
Abstract
Atherosclerosis is a multifactorial vascular disease that develops in the course of a lifetime. Numerous risk factors for atherosclerosis have been identified, mostly inflicting pro-inflammatory effects. Vessel injury, such as occurring during erosion or rupture of atherosclerotic lesions triggers blood coagulation, in attempt to maintain hemostasis (protect against bleeding). However, thrombo-inflammatory mechanisms may drive blood coagulation such that thrombosis develops, the key process underlying myocardial infarction and ischemic stroke (not due to embolization from the heart). In the blood coagulation system, platelets and coagulation proteins are both essential elements. Hyperreactivity of blood coagulation aggravates atherosclerosis in preclinical models. Pharmacologic inhibition of blood coagulation, either with platelet inhibitors, or better documented with anticoagulants, or both, limits the risk of thrombosis and may potentially reverse atherosclerosis burden, although the latter evidence is still based on animal experimentation.
Patients at risk of atherothrombotic complications should receive a single antiplatelet agent (acetylsalicylic acid, ASA, or clopidogrel); those who survived an atherothrombotic event will be prescribed temporary dual antiplatelet therapy (ASA plus a P2Y12 inhibitor) in case of myocardial infarction (6-12 months), or stroke (<6 weeks), followed by a single antiplatelet agent indefinitely. High risk for thrombosis patients (such as those with peripheral artery disease) benefit from a combination of an anticoagulant and ASA. The price of gained efficacy is always increased risk of (major) bleeding; while tailoring therapy to individual needs may limit the risks to some extent, new generations of agents that target less critical elements of hemostasis and coagulation mechanisms are needed to maintain efficacy while reducing bleeding risks.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Anticoagulants
- Antiplatelet therapy
- Aspirin
- Atherosclerosis
- Atherothrombosis
- Clopidogrel
- Coagulation
- Platelets
- Thrombosis
1 Atherogenesis and the Role of Blood Coagulation Components
Atherosclerosis is a multifactorial vascular disease that develops in the course of a lifetime. Numerous risk factors for atherosclerosis have been identified, mostly inflicting pro-inflammatory effects, hence, the term “chronic inflammatory disease” (Ross 1999). The blood coagulation system has a primary role in maintaining hemostasis, preventing fatal bleeding (Spronk et al. 2003a). A second function relates to wound healing in a broader sense. Blood coagulation becomes activated in response to any vascular injury causing contact between the vascular matrix and/or cells with blood. This clotting process likely is meant to seal the wound surface and help to repair the underlying wound bed. Platelets, coagulation, and fibrinolysis factors, as well as fibrin are key elements in this wound healing process, and the administration of some of these components (platelets, fibrinogen) is also studied in clinical trials on wound healing of the skin (Hoffman 2018; Opneja et al. 2019). In the vasculature, pro-atherogenic changes of the vascular endothelium, involving endothelial cell (EC) activation, dysfunction, or damage (exposing subendothelial matrix), trigger the coagulation system in an attempt to vascular wound healing.
1.1 Blood Coagulation: Impact on Vascular Endothelial Cells
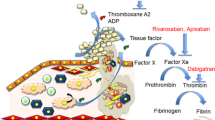
The blood coagulation system consists of different pathways that together provide the hemostatic plug or, in pathological situations, the thrombus. Platelets and coagulation proteins act in concert to build the fibrin-platelet clot on top of the damaged vessel wall, while the fibrinolytic system helps to limit clot formation and acts to restore blood flow upon (partial) clot lysis.
In atherogenesis, early inflammatory endothelial perturbation (Gimbrone Jr. and Garcia-Cardena 2016) may trigger activation of platelets that together with leukocyte populations provide a first thrombo-inflammatory response to injury (Fig. 1) (Messner and Bernhard 2014). Activation of platelets (but also other cells) yields extracellular vesicles (EVs) that promote thrombo-inflammatory reactions, amplifying fibrin formation (Badimon et al. 2017). Extracellular vesicles are particles that are naturally released from nearly all kinds of cells; carry a cargo of proteins, RNA, and lipids from the parent cell; and are thus considered to be key components in cell-cell communication. Repeated and/or ongoing inflammatory pressure (endogenous factors like oxidized lipoproteins, glycated end products, homocysteine, etc. and exogenous factors like smoking and other sources of particulate matter) challenges the vascular endothelium to become activated and permeable for inflammatory cells, lipids, and other toxic components (Mozaffarian et al. 2008).

Interactions of platelets, leucocytes, coagulation factors, and vascular endothelial cells in atherogenesis and atherothrombosis. Under physiologic conditions, thrombin (IIa) supports endothelial cell integrity through endothelial cell-mediated APC and TAFIa generation. PARs are expressed at the endothelial cell (EC) surface to mediate cytoprotective effects of APC through activation of PAR1. APC is activated by the thrombin-thrombomodulin (TM) complex, which also converts TAFI into activated TAFI (TAFIa), which has anti-inflammatory properties. Under pro-inflammatory conditions, several pro-inflammatory cytokines alter PAR expression patterns and downregulate the protective cellular receptors like TM and EPCR. The result is a shift to prothrombotic and offensive functions of PAR-EC interactions. Thrombin-induced PAR1 activation in combination with endogenous factors like smoking and oxidized lipoproteins challenges the vascular endothelium to become permeable for inflammatory cells. Damage of the endothelial layer results in exposure of highly reactive subendothelial proteins, such as collagen. Under arterial shear stress, vWF bound to collagen enables platelet adhesion via the glycoprotein (GP)Ib-IX-V complex. Activation of platelets yields extracellular vesicles that promote thrombo-inflammatory reactions, including attraction of leukocytes. Activated platelets deposit chemokines, such as CXCL4 and CCL5, on the inflamed endothelium, thereby recruiting leucocytes, which bind to platelets adhering to the (sub)endothelium. Platelets and leukocytes secrete pro-inflammatory and pro-angiogenic factors that further support atherogenesis. Traces of thrombin activate platelets through PAR1 and PAR4. These activated platelets provide a procoagulant phosphatidylserine (PS)-rich surface on which coagulation factors can gather, ultimately leading to the conversion of prothrombin (II) to thrombin (IIa). Thrombin-induced PAR activation induces inflammatory damage to the vascular endothelium. EC endothelial cells, APC activated protein C, PAR protease-activated receptor, EPCR endothelial cell protein C receptor, TM thrombomodulin, TAFI thrombin-activatable fibrinolysis inhibitor, GP1b-IX-V glycoprotein Ib-IX-V, vWF von Willebrand factor, CXCL4 chemokine ligand 4, CCL5 chemokine ligand 5, PS phosphatidylserine, EV extracellular vesicle
Disruption of the endothelial cell barrier is induced or aggravated by pro-inflammatory cytokines, metalloproteases, and cellular enzymes like elastase and trypsin as well as by the influence of coagulation serine proteases like factor Xa and thrombin, acting on protease-activated receptors (PARs) (Coughlin 2005; Posma et al. 2016; Ruf 2018). Physiologically, PARs are expressed at the endothelial cell surface to mediate endothelial protective effects of activated protein C (APC), through activation of PAR1 (Mosnier et al. 2012). Besides activation of protein C, the thrombin-thrombomodulin (TM) complex also converts plasma pro-carboxypeptidase B2 (proCPB2 or thrombin-activatable fibrinolysis inhibitor, TAFI). Activated TAFI (TAFIa) inhibits plasmin formation and hence stabilizes fibrin clots by inhibiting plasmin generation and fibrinolysis (Fujiwara et al. 2012; Morser et al. 2010; Myles et al. 2003; Shao et al. 2015). TAFIa also has anti-inflammatory properties including the inactivation of pro-inflammatory mediators like bradykinin, anaphylatoxins C3a and C5a, and thrombin-cleaved OPN (Myles et al. 2003; Naito et al. 2013; Nishimura et al. 2007; Relja et al. 2013). This way, under physiologic conditions thrombin supports endothelial cell integrity through endothelial cell-mediated APC and TAFIa generation.
Under inflammatory pressure, pro-inflammatory cytokines alter PAR expression patterns and downregulate protective cellular receptors like TM and endothelial cell protein C receptor (EPCR) (Esmon 1995, 2014). The result is a shift from anticoagulant (and protective) to prothrombotic (and offensive) functions of serine protease-PAR-EC interactions (Fig. 1). In the case of PAR-1, the shift from protective (APC induced) to inflammatory (thrombin induced) is referred to as “biased” signaling (Griffin et al. 2015).
1.2 Platelets and Extracellular Vesicles
Mouse studies provide mechanistic insight in the pro-atherogenic functions of blood components including platelets, EVs, and coagulation proteins. Platelets and platelet-derived EVs are important messengers of various mediators including microRNAs that modify inflammation and atherogenesis. A range of studies provided evidence for involvement of EVs on various key pathways in atherogenesis, including inflammation, calcification, and cell trafficking (Badimon et al. 2017; Paone et al. 2019; Alique et al. 2018; Bouchareychas and Raffai 2018; Miller et al. 2016). Ongoing research is addressing ways to utilize some of these functions of EVs including transportation of microRNA for potential therapeutic purposes in atherosclerosis (Yin et al. 2015). Majority of EVs are platelet-/megakaryocyte-derived and contain cytokines, RNA species, coagulation factors, etc. EVs can interact with, e.g., leukocytes and regulate activity (Vasina et al. 2013).
Platelets roll and adhere to activated endothelium (Schulz and Massberg 2012). Platelets promote the differentiation and activation of leukocytes (Lievens and von Hundelshausen 2011). Activated platelets deposit chemokines, mainly chemokine ligand 4 (CXCL4) and chemokine ligand 5 (CCL5), on the inflamed endothelium, thereby recruiting leukocytes and exacerbating atherosclerosis (von Hundelshausen et al. 2005; Huo et al. 2003). Also, secreted chemokines attract leukocytes, which bind to platelets adhering to the (sub) endothelium. Platelets and leukocytes secrete pro-inflammatory and pro-angiogenic factors that further support atherogenesis (Semple et al. 2011). Multiple (receptor) interactions between platelets-leukocytes and platelets-vessel wall have been observed in inflammatory conditions such as atherosclerosis (Koupenova et al. 2018; Koenen 2016). Platelet-neutrophil interactions and the release of neutrophil activation products including neutrophil extracellular traps (NETs) may also trigger coagulation.
Cross talk between platelets and coagulation is another important mechanism that operates at different levels. Traces of thrombin activate platelets through PAR1, with PAR4 activation as a delayed, secondary mechanism. Activated platelets provide a phosphatidylserine (PS)-rich surface on which coagulation reactions take place (tenase and prothrombinase complex formation). Upon induction with strong agonists such as a combination of thrombin and collagen, PS exposure allows binding of Gla domain-containing coagulation factors which enhances the activity of coagulation factor complexes. This procoagulant platelet response is supported by the secretion of coagulation factors (prothrombin, Factor V, Factor 8 transcript, Factor XIII, and fibrinogen) and modified by anticoagulation factors (e.g., antithrombin, tissue factor pathway inhibitor (TFPI), protein S). Platelet-derived factor V may be an important determinant of local prothrombinase formation, driving atherogenesis (Ren et al. 2017). Additional factors that are secreted by platelets are tissue factor (TF) (the origin still somewhat controversial) and protein disulfide-isomerase (PDI) that acts to de-encrypt TF in an active conformation. Polyphospate (PolyP) is secreted that may activate factor XII. Platelet-bound factor XI drives coagulation in an angiotensin-dependent manner (Kossmann et al. 2017). Another mechanism that bridges platelets and coagulation is by binding coagulation factors via the glycoprotein (Gp) complexes GPIb-V-IX, GPIIb/IIIa, and GPVI (Swieringa et al. 2018). Finally, platelets alter fibrin structure (via regulating local thrombin concentration) and mediate clot contraction. This process involves different platelet populations, including procoagulant platelets that primarily stimulate fibrin formation in the periphery of the clot, associated with clot contraction (Nechipurenko et al. 2019). In the course of this process, thrombin-activated platelets become fragmented, a process that limits further dissemination of activated platelets (Kim et al. 2019).
1.3 Antiplatelet Agents and Atherosclerosis
From the described thrombo-inflammatory mechanisms, one may infer that platelet inhibition with antiplatelet medication could potentially attenuate atherogenesis and atherosclerosis. In addition to the inhibition of thromboxane A2 (TXA2) production, aspirin (acetylsalicylic acid (ASA) being the active component) increases platelet nitric oxide (NO) synthesis, protects NO from its inactivation, and improves endothelial dysfunction (Russo et al. 2017). Aspirin also has anti-inflammatory effects, but whether the doses used to prevent platelet aggregation are sufficient to produce meaningful anti-inflammatory effects in humans remains uncertain. In mice, low-dose aspirin improves vascular inflammation and stabilizes plaques (Cyrus et al. 2002) or limits plaque severity, maybe related to reduced fractalkine levels (Liu et al. 2010). In humans, aspirin has been shown to reduce the levels of pro-inflammatory cytokines including interleukin (Il)-6 and monocyte colony-stimulating factor (Ikonomidis et al. 1999) and to protect the endothelium against inflammatory challenge (Kharbanda et al. 2002). In spite of these and other possible protective effects, the net effect on human atherosclerosis remains uncertain (Tousoulis et al. 2016). With regard to thrombosis, literature points to effects of aspirin on fibrin clot formation and stability that may contribute to its antithrombotic action (Gurbel et al. 2019).
Evidence for a role of the P2Y12 receptor in atherogenesis comes from studies in mice with a double apoE and P2Y12 deletion genotype that show reduced lesion area, increased fibrous content at the plaque site, and decreased monocyte/macrophage infiltration of the lesions in the double knockout animals as compared to control apoE−/− mice (Li et al. 2012; West et al. 2014). The P2Y12 inhibitor clopidogrel reduced levels of p-selectin, e-selectin, monocyte chemoattractant protein-1, and platelet-derived growth factor β, reduced macrophage and T-cell infiltration in atherosclerotic lesions, and delayed the development and progression of de novo atherosclerosis (Heim et al. 2016).
In preclinical models clopidogrel attenuated atherosclerosis (Heim et al. 2016; Afek et al. 2009; Takeda et al. 2012), although these protective effects were not evident in other studies (Schulz and Massberg 2012; West et al. 2014). Similarly, ticagrelor reduced the initiation of atherosclerosis in apoE−/− mice (Schirmer et al. 2012) although this effect was not observed by West et al. (West et al. 2014). Discrepancies between the positive and negative studies may be related to drug dose, timing, and duration of treatment (Nylander and Schulz 2016). Furthermore, stabilization and reduced necrotic core were seen in mice with established plaques at 20 weeks of age, for ticagrelor (Buchheiser et al. 2011).
1.4 Coagulation Proteases
Coagulation proteins are for the large part synthesized in the liver and secreted in blood after posttranslational modification. Many of the proteins are zymogens (or proenzymes) that require limited proteolysis to become fully active. It requires one or more triggers like tissue factor (TF) and amplifiers like thrombin, to activate a cascade response yielding fibrin formation at the sites of injury. The liver also makes a series of anticoagulant proteins including antithrombin and protein C that act to dampen the coagulation cascade physiologically. A fairly large number of coagulation proteins are also expressed outside the liver; this can be constitutive (like protein S that is for 50% synthesized in vascular EC; tissue factor pathway inhibitor (TFPI) that is partially liver, partially megakaryocyte derived; factor VIII from liver sinusoidal endothelial cells; TF expressed in fibroblasts of the arterial adventitia) or inducible under conditions of inflammation: an example is production of TF in blood leukocytes (monocytes/macrophages, neutrophils) and possibly platelets, vascular smooth muscle cells (VSMC), and other cell types (Grover and Mackman 2018). Other proteins like factor VII (Wilcox et al. 2003) and factor X (Sanada et al. 2017) can be expressed by VSMC and fibroblasts. Proteins like factor XI, XII, and prothrombin have been demonstrated in extravascular localizations including atherosclerotic vessels, but whether these are deposited or locally synthesized remains unknown (Borissoff et al. 2010; Wilcox 1994; Soardi et al. 1961). Of interest, coagulation enzymes including thrombin and aPC participate in the control of hematopoiesis in the bone marrow (Nguyen et al. 2018), which may be relevant for the response to stress situations including inflammation.
Within atherosclerotic lesions, coagulation proteins like TF and factors VII and X contribute to form catalytic complexes driving thrombin and fibrin formation (Borissoff et al. 2010). The local formation of fibrin and its split products are modifiers of angiogenesis and cell trafficking within the plaques (Fay 2004; Binder et al. 2017; Badimon and Vilahur 2014; Spronk et al. 2018). The important role of plasminogen activator (receptor)-plasmin and plasminogen activator 1 (PAI-1) system in regulating controlled proteolysis in atherosclerosis, with impact on inflammation (Foley 2017), falls beyond the scope of this chapter.
In general, hypercoagulability and/or thrombophilia has a modest but detectable effect on atherosclerosis, at least coronary artery disease (CAD), peripheral artery disease (PAD), and other manifestations of atherosclerosis (Borissoff et al. 2012; Kleinegris et al. 2013; Lowe and Rumley 2014). This association is not unequivocal, and the lack of protective effect of specific coagulation deficiencies such as in hemophilia, which is not associated with reduced burden of atherosclerosis, argues against very strong influence of coagulation activity in human atherogenesis (Biere-Rafi et al. 2012; Kamphuisen and ten Cate 2014). Nevertheless, evidence from preclinical models is quite striking (Borissoff et al. 2011). Coagulation proteases tend to aggravate atherogenesis toward atherosclerosis in mouse models of atherosclerosis, mostly apoE−/− mice, under pressure of a Western-type diet. In these mice, any hypercoagulable effect introduced by backcrossing apoE−/− mice on a specific mouse with increased procoagulant tendency such as factor V Leiden, or TMPro/pro mutation, results in worsening atherogenesis. In contrast, mice with a less procoagulant phenotype like FXI−/−, FVII+/−, or some of the FVIII−/− traits tend to be protected against atherosclerosis progression (Borissoff et al. 2011; Shnerb Ganor et al. 2016; Mackman 2016).
Application of direct oral anticoagulants that inhibit thrombin (dabigatran) or factor Xa (rivaroxaban) slows down atherosclerosis in apoE−/− mice. At least five of such studies document protection against progression of atherosclerosis, including improved plaque stability, while on dabigatran (Lee et al. 2012; Kadoglou et al. 2012; Borissoff et al. 2013; Pingel et al. 2014; Preusch et al. 2015). One protective mechanism may involve the dabigatran-mediated attenuation of pro-inflammatory M1 macrophages in the vessel wall, observed in Lldr−/− mice (Feldmann et al. 2019).
Detrimental effects of dabigatran exposure have also been published; in a diabetic rat model, dabigatran exposure caused increased platelet reactivity, increased coronary lipid deposition, as well as increased PAR4 expression in vessels (Scridon et al. 2019).
Rivaroxaban, the direct factor Xa inhibitor, also attenuates atherosclerosis in apoE−/− mice (Zhou et al. 2011; Hara et al. 2015) or even reverses existing atherosclerosis in apoE−/− mice (Posthuma et al. 2019). Overall, most studies suggest that applying direct oral anticoagulants (DOAC) in atherogenic mice slows down but also stabilizes atherosclerotic lesions and alter the plaque phenotype toward more (Borissoff et al. 2013) or sometimes less stability features (Seehaus et al. 2009), the latter depending on age and sex and probably additional factors like diet. The observed regression on rivaroxaban similarly showed diminished instability markers, but the actual mechanisms explaining diminished plaque volume on anticoagulation remain to be determined (Posthuma et al. 2019).
These proof-of-concept studies support important roles for coagulation proteases FXa and thrombin in driving atherogenesis and altering the phenotype in different directions. Inhibiting factor Xa to actually reverse atherosclerosis in mice raises many new questions regarding the underlying mechanisms of factor Xa-mediated cell signaling and its impact on plaque progression/regression. The protection is thought to involve reducing the impact of FXa and thrombin on PAR1 and 2 activation, respectively. The importance of these PARs in regulating atherosclerosis is emerging; recently, protection against atherosclerosis in PAR2−/− × apoE−/− mice was shown, associated with reduced activity of nuclear factor-κB (NFκB)-regulated inflammation (Hara et al. 2018). However, the importance of coagulation proteases as compared to other possible ligands remains to be demonstrated.
2 From Atherosclerosis to Atherothrombosis
During atherogenesis there are different possible stages of atherothrombosis development that could be characterized by combinations of acute or acute on chronic thrombus formation. The occurrence of atherothrombosis is not a single event in time; in series of autopsy studies, it was convincingly demonstrated that coronary thrombi of all stages of development can be found in patients that suddenly died (Kramer et al. 2010). These studies demonstrate that atherothrombosis is part of an ongoing process with temporary, sometimes partial occlusions, subsequent remodeling of thrombus and vessel, gradually changing the vessel wall morphology, and affecting the lumen (Mastenbroek et al. 2015).
Atherogenesis and atherosclerosis may go through several stages of disease. A first and early stage relates to “perturbation” of the vascular endothelium (Nawroth et al. 1984; de Groot et al. 1987), referring to a state of endothelial cell activation associated with a disturbance of the anticoagulant/procoagulant balance, in a pro-inflammatory and thrombotic direction. It could be imagined that this perturbation may in fact be a first trigger in specific types of arterial thrombosis that may form in absence of visible atherosclerosis with sensitive imaging of coronary arteries. A second, more frequent scenario is the formation of a thrombus based on a damaged atherosclerotic plaque. Since long, two fundamentally different scenarios, plaque erosion versus plaque rupture, are recognized (Fuster et al. 1992a, b; Arbab-Zadeh et al. 2012).
While plaque rupture was previously recognized as the dominant pathophysiologic mechanism, recent studies suggest a gradual shift toward less plaque rupture and persistently frequent plaque erosion. One hypothesis for this change in time is declining impact of smoking and increased attention for and management of cardiovascular risk factors (e.g., statins, smoking cessation) that might translate into vascular protective effects and a more stable plaque phenotype (Quillard et al. 2017; Pasterkamp et al. 2017). Currently, about 30% of ST-elevation myocardial infarction (STEMI) is thought to result from eroded plaque lesions (Libby 2013).
Although known for a long time and recognized also as a predilection site for atherothrombosis in the aforementioned autopsy studies, erosion probably triggers fundamentally different mechanisms than rupture of a vulnerable cap (Quillard et al. 2017). Plasma from patients with STEMI showed higher levels of epidermal growth factor and thrombospondin-1 in patients with intact fibrotic cap versus those with ruptured cap lesions, while interferon-inducible T-cell alpha chemoattractant (I-TAC) was lower in coronary blood from intact cap lesion subjects (Chandran et al. 2017). In thrombectomy specimens from these patients, I-TAC mRNA expression levels were markedly increased in patients with eroded lesions. Eroded lesions are characterized by fewer inflammatory cells, abundant extracellular matrix, and the presence of neutrophil extracellular traps (NETs). Eroded plaques contain more myeloperoxidase (MPO)-positive inflammatory cells; MPO is a marker of NETs. Patients with plaque erosion have more MPO in peripheral blood than those with plaque fissure (Ferrante et al. 2010). In addition, components from extracellular matrix including hyaluronan that interact with prohemostatic proteins like fibrinogen, fibrin, and fibronectin are important erosion determinants (Pedicino et al. 2018; Wight 2018). Inflammation and NETs may be complimentary mechanisms to trigger blood coagulation toward thrombus formation. Ruptured lesions typically express inflammatory cells, matrix containing oxidized lipids, and subendothelial proteins from the matrix and on inflammatory cells, including procoagulant tissue factor, factors VII and X, as well as platelets localizing and activating proteins including von Willebrand factor (vWF), collagen, and thrombospondin (Quillard et al. 2017; Pasterkamp et al. 2017). If, indeed, there are fundamental differences in the phenotypes of eroded versus ruptured lesions, it may be anticipated that thrombus formation also follows different pathways. On the other hand, it has been suggested that the main difference between eroded and ruptured plaque is the absence of direct contact between blood components and the necrotic core (Badimon and Vilahur 2014). Better insight into the mechanisms involved in atherothrombosis related to erosion or rupture of plaques is important in order to tailor antithrombotic protection in a more mechanistically founded direction. As will be discussed below, current antithrombotic management does not yet consider such differences.
3 Antithrombotic Therapy: Clinical Principles and Applications
3.1 Single Antiplatelet Agents: Mode of Action and Side Effects
Aspirin is the common name for acetylsalicylic acid, a compound that acts by inhibiting prostaglandin synthesis in different cells; the relevant antithrombotic effect is thought to be mostly based on inhibition of the cyclooxygenase-1 production of thromboxane A2 in platelets (Gresele 2002). It is prescribed at doses between ±75 and 325 mg od, mostly related to regional differences (higher doses more common in the USA than Europe). Its intake results in irreversible inhibition of platelet activation and aggregation. To achieve continued effect, daily intake of aspirin is needed to suppress newly formed platelets, about 10% per day.
Clopidogrel is a prodrug that needs metabolization in the liver (Coukell and Markham 1997). It is ingested as a single dose of 75 mg/day, and the active metabolite shows a large interindividual variation in blood. This variability is partly explained by genetic polymorphisms encoding cytochrome P450 (CYP) 2C19, the hepatic enzyme involved in biotransformation of the prodrug clopidogrel to its active metabolite (Shuldiner et al. 2009). The active metabolite interferes with the binding of ADP to the P2Y12 receptor. This pharmacodynamic variation translates into variation in clopidogrel effectiveness after percutaneous coronary intervention (PCI) (Mega et al. 2010; Sibbing et al. 2009). The interindividual variation in response to clopidogrel, commonly known as clopidogrel “resistance,” was first identified in patients with coronary disease, occurring up to 25% of cases in platelet function testing (depending on the test used). Given the association between clopidogrel high on-treatment platelet reactivity (HTPR) and increased incidences of major adverse cardiovascular events (MACE), several studies addressed the question whether dose adjustment based on platelet function testing would correct this problem. Unfortunately, none of the studies demonstrated that test-based adjusted clopidogrel dosing could improve clinical efficacy of such interventions (Price et al. 2011; Collet et al. 2012). In part, the variation in clopidogrel activity is “corrected” for by the development of more potent P2Y12 inhibitors with a more predictable pharmacodynamic profile including prasugrel, ticagrelor, and cangrelor.
The principal side effect of aspirin is bleeding, and the regular use of aspirin increases the risk of particularly gastrointestinal bleeding twofold (Garcia Rodriguez et al. 2016). Risk factors for bleeding should therefore be taken into account, including (recent) GI ulcer (H. pylori infection may be additional factor), old age, and use of interacting medication including other antiplatelet therapy (APT), NSAIDs, COX-2 selective inhibitors, oral anticoagulants, and corticosteroids. In individual decision-making, balancing the pros (risk of atherothrombotic complications) and the cons (mostly bleeding) needs to be done, and certain decision support tools like the app “Aspirin Guide” can be helpful in this regard. In general, the risk of GI bleeding with aspirin can be effectively reduced with proton pump inhibitors (PPI), better than with histamine-2 receptor antagonists (Mo et al. 2015; Szabo et al. 2017). Recently, a fixed-dose combination of aspirin and omeprazole (Yosprala) was approved by the FDA, hoping that the simultaneous intake of these agents would improve adherence by reducing gastric side effects (Veltri 2018). Although clopidogrel does not interfere with prostaglandins in gastric mucosal tissue, its use is also associated with an increased risk of bleeding, of which GI bleeds are the most common type. The standard use of PPI in patients on clopidogrel has been disputed; in fact there is evidence that its concomitant use is associated with an increased risk of MACE (Bundhun et al. 2017). Compared to clopidogrel, the newer and more potent P2Y12 inhibitors prasugrel and ticagrelor are associated with an increase in the risk of major bleeding. These agents are generally not prescribed as single APT, although ticagrelor may be used as single agent in selected PAD patients (Hiatt et al. 2017).
3.2 Primary Prevention in the Population; Selecting the Right Subject?
In patients with atherosclerotic vascular disease, antithrombotic medication has been applied since the late 1950s of the past century. Traditionally, platelets were regarded key players in atherothrombosis; hence much focus has been put on antiplatelet agents, initially mostly aspirin. Given the efficacy/safety profile, primary prevention with aspirin has been extensively studied. Recent studies and meta-analyses of decades of large trials refute a major role for aspirin in primary prevention. Exceptions may be subjects with diabetes in whom aspirin showed to reduce the incidence of major vascular events including myocardial infarction, at a price of increased major bleeding risk (Group ASC et al. 2018). Otherwise, primary prevention with aspirin in apparently healthy subjects may require additional risk factors like coronary calcification to yield sufficient net clinical benefit. A recent discussion paper on the pros and cons of aspirin for primary prevention in elderly subjects shows the jury is still out (McNeil et al. 2018a, b, c; Fernandes et al. 2019).
3.3 Primary Prevention in Subjects with Atherosclerosis
Primary prevention (of MACE) with aspirin or other APT is warranted in all subjects with symptomatic atherosclerotic disease, including angina, or symptomatic PAD. Here, the risk/benefit ratio is clearly in favor of indefinite APT. In patients with any evidence of coronary artery disease, indefinite single APT with aspirin is recommended. This policy is adjusted in case of emerging interventions like PCI; see further.
Remarkably, aspirin was not better than placebo in patients with asymptomatic lower extremity artery disease (LEAD), in spite of the fact that their mortality is comparable to patients with symptomatic disease (Fowkes et al. 2010). In symptomatic PAD, there is a certain preference for the P2Y12 inhibitor clopidogrel over aspirin, as the PAD subgroup in the CAPRIE trial that compared aspirin with clopidogrel in subjects at high risk for cardiovascular complications showed better antithrombotic efficacy for clopidogrel at comparable bleeding risk (Table 1) (Committee 1996). However, both aspirin and clopidogrel are still used for this indication. Stronger acting APT like ticagrelor may be used as alternative in patients with PAD in case of failure, allergy, or “resistance” to aspirin or clopidogrel, based on the EUCLID trial showing non-inferiority of ticagrelor versus clopidogrel (Hiatt et al. 2017). Table 1 presents an overview of different therapeutic strategies in symptomatic patients with stable CAD or PAD, referring to the large trials supporting these strategies.
The principle of combining APT was established in the CURE trial, comparing the efficacy and safety of aspirin plus clopidogrel with aspirin alone in the secondary prevention of MACE in patients with coronary disease (Mehta et al. 2001). Since then, dual antiplatelet therapy (DAPT) has become a cornerstone treatment in secondary prevention following PCI (see further). However, the concept also triggered studies as to the potential benefits DAPT would offer in patients with high-risk cardiovascular disease. Large trials like CHARISMA (Bhatt et al. 2006) tested this concept, the outcome of which was however negative in failing to show improved efficacy while increasing the bleeding risk. Consequently, DAPT is not recommended in any patient for primary prevention of MACE, unless there are subject-specific reasons for this more potent combination.
In PAD, in general, combined antiplatelet therapy does not add benefit to the patient and increases bleeding risk; for this reason, it is only applied for short-term use, e.g., after endovascular interventions (Hess et al. 2017). The use of oral anticoagulants (mostly vitamin K antagonists) in therapeutic intensity is not indicated in patients with PAD, except for those that underwent venous bypass grafting (Dutch Bypass Oral Anticoagulants or Aspirin (BOA) Study Group 2000). The most recent regimen studied in patients with high-risk vascular disease, including PAD, is the combination of an anticoagulant rivaroxaban 2.5 mg bd plus low-dose aspirin, which reduced cardiovascular mortality as well as major acute limb events in patients with PAD (patients with LEAD or carotid artery disease) (Anand et al. 2018). From a mechanistic perspective, targeting platelets and the plasmatic coagulation system seems rational, given the postulated pathophysiologic mechanisms discussed above. Moreover, targeting FXa, herewith also inhibiting the formation of thrombin, reduces the potential interactions between these proteases and cellular PARs. Thus, vascular protective effects may be an additional consequence of a strategy that includes an anticoagulant (although the clinical evidence is still weak).
3.4 Secondary Prevention of Atherothrombosis in Patients with Arterial Vascular Disease
Secondary prevention with APT, following myocardial infarction or ischemic stroke (not due to cardiac embolism, thus including a spectrum of non-embolic strokes), is straightforward and based on class 1A evidence.
Combined APT (dual (DAPT) antiplatelet therapy) is indicated in all settings of acute coronary syndrome, with or without PCI with stent placement (DAPT for 6–12 months, longer in selected cases) (Roffi et al. 2016; Authors/Task Force et al. 2014; Amsterdam et al. 2014). In acute ischemic stroke, DAPT is only indicated for a very limited duration, up to 6 weeks, due to the observed increased bleeding risk (Wang et al. 2013; Johnston et al. 2018). Following these time windows, single APT will be continued indefinitely, comprising of aspirin in most patients with CAD and aspirin or, more commonly, clopidogrel in patients after atherothrombotic stroke (Hackam and Spence 2019).
In most combined APT regimens, aspirin remains an element; in patients with CAD, DAPT comprises aspirin plus a P2Y12 inhibitor, prasugrel or ticagrelor (unless contraindicated clopidogrel, a weaker P2Y12 inhibitor, is used in combination with aspirin). Although the thienopyridine prasugrel is a prodrug like clopidogrel, it only requires a single oxidation step to form its active metabolite, and it seems to be not affected by genetic variations in CYP enzymes. Compared to clopidogrel its use is associated with an increase in the risk of major bleeding, especially among those with age ≥75 years or body weight <60 kg, and is contraindicated in patients with prior stroke (Wiviott et al. 2007). Ticagrelor, an oral, direct acting, reversible P2Y12 receptor antagonist, provides faster and greater platelet inhibition with less patient-to-patient variation (Wallentin et al. 2009).
Despite intensification of APT in recent years, an approximately 10% risk for recurrent ischemic events at 1 year after coronary events still remains (Wiviott et al. 2007; Wallentin et al. 2009). With blockade of the TXA2 pathway and the P2Y12 receptor, platelets can still be activated by thrombin, via the PAR1 and PAR4 receptor on their surface (Olie et al. 2019). Vorapaxar is an orally administered, competitive PAR1 antagonist that blocks thrombin-mediated platelet activation via PAR1, without inhibiting other modes of thrombin activity, such as fibrin formation, protein C activation, and PAR4 activation. On top of standard antiplatelet therapy (consisting of aspirin in almost all cases and a thienopyridine or dipyridamole in a significant proportion), addition of vorapaxar led to significant reduction in rates of ischemic cardiovascular events in patients with stable CAD or PAD, but at the price of increased major bleeding, limiting its use in clinical practice (Morrow et al. 2012). Non-platelet-mediated effects of PAR inhibition on the vascular endothelium have been speculated on, since PAR1 is also present on endothelial cells and VSMCs, where it mediates mitogenic effects (Posma et al. 2016). Thus, PAR1 inhibition might be effective in reducing vascular remodeling and consecutive progression of atherosclerosis.
In patients with atherosclerotic disease, oral anticoagulants were not routinely applied until recently. One reason for not using anticoagulants was the assumption that atherothrombosis primarily is a platelet-dependent phenomenon, given the efficacy data of all APT trials. A second reason to refrain from anticoagulants was the concern of cholesterol embolization that could follow anticoagulant-associated instability and plaque rupture; however, the evidence for a causal effect of anticoagulants is poor.
Early trials with vitamin K antagonists (van Bergen et al. 1994; Smith et al. 1990) in CAD patients had provided proof of principle that inhibition of coagulation may be of additional benefit in atherothrombotic disease, although in daily practice, their use was practically abandoned due to markedly increased bleeding and more effective protection by DAPT. Nonetheless, after the introduction of direct oral anticoagulants (DOACs), the role of these anticoagulants in secondary prevention of ischemic events was re-evaluated in several trials (Olie et al. 2018). Addition of low-dose rivaroxaban (2.5 mg twice daily) on top of APT reduced major adverse cardiac events both in patients with acute coronary syndrome (in the ATLAS-ACS-2 TIMI 51 trial (Mega et al. 2012)) and in patients with stable CAD or PAD (in the COMPASS trial (Eikelboom et al. 2017)). In COMPASS also cardiovascular mortality was reduced in patients receiving the combination of vascular doses of rivaroxaban (2.5 mg bd) and aspirin as compared to either agent alone, which means a breakthrough in the efficacy of antithrombotic management (Coppens et al. 2019).
4 The Effects of Antithrombotic Therapy on the Vessel Wall and Atherogenesis: Clinical Relevance?
Platelets are pivotal in atherogenesis, but the impact of antiplatelet agents beyond inhibition of platelet activation and aggregation (including the procoagulant role platelets play in catalyzing phospholipid-dependent coagulation reactions) remains controversial. As discussed, low-dose aspirin may have local anti-inflammatory effects in the vessel wall, but evidence for diminished atherosclerosis related to aspirin intake is not present.
Human studies revealed several anti-inflammatory effects and reduction in platelet CD40 ligand and CD62, respectively, associated with clopidogrel (less evident in those with clopidogrel “resistance”) and prasugrel, both thienopyridines (summarized in (Nylander and Schulz 2016)). Ticagrelor had anti-inflammatory effects including reduced formation of platelet-neutrophil aggregates in inflammation models and more profound reduction in Il-6 in a human sepsis model as compared to clopidogrel, suggesting that stronger inhibition of the P2Y12 receptor (ticagrelor) may provide greater anti-inflammatory effect (Nylander and Schulz 2016). Off-target effects of ticagrelor on endothelial function and vascular biomarkers have been speculated on, but a recent study comparing ticagrelor to prasugrel and clopidogrel found no evidence for any non-platelet-mediated effects in post-acute coronary syndrome patients (Ariotti et al. 2018).
The previous generation of oral anticoagulants, the vitamin K antagonists (VKA), has defined effects on the vasculature due to inhibition of carboxylation of specific vitamin K-dependent proteins like matrix Gla protein, a protein involved in the inhibition of both medial and intimal calcification. Ample mouse studies support these effects, where VKA rapidly induces calcification that can be partially prevented by additional vitamin K administration (Spronk et al. 2003b). Whether the VKA achieved inhibition of FXa and thrombin generation also in part affects atherosclerosis remains difficult to dissect, because of the overwhelming effect of calcification. Whether VKA affects plaque stability in humans is still uncertain. Observational studies suggest more plaque instability in patients on VKA due to intraplaque hemorrhage particularly upon prolonged exposure and higher intensity of anticoagulation (Mujaj et al. 2018; Li et al. 2014). However, in the Rotterdam study, the effects were comparable for anticoagulants (VKA) and aspirin, so the specificity and causal contribution of anticoagulation versus antiplatelet effects remain unproven. More direct evidence for effects of inhibiting coagulation protease needs to come from studies with DOACs. Two studies that either randomized patients to rivaroxaban or warfarin (Lee et al. 2018) or performed propensity score matching yielding three populations that used DOAC, warfarin, or no anticoagulants (Plank et al. 2018) were recently published. Both studies show diminished calcification and less instability features in patients on DOAC versus VKA. These somewhat preliminary data seem to point to a possibly favorable impact of DOAC (hence, FXa/thrombin inhibition) on atherosclerosis and the vessel wall.
5 Novel Antiplatelet and Anticoagulant Targets
In spite of the available array of antithrombotic agents, there is still a need for further improvement. One of the critical downsides of all antithrombotic agents is a risk of bleeding, linked to the potency of the drug. Thus, more effective antiplatelet agents like prasugrel or ticagrelor will generally inflict more bleeding risk than the less potent P2Y12 inhibitor clopidogrel. Combinations of APT like in DAPT or even triple therapy will also show increased bleeding risk as compared to single agents.
Although the current class of DOAC has a better safety profile with regard to intracranial bleeding as compared to VKA, there is still a substantial risk of other clinically relevant bleeding complications, including gastrointestinal bleeds. Combined anticoagulant and APT therapy also has increased bleeding potential, and even the relatively low doses of rivaroxaban in the COMPASS regimen, combined with aspirin, increase the risk of major bleeding (Eikelboom et al. 2017).
With existing antiplatelet drugs, a therapeutic ceiling seems to be reached, whereby increased potency is offset by elevated bleeding risk. Ongoing research on developing new antiplatelet drugs therefore focuses on identifying targets that inhibit thrombosis while maintaining hemostasis. Developing thrombi at sites of endothelial injury are now known to be composed of two distinct regions: the hemostatic plug (composed of highly activated platelets and rich in fibrin) and the propagating platelet thrombus (composed of platelets in a low activation state) (Stalker et al. 2013). The latter region of the propagating thrombus is regulated by phosphatidylinositol 3-kinase-β (PI3Kβ), glycoprotein (GP)IIb/IIIa outside-in signaling, and activation of protein disulfide-isomerase (PDI). Inhibition of these factors that regulate thrombus propagation seems to protect against thrombotic occlusion while preserving hemostasis. Therefore, novel agents include PI3Kβ inhibitors, PDI inhibitors, conformation-specific targeting of activated GPIIb/IIIa, and selective inhibition of GPIIb/IIIa outside-in signaling (McFadyen et al. 2018).
Other candidate drugs include inhibitors of the GPIb-vWF axis, novel PAR1 and PAR4 inhibitors, and blockade of platelet GPVI-mediated adhesion pathways. The GPIb-IX-V receptor binds to vWF during injury and under conditions of high shear stress, allowing early platelet adhesion to the subendothelium. Therefore, various inhibitors of this axis have been developed. Although two antibodies against vWF (ARC1779 and caplacizumab) have demonstrated to have antithrombotic effects, their development has been halted owing to an increased incidence of bleeding (Markus et al. 2011; Muller et al. 2013). However, additional agents, directly targeting GPIb or the vWF binding domain, are under development. As discussed, the use of the currently available PAR-1 antagonist vorapaxar is limited by substantially increased rates of bleeding. Besides orthosteric antagonists like vorapaxar, another class of PAR1 inhibitors, called paramodulins, has been developed. These target the cytoplasmic face of PAR1, contrary to blocking the ligand-binding site like vorapaxar, which inhibits all signaling downstream of the PAR1 receptor. This may allow paramodulins to selectively block platelet and endothelial cell activation mediated by PAR1 while maintaining the cytoprotective signaling pathways in endothelial cells (Aisiku et al. 2015). As thrombin activates platelets via both PAR1 and PAR4, also the PAR4 receptor is currently under investigation as a target for platelet antagonism (McFadyen et al. 2018). Another promising target is the interaction between GPVI and collagen. The observation that GPVI is platelet-specific, in combination with the fact that patients with GPVI deficiency usually suffer from only a mild bleeding phenotype, has led to strategies targeting GPVI. Phase II trials, studying the anti-GPVI agent Revacept in patients with stable CAD and symptomatic carotid stenosis, are currently underway (Majithia and Bhatt 2019).
In recent years, the contribution of the proteins of the contact system (factors VIII, IX, XI, and XII, prekallikrein, and high-molecular-weight kininogen) to the process of atherothrombosis has gained more attention. Factor XII-deficient humans have a normal hemostatic capacity, while animal models have revealed an important role of factor XIIa-driven coagulation in arterial thrombosis (Kuijpers et al. 2014). Furthermore, factor XIIa contributes to inflammation through the activation of the inflammatory bradykinin-producing kallikrein-kinin system (Nickel et al. 2017; Long et al. 2016). Thus, pharmacological inhibition of factor XII(a) may not only be a safer therapeutic strategy (by inhibition of thrombosis while preserving hemostasis) but also has additional beneficial anti-inflammatory and anti-atherogenic effects. Currently, factor XII(a) and its activator polyphosphate are being studied as potential targets for prevention of thrombosis. However, factor XIIa also stimulated the fibrinolytic pathway (Long et al. 2016), and inhibition may thus have potential prothrombotic side effects. Moreover, when thrombosis is initiated by TF exposure, small amounts of thrombin generated by extrinsic tenase have the potential to activate FXI, thereby bypassing FXII inhibition. Therefore, FXI inhibition may be a better target than FXII inhibition. Furthermore, besides attenuation of coagulation, factor XI deprivation has also been shown to slow down atherogenesis in apoE/factor XI double knockout mice (Shnerb Ganor et al. 2016). Several potential strategies to target FXI are currently under investigation, including antisense oligonucleotides (ASOs) that reduce hepatic synthesis of FXI, monoclonal antibodies that suppress FXIa generation and inhibit FXIa activity, and aptamers that block the binding site and small molecules that bind reversibly to the active site of FXIa and inhibits its activity (Weitz and Chan 2019). Clinical phase 2 studies with FXI-directed ASOs, monoclonal antibodies against FXIa, and an oral FXIa inhibitor have been performed (Buller et al. 2015) or are currently underway (Weitz and Chan 2019).
References
Afek A, Kogan E, Maysel-Auslender S, Mor A, Regev E, Rubinstein A et al (2009) Clopidogrel attenuates atheroma formation and induces a stable plaque phenotype in apolipoprotein E knockout mice. Microvasc Res 77(3):364–369
Aisiku O, Peters CG, de Ceunynck K, Ghosh CC, Dilks JR, Fustolo-Gunnink SF et al (2015) Parmodulins inhibit thrombus formation without inducing endothelial injury caused by vorapaxar. Blood 125(12):1976–1985
Alique M, Ramirez-Carracedo R, Bodega G, Carracedo J, Ramirez R (2018) Senescent microvesicles: a novel advance in molecular mechanisms of atherosclerotic calcification. Int J Mol Sci 19(7):2003
Amsterdam EA, Wenger NK, Brindis RG, Casey DE Jr, Ganiats TG, Holmes DR Jr et al (2014) 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol 64(24):e139–e228
Anand SS, Bosch J, Eikelboom JW, Connolly SJ, Diaz R, Widimsky P et al (2018) Rivaroxaban with or without aspirin in patients with stable peripheral or carotid artery disease: an international, randomised, double-blind, placebo-controlled trial. Lancet 391:219–229
Arbab-Zadeh A, Nakano M, Virmani R, Fuster V (2012) Acute coronary events. Circulation 125(9):1147–1156
Ariotti S, Ortega-Paz L, van Leeuwen M, Brugaletta S, Leonardi S, Akkerhuis KM et al (2018) Effects of ticagrelor, prasugrel, or clopidogrel on endothelial function and other vascular biomarkers: a randomized crossover study. JACC Cardiovasc Interv 11(16):1576–1586
Authors/Task Force M, Windecker S, Kolh P, Alfonso F, Collet JP, Cremer J et al (2014) 2014 ESC/EACTS guidelines on myocardial revascularization: the Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS) developed with the special contribution of the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur Heart J 35(37):2541–2619
Badimon L, Vilahur G (2014) Thrombosis formation on atherosclerotic lesions and plaque rupture. J Intern Med 276(6):618–632
Badimon L, Suades R, Arderiu G, Pena E, Chiva-Blanch G, Padro T (2017) Microvesicles in atherosclerosis and angiogenesis: from bench to bedside and reverse. Front Cardiovasc Med 4:77
Bhatt DL, Fox KA, Hacke W, Berger PB, Black HR, Boden WE et al (2006) Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 354(16):1706–1717
Biere-Rafi S, Tuinenburg A, Haak BW, Peters M, Huijgen R, De Groot E et al (2012) Factor VIII deficiency does not protect against atherosclerosis. J Thromb Haemost 10(1):30–37
Binder V, Bergum B, Jaisson S, Gillery P, Scavenius C, Spriet E et al (2017) Impact of fibrinogen carbamylation on fibrin clot formation and stability. Thromb Haemost 117(5):899–910
Borissoff JI, Heeneman S, Kilinc E, Kassak P, van Oerle R, Winckers K et al (2010) Early atherosclerosis exhibits an enhanced procoagulant state. Circulation 122(8):821–830
Borissoff JI, Spronk HM, ten Cate H (2011) The hemostatic system as a modulator of atherosclerosis. N Engl J Med 364(18):1746–1760
Borissoff JI, Joosen IA, Versteylen MO, Spronk HM, ten Cate H, Hofstra L (2012) Accelerated in vivo thrombin formation independently predicts the presence and severity of CT angiographic coronary atherosclerosis. JACC Cardiovasc Imaging 5(12):1201–1210
Borissoff JI, Otten JJ, Heeneman S, Leenders P, van Oerle R, Soehnlein O et al (2013) Genetic and pharmacological modifications of thrombin formation in apolipoprotein e-deficient mice determine atherosclerosis severity and atherothrombosis onset in a neutrophil-dependent manner. PLoS One 8(2):e55784
Bouchareychas L, Raffai RL (2018) Apolipoprotein E and atherosclerosis: from lipoprotein metabolism to MicroRNA control of inflammation. J Cardiovasc Dev Dis 5(2):30
Buchheiser A, Ebner A, Burghoff S, Ding Z, Romio M, Viethen C et al (2011) Inactivation of CD73 promotes atherogenesis in apolipoprotein E-deficient mice. Cardiovasc Res 92(2):338–347
Buller HR, Bethune C, Bhanot S, Gailani D, Monia BP, Raskob GE et al (2015) Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med 372(3):232–240
Bundhun PK, Teeluck AR, Bhurtu A, Huang WQ (2017) Is the concomitant use of clopidogrel and proton pump inhibitors still associated with increased adverse cardiovascular outcomes following coronary angioplasty?: a systematic review and meta-analysis of recently published studies (2012–2016). BMC Cardiovasc Disord 17(1):3
Chandran S, Watkins J, Abdul-Aziz A, Shafat M, Calvert PA, Bowles KM et al (2017) Inflammatory differences in plaque erosion and rupture in patients with ST-segment elevation myocardial infarction. J Am Heart Assoc 6(5):e005868
Collet JP, Cuisset T, Range G, Cayla G, Elhadad S, Pouillot C et al (2012) Bedside monitoring to adjust antiplatelet therapy for coronary stenting. N Engl J Med 367(22):2100–2109
Committee CS (1996) A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE steering committee. Lancet 348(9038):1329–1339
Coppens M, Weitz JI, Eikelboom JWA (2019) Synergy of dual pathway inhibition in chronic cardiovascular disease. Circ Res 124(3):416–425
Coughlin SR (2005) Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost 3(8):1800–1814
Coukell AJ, Markham A (1997) Clopidogrel. Drugs 54(5):745–750; Discussion 51
Cyrus T, Sung S, Zhao L, Funk CD, Tang S, Pratico D (2002) Effect of low-dose aspirin on vascular inflammation, plaque stability, and atherogenesis in low-density lipoprotein receptor-deficient mice. Circulation 106(10):1282–1287
de Groot PG, Reinders JH, Sixma JJ (1987) Perturbation of human endothelial cells by thrombin or PMA changes the reactivity of their extracellular matrix towards platelets. J Cell Biol 104(3):697–704
Dutch Bypass Oral Anticoagulants or Aspirin (BOA) Study Group (2000) Efficacy of Oral anticoagulants compared with aspirin after infrainguinal bypass surgery (The Dutch Bypass Oral Anticoagulants or Aspirin Study): a randomised trial. Lancet 355(9201):346–351
Eikelboom JW, Connolly SJ, Bosch J, Dagenais GR, Hart RG, Shestakovska O et al (2017) Rivaroxaban with or without aspirin in stable cardiovascular disease. N Engl J Med 377:1319–1330
Esmon CT (1995) Inflammation and thrombosis: the impact of inflammation on the protein C anticoagulant pathway. Haematologica 80(2 Suppl):49–56
Esmon CT (2014) Targeting factor Xa and thrombin: impact on coagulation and beyond. Thromb Haemost 111(4):625–633
Fay WP (2004) Plasminogen activator inhibitor 1, fibrin, and the vascular response to injury. Trends Cardiovasc Med 14(5):196–202
Feldmann K, Grandoch M, Kohlmorgen C, Valentin B, Gerfer S, Nagy N et al (2019) Decreased M1 macrophage polarization in dabigatran-treated Ldlr-deficient mice: implications for atherosclerosis and adipose tissue inflammation. Atherosclerosis 287:81–88
Fernandes A, McEvoy JW, Halvorsen S (2019) Doctor, should I keep taking an aspirin a day? N Engl J Med 380(20):1967–1970
Ferrante G, Nakano M, Prati F, Niccoli G, Mallus MT, Ramazzotti V et al (2010) High levels of systemic myeloperoxidase are associated with coronary plaque erosion in patients with acute coronary syndromes: a clinicopathological study. Circulation 122(24):2505–2513
Foley JH (2017) Plasmin(ogen) at the nexus of fibrinolysis, inflammation, and complement. Semin Thromb Hemost 43(2):135–142
Fowkes FG, Price JF, Stewart MC, Butcher I, Leng GC, Pell AC et al (2010) Aspirin for prevention of cardiovascular events in a general population screened for a low ankle brachial index: a randomized controlled trial. JAMA 303(9):841–848
Fujiwara A, Taguchi O, Takagi T, D'Alessandro-Gabazza CN, Boveda-Ruiz D, Toda M et al (2012) Role of thrombin-activatable fibrinolysis inhibitor in allergic bronchial asthma. Lung 190(2):189–198
Fuster V, Badimon L, Badimon JJ, Chesebro JH (1992a) The pathogenesis of coronary artery disease and the acute coronary syndromes (1). N Engl J Med 326(4):242–250
Fuster V, Badimon L, Badimon JJ, Chesebro JH (1992b) The pathogenesis of coronary artery disease and the acute coronary syndromes (2). N Engl J Med 326(5):310–318
Garcia Rodriguez LA, Martin-Perez M, Hennekens CH, Rothwell PM, Lanas A (2016) Bleeding risk with long-term low-dose aspirin: a systematic review of observational studies. PLoS One 11(8):e0160046
Gimbrone MA Jr, Garcia-Cardena G (2016) Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res 118(4):620–636
Gresele P (2002) Platelets in thrombotic and non-thrombotic disorders: pathophysiology, pharmacology, and therapeutics. Cambridge University Press, Cambridge
Griffin JH, Zlokovic BV, Mosnier LO (2015) Activated protein C: biased for translation. Blood 125(19):2898–2907
Group ASC, Bowman L, Mafham M, Wallendszus K, Stevens W, Buck G et al (2018) Effects of aspirin for primary prevention in persons with diabetes mellitus. N Engl J Med 379(16):1529–1539
Grover SP, Mackman N (2018) Tissue factor: an essential mediator of hemostasis and trigger of thrombosis. Arterioscler Thromb Vasc Biol 38(4):709–725
Gurbel PA, Fox KAA, Tantry US, Ten Cate H, Weitz JI (2019) Combination antiplatelet and oral anticoagulant therapy in patients with coronary and peripheral artery disease. Circulation 139(18):2170–2185
Hackam DG, Spence JD (2019) Antiplatelet therapy in ischemic stroke and transient ischemic attack. Stroke 50(3):773–778
Hara T, Fukuda D, Tanaka K, Higashikuni Y, Hirata Y, Nishimoto S et al (2015) Rivaroxaban, a novel oral anticoagulant, attenuates atherosclerotic plaque progression and destabilization in ApoE-deficient mice. Atherosclerosis 242(2):639–646
Hara T, Phuong PT, Fukuda D, Yamaguchi K, Murata C, Nishimoto S et al (2018) Protease-activated receptor-2 plays a critical role in vascular inflammation and atherosclerosis in apolipoprotein E-deficient mice. Circulation 138(16):1706–1719
Heim C, Gebhardt J, Ramsperger-Gleixner M, Jacobi J, Weyand M, Ensminger SM (2016) Clopidogrel significantly lowers the development of atherosclerosis in ApoE-deficient mice in vivo. Heart Vessel 31(5):783–794
Hess CN, Norgren L, Ansel GM, Capell WH, Fletcher JP, Fowkes FGR et al (2017) A structured review of antithrombotic therapy in peripheral artery disease with a focus on revascularization: a TASC (InterSociety Consensus for the Management of Peripheral Artery Disease) initiative. Circulation 135(25):2534–2555
Hiatt WR, Fowkes FG, Heizer G, Berger JS, Baumgartner I, Held P et al (2017) Ticagrelor versus clopidogrel in symptomatic peripheral artery disease. N Engl J Med 376(1):32–40
Hoffman M (2018) The tissue factor pathway and wound healing. Semin Thromb Hemost 44(2):142–150
Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S et al (2003) Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med 9(1):61–67
Ikonomidis I, Andreotti F, Economou E, Stefanadis C, Toutouzas P, Nihoyannopoulos P (1999) Increased proinflammatory cytokines in patients with chronic stable angina and their reduction by aspirin. Circulation 100(8):793–798
Johnston SC, Easton JD, Farrant M, Barsan W, Conwit RA, Elm JJ et al (2018) Clopidogrel and aspirin in acute ischemic stroke and high-risk TIA. N Engl J Med 379(3):215–225
Kadoglou NP, Moustardas P, Katsimpoulas M, Kapelouzou A, Kostomitsopoulos N, Schafer K et al (2012) The beneficial effects of a direct thrombin inhibitor, dabigatran etexilate, on the development and stability of atherosclerotic lesions in apolipoprotein E-deficient mice: dabigatran etexilate and atherosclerosis. Cardiovasc Drugs Ther 26(5):367–374
Kamphuisen PW, ten Cate H (2014) Cardiovascular risk in patients with hemophilia. Blood 123(9):1297–1301
Kharbanda RK, Walton B, Allen M, Klein N, Hingorani AD, MacAllister RJ et al (2002) Prevention of inflammation-induced endothelial dysfunction: a novel vasculo-protective action of aspirin. Circulation 105(22):2600–2604
Kim OV, Nevzorova TA, Mordakhanova ER, Ponomareva AA, Andrianova IA, Le Minh G et al (2019) Fatal dysfunction and disintegration of thrombin-stimulated platelets. Haematologica 104(9):1866–1878
Kleinegris MC, ten Cate H, ten Cate-Hoek AJ (2013) D-dimer as a marker for cardiovascular and arterial thrombotic events in patients with peripheral arterial disease. A systematic review. Thromb Haemost 110(2):233–243
Koenen RR (2016) The prowess of platelets in immunity and inflammation. Thromb Haemost 116(4):605–612
Kossmann S, Lagrange J, Jackel S, Jurk K, Ehlken M, Schonfelder T et al (2017) Platelet-localized FXI promotes a vascular coagulation-inflammatory circuit in arterial hypertension. Sci Transl Med 9(375):eaah4923
Koupenova M, Clancy L, Corkrey HA, Freedman JE (2018) Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res 122(2):337–351
Kramer MC, Rittersma SZ, de Winter RJ, Ladich ER, Fowler DR, Liang YH et al (2010) Relationship of thrombus healing to underlying plaque morphology in sudden coronary death. J Am Coll Cardiol 55(2):122–132
Kuijpers MJ, van der Meijden PE, Feijge MA, Mattheij NJ, May F, Govers-Riemslag J et al (2014) Factor XII regulates the pathological process of thrombus formation on ruptured plaques. Arterioscler Thromb Vasc Biol 34(8):1674–1680
Lee IO, Kratz MT, Schirmer SH, Baumhakel M, Bohm M (2012) The effects of direct thrombin inhibition with dabigatran on plaque formation and endothelial function in apolipoprotein E-deficient mice. J Pharmacol Exp Ther 343(2):253–257
Lee J, Nakanishi R, Li D, Shaikh K, Shekar C, Osawa K et al (2018) Randomized trial of rivaroxaban versus warfarin in the evaluation of progression of coronary atherosclerosis. Am Heart J 206:127–130
Li D, Wang Y, Zhang L, Luo X, Li J, Chen X et al (2012) Roles of purinergic receptor P2Y, G protein-coupled 12 in the development of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 32(8):e81–e89
Li X, Vink A, Niessen HW, Kers J, de Boer OJ, Ploegmakers HJ et al (2014) Total burden of intraplaque hemorrhage in coronary arteries relates to the use of coumarin-type anticoagulants but not platelet aggregation inhibitors. Virchows Arch 465(6):723–729
Libby P (2013) Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med 368(21):2004–2013
Lievens D, von Hundelshausen P (2011) Platelets in atherosclerosis. Thromb Haemost 106(5):827–838
Liu H, Jiang D, Zhang S, Ou B (2010) Aspirin inhibits fractalkine expression in atherosclerotic plaques and reduces atherosclerosis in ApoE gene knockout mice. Cardiovasc Drugs Ther 24(1):17–24
Long AT, Kenne E, Jung R, Fuchs TA, Renne T (2016) Contact system revisited: an interface between inflammation, coagulation, and innate immunity. J Thromb Haemost 14(3):427–437
Lowe G, Rumley A (2014) The relevance of coagulation in cardiovascular disease: what do the biomarkers tell us? Thromb Haemost 112(5):860–867
Mackman N (2016) The clot thickens in atherosclerosis. Arterioscler Thromb Vasc Biol 36(3):425–426
Majithia A, Bhatt DL (2019) Novel antiplatelet therapies for atherothrombotic diseases. Arterioscler Thromb Vasc Biol 39(4):546–557
Markus HS, McCollum C, Imray C, Goulder MA, Gilbert J, King A (2011) The von Willebrand inhibitor ARC1779 reduces cerebral embolization after carotid endarterectomy: a randomized trial. Stroke 42(8):2149–2153
Mastenbroek TG, van Geffen JP, Heemskerk JW, Cosemans JM (2015) Acute and persistent platelet and coagulant activities in atherothrombosis. J Thromb Haemost 13(Suppl 1):S272–S280
McFadyen JD, Schaff M, Peter K (2018) Current and future antiplatelet therapies: emphasis on preserving haemostasis. Nat Rev Cardiol 15(3):181–191
McNeil JJ, Nelson MR, Woods RL, Lockery JE, Wolfe R, Reid CM et al (2018a) Effect of aspirin on all-cause mortality in the healthy elderly. N Engl J Med 379(16):1519–1528
McNeil JJ, Wolfe R, Woods RL, Tonkin AM, Donnan GA, Nelson MR et al (2018b) Effect of aspirin on cardiovascular events and bleeding in the healthy elderly. N Engl J Med 379(16):1509–1518
McNeil JJ, Woods RL, Nelson MR, Reid CM, Kirpach B, Wolfe R et al (2018c) Effect of aspirin on disability-free survival in the healthy elderly. N Engl J Med 379(16):1499–1508
Mega JL, Simon T, Collet JP, Anderson JL, Antman EM, Bliden K et al (2010) Reduced-function CYP2C19 genotype and risk of adverse clinical outcomes among patients treated with clopidogrel predominantly for PCI: a meta-analysis. JAMA 304(16):1821–1830
Mega JL, Braunwald E, Wiviott SD, Bassand JP, Bhatt DL, Bode C et al (2012) Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med 366(1):9–19
Mehta SR, Yusuf S, Peters RJ, Bertrand ME, Lewis BS, Natarajan MK et al (2001) Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 358(9281):527–533
Messner B, Bernhard D (2014) Smoking and cardiovascular disease: mechanisms of endothelial dysfunction and early atherogenesis. Arterioscler Thromb Vasc Biol 34(3):509–515
Miller VM, Lahr BD, Bailey KR, Hodis HN, Mulvagh SL, Jayachandran M (2016) Specific cell-derived microvesicles: linking endothelial function to carotid artery intima-media thickness in low cardiovascular risk menopausal women. Atherosclerosis 246:21–28
Mo C, Sun G, Lu ML, Zhang L, Wang YZ, Sun X et al (2015) Proton pump inhibitors in prevention of low-dose aspirin-associated upper gastrointestinal injuries. World J Gastroenterol 21(17):5382–5392
Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP et al (2012) Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med 366(15):1404–1413
Morser J, Gabazza EC, Myles T, Leung LL (2010) What has been learnt from the thrombin-activatable fibrinolysis inhibitor-deficient mouse? J Thromb Haemost 8(5):868–876
Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH (2012) Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood 120(26):5237–5246
Mozaffarian D, Wilson PW, Kannel WB (2008) Beyond established and novel risk factors: lifestyle risk factors for cardiovascular disease. Circulation 117(23):3031–3038
Mujaj B, Bos D, Muka T, Lugt AV, Ikram MA, Vernooij MW et al (2018) Antithrombotic treatment is associated with intraplaque haemorrhage in the atherosclerotic carotid artery: a cross-sectional analysis of the Rotterdam study. Eur Heart J 39(36):3369–3376
Muller O, Bartunek J, Hamilos M, Berza CT, Mangiacapra F, Ntalianis A et al (2013) von Willebrand factor inhibition improves endothelial function in patients with stable angina. J Cardiovasc Transl Res 6(3):364–370
Myles T, Nishimura T, Yun TH, Nagashima M, Morser J, Patterson AJ et al (2003) Thrombin activatable fibrinolysis inhibitor, a potential regulator of vascular inflammation. J Biol Chem 278(51):51059–51067
Naito M, Taguchi O, Kobayashi T, Takagi T, D’Alessandro-Gabazza CN, Matsushima Y et al (2013) Thrombin-activatable fibrinolysis inhibitor protects against acute lung injury by inhibiting the complement system. Am J Respir Cell Mol Biol 49(4):646–653
Nawroth PP, Stern DM, Kaplan KL, Nossel HL (1984) Prostacyclin production by perturbed bovine aortic endothelial cells in culture. Blood 64(4):801–806
Nechipurenko DY, Receveur N, Yakimenko AO, Shepelyuk TO, Yakusheva AA, Kerimov RR et al (2019) Clot contraction drives the translocation of procoagulant platelets to thrombus surface. Arterioscler Thromb Vasc Biol 39(1):37–47
Nguyen TS, Lapidot T, Ruf W (2018) Extravascular coagulation in hematopoietic stem and progenitor cell regulation. Blood 132(2):123–131
Nickel KF, Long AT, Fuchs TA, Butler LM, Renne T (2017) Factor XII as a therapeutic target in thromboembolic and inflammatory diseases. Arterioscler Thromb Vasc Biol 37(1):13–20
Nishimura T, Myles T, Piliponsky AM, Kao PN, Berry GJ, Leung LL (2007) Thrombin-activatable procarboxypeptidase B regulates activated complement C5a in vivo. Blood 109(5):1992–1997
Nylander S, Schulz R (2016) Effects of P2Y12 receptor antagonists beyond platelet inhibition – comparison of ticagrelor with thienopyridines. Br J Pharmacol 173(7):1163–1178
Olie RH, van der Meijden PEJ, Ten Cate H (2018) The coagulation system in atherothrombosis: implications for new therapeutic strategies. Res Pract Thromb Haemost 2(2):188–198
Olie RH, van der Meijden PEJ, Spronk HMH, van Oerle R, Barvik S, Bonarjee VVS et al (2019) Effects of the PAR-1 antagonist vorapaxar on platelet activation and coagulation biomarkers in patients with stable coronary artery disease. TH Open 3(3):e259–ee62
Opneja A, Kapoor S, Stavrou EX (2019) Contribution of platelets, the coagulation and fibrinolytic systems to cutaneous wound healing. Thromb Res 179:56–63
Paone S, Baxter AA, Hulett MD, Poon IKH (2019) Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell Mol Life Sci 76(6):1093–1106
Pasterkamp G, den Ruijter HM, Libby P (2017) Temporal shifts in clinical presentation and underlying mechanisms of atherosclerotic disease. Nat Rev Cardiol 14(1):21–29
Pedicino D, Vinci R, Giglio AF, Pisano E, Porto I, Vergallo R et al (2018) Alterations of hyaluronan metabolism in acute coronary syndrome: implications for plaque erosion. J Am Coll Cardiol 72(13):1490–1503
Pingel S, Tiyerili V, Mueller J, Werner N, Nickenig G, Mueller C (2014) Thrombin inhibition by dabigatran attenuates atherosclerosis in ApoE deficient mice. Arch Med Sci 10(1):154–160
Plank F, Beyer C, Friedrich G, Stuhlinger M, Hintringer F, Dichtl W et al (2018) Influence of vitamin K antagonists and direct oral anticoagulation on coronary artery disease: a CTA analysis. Int J Cardiol 260:11–15
Posma JJ, Posthuma JJ, Spronk HM (2016) Coagulation and non-coagulation effects of thrombin. J Thromb Haemost 14(10):1908–1916
Posthuma JJ, Posma JJN, van Oerle R, Leenders P, van Gorp RH, Jaminon AMG et al (2019) Targeting coagulation factor Xa promotes regression of advanced atherosclerosis in apolipoprotein-E deficient mice. Sci Rep 9(1):3909
Preusch MR, Ieronimakis N, Wijelath ES, Cabbage S, Ricks J, Bea F et al (2015) Dabigatran etexilate retards the initiation and progression of atherosclerotic lesions and inhibits the expression of oncostatin M in apolipoprotein E-deficient mice. Drug Des Devel Ther 9:5203–5211
Price MJ, Berger PB, Teirstein PS, Tanguay JF, Angiolillo DJ, Spriggs D et al (2011) Standard- vs high-dose clopidogrel based on platelet function testing after percutaneous coronary intervention: the GRAVITAS randomized trial. JAMA 305(11):1097–1105
Quillard T, Franck G, Mawson T, Folco E, Libby P (2017) Mechanisms of erosion of atherosclerotic plaques. Curr Opin Lipidol 28(5):434–441
Relja B, Lustenberger T, Puttkammer B, Jakob H, Morser J, Gabazza EC et al (2013) Thrombin-activatable fibrinolysis inhibitor (TAFI) is enhanced in major trauma patients without infectious complications. Immunobiology 218(4):470–476
Ren M, Li R, Chen N, Pang N, Li Y, Deng X et al (2017) Platelet-derived factor V is a critical mediator of arterial thrombosis. J Am Heart Assoc 6(4):e006345
Roffi M, Patrono C, Collet JP, Mueller C, Valgimigli M, Andreotti F et al (2016) 2015 ESC guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: Task Force for the Management of acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology (ESC). Eur Heart J 37(3):267–315
Ross R (1999) Atherosclerosis – an inflammatory disease. N Engl J Med 340(2):115–126
Ruf W (2018) Proteases, protease-activated receptors, and atherosclerosis. Arterioscler Thromb Vasc Biol 38(6):1252–1254
Russo I, Penna C, Musso T, Popara J, Alloatti G, Cavalot F et al (2017) Platelets, diabetes and myocardial ischemia/reperfusion injury. Cardiovasc Diabetol 16(1):71
Sanada F, Muratsu J, Otsu R, Shimizu H, Koibuchi N, Uchida K et al (2017) Local production of activated factor X in atherosclerotic plaque induced vascular smooth muscle cell senescence. Sci Rep 7(1):17172
Schirmer SH, Kratz MT, Kazakov A, Nylander S, Baumhaekel M, Laufs U et al (2012) 1357 - inhibition of the adenosine diphosphate receptor P2Y12 reduces atherosclerotic plaque size in hypercholesterolemic ApoE−/− mice. Eur Heart J 33(suppl_1):19–338
Schulz C, Massberg S (2012) Platelets in atherosclerosis and thrombosis. Handb Exp Pharmacol 210:111–133
Scridon A, Marginean A, Hutanu A, Chinezu L, Gheban D, Perian M et al (2019) Vascular protease-activated receptor 4 upregulation, increased platelet aggregation, and coronary lipid deposits induced by long-term dabigatran administration – results from a diabetes animal model. J Thromb Haemost 17(3):538–550
Seehaus S, Shahzad K, Kashif M, Vinnikov IA, Schiller M, Wang H et al (2009) Hypercoagulability inhibits monocyte transendothelial migration through protease-activated receptor-1-, phospholipase-Cbeta-, phosphoinositide 3-kinase-, and nitric oxide-dependent signaling in monocytes and promotes plaque stability. Circulation 120(9):774–784
Semple JW, Italiano JE Jr, Freedman J (2011) Platelets and the immune continuum. Nat Rev Immunol 11(4):264–274
Shao Z, Nishimura T, Leung LL, Morser J (2015) Carboxypeptidase B2 deficiency reveals opposite effects of complement C3a and C5a in a murine polymicrobial sepsis model. J Thromb Haemost 13(6):1090–1102
Shnerb Ganor R, Harats D, Schiby G, Gailani D, Levkovitz H, Avivi C et al (2016) Factor XI deficiency protects against atherogenesis in apolipoprotein E/factor XI double knockout mice. Arterioscler Thromb Vasc Biol 36(3):475–481
Shuldiner AR, O’Connell JR, Bliden KP, Gandhi A, Ryan K, Horenstein RB et al (2009) Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 302(8):849–857
Sibbing D, Stegherr J, Latz W, Koch W, Mehilli J, Dorrler K et al (2009) Cytochrome P450 2C19 loss-of-function polymorphism and stent thrombosis following percutaneous coronary intervention. Eur Heart J 30(8):916–922
Smith P, Arnesen H, Holme I (1990) The effect of warfarin on mortality and reinfarction after myocardial infarction. N Engl J Med 323(3):147–152
Soardi F, Nicrosini F, Del Favero A, Pasotti C (1961) Experimental cholesterin atherosclerosis in the rabbit: action of a duodenal heparinoid. III. Modifications in the factors of hemocoagulation, plasmin and plasminogen, and in the thromboplastin and fibrinolytic activity of the aortic wall. Farmaco Prat 16:560–568
Spronk HM, Govers-Riemslag JW, ten Cate H (2003a) The blood coagulation system as a molecular machine. BioEssays 25(12):1220–1228
Spronk HM, Soute BA, Schurgers LJ, Thijssen HH, de Mey JG, Vermeer C (2003b) Tissue-specific utilization of menaquinone-4 results in the prevention of arterial calcification in warfarin-treated rats. J Vasc Res 40(6):531–537
Spronk HMH, Padro T, Siland JE, Prochaska JH, Winters J, van der Wal AC et al (2018) Atherothrombosis and thromboembolism: position paper from the Second Maastricht Consensus Conference on Thrombosis. Thromb Haemost 118(2):229–250
Stalker TJ, Traxler EA, Wu J, Wannemacher KM, Cermignano SL, Voronov R et al (2013) Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood 121(10):1875–1885
Swieringa F, Spronk HMH, Heemskerk JWM, van der Meijden PEJ (2018) Integrating platelet and coagulation activation in fibrin clot formation. Res Pract Thromb Haemost 2(3):450–460
Szabo IL, Matics R, Hegyi P, Garami A, Illes A, Sarlos P et al (2017) PPIs prevent aspirin-induced gastrointestinal bleeding better than H2RAs. A systematic review and meta-analysis. J Gastrointestin Liver Dis 26(4):395–402
Takeda M, Yamashita T, Shinohara M, Sasaki N, Tawa H, Nakajima K et al (2012) Beneficial effect of anti-platelet therapies on atherosclerotic lesion formation assessed by phase-contrast X-ray CT imaging. Int J Cardiovasc Imaging 28(5):1181–1191
Tousoulis D, Oikonomou E, Economou EK, Crea F, Kaski JC (2016) Inflammatory cytokines in atherosclerosis: current therapeutic approaches. Eur Heart J 37(22):1723–1732
van Bergen PFMM et al (1994) Effect of long-term oral anticoagulant treatment on mortality and cardiovascular morbidity after myocardial infarction. Anticoagulants in the Secondary Prevention of Events in Coronary Thrombosis (ASPECT) Research Group. Lancet 343(8896):499–503
Vasina EM, Cauwenberghs S, Staudt M, Feijge MA, Weber C, Koenen RR et al (2013) Aging- and activation-induced platelet microparticles suppress apoptosis in monocytic cells and differentially signal to proinflammatory mediator release. Am J Blood Res 3(2):107–123
Veltri KT (2018) Yosprala: a fixed dose combination of aspirin and omeprazole. Cardiol Rev 26(1):50–53
von Hundelshausen P, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE et al (2005) Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood 105(3):924–930
Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C et al (2009) Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 361(11):1045–1057
Wang Y, Wang Y, Zhao X, Liu L, Wang D, Wang C et al (2013) Clopidogrel with aspirin in acute minor stroke or transient ischemic attack. N Engl J Med 369(1):11–19
Weitz JI, Chan NC (2019) Advances in antithrombotic therapy. Arterioscler Thromb Vasc Biol 39(1):7–12
West LE, Steiner T, Judge HM, Francis SE, Storey RF (2014) Vessel wall, not platelet, P2Y12 potentiates early atherogenesis. Cardiovasc Res 102(3):429–435
Wight TN (2018) A role for extracellular matrix in atherosclerotic plaque erosion. J Am Coll Cardiol 72(13):1504–1505
Wilcox JN (1994) Thrombotic mechanisms in atherosclerosis. Coron Artery Dis 5(3):223–229
Wilcox JN, Noguchi S, Casanova J (2003) Extrahepatic synthesis of factor VII in human atherosclerotic vessels. Arterioscler Thromb Vasc Biol 23(1):136–141
Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S et al (2007) Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 357(20):2001–2015
Yin M, Loyer X, Boulanger CM (2015) Extracellular vesicles as new pharmacological targets to treat atherosclerosis. Eur J Pharmacol 763(Pt A):90–103
Zhou Q, Bea F, Preusch M, Wang H, Isermann B, Shahzad K et al (2011) Evaluation of plaque stability of advanced atherosclerotic lesions in apo E-deficient mice after treatment with the oral factor Xa inhibitor rivaroxaban. Mediat Inflamm 2011:432080
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this chapter

Cite this chapter
Olie, R.H., van der Meijden, P.E.J., Spronk, H.M.H., ten Cate, H. (2020). Antithrombotic Therapy: Prevention and Treatment of Atherosclerosis and Atherothrombosis. In: von Eckardstein, A., Binder, C.J. (eds) Prevention and Treatment of Atherosclerosis . Handbook of Experimental Pharmacology, vol 270. Springer, Cham. https://doi.org/10.1007/164_2020_357
Download citation
DOI: https://doi.org/10.1007/164_2020_357
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-86075-2
Online ISBN: 978-3-030-86076-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)