boring L, Gosling J, Cleary M, Charo IF (1998) Decreased lesion formation in CCR2(−/−) mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 394:894–897. https://doi.org/10.1038/29788
Bouabdallaoui N, Tardif JC, Waters DD et al (2020) Time-to-treatment initiation of colchicine and cardiovascular outcomes after myocardial infarction in the colchicine cardiovascular outcomes trial (COLCOT). Eur Heart J 41:4092–4099. https://doi.org/10.1093/eurheartj/ehaa659
Boyman O, Sprent J (2012) The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol 12:180–190
Braunersreuther V, Zernecke A, Arnaud C et al (2007) Ccr5 but not Ccr1 deficiency reduces development of diet-induced atherosclerosis in mice. Arterioscler Thromb Vasc Biol 27:373–379. https://doi.org/10.1161/01.ATV.0000253886.44609.ae
Brunstein CG, Miller JS, Cao Q et al (2011) Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood 117:1061–1070. https://doi.org/10.1182/blood-2010-07-293795
Buono C, Binder CJ, Stavrakis G et al (2005) T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Arterioscler Thromb Vasc Biol 39(7):1379–1389
Burioni R, Canducci F, Saita D et al (2009) Antigen-driven evolution of B lymphocytes in coronary atherosclerotic plaques. J Immunol 183:2537–2544. https://doi.org/10.4049/jimmunol.0901076
Burmester GR, Gordon KB, Rosenbaum JT et al (2020) Long-term safety of adalimumab in 29,967 adult patients from global clinical trials across multiple indications: an updated analysis. Adv Ther 37:364–380. https://doi.org/10.1007/s12325-019-01145-8
Butcher MJ, Filipowicz AR, Waseem TC et al (2016) Atherosclerosis-driven Treg plasticity results in formation of a dysfunctional subset of plastic IFNγ+ Th1/Tregs. Circ Res 119:1190–1203. https://doi.org/10.1161/CIRCRESAHA.116.309764
Caligiuri G, Khallou-Laschet J, Vandaele M et al (2007) Phosphorylcholine-targeting immunization reduces atherosclerosis. J Am Coll Cardiol 50:540–546. https://doi.org/10.1016/J.JACC.2006.11.054
Canault M, Peiretti F, Kopp F et al (2006) The TNF alpha converting enzyme (TACE/ADAM17) is expressed in the atherosclerotic lesions of apolipoprotein E-deficient mice: possible contribution to elevated plasma levels of soluble TNF alpha receptors. Atherosclerosis 187:82–91. https://doi.org/10.1016/j.atherosclerosis.2005.08.031
Cardilo-Reis L, Gruber S, Schreier SM et al (2012) Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol Med 4:1072–1086. https://doi.org/10.1002/emmm.201201374
Centa M, Jin H, Hofste L et al (2019) Germinal center-derived antibodies promote atherosclerosis plaque size and stability. Circulation 139:2466. https://doi.org/10.1161/CIRCULATIONAHA.118.038534
Chan CT, Sobey CG, Lieu M et al (2015) Obligatory role for B cells in the development of angiotensin II-dependent hypertension. Hypertension 66:1023–1033. https://doi.org/10.1161/HYPERTENSIONAHA.115.05779
Chatzigeorgiou A, Seijkens T, Zarzycka B et al (2014) Blocking CD40-TRAF6 signaling is a therapeutic target in obesity-associated insulin resistance. Proc Natl Acad Sci U S A 111:2686–2691. https://doi.org/10.1073/pnas.1400419111
Chen S, Wang Y, Pan Y et al (2020) Novel role for Tranilast in regulating NLRP3 ubiquitination, vascular inflammation, and atherosclerosis. J Am Heart Assoc 9:e015513. https://doi.org/10.1161/JAHA.119.015513
Cheng X, Yu X, Ding Y et al (2008) The Th17/Treg imbalance in patients with acute coronary syndrome. Clin Immunol 127:89–97. https://doi.org/10.1016/J.CLIM.2008.01.009
Choi HK, Hernán MA, Seeger JD et al (2002) Methotrexate and mortality in patients with rheumatoid arthritis: a prospective study. Lancet 359:1173–1177. https://doi.org/10.1016/S0140-6736(02)08213-2
Chyu KY, Zhao X, Reyes OS et al (2005) Immunization using an Apo B-100 related epitope reduces atherosclerosis and plaque inflammation in hypercholesterolemic apo E (−/−) mice. Biochem Biophys Res Commun 338:1982–1989. https://doi.org/10.1016/j.bbrc.2005.10.141
Cipriani S, Francisci D, Mencarelli A et al (2013) Efficacy of the CCR5 antagonist maraviroc in reducing early, ritonavir-induced atherogenesis and advanced plaque progression in mice. Circulation 127:2114–2124. https://doi.org/10.1161/CIRCULATIONAHA.113.001278
Clarke MCH, Talib S, Figg NL, Bennett MR (2010) Vascular smooth muscle cell apoptosis induces interleukin-1-directed inflammation: effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ Res 106:363–372. https://doi.org/10.1161/CIRCRESAHA.109.208389
Clement M, Guedj K, Andreata F et al (2015) Control of the T follicular helper–germinal center B-cell Axis by CD8 + regulatory T cells limits atherosclerosis and tertiary lymphoid organ development. Circulation 131:560–570. https://doi.org/10.1161/CIRCULATIONAHA.114.010988
Coll RC, Robertson AAB, Chae JJ et al (2015) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21:248–257. https://doi.org/10.1038/nm.3806
Combadière C, Potteaux S, Rodero M et al (2008) Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6Chi and Ly6Clo monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 117:1649–1657. https://doi.org/10.1161/CIRCULATIONAHA.107.745091
Curtis JR, John A, Baser O (2012) Dyslipidemia and changes in lipid profiles associated with rheumatoid arthritis and initiation of anti-tumor necrosis factor therapy. Arthritis Care Res 64:1282–1291. https://doi.org/10.1002/acr.21693
Czerkinsky C, Holmgren J (2012) Mucosal delivery routes for optimal immunization: targeting immunity to the right tissues. Curr Top Microbiol Immunol 354:1–18
Danesh J, Kaptoge S, Mann AG et al (2008) Long-term interleukin-6 levels and subsequent risk of coronary heart disease: two new prospective studies and a systematic review. PLoS Med 5:0600–0610. https://doi.org/10.1371/journal.pmed.0050078
Del Porto F, Laganà B, Lai S et al (2007) Response to anti-tumour necrosis factor alpha blockade is associated with reduction of carotid intima-media thickness in patients with active rheumatoid arthritis. Rheumatology 46:1111–1115. https://doi.org/10.1093/rheumatology/kem089
Devlin CM, Kuriakose G, Hirsch E, Tabas I (2002) Genetic alterations of IL-1 receptor antagonist in mice affect plasma cholesterol level and foam cell lesion size. Proc Natl Acad Sci U S A 99:6280–6285. https://doi.org/10.1073/pnas.092324399
Dewberry R, Holden H, Crossman D, Francis S (2000) Interleukin-1 receptor antagonist expression in human endothelial cells and atherosclerosis. Arterioscler Thromb Vasc Biol 20:2394–2400. https://doi.org/10.1161/01.ATV.20.11.2394
Di Ianni M, Falzetti F, Carotti A et al (2011) Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 117:3921–3928. https://doi.org/10.1182/blood-2010-10-311894
Dinh TN, Kyaw TS, Kanellakis P et al (2012) Cytokine therapy with interleukin-2/anti-interleukin-2 monoclonal antibody complexes expands CD4+CD25+Foxp3+ regulatory T cells and attenuates development and progression of atherosclerosis. Circulation 126:1256–1266. https://doi.org/10.1161/CIRCULATIONAHA.112.099044
Douna H, Amersfoort J, Schaftenaar FH et al (2019) Bidirectional effects of IL-10+ regulatory B cells in Ldlr−/− mice. Atherosclerosis 280:118–125. https://doi.org/10.1016/J.ATHEROSCLEROSIS.2018.11.019
Drobni ZD, Alvi RM, Taron J et al (2020) Association between immune checkpoint inhibitors with cardiovascular events and atherosclerotic plaque. Circulation 142:2299–2311. https://doi.org/10.1161/CIRCULATIONAHA.120.049981
Duewell P, Kono H, Rayner KJ et al (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464:1357–1361. https://doi.org/10.1038/nature08938
Dunér P, Mattisson IY, Fogelstrand P et al (2021) Antibodies against apo B100 peptide 210 inhibit atherosclerosis in apoE−/− mice. Sci Rep 11:9022. https://doi.org/10.1038/s41598-021-88430-1
Elhage R, Maret A, Pieraggi MT et al (1998) Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation 97:242–244. https://doi.org/10.1161/01.CIR.97.3.242
Elhage R, Clamens S, Besnard S et al (2001) Involvement of interleukin-6 in atherosclerosis but not in the prevention of fatty streak formation by 17β-estradiol in apolipoprotein E-deficient mice. Atherosclerosis 156:315–320. https://doi.org/10.1016/S0021-9150(00)00682-1
Everett BM, Pradhan AD, Solomon DH et al (2013) Rationale and design of the cardiovascular inflammation reduction trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J 166:199–207. https://doi.org/10.1016/j.ahj.2013.03.018
Ewing MM, Karper JC, Abdul S et al (2013) T-cell co-stimulation by CD28–CD80/86 and its negative regulator CTLA-4 strongly influence accelerated atherosclerosis development. Int J Cardiol 168:1965–1974. https://doi.org/10.1016/J.IJCARD.2012.12.085
Ference BA, Ginsberg HN, Graham I et al (2017) Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 38:2459–2472. https://doi.org/10.1093/eurheartj/ehx144
Fisman EZ, Benderly M, Esper RJ et al (2006) Interleukin-6 and the risk of future cardiovascular events in patients with angina pectoris and/or healed myocardial infarction. Am J Cardiol 98:14–18. https://doi.org/10.1016/j.amjcard.2006.01.045
Foks AC, Frodermann V, ter Borg M et al (2011) Differential effects of regulatory T cells on the initiation and regression of atherosclerosis. Atherosclerosis 218:53–60. https://doi.org/10.1016/j.atherosclerosis.2011.04.029
Foks AC, Lichtman AH, Kuiper J (2015) Treating atherosclerosis with regulatory T cells. Arterioscler Thromb Vasc Biol 35:280–287. https://doi.org/10.1161/ATVBAHA.114.303568
França CN, Izar MCO, Hortêncio MNS et al (2017) Monocyte subtypes and the CCR2 chemokine receptor in cardiovascular disease. Clin Sci 131:1215–1224
Francisci D, Pirro M, Schiaroli E et al (2019) Maraviroc intensification modulates atherosclerotic progression in HIV-suppressed patients at high cardiovascular risk. A randomized, crossover pilot study. Open Forum Infect Dis 6:1–7. https://doi.org/10.1093/ofid/ofz112
Fredrikson GN, Söderberg I, Lindholm M et al (2003) Inhibition of atherosclerosis in ApoE-null mice by immunization with ApoB-100 peptide sequences. Arterioscler Thromb Vasc Biol 23:879–884. https://doi.org/10.1161/01.ATV.0000067937.93716.DB
Fredrikson GN, Andersson L, Söderberg I et al (2005) Atheroprotective immunization with MDA-modified apo B-100 peptide sequences is associated with activation of Th2 specific antibody expression. Autoimmunity 38:171–179. https://doi.org/10.1080/08916930500050525
Fredrikson GN, Björkbacka H, Söderberg I et al (2008) Treatment with apo B peptide vaccines inhibits atherosclerosis in human apo B-100 transgenic mice without inducing an increase in peptide-specific antibodies. J Intern Med 264:563–570. https://doi.org/10.1111/j.1365-2796.2008.01995.x
Freigang S, Hörkkö S, Miller E et al (1998) Immunization of LDL receptor–deficient mice with homologous malondialdehyde-modified and native LDL reduces progression of atherosclerosis by mechanisms other than induction of high titers of antibodies to oxidative neoepitopes. Arterioscler Thromb Vasc Biol 18:1972–1982. https://doi.org/10.1161/01.ATV.18.12.1972
Freigang S, Ampenberger F, Weiss A et al (2013) Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat Immunol 14:1045–1053. https://doi.org/10.1038/ni.2704
Freitag D, Butterworth AS, Willeit P et al (2015) Cardiometabolic effects of genetic upregulation of the interleukin 1 receptor antagonist: a Mendelian randomisation analysis. Lancet Diabetes Endocrinol 3:243–253. https://doi.org/10.1016/S2213-8587(15)00034-0
Fröbert O, Götberg M, Angerås O et al (2017) Design and rationale for the influenza vaccination after myocardial infarction (IAMI) trial. A registry-based randomized clinical trial. Am Heart J 189:94–102. https://doi.org/10.1016/j.ahj.2017.04.003
Galea J, Armstrong J, Gadsdon P et al (1996) Interleukin-1β in coronary arteries of patients with ischemic heart disease. Arterioscler Thromb Vasc Biol 16:1000–1006. https://doi.org/10.1161/01.ATV.16.8.1000
Gardner SE, Humphry M, Bennett MR, Clarke MCH (2015) Senescent vascular smooth muscle cells drive inflammation through an interleukin-1α-dependent senescence-associated secretory phenotype. Arterioscler Thromb Vasc Biol 35:1963–1974. https://doi.org/10.1161/ATVBAHA.115.305896
George J, Afek A, Gilburd B et al (1998) Hyperimmunization of apo-E-deficient mice with homologous malondialdehyde low-density lipoprotein suppresses early atherogenesis. Atherosclerosis 138:147–152. https://doi.org/10.1016/S0021-9150(98)00015-X
Gilbert J, Lekstrom-Himes J, Donaldson D et al (2011) Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am J Cardiol 107:906–911. https://doi.org/10.1016/j.amjcard.2010.11.005
Gisterå A, Hansson GK (2017) The immunology of atherosclerosis. Nat Rev Nephrol 13:368–380
Gisterå A, Hermansson A, Strodthoff D et al (2017) Vaccination against T-cell epitopes of native ApoB100 reduces vascular inflammation and disease in a humanized mouse model of atherosclerosis. J Intern Med 281:383–397. https://doi.org/10.1111/joim.12589
Gomez D, Baylis RA, Durgin BG et al (2018) Interleukin-1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat Med 24:1418–1429. https://doi.org/10.1038/s41591-018-0124-5
Gonen A, Hansen LF, Turner WW et al (2014) Atheroprotective immunization with malondialdehyde-modified LDL is hapten specific and dependent on advanced MDA adducts: implications for development of an atheroprotective vaccine. J Lipid Res 55:2137–2155. https://doi.org/10.1194/jlr.M053256
González P, Alvarez R, Batalla A et al (2001) Genetic variation at the chemokine receptors CCR5/CCR2 in myocardial infarction. Genes Immun 2:191–195. https://doi.org/10.1038/sj.gene.6363760
Gonzalez-Juanatey C, Llorca J, Sanchez Andrade A et al (2006) Short-term adalimumab therapy improves endothelial function in patients with rheumatoid arthritis refractory to infliximab. Clin Exp Rheumatol 24(3):309–312
Gonzalez-Juanatey C, Vazquez-Rodriguez TR, Miranda-Filloy JA et al (2012) Anti-TNF-alpha-adalimumab therapy is associated with persistent improvement of endothelial function without progression of carotid intima-media wall thickness in patients with rheumatoid arthritis refractory to conventional therapy. Mediators Inflamm 2012:674265. https://doi.org/10.1155/2012/674265
Grasset EK, Duhlin A, Agardh HE et al (2015) Sterile inflammation in the spleen during atherosclerosis provides oxidation-specific epitopes that induce a protective B-cell response. Proc Natl Acad Sci 112(16):E2030–E2038. https://doi.org/10.1073/pnas.1421227112
Grievink HW, Gal P, Ozsvar Kozma M et al (2020) The effect of a 13-valent conjugate pneumococcal vaccine on circulating antibodies against oxidized LDL and phosphorylcholine in man, a randomized placebo-controlled clinical trial. Biology (Basel) 9:1–10. https://doi.org/10.3390/biology9110345
Griffin DO, Holodick NE, Rothstein TL (2011) Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70. J Exp Med 208:67–80. https://doi.org/10.1084/jem.20101499
Groß O, Yazdi AS, Thomas CJ et al (2012) Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of Caspase-1. Immunity 36:388–400. https://doi.org/10.1016/j.immuni.2012.01.018
Gruber S, Hendrikx T, Tsiantoulas D et al (2016) sialic acid-binding immunoglobulin-like lectin G promotes atherosclerosis and liver inflammation by suppressing the protective functions of B-1 cells. Cell Rep 14:2348–2361. https://doi.org/10.1016/j.celrep.2016.02.027
Grundtman C, Jakic B, Buszko M et al (2015) Mycobacterial heat shock protein 65 (mbHSP65)-induced atherosclerosis: preventive oral tolerization and definition of atheroprotective and atherogenic mbHSP65 peptides. Atherosclerosis 242:303–310. https://doi.org/10.1016/j.atherosclerosis.2015.06.044
Hamaguchi Y, Uchida J, Cain DW et al (2005) The peritoneal cavity provides a protective niche for B1 and conventional B lymphocytes during anti-CD20 immunotherapy in mice. J Immunol 174:4389–4399. https://doi.org/10.4049/JIMMUNOL.174.7.4389
Hamze M, Desmetz C, Berthe ML et al (2013) Characterization of resident B cells of vascular walls in human atherosclerotic patients. J Immunol 191:3006–3016. https://doi.org/10.4049/jimmunol.1202870
Han S, Liu P, Zhang W et al (2007) The opposite-direction modulation of CD4+CD25+ Tregs and T helper 1 cells in acute coronary syndromes. Clin Immunol 124:90–97. https://doi.org/10.1016/J.CLIM.2007.03.546
Hassan S, Milman U, Feld J et al (2016) Effects of anti-TNF-α treatment on lipid profile in rheumatic diseases: an analytical cohort study. Arthritis Res Ther 18:261. https://doi.org/10.1186/s13075-016-1148-1
Hennessy T, Soh L, Bowman M et al (2019) The low dose colchicine after myocardial infarction (LoDoCo-MI) study: a pilot randomized placebo controlled trial of colchicine following acute myocardial infarction. Am Heart J 215:62–69. https://doi.org/10.1016/j.ahj.2019.06.003
Herbin O, Ait-Oufella H, Yu W et al (2012) Regulatory T-cell response to apolipoprotein B100–derived peptides reduces the development and progression of atherosclerosis in mice. Arterioscler Thromb Vasc Biol 32:605–612. https://doi.org/10.1161/ATVBAHA.111.242800
Herold KC, Gitelman SE, Willi SM et al (2013) Teplizumab treatment may improve C-peptide responses in participants with type 1 diabetes after the new-onset period: a randomised controlled trial. Diabetologia 56:391–400. https://doi.org/10.1007/s00125-012-2753-4
Herold KC, Bundy BN, Long SA et al (2019) An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med 381:603–613. https://doi.org/10.1056/nejmoa1902226
Hilgendorf I, Theurl I, Gerhardt LMS et al (2014) Innate response activator B cells aggravate atherosclerosis by stimulating T Helper-1 adaptive Immunity. Clinical perspective. Circulation 129:1677–1687. https://doi.org/10.1161/CIRCULATIONAHA.113.006381
Hofmann K, Clauder AK, Manz RA (2018) Targeting B cells and plasma cells in autoimmune diseases. Front Immunol 9:835
Hollan I, Meroni PL, Ahearn JM et al (2013) Cardiovascular disease in autoimmune rheumatic diseases. Autoimmun Rev 12:1004–1015
Hsieh MJ, Lee CH, Tsai ML et al (2020) Biologic agents reduce cardiovascular events in rheumatoid arthritis not responsive to tumour necrosis factor inhibitors: a national cohort study. Can J Cardiol 36:1739–1746. https://doi.org/10.1016/j.cjca.2020.01.003
Hsue PY, Scherzer R, Grunfeld C et al (2014) Depletion of B-cells with rituximab improves endothelial function and reduces inflammation among individuals with rheumatoid arthritis. J Am Heart Assoc 3. https://doi.org/10.1161/JAHA.114.001267
Huan T, Zhang B, Wang Z et al (2013) A systems biology framework identifies molecular underpinnings of coronary heart disease. Arterioscler Thromb Vasc Biol 33:1427. https://doi.org/10.1161/ATVBAHA.112.300112
Huang Y, Jiang H, Chen Y et al (2018) Tranilast directly targets <scp>NLRP</scp> 3 to treat inflammasome-driven diseases. EMBO Mol Med 10(4):e8689. https://doi.org/10.15252/emmm.201708689
Huang R, Zhao SR, Li Y et al (2020) Association of tumor necrosis factor-α gene polymorphisms and coronary artery disease susceptibility: a systematic review and meta-analysis. BMC Med Genet 21:29. https://doi.org/10.1186/s12881-020-0952-2
Iribarren C, Rahmaoui A, Long AA et al (2017a) Cardiovascular and cerebrovascular events among patients receiving omalizumab: results from EXCELS, a prospective cohort study in moderate to severe asthma. J Allergy Clin Immunol 139:1489–1495. https://doi.org/10.1016/j.jaci.2016.07.038
Iribarren C, Rothman KJ, Bradley MS et al (2017b) Cardiovascular and cerebrovascular events among patients receiving omalizumab: pooled analysis of patient-level data from 25 randomized, double-blind, placebo-controlled clinical trials. J Allergy Clin Immunol 139:1678–1680. https://doi.org/10.1016/j.jaci.2016.12.953
Isoda K, Sawada S, Ishigami N et al (2004) Lack of interleukin-1 receptor antagonist modulates plaque composition in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 24:1068–1073. https://doi.org/10.1161/01.ATV.0000127025.48140.a3
Jackson SW, Scharping NE, Jacobs HM et al (2016) Cutting edge: BAFF overexpression reduces atherosclerosis via TACI-dependent B cell activation. J Immunol 197:4529–4534. https://doi.org/10.4049/jimmunol.1601198
Jacobsson LTH, Turesson C, Gülfe A et al (2005) Treatment with tumor necrosis factor blockers is associated with a lower incidence of first cardiovascular events in patients with rheumatoid arthritis. J Rheumatol 32:1213–1218
Jin Y, Kang EH, Brill G et al (2018) Cardiovascular (CV) risk after initiation of abatacept versus TNF inhibitors in rheumatoid arthritis patients with and without baseline CV disease. J Rheumatol 45:1240–1248. https://doi.org/10.3899/jrheum.170926
Jing H, Yong L, Haiyan L et al (2011) Oral administration of Lactococcus lactis delivered heat shock protein 65 attenuates atherosclerosis in low-density lipoprotein receptor-deficient mice. Vaccine 29:4102–4109. https://doi.org/10.1016/j.vaccine.2011.03.105
Jostock T, Müllberg J, Özbek S et al (2001) Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem 268:160–167. https://doi.org/10.1046/j.1432-1327.2001.01867.x
Jun HL, Hee JU, Park JW et al (2009) Interleukin-1β promotes the expression of monocyte chemoattractant protein-1 in human aorta smooth muscle cells via multiple signaling pathways. Exp Mol Med 41:757–764. https://doi.org/10.3858/emm.2009.41.10.082
Kang EH, Jin Y, Brill G et al (2018) Comparative cardiovascular risk of abatacept and tumor necrosis factor inhibitors in patients with rheumatoid arthritis with and without diabetes mellitus: a multidatabase cohort study. J Am Heart Assoc 7:e007393. https://doi.org/10.1161/JAHA.117.007393
Karnell JL, Rieder SA, Ettinger R, Kolbeck R (2019) Targeting the CD40-CD40L pathway in autoimmune diseases: humoral immunity and beyond. Adv Drug Deliv Rev 141:92–103
Kasahara K, Sasaki N, Yamashita T et al (2014) CD3 antibody and IL-2 complex combination therapy inhibits atherosclerosis by augmenting a regulatory immune response. J Am Heart Assoc 3. https://doi.org/10.1161/JAHA.113.000719
Kelley N, Jeltema D, Duan Y, He Y (2019) The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci 20:3328
Kerekes G, Soltész P, Dér H et al (2009) Effects of rituximab treatment on endothelial dysfunction, carotid atherosclerosis, and lipid profile in rheumatoid arthritis. Clin Rheumatol 28:705–710. https://doi.org/10.1007/s10067-009-1095-1
Khamis RY, Hughes AD, Caga-anan M et al (2016) EBioMedicine high serum immunoglobulin G and M levels predict freedom from adverse cardiovascular events in hypertension : a nested case-control substudy of the Anglo-Scandinavian cardiac outcomes trial. EBioMedicine 9:372–380. https://doi.org/10.1016/j.ebiom.2016.06.012
Kim DG, Lee J, Seo WJ et al (2019) Rituximab protects against development of atherosclerotic cardiovascular disease after kidney transplantation: a propensity-matched study. Sci Rep 9:1–8. https://doi.org/10.1038/s41598-019-52942-8
Kimura T, Tse K, McArdle S et al (2017) Atheroprotective vaccination with MHC-II-restricted ApoB peptides induces peritoneal IL-10-producing CD4 T cells. Am J Physiol Circ Physiol 312:H781–H790. https://doi.org/10.1152/ajpheart.00798.2016
Kimura T, Kobiyama K, Winkels H et al (2018) Regulatory CD4+ T cells recognize major histocompatibility complex class II molecule-restricted peptide epitopes of apolipoprotein B. Circulation 138:1130–1143. https://doi.org/10.1161/CIRCULATIONAHA.117.031420
Kirii H, Niwa T, Yamada Y et al (2003) Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 23:656–660. https://doi.org/10.1161/01.ATV.0000064374.15232.C3
Kita T, Yamashita T, Sasaki N et al (2014) Regression of atherosclerosis with anti-CD3 antibody via augmenting a regulatory T-cell response in mice. Cardiovasc Res 102:107–117. https://doi.org/10.1093/cvr/cvu002
Klatzmann D, Abbas AK (2015) The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol 15:283–294. https://doi.org/10.1038/nri3823
Kleveland O, Kunszt G, Bratlie M et al (2016) Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: a double-blind, randomized, placebo-controlled phase 2 trial. Eur Heart J 37:2406–2413. https://doi.org/10.1093/eurheartj/ehw171
Klingenberg R, Lebens M, Hermansson A et al (2010) Intranasal immunization with an apolipoprotein B-100 fusion protein induces antigen-specific regulatory T cells and reduces atherosclerosis. Arterioscler Thromb Vasc Biol 30:946–952. https://doi.org/10.1161/ATVBAHA.109.202671
Klingenberg R, Ketelhuth DFJ, Strodthoff D et al (2012) Subcutaneous immunization with heat shock protein-65 reduces atherosclerosis in Apoe −/− mice. Immunobiology 217:540–547. https://doi.org/10.1016/j.imbio.2011.06.006
Klingenberg R, Gerdes N, Badeau RM et al (2013) Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J Clin Invest 123:1323–1334. https://doi.org/10.1172/JCI63891
Koenen RR, Weber C (2010) Therapeutic targeting of chemokine interactions in atherosclerosis. Nat Rev Drug Discov 9:141–153. https://doi.org/10.1038/nrd3048
Kortelainen ML, Porvari K (2014) Adventitial macrophage and lymphocyte accumulation accompanying early stages of human coronary atherogenesis. Cardiovasc Pathol 23:193–197. https://doi.org/10.1016/j.carpath.2014.03.001
Kounis NG, Hahalis G (2016) Serum IgE levels in coronary artery disease. Atherosclerosis 251:498–500. https://doi.org/10.1016/j.atherosclerosis.2016.05.045
Kuziel WA, Dawson TC, Quinones M et al (2003) CCR5 deficiency is not protective in the early stages of atherogenesis in apoE knockout mice. Atherosclerosis 167:25–32. https://doi.org/10.1016/S0021-9150(02)00382-9
Kyaw T, Tay C, Khan A et al (2010) Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol 185:4410–4419. https://doi.org/10.4049/jimmunol.1000033
Kyaw T, Tay C, Hosseini H et al (2012) Depletion of B2 but not B1a B cells in BAFF receptor-deficient ApoE−/− mice attenuates atherosclerosis by potently ameliorating arterial inflammation. PLoS One 7:e29371. https://doi.org/10.1371/journal.pone.0029371
Kyaw T, Loveland P, Kanellakis P et al (2021) Alarmin-activated B cells accelerate murine atherosclerosis after myocardial infarction via plasma cell-immunoglobulin-dependent mechanisms. Eur Heart J 42:938–947. https://doi.org/10.1093/eurheartj/ehaa995
Kyrklund M, Bildo M, Akhi R et al (2020) Humoral immune response to heat shock protein 60 of Aggregatibacter actinomycetemcomitans and cross-reactivity with malondialdehyde acetaldehyde-modified LDL. PLoS One 15:e0230682. https://doi.org/10.1371/journal.pone.0230682
Lalezari J, Yadavalli GK, Para M et al (2008) Safety, pharmacokinetics, and antiviral activity of HGS004, a novel fully human IgG4 monoclonal antibody against CCR5, in HIV-1–infected patients. J Infect Dis 197:721–727. https://doi.org/10.1086/527327
Lameijer M, Binderup T, Van Leent MMT et al (2018) Efficacy and safety assessment of a TRAF6-targeted nanoimmunotherapy in atherosclerotic mice and non-human primates. Nat Biomed Eng 2:279–292. https://doi.org/10.1038/s41551-018-0221-2
Langevitz P, Livneh A, Neumann L et al (2001) Prevalence of ischemic heart disease in patients with familial Mediterranean fever. Isr Med Assoc J 3:9–12
Lefkou E, Fragakis N, Ioannidou E et al (2010) Increased levels of proinflammatory cytokines in children with family history of coronary artery disease. Clin Cardiol 33:E6. https://doi.org/10.1002/clc.20434
Lehrer-Graiwer J, Singh P, Abdelbaky A et al (2015) FDG-PET imaging for oxidized LDL in stable atherosclerotic disease: a phase II study of safety, tolerability, and anti-inflammatory activity. JACC Cardiovasc Imaging 8:493–494. https://doi.org/10.1016/J.JCMG.2014.06.021
Lewis MJ, Malik TH, Ehrenstein MR et al (2009) Immunoglobulin M is required for protection against atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 120:417–426. https://doi.org/10.1161/CIRCULATIONAHA.109.868158
Li J, McArdle S, Gholami A et al (2016) CCR5+T-bet+FoxP3+ effector CD4 T cells drive atherosclerosis. Circ Res 118:1540–1552. https://doi.org/10.1161/CIRCRESAHA.116.308648
Libby P (2021) The changing landscape of atherosclerosis. Nature 592:524–533. https://doi.org/10.1038/s41586-021-03392-8
Lutgens E, Cleutjens KBJM, Heeneman S et al (2000) Both early and delayed anti-CD40L antibody treatment induces a stable plaque phenotype. Proc Natl Acad Sci U S A 97:7464–7469. https://doi.org/10.1073/pnas.97.13.7464
Lutgens E, Lievens D, Beckers L et al (2010) Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med 207:391–404. https://doi.org/10.1084/jem.20091293
Ma K, Lv S, Liu B et al (2013) CTLA4-IgG ameliorates homocysteine-accelerated atherosclerosis by inhibiting T-cell overactivation in apoE−/− mice. Cardiovasc Res 97:349–359. https://doi.org/10.1093/cvr/cvs330
Makino Y, Cook DN, Smithies O et al (2002) Impaired T cell function in RANTES-deficient mice. Clin Immunol 102:302–309. https://doi.org/10.1006/clim.2001.5178
Mantani PT, Ljungcrantz I, Andersson L et al (2014) Circulating CD40+ and CD86+ B cell subsets demonstrate opposing associations with risk of stroke. Arterioscler Thromb Vasc Biol 34:211–218. https://doi.org/10.1161/ATVBAHA.113.302667
Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A et al (2014) Therapy of type 1 diabetes with CD4+CD25highCD127-regulatory T cells prolongs survival of pancreatic islets - results of one year follow-up. Clin Immunol 153:23–30. https://doi.org/10.1016/j.clim.2014.03.016
Maron R, Sukhova G, Faria AM et al (2002) Mucosal administration of heat shock protein-65 decreases atherosclerosis and inflammation in aortic arch of low-density lipoprotein receptor-deficient mice. Circulation 106:1708–1715. https://doi.org/10.1161/01.CIR.0000029750.99462.30
Mathew JM, H-Voss J, LeFever A et al (2018) A phase i clinical trial with ex vivo expanded recipient regulatory t cells in living donor kidney transplants. Sci Rep 8:7428. https://doi.org/10.1038/s41598-018-25574-7
Matsumura T, Kugiyama K, Sugiyama S et al (1999) Suppression of atherosclerotic development in Watanabe heritable hyperlipidemic rabbits treated with an oral antiallergic drug, tranilast. Circulation 99:919–924. https://doi.org/10.1161/01.CIR.99.7.919
McGeough MD, Wree A, Inzaugarat ME et al (2017) TNF regulates transcription of NLRP3 inflammasome components and inflammatory molecules in cryopyrinopathies. J Clin Invest 127:4488–4497. https://doi.org/10.1172/JCI90699
McInnes IB, Thompson L, Giles JT et al (2015) Effect of interleukin-6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo-controlled study. Ann Rheum Dis 74:694–702. https://doi.org/10.1136/annrheumdis-2013-204345
Mendall MA, Patel P, Asante M et al (1997) Relation of serum cytokine concentrations to cardiovascular risk factors and coronary heart disease. Heart 78:273–277. https://doi.org/10.1136/hrt.78.3.273
Merhi-Soussi F, Kwak BR, Magne D et al (2005) Interleukin-1 plays a major role in vascular inflammation and atherosclerosis in male apolipoprotein E-knockout mice. Cardiovasc Res 66:583–593. https://doi.org/10.1016/j.cardiores.2005.01.008
Mor A, Luboshits G, Planer D et al (2006) Altered status of CD4+CD25+ regulatory T cells in patients with acute coronary syndromes. Eur Heart J 27:2530–2537. https://doi.org/10.1093/eurheartj/ehl222
Mor A, Planer D, Luboshits G et al (2007) Role of naturally occurring CD4+CD25+ regulatory T cells in experimental atherosclerosis. Arterioscler Thromb Vasc Biol 27:893–900. https://doi.org/10.1161/01.ATV.0000259365.31469.89
Morris-Rosenfeld S, Lipinski MJ, McNamara CA (2014) Understanding the role of B cells in atherosclerosis: potential clinical implications. Expert Rev Clin Immunol 10:77–89. https://doi.org/10.1586/1744666X.2014.857602
Morton AC, Rothman AMK, Greenwood JP et al (2015) The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: the MRC-ILA heart study. Eur Heart J 36:377–384. https://doi.org/10.1093/eurheartj/ehu272
Newland SA, Mohanta S, Clément M et al (2017) Type-2 innate lymphoid cells control the development of atherosclerosis in mice. Nat Commun 8:15781. https://doi.org/10.1038/ncomms15781
Nidorf M, Thompson PL (2007) Effect of colchicine (0.5 mg twice daily) on high-sensitivity C-reactive protein independent of aspirin and atorvastatin in patients with stable coronary artery disease. Am J Cardiol 99:805–807. https://doi.org/10.1016/j.amjcard.2006.10.039
Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL (2013) Low-dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol 61:404–410. https://doi.org/10.1016/j.jacc.2012.10.027
Nidorf SM, Fiolet ATL, Eikelboom JW et al (2019) The effect of low-dose colchicine in patients with stable coronary artery disease: the LoDoCo2 trial rationale, design, and baseline characteristics. Am Heart J 218:46–56. https://doi.org/10.1016/j.ahj.2019.09.011
Nidorf SM, Fiolet ATL, Mosterd A et al (2020) Colchicine in patients with chronic coronary disease. N Engl J Med 383:1838–1847. https://doi.org/10.1056/NEJMoa2021372
Nilsson J, Calara F, Regnstrom J et al (1997) Immunization with homologous oxidized low density lipoprotein reduces neointimal formation after balloon injury in hypercholesterolemic rabbits. J Am Coll Cardiol 30:1886–1891. https://doi.org/10.1016/S0735-1097(97)00366-5
Novikova DS, Popkova TV, Lukina GV et al (2016) The effects of rituximab on lipids, arterial stiffness and carotid intima-media thickness in rheumatoid arthritis. J Korean Med Sci 31:202–207. https://doi.org/10.3346/jkms.2016.31.2.202
Nurmohamed MT (2009) Cardiovascular risk in rheumatoid arthritis. Autoimmun Rev 8:663–667
Nus M, Sage AP, Lu Y et al (2017) Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat Med 23:601. https://doi.org/10.1038/nm.4315
Ohta H, Wada H, Niwa T et al (2005) Disruption of tumor necrosis factor-α gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis 180:11–17. https://doi.org/10.1016/j.atherosclerosis.2004.11.016
Ozen G, Pedro S, Michaud K (2020) The risk of cardiovascular events associated with disease-modifying antirheumatic drugs in rheumatoid arthritis. J Rheumatol 48(5):648–655. https://doi.org/10.3899/jrheum.200265
Palinski W, Miller E, Witztum JL (1995) Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherogenesis. Proc Natl Acad Sci USA 92:821–825. https://doi.org/10.1073/PNAS.92.3.821
Pattison JM, Nelson PJ, Huie P et al (1996) RANTES chemokine expression in transplant-associated accelerated atherosclerosis. J Heart Lung Transplant 15:1194–1199
Poels K, van Leent MMT, Reiche ME et al (2020) Antibody-mediated inhibition of CTLA4 aggravates atherosclerotic plaque inflammation and progression in hyperlipidemic mice. Cell 9:1987. https://doi.org/10.3390/cells9091987
Porsch F, Binder CJ (2019) Impact of B-cell–targeted therapies on cardiovascular disease. Arterioscler Thromb Vasc Biol 39:1705–1714. https://doi.org/10.1161/ATVBAHA.119.311996
Poulsen CB, Al-Mashhadi AL, Von Wachenfeldt K et al (2016) Treatment with a human recombinant monoclonal IgG antibody against oxidized LDL in atherosclerosis-prone pigs reduces cathepsin S in coronary lesions. Int J Cardiol 215:506–515. https://doi.org/10.1016/j.ijcard.2016.03.222
Pradhan AD (2021) Time to commence or time out for colchicine in secondary prevention of cardiovascular disease? Eur Heart J. https://doi.org/10.1093/eurheartj/ehab210
Primdahl J, Clausen J, Hørslev-Petersen K (2013) Results from systematic screening for cardiovascular risk in outpatients with rheumatoid arthritis in accordance with the EULAR recommendations. Ann Rheum Dis 72:1771–1776. https://doi.org/10.1136/annrheumdis-2013-203688
Proto JD, Doran AC, Gusarova G et al (2018) Regulatory T cells promote macrophage efferocytosis during inflammation resolution. Immunity 49:666–677. https://doi.org/10.1016/J.IMMUNI.2018.07.015
Protogerou AD, Zampeli E, Fragiadaki K et al (2011) A pilot study of endothelial dysfunction and aortic stiffness after interleukin-6 receptor inhibition in rheumatoid arthritis. Atherosclerosis 219:734–736. https://doi.org/10.1016/j.atherosclerosis.2011.09.015
Provan SA, Berg IJ, Hammer HB et al (2015) The impact of newer biological disease modifying anti-rheumatic drugs on cardiovascular risk factors: a 12-month longitudinal study in rheumatoid arthritis patients treated with rituximab, abatacept and tociliziumab. PLoS One 10:e0130709. https://doi.org/10.1371/journal.pone.0130709
Que X, Hung MY, Yeang C et al (2018) Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 558:301–306. https://doi.org/10.1038/s41586-018-0198-8
Quinones MP, Martinez HG, Jimenez F et al (2007) CC chemokine receptor 5 influences late-stage atherosclerosis. Atherosclerosis 195:e92–e103. https://doi.org/10.1016/j.atherosclerosis.2007.03.026
Radocha J, van de Donk NWCJ, Weisel K (2021) Monoclonal antibodies and antibody drug conjugates in multiple myeloma. Cancers (Basel) 13
Rajamaki K, Lappalainen J, Oorni K et al (2010) Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One 5:7. https://doi.org/10.1371/journal.pone.0011765
Rauch M, Tussiwand R, Bosco N, Rolink AG (2009) Crucial role for BAFF-BAFF-R signaling in the survival and maintenance of mature B cells. PLoS One 4:e5456. https://doi.org/10.1371/journal.pone.0005456
Reiss AB, Carsons SE, Anwar K et al (2008) Atheroprotective effects of methotrexate on reverse cholesterol transport proteins and foam cell transformation in human THP-1 monocyte/macrophages. Arthritis Rheum 58:3675–3683. https://doi.org/10.1002/art.24040
Ren S, Newby D, Li SC et al (2015) Effect of the adult pneumococcal polysaccharide vaccine on cardiovascular disease: a systematic review and meta-analysis. Open Hear 2:e000247. https://doi.org/10.1136/openhrt-2015-000247
Ren S, Hure A, Peel R et al (2016) Rationale and design of a randomized controlled trial of pneumococcal polysaccharide vaccine for prevention of cardiovascular events: the Australian study for the prevention through immunization of cardiovascular events (AUSPICE). Am Heart J 177:58–65. https://doi.org/10.1016/j.ahj.2016.04.003
Ridker PM, Hennekens CH, Buring JE, Rifai N (2000a) C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 342:836–843. https://doi.org/10.1056/NEJM200003233421202
Ridker PM, Rifai N, Stampfer MJ, Hennekens CH (2000b) Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation 101:1767–1772. https://doi.org/10.1161/01.CIR.101.15.1767
Ridker PM, Danielson E, Fonseca FA et al (2009) Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet 373:1175–1182. https://doi.org/10.1016/S0140-6736(09)60447-5
Ridker PM, Everett BM, Thuren T et al (2017) Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377:1119–1131. https://doi.org/10.1056/NEJMoa1707914
Ridker PM, Libby P, MacFadyen JG et al (2018a) Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: analyses from the Canakinumab anti-inflammatory thrombosis outcomes study (CANTOS). Eur Heart J 39:3499–3507. https://doi.org/10.1093/eurheartj/ehy310
Ridker PM, MacFadyen JG, Everett BM et al (2018b) Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet 391:319–328. https://doi.org/10.1016/S0140-6736(17)32814-3
Ridker PM, MacFadyen JG, Thuren T, Libby P (2020) Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1b inhibition with canakinumab: further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur Heart J 41:2153–2163. https://doi.org/10.1093/eurheartj/ehz542
Rosenfeld SM, Perry HM, Gonen A et al (2015) B-1b cells secrete atheroprotective IgM and attenuate atherosclerosis. Circ Res 117
Roubille C, Richer V, Starnino T et al (2015) The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta-analysis. Ann Rheum Dis 74:480–489
Rus FG, Niculescu F, Vlaicu R (1991) Tumor necrosis factor-alpha in human arterial wall with atherosclerosis. Atherosclerosis 89:247–254. https://doi.org/10.1016/0021-9150(91)90066-C
Rus HG, Vlaicu R, Niculescu F (1996) Interleukin-6 and interleukin-8 protein and gene expression in human arterial atherosclerotic wall. Atherosclerosis 127:263–271. https://doi.org/10.1016/S0021-9150(96)05968-0
Saag KG, Gim GT, Patkar NM et al (2008) American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Care Res 59:762–784
Sage AP, Tsiantoulas D, Baker L et al (2012) BAFF receptor deficiency reduces the development of atherosclerosis in mice--brief report. Arterioscler Thromb Vasc Biol 32:1573–1576. https://doi.org/10.1161/ATVBAHA.111.244731
Sage AP, Nus M, Baker LL et al (2015) Regulatory B cell-specific interleukin-10 is dispensable for atherosclerosis development in mice. Arterioscler Thromb Vasc Biol 35:1770–1773. https://doi.org/10.1161/ATVBAHA.115.305568
Sage AP, Nus M, Bagchi Chakraborty J et al (2017) X-box binding protein-1 dependent plasma cell responses limit the development of atherosclerosis. Circ Res 121:270–281. https://doi.org/10.1161/CIRCRESAHA.117.310884
Sage AP, Tsiantoulas D, Binder CJ, Mallat Z (2018) The role of B cells in atherosclerosis. Nat Rev Cardiol 16(3):180–196. https://doi.org/10.1038/s41569-018-0106-9
Saigusa R, Winkels H, Ley KT (2020) Cell subsets and functions in atherosclerosis. Nat Rev Cardiol 17:387. https://doi.org/10.1038/s41569-020-0352-5
Sánchez-Trujillo L, Jerjes-Sanchez C, Rodriguez D et al (2019) Phase II clinical trial testing the safety of a humanised monoclonal antibody anti-CD20 in patients with heart failure with reduced ejection fraction, ICFEr-RITU2: study protocol. BMJ Open 9:e022826. https://doi.org/10.1136/bmjopen-2018-022826
Sattar N, Crompton P, Cherry L et al (2007) Effects of tumor necrosis factor blockade on cardiovascular risk factors in psoriatic arthritis: a double-blind, placebo-controlled study. Arthritis Rheum 56:831–839. https://doi.org/10.1002/art.22447
Schaheen B, Downs EA, Serbulea V et al (2016) B-cell depletion promotes aortic infiltration of immunosuppressive cells and is protective of experimental aortic aneurysm. Arterioscler Thromb Vasc Biol 36:2191–2202. https://doi.org/10.1161/ATVBAHA.116.307559
Schieffer B, Schieffer E, Hilfiker-Kleiner D et al (2000) Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques. Circulation 101:1372–1378. https://doi.org/10.1161/01.CIR.101.12.1372
Schieffer B, Selle T, Hilfiker A et al (2004) Impact of interleukin-6 on plaque development and morphology in experimental atherosclerosis. Circulation 110:3493–3500. https://doi.org/10.1161/01.CIR.0000148135.08582.97
Schiopu A, Bengtsson J, Söderberg I et al (2004) Recombinant human antibodies against aldehyde-modified apolipoprotein B-100 peptide sequences inhibit atherosclerosis. Circulation 110:2047–2052. https://doi.org/10.1161/01.CIR.0000143162.56057.B5
Schiopu A, Frendéus B, Jansson B et al (2007) Recombinant antibodies to an oxidized low-density lipoprotein epitope induce rapid regression of atherosclerosis in Apobec-1−/−/low-density lipoprotein receptor−/−mice. J Am Coll Cardiol 50:2313–2318. https://doi.org/10.1016/J.JACC.2007.07.081
Schönbeck U, Sukhova GK, Shimizu K et al (2000) Inhibition of CD40 signaling limits evolution of established atherosclerosis in mice. Proc Natl Acad Sci U S A 97:7458–7463. https://doi.org/10.1073/pnas.97.13.7458
Schuett H, Oestreich R, Waetzig GH et al (2012) Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler Thromb Vasc Biol 32:281–290. https://doi.org/10.1161/ATVBAHA.111.229435
Seijkens TTP, van Tiel CM, Kusters PJH et al (2018) Targeting CD40-induced TRAF6 signaling in macrophages reduces atherosclerosis. J Am Coll Cardiol 71:527–542. https://doi.org/10.1016/j.jacc.2017.11.055
Seino Y, Ikeda U, Ikeda M et al (1994) Interleukin 6 gene transcripts are expressed in human atherosclerotic lesions. Cytokine 6:87–91. https://doi.org/10.1016/1043-4666(94)90013-2
Shah ASV, Stelzle D, Ken Lee K et al (2018) Global burden of atherosclerotic cardiovascular disease in people living with HIV systematic review and meta-analysis. Circulation 138:1100–1112
Sharma M, Schlegel MP, Afonso MS et al (2020) Regulatory T cells license macrophage pro-resolving functions during atherosclerosis regression. Circ Res 127:335–353. https://doi.org/10.1161/CIRCRESAHA.119.316461
Sharpe AH, Freeman GJ (2002) The B7-CD28 superfamily. Nat Rev Immunol 2:116–126
Shaw PX, Hörkkö S, Chang MK et al (2000) Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J Clin Invest 105:1731–1740. https://doi.org/10.1172/JCI8472
Shiri-Sverdlov R, Dos Reis IM, Oligschlaeger Y et al (2021) The influence of a conjugated pneumococcal vaccination on plasma antibody levels against oxidized low-density lipoprotein in metabolic disease patients: a single-arm pilot clinical trial. Antioxidants 10:1–12. https://doi.org/10.3390/antiox10010129
Simons KH, de Jong A, Jukema JW et al (2019) T cell co-stimulation and co-inhibition in cardiovascular disease: a double-edged sword. Nat Rev Cardiol 2019:1–19. https://doi.org/10.1038/s41569-019-0164-7
Singh JA, Furst DE, Bharat A et al (2012) 2012 update of the 2008 American college of rheumatology recommendations for the use of disease-modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis. Arthritis Care Res 64:625–639. https://doi.org/10.1002/acr.21641
Sode J, Vogel U, Bank S et al (2014) Anti-TNF treatment response in rheumatoid arthritis patients is associated with genetic variation in the NLRP3-inflammasome. PLoS One 9:e100361. https://doi.org/10.1371/journal.pone.0100361
Song L, Schindler C (2004) IL-6 and the acute phase response in murine atherosclerosis. Atherosclerosis 177:43–51. https://doi.org/10.1016/j.atherosclerosis.2004.06.018
Steffens S, Burger F, Pelli G et al (2006) Short-term treatment with anti-CD3 antibody reduces the development and progression of atherosclerosis in mice. Circulation 114:1977–1984. https://doi.org/10.1161/CIRCULATIONAHA.106.627430
Strang AC, Bisoendial RJ, Kootte RS et al (2013) Pro-atherogenic lipid changes and decreased hepatic LDL receptor expression by tocilizumab in rheumatoid arthritis. Atherosclerosis 229:174–181. https://doi.org/10.1016/j.atherosclerosis.2013.04.031
Strom AC, Cross AJ, Cole JE et al (2015) B regulatory cells are increased in hypercholesterolaemic mice and protect from lesion development via IL-10. Thromb Haemost 114:835–847. https://doi.org/10.1160/TH14-12-1084
Szalai C, Duba J, Prohászka Z et al (2001) Involvement of polymorphisms in the chemokine system in the susceptibility for coronary artery disease (CAD). Coincidence of elevated Lp (a) and MCP-1-2518 G/G genotype in CAD patients. Atherosclerosis 158:233–239. https://doi.org/10.1016/S0021-9150(01)00423-3
Tabas I, Glass CK (2013) Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science 339:166–172
Tam LS, Shang Q, Kun EW et al (2014) The effects of golimumab on subclinical atherosclerosis and arterial stiffness in ankylosing spondylitis-a randomized, placebo-controlled pilot trial. Rheumatol (United Kingdom) 53:1065–1074. https://doi.org/10.1093/rheumatology/ket469
Tardif JC, Kouz S, Waters DD et al (2019) Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med 381:2497–2505. https://doi.org/10.1056/NEJMoa1912388
Tattersall MC, Guo M, Korcarz CE et al (2015) Asthma predicts cardiovascular disease events: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol 35:1520–1525. https://doi.org/10.1161/ATVBAHA.115.305452
Tay C, Liu Y-H, Kanellakis P et al (2018) Follicular B cells promote atherosclerosis via T cell–mediated differentiation into plasma cells and secreting pathogenic immunoglobulin G. Arterioscler Thromb Vasc Biol 38:5. https://doi.org/10.1161/ATVBAHA.117.310678
Trzonkowski P, Bieniaszewska M, Juścińska J et al (2009) First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol 133:22–26. https://doi.org/10.1016/j.clim.2009.06.001
Tse K, Gonen A, Sidney J et al (2013) Atheroprotective vaccination with MHC-II restricted peptides from ApoB-100. Front Immunol 4:493. https://doi.org/10.3389/fimmu.2013.00493
Tsiantoulas D, Bot I, Ozsvar-Kozma M et al (2017) Increased plasma IgE accelerate atherosclerosis in secreted IgM deficiency. Circ Res 120:78–84. https://doi.org/10.1161/CIRCRESAHA.116.309606
Tsiantoulas D, Sage AP, Göderle L et al (2018) B cell–activating factor neutralization aggravates atherosclerosis. Circulation 138:2263–2273. https://doi.org/10.1161/CIRCULATIONAHA.117.032790
Upadhye A, Srikakulapu P, Gonen A et al (2019) Diversification and CXCR4-dependent establishment of the bone marrow B-1a cell Pool governs atheroprotective IgM production linked to human coronary atherosclerosis. Circ Res 125:e55–e70. https://doi.org/10.1161/CIRCRESAHA.119.315786
Upadhye A, Sturek JM, McNamara CA (2020) 2019 Russell Ross memorial lecture in vascular biology. Arterioscler Thromb Vasc Biol 40:309–322. https://doi.org/10.1161/ATVBAHA.119.313064
Vaidya K, Arnott C, Martínez GJ et al (2018) Colchicine therapy and plaque stabilization in patients with acute coronary syndrome: a CT coronary angiography study. JACC Cardiovasc Imaging 11:305–316. https://doi.org/10.1016/j.jcmg.2017.08.013
van den Berg VJ, Vroegindewey MM, Kardys I et al (2019) Anti-oxidized LDL antibodies and coronary artery disease: a systematic review. Antioxidants 8:10
Van Der Heijden T, Kritikou E, Venema W et al (2017) NLRP3 Inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler Thromb Vasc Biol 37:1457–1461. https://doi.org/10.1161/ATVBAHA.117.309575
Van Gool F, Molofsky AB, Morar MM et al (2014) Interleukin-5-producing group 2 innate lymphoid cells control eosinophilia induced by interleukin-2 therapy. Blood 124:3572–3576. https://doi.org/10.1182/blood-2014-07-587493
Van Puijvelde GHM, Hauer AD, De Vos P et al (2006) Induction of oral tolerance to oxidized low-density lipoprotein ameliorates atherosclerosis. Circulation 114:1968–1976. https://doi.org/10.1161/CIRCULATIONAHA.106.615609
Van Puijvelde GHM, Van Es T, Van Wanrooij EJA et al (2007) Induction of oral tolerance to HSP60 or an HSP60-peptide activates t cell regulation and reduces atherosclerosis. Arterioscler Thromb Vasc Biol 27:2677–2683. https://doi.org/10.1161/ATVBAHA.107.151274
Veillard NR, Kwak B, Pelli G et al (2004) Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ Res 94:253–261. https://doi.org/10.1161/01.RES.0000109793.17591.4E
Vromman A, Ruvkun V, Shvartz E et al (2019) Stage-dependent differential effects of interleukin-1 isoforms on experimental atherosclerosis. Eur Heart J 40:2482–2491. https://doi.org/10.1093/eurheartj/ehz008
Wæhre T, Yndestad A, Smith C et al (2004) Increased expression of interleukin-1 in coronary artery disease with downregulatory effects of HMG-CoA reductase inhibitors. Circulation 109:1966–1972. https://doi.org/10.1161/01.CIR.0000125700.33637.B1
Wang X, Feuerstein GZ, Gu JL et al (1995) Interleukin-1β induces expression of adhesion molecules in human vascular smooth muscle cells and enhances adhesion of leukocytes to smooth muscle cells. Atherosclerosis 115:89–98. https://doi.org/10.1016/0021-9150(94)05503-B
Wang J, Cheng X, Xiang M-X et al (2011) IgE stimulates human and mouse arterial cell apoptosis and cytokine expression and promotes atherogenesis in Apoe−/− mice. J Clin Invest 121:3564–3577. https://doi.org/10.1172/JCI46028
Wessels JAM, Huizinga TWJ, Guchelaar HJ (2008) Recent insights in the pharmacological actions of methotrexate in the treatment of rheumatoid arthritis. Rheumatology 47:249–255
Westlake SL, Colebatch AN, Baird J et al (2010) The effect of methotrexate on cardiovascular disease in patients with rheumatoid arthritis: a systematic literature review. Rheumatology 49:295–307. https://doi.org/10.1093/rheumatology/kep366
Wezel A, Lagraauw HM, van der Velden D et al (2015) Mast cells mediate neutrophil recruitment during atherosclerotic plaque progression. Atherosclerosis 241:289–296. https://doi.org/10.1016/j.atherosclerosis.2015.05.028
Wick G, Jakic B, Buszko M et al (2014) The role of heat shock proteins in atherosclerosis. Nat Rev Cardiol 11:516–529
Wigren M, Kolbus D, Dunér P et al (2011) Evidence for a role of regulatory T cells in mediating the atheroprotective effect of apolipoprotein B peptide vaccine. J Intern Med 269:546–556. https://doi.org/10.1111/j.1365-2796.2010.02311.x
Wigren M, Björkbacka H, Andersson L et al (2012) Low levels of circulating CD4+FoxP3+ T cells are associated with an increased risk for development of myocardial infarction but not for stroke. Arterioscler Thromb Vasc Biol 32:2000–2004. https://doi.org/10.1161/ATVBAHA.112.251579
Wilcox JN, Nelken NA, Coughlin SR et al (1994) Local expression of inflammatory cytokines in human atherosclerotic plaques. J Atheroscler Thromb 1:S10–S13. https://doi.org/10.5551/jat1994.1.Supplemment1_S10
Winter C, Silvestre-Roig C, Ortega-Gomez A et al (2018) Chrono-pharmacological targeting of the CCL2-CCR2 Axis ameliorates atherosclerosis. Cell Metab 28:175–182. https://doi.org/10.1016/j.cmet.2018.05.002
Wolf D, Ley K (2019) Immunity and inflammation in atherosclerosis. Circ Res 124:315–327
Wolf D, Gerhardt T, Winkels H et al (2020) Pathogenic autoimmunity in atherosclerosis evolves from initially protective apolipoprotein B 100 –reactive CD4 + T-regulatory cells. Circulation 142:1279–1293. https://doi.org/10.1161/CIRCULATIONAHA.119.042863
Wu LC, Zarrin AA (2014) The production and regulation of IgE by the immune system. Nat Rev Immunol 14:247–259
Xiao N, Yin M, Zhang L et al (2009) Tumor necrosis factor-alpha deficiency retards early fatty-streak lesion by influencing the expression of inflammatory factors in apoE-null mice. Mol Genet Metab 96:239–244. https://doi.org/10.1016/j.ymgme.2008.11.166
Yang M, Sun L, Wang S et al (2010) Novel function of B cell-activating factor in the induction of IL-10-producing regulatory B cells. J Immunol 184:3321–3325. https://doi.org/10.4049/jimmunol.0902551
Yu A, Zhu L, Altman NH, Malek TR (2009) A low Interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity 30:204–217. https://doi.org/10.1016/j.immuni.2008.11.014
Zakai NA, Katz R, Jenny NS et al (2007) Inflammation and hemostasis biomarkers and cardiovascular risk in the elderly: the cardiovascular health study. J Thromb Haemost 5:1128–1135. https://doi.org/10.1111/j.1538-7836.2007.02528.x
Zelová H, Hošek J (2013) TNF-α signalling and inflammation: interactions between old acquaintances. Inflamm Res 62:641–651
Zernecke A, Weber C (2014) Chemokines in atherosclerosis: proceedings resumed. Arterioscler Thromb Vasc Biol 34:742–750. https://doi.org/10.1161/ATVBAHA.113.301655
Zhang J, Xie F, Yun H et al (2016) Comparative effects of biologics on cardiovascular risk among older patients with rheumatoid arthritis. Ann Rheum Dis 75:1813–1818. https://doi.org/10.1136/annrheumdis-2015-207870
Zhang X, Li J, Luo S et al (2020) IgE contributes to atherosclerosis and obesity by affecting macrophage polarization, macrophage protein network, and foam cell formation. Arterioscler Thromb Vasc Biol 40:597–610. https://doi.org/10.1161/ATVBAHA.119.313744
Zhao TX, Kostapanos M, Griffiths C et al (2018) Low-dose interleukin-2 in patients with stable ischaemic heart disease and acute coronary syndromes (LILACS): protocol and study rationale for a randomised, double-blind, placebo-controlled, phase I/II clinical trial. BMJ Open 8:e022452. https://doi.org/10.1136/bmjopen-2018-022452
Zhao T, Sriranjan R, Lu Y et al (2020a) Low dose interleukin-2 in patients with stable ischaemic heart disease and acute coronary syndrome (LILACS). Eur Heart J 41:2. https://doi.org/10.1093/ehjci/ehaa946.1735
Zhao TX, Newland SA, Mallat Z (2020b) 2019 ATVB plenary lecture: Interleukin-2 therapy in cardiovascular disease: the potential to regulate innate and adaptive immunity. Arterioscler Thromb Vasc Biol 40:853–864. https://doi.org/10.1161/ATVBAHA.119.312287
Zhao TX, Ur-Rahman MA, Sage AP et al (2021) Rituximab in patients with acute ST-elevation myocardial infarction (RITA-MI): an experimental medicine safety study. Cardiovasc Res. https://doi.org/10.1093/cvr/cvab113
Zhou X, Caligiuri G, Hamsten A et al (2001) LDL immunization induces T-cell–dependent antibody formation and protection against atherosclerosis. Arterioscler Thromb Vasc Biol 21:108–114. https://doi.org/10.1161/01.ATV.21.1.108
Zirlik A, Maier C, Gerdes N et al (2007) CD40 ligand mediates inflammation independently of CD40 by interaction with Mac-1. Circulation 115:1571–1580. https://doi.org/10.1161/CIRCULATIONAHA.106.683201
Zouggari Y, Ait-Oufella H, Bonnin P et al (2013) B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med 19:1273–1280. https://doi.org/10.1038/nm.3284
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Deroissart, J., Porsch, F., Koller, T., Binder, C.J. (2021). Anti-inflammatory and Immunomodulatory Therapies in Atherosclerosis. In: von Eckardstein, A., Binder, C.J. (eds) Prevention and Treatment of Atherosclerosis . Handbook of Experimental Pharmacology, vol 270. Springer, Cham. https://doi.org/10.1007/164_2021_505
Download citation
DOI: https://doi.org/10.1007/164_2021_505
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-86075-2
Online ISBN: 978-3-030-86076-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)