
Abstract
Nonalcoholic fatty liver disease (NAFLD) is considered the hepatic manifestation of the metabolic syndrome (MetS) and comprises one of the largest health threats of the twenty-first century. In this chapter, we review the current state of knowledge of NAFLD and underline the striking similarities with atherosclerosis. We first describe current epidemiological data showing the staggering increase of NAFLD numbers and its related clinical and economic costs. We then provide an overview of pathophysiological hepatic processes in NAFLD and highlight the systemic aspects of NAFLD that point toward metabolic crosstalk between organs as an important cause of metabolic disease. Finally, we end by highlighting the currently investigated therapeutic approaches for NAFLD, which also show strong similarities with a range of treatment options for atherosclerosis.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Epidemiology
1.1 Definition, Prevalence, and Incidence of NAFLD
Being the most prevalent chronic liver disease worldwide (Li et al. 2019), nonalcoholic fatty liver disease (NAFLD) covers a diseases spectrum, initiating with hepatic steatosis which is defined by the presence of ≥5% hepatic fat (referred to steatosis) in the absence of any secondary cause of hepatic steatosis such as chronic viral hepatitis and alcohol consumption (21 drinks/week in men and 14 drinks/week in women) (Chalasani et al. 2012, 2018). In a second, more advanced stage, hepatic steatosis may advance into nonalcoholic steatohepatitis (NASH), which is characterized by a combination of hepatic steatosis and inflammation in the presence or absence of fibrosis. Finally, NASH can progress into advanced-stage liver diseases such as cirrhosis and hepatocellular carcinoma (HCC) (Angulo 2002; Anstee and Day 2013; Calzadilla Bertot and Adams 2016; Chalasani et al. 2018; Fazel et al. 2016; Jou et al. 2008; Sayiner et al. 2016; Younossi and Henry 2016; Younossi et al. 2018a). Being the hepatic component of the metabolic syndrome (MetS) (Chalasani et al. 2012), NAFLD is commonly associated with other metabolic disorders such as obesity, which is also linked to the development of cardiovascular diseases such as atherosclerosis (Chalasani et al. 2018; Paoletti et al. 2006). Considering these links between NAFLD, obesity, and atherosclerosis, it is no surprise that NAFLD has become the most prevalent liver disease worldwide (Li et al. 2019).
Indeed, in the last three decades, the prevalence of NAFLD has increased at a constant rate. Numbers from a study from Younossi et al. showed the evolution of NAFLD prevalence in the United States from 1988 to 2008 ranging from 5.51% (1988–1994) to 9.84% (1999–2004) and 11.01% (2005–2008), indicating a twofold increase over two decades (Younossi et al. 2011a). At a global level, it is currently estimated that NAFLD affects about 25% of the general population. At the other hand, NASH has been calculated at 2–5% of the general population and so far represents the minority of NAFLD patients (10–20%) (Younossi et al. 2016a). However, while hepatic steatosis seems less harmful for the liver, patients suffering from steatosis are at increased risk for cardiac-related death (Targher et al. 2016). Therefore, steatotic patients should be monitored even at an early stage of the disease. For the future, Estes et al. predicted the NAFLD population to increase with 21% in 2030, expecting a staggering 100.9 million NAFLD patients, of which 27.00 million patients would also suffer from NASH, the latter indicating a 63% increase in prevalence compared to current numbers (Estes et al. 2018).
From a regional perspective, the pooled incidence of NAFLD in the West (being Europe and Northern America) was estimated to be 28 per 1,000 persons per year (Chalasani et al. 2018; Younossi et al. 2016b). As mentioned previously, NAFLD has been reported as the most common liver disease in the United States (Bellentani and Marino 2009). However, within the Asian population, recent metadata also indicated the global incidence rate of NAFLD at 50.9 cases per 1,000 individuals per year (Li et al. 2019). Indeed, while it was initially perceived as a “Western disease,” NAFLD is now highly prevalent in all continents with the highest rates reported in South America (31%) and the Middle East (32%), followed by Asia (27%), the United States (24%), and Europe (23%), while being less common in Africa (14%) (Younossi et al. 2016b).
Taken these numbers into account, and especially those indicating the exponential increase of NASH patients, it is clear that NAFLD poses one of the largest burdens on current healthcare systems, emphasizing the urge for early and fast treatment to prevent further escalation of this disease.
1.2 Association with Other Diseases
NAFLD has been reported to be strongly linked to obesity, with a prevalence as high as 80% in obese patients and only 16% in individuals with a normal BMI and without metabolic risk factors (Bellentani et al. 2000; Williams et al. 2011). Relevantly, obesity seems to play a role in both the initial process leading to simple steatosis but also to its progression toward NASH (Polyzos et al. 2017). Indeed, it has been demonstrated that the risk of NASH development is lower in lean than overweight/obese individuals (Sookoian and Pirola 2018). Next to NASH, also patients suffering from hepatic fibrosis tend to be rather obese than non-obese (86% vs. 27%, respectively) (Fassio et al. 2004).
Besides obesity, it has been suggested that NAFLD is also tightly linked to cardiovascular diseases (Patil and Sood 2017). Recently, clinical observations indicated that NASH increases atherosclerosis and cardiovascular risks by local overexpression of inflammatory mediators, endothelial damage, and regulators of blood pressure (Targher et al. 2009, 2010). Others confirmed that NAFLD is independently associated with atherosclerosis progression (Targher et al. 2010). Additionally, other studies demonstrated that NAFLD patients have impaired flow-mediated vasodilatation (Villanova et al. 2005), increased carotid artery intimal-medial thickness and an increased prevalence of carotid atherosclerotic plaques compared to healthy subjects (Sookoian and Pirola 2008), independently of obesity and other established risk factors. These observations therefore emphasize the link between NAFLD and atherosclerosis.
Additionally, NAFLD has also been suggested as a risk factor for gastrointestinal tract malignancies, such as colorectal cancer (Lindenmeyer and McCullough 2018; Muhidin et al. 2012; Wong et al. 2011). Also, NAFLD is also associated with chronic kidney disease (CKD) (Musso et al. 2014), which is defined by the presence of kidney damage or a reduced glomerular filtration rate for 3 months or more (Levey et al. 2005). Specifically, it has been suggested that the prevalence of CKD in NAFLD patients is between 4 and 40% (Marcuccilli and Chonchol 2016). This association has been partly explained by the NAFLD-associated microvascular alterations that also affect the kidney (Musso et al. 2016). Moreover these microvascular alterations have been linked to cerebrovascular disease, potentially contributing to cognitive impairment (Lombardi et al. 2019). Finally, polycystic ovary syndrome (PCOS), a condition that leads to the production of higher-than-normal amounts of male hormones in women, is also associated with NAFLD (Vassilatou 2014; Wu et al. 2018a). However, the clinical significance and its pathophysiological basis remain to be further investigated.
1.3 Clinical, Economic, and Social Burden of NAFLD
In spite of its increased prevalence over the last decades, to our knowledge, the clinical burden of NAFLD has not been characterized in detail (Boursier et al. 2018; Lam et al. 2016; Mullerova et al. 2019; Welte et al. 2012). Notwithstanding that all stages of NAFLD contribute to its clinical burden, the more progressive stages of NALFD are expected to have the largest impact (Sayiner et al. 2016). As a progressive form of NAFLD, NASH is currently the second leading cause for liver transplantation in the United States and even the leading cause for liver transplantation in females (Noureddin et al. 2018). Furthermore, it is known that NAFLD has become one of the leading causes for cirrhosis (Kadayifci et al. 2008), the end-stage liver disease which is associated with high risks for development of bacterial infections leading to hospitalization (Albillos et al. 2014; Li et al. 2018; Singal et al. 2014). Additionally, the presence of advanced fibrosis (stage ≥2) in NAFLD has been directly associated with liver-related mortality (Angulo et al. 2015; Younossi et al. 2011b). There is also accumulating evidence that NAFLD is an important risk factor for hepatocellular carcinoma (HCC), which is the fifth most common type of cancer and third most common cause of cancer mortality (El-Serag and Rudolph 2007; Piscaglia et al. 2016; Younossi et al. 2015a). Translating the latter described observations into exact numbers, a recent study showed that in 2015, 28,000 deaths (2.2% of all deaths in the NAFLD population) were related to cirrhosis, HCC, or liver transplantation, while 162,560 deaths (accounting for 12.8% of all NAFLD deaths) were due to cardiovascular diseases (Angulo et al. 2015; Ekstedt et al. 2015). Indeed, these data point toward an important role for cardiovascular diseases in NAFLD-related mortality. In line with the previously estimated increased prevalence of NAFLD in the future, reports have indicated that the total number of deaths resulting from NAFLD will increase 44%, reaching 1.83 million deaths at an annual basis by 2030 (Estes et al. 2018). Regarding to the healthcare expenditure, Lam et al. demonstrated that more frequent clinical visits are associated with improved outcomes in pediatric NAFLD patients, substantiating the importance of frequent monitoring and follow-up to manage NAFLD progression (Lam et al. 2016). Moreover, Boursier et al. evaluated the hospitalization of NAFLD-/NASH-related end-stage liver disease (ESLD) patients by a 7-year follow-up study in France. This report described that ESLD patients experience more hospitalization per year (over 400%), which are longer (400% increase in length) and are associated with a 300% increase in hospitalization costs (Boursier et al. 2018). These numbers therefore indicate the gigantic investments that are required to manage NAFLD.
In line with the increasing prevalence of NAFLD, the economic costs related to NAFLD have also been predicted to rise in the future. In the United States alone, approximately 103 billion dollars are annually spent on NAFLD-related costs, while in European countries (i.e., France, the United Kingdom, Germany, and Italy), these costs account for approximately €35 billion (Abdelmalek 2016). Of particular concern is the rising prevalence of obesity as well as the increase in general healthcare costs, which contributes to a tenfold increase in the current economic burden of NAFLD by 2025 (Abdelmalek 2016; Younossi et al. 2016a). Several studies also investigated the relationship between NAFLD patients and healthcare utilization and associated costs. These studies demonstrated that the number of outpatient visits for patients with NAFLD significantly doubled over time and underlined the fact that, between 2005 and 2010, the healthcare costs of inpatients and outpatients were increased 5% and 10%, respectively (Baumeister et al. 2008; Younossi et al. 2014, 2015b). Additionally, a study of the hepatology clinics in the West Suffolk area of the United Kingdom showed that the total annual hepatology budget for these specialized clinics was £130,000, including £58,000 for resources and £72,000 for clinic attendances. Moreover, the latest research estimating the economic burden of NASH patients by using a Markov decision analytic model demonstrated that lifetime costs of all NASH patients was approximately $222.6 billion in the United States in 2017, with $95.4 billion reflecting the advanced NASH population (Younossi et al. 2019).
From an individual perspective, NALFD patients have to contend with a range of symptoms such as fatigue, decreased physical activity, and emotional health impairment which affect their quality of life (health-related quality of life (HR-QOL)) (Golabi et al. 2016; Loria et al. 2013; Younossi and Henry 2015, 2016). Several studies have demonstrated that NAFLD patients had poorer HR-QOL compared to other chronic liver diseases and also showed that NAFLD-related fatigue associated with impairments in physical functioning (Afendy et al. 2009; Dan et al. 2007; Newton et al. 2008). Potential explanations for the decrease of HR-QOL in NAFLD patients that have been raised are related to obesity and psychological processes as well as psychiatric issues such as depression and anxiety (Stewart and Levenson 2012; Surdea-Blaga and Dumitrascu 2011; Weinstein et al. 2011). Indeed, several studies have demonstrated that depressive disorders, as well as anxiety disorders, are more frequent in patients with NAFLD/NASH and are associated with more advanced liver histological abnormalities, such as severe hepatocyte ballooning (Elwing et al. 2006; Macavei et al. 2016; Stewart et al. 2015; Weinstein et al. 2011).
Altogether, these numbers substantiate the impact of NAFLD on healthcare, economy, and also the daily life of individual patients. To prevent further escalation of the disease, it is therefore of utmost importance to increase the understanding of the disease to find approaches to diagnose and treat (and if possible prevent) the progression of NAFLD.
2 Pathophysiology of NAFLD
As NAFLD comprises a spectrum of diseases, multiple pathophysiological processes are involved including dysregulation of lipid metabolism, increased hepatic inflammation, and the presence of hepatic fibrosis. However, how patients with steatosis develop inflammation is still unclear, leaving a blind spot in the understanding of how NASH exactly arises. Nevertheless, scientists have succeeded in unraveling several disease processes in NALFD, which appear to show striking similarities with disease processes described in atherosclerosis. Furthermore, increasing evidence links different metabolic organs to NAFLD development, emphasizing the presence of metabolic crosstalk during NAFLD.
2.1 Intrahepatic Disturbances During NAFLD
The liver constitutes a key role in regulating whole body metabolism, which involves a complex interplay between different hepatic cell types ranging from hepatocytes as parenchymal cells to Kupffer cells (KCs), stellate cells, and liver sinusoidal endothelial cells (LSECs) among other cell types. As such, each of these cell types is influenced by pathophysiological processes during NAFLD, eventually leading to hepatic disturbances at whole organ level.
2.1.1 Lipo- and Glucotoxicity
Due to low physical activity and increased consumption of fats, lipotoxicity has arisen as one of the main players to contribute to NAFLD pathogenesis (Ibrahim et al. 2011). Lipotoxicity is defined by the excess generation of cytosolic lipids (mainly triglycerides and subtypes of free fatty acids) that have direct adverse effects on metabolic pathways of the cell (Schaffer 2016). Under normal conditions, triglycerides and free fatty acids are stored in adipose tissue, where they can be employed as energy source during periods of energy deprivation or during extreme exercise (Muro et al. 2014). However, in obesity, when the storage capacity of adipose tissue is exceeded, free fatty acids accumulate in ectopic organs, including the liver (but also in the arteries). As such, hepatic steatosis (Wojcik-Cichy et al. 2018) develops, resulting in the formation of adverse metabolites that hamper normal cellular physiology. For example, an excess of free fatty acids such as palmitic or stearic acids induces the generation of toxic metabolites, leading to caspase-dependent apoptosis of hepatocytes (Kakisaka et al. 2012; Pfaffenbach et al. 2010) but also of cardiomyocytes (Drosatos and Schulze 2013; Zou et al. 2017) and endothelial cells (Artwohl et al. 2008; Chinen et al. 2007). Indeed, while free fatty acid-induced apoptosis of hepatocytes is a key feature of lipotoxicity in the context of NAFLD (Trauner et al. 2010), other reports have shown that excess palmitate induces endoplasmic reticulum stress and apoptosis in the context of atherosclerosis as well (Erbay et al. 2009). Therefore, free fatty acid-induced apoptosis of parenchymal cells appears to be a shared mechanism between NAFLD and atherosclerosis development.
Besides inducing apoptosis, free fatty acid influx into hepatocytes also influences the function of key enzymes and nuclear receptors involved in hepatic de novo lipogenesis, fatty acid oxidation, and cholesterol metabolism, thereby further disturbing hepatic lipid metabolism. Indeed, expression levels of acetyl-coenzyme-A carboxylase 1, a key enzyme in fatty acid metabolism (Barber et al. 2005), were shown to decrease in advanced stages of NASH compared to individuals with steatosis (Nagaya et al. 2010). Moreover, in the transcriptional levels, liver X receptor (LXR), a nuclear receptor involved with regulation of cholesterol, fatty acid, and glucose metabolism (Kalaany and Mangelsdorf 2006), correlated with intrahepatic inflammation and fibrosis in NAFLD patients (Ahn et al. 2014; Ni et al. 2017). In line, macrophage-targeted delivery of LXR agonist inside the atherosclerotic plaque reduced atherosclerosis progression (Guo et al. 2018), pointing toward a key function of LXR in both NAFLD and atherosclerosis development.
Lipotoxic responses also affect LSECs, a type of non-parenchymal cell that is specifically involved in maintaining hepatic vascular tone and quiescence of hepatic stellate cells that are responsible for the fibrotic response. Upon treatment with oxidized lipids (Zhang et al. 2014) or palmitic acid (Matsumoto et al. 2018), LSCEs directly or indirectly triggered the release of reactive oxygen species (ROS) production (Peters et al. 2018), which influences mechanisms related to inflammation and fibrosis (Ni et al. 2017).
Increased hepatic fat accumulation has also been associated with reduced levels of high-density lipoproteins (HDL) and increased levels of total plasma cholesterol, low-density lipoproteins (LDL), and very low-density lipoprotein particles (Koruk et al. 2003), the latter being involved with hepatic lipid export. Moreover, besides triglycerides and fatty acids, it has become evident that cholesterol is a key player in inducing hepatic inflammatory responses (Caballero et al. 2009; Plat et al. 2014; van Rooyen et al. 2011). Indeed, in the context of obesity-associated diseases, it was shown that cholesterol levels are associated with hepatic inflammation (Musso et al. 2003; Puri et al. 2007) and atherosclerosis (Ference et al. 2017) in humans. In agreement with these data, it has previously been shown that omitting cholesterol from the diet was able to prevent liver inflammation in hyperlipidemic and atherosclerosis-prone mice (Wouters et al. 2008), pointing toward cholesterol as a significant risk factor for early onset of NASH and progression of atherosclerosis.
In addition to lipotoxicity, glucotoxicity is a metabolic condition linked to increased intake of dietary sugars, resulting in hyperglycemia which may cause hepatotoxic effects by increasing steatosis (Mota et al. 2016). For instance, it was shown that high carbohydrate intake plays a role in de novo lipogenesis and hepatic steatosis (Ackerman et al. 2005; Neuschwander-Tetri et al. 2012), presumably via activation of lipogenic enzymes such as fatty acid synthase and stearoyl-CoA desaturase-1 (Maslak et al. 2015). In addition, high-fructose intake was shown to correlate with the severity of fibrosis in NAFLD patients (Basaranoglu et al. 2015; Mota et al. 2016) and carbohydrate intake associated with the progression of coronary atherosclerosis (Mozaffarian et al. 2004). Glucotoxic and lipotoxic products, including free fatty acids, cholesterol, and ceramides, among others (Han et al. 2008), are also involved in the activation of cellular stress responses. For instance, it has been shown that saturated long-chain fatty acids can disturb metabolic fluxes, thereby increasing the production of harmful lipid intermediates (Kakisaka et al. 2012). These intermediates can promote the release of ROS, leading to oxidative stress and hence the progression from steatosis to NASH (Matsuzawa et al. 2007; van Herpen and Schrauwen-Hinderling 2008) and the development of atherosclerosis (Nowak et al. 2017).
Overall, these evidences show that triglycerides, fatty acids, and cholesterol overload disturb essential processes in the liver that result in NAFLD features, placing lipids at the center of NAFLD development. Moreover, as these processes show striking similarities with disturbances present in atherosclerosis, lipotoxicity is a denominator linking NAFLD to atherosclerosis.
2.1.2 Oxidative Stress and Mitochondrial Dysfunction
As mentioned in the previous paragraph, part of the lipotoxic response involves the generation of ROS, resulting in oxidative stress. Oxidative stress comprises a state during which there is an imbalance between generation of ROS at one hand and an inability to detoxify (i.e., via antioxidant mechanisms) these oxygenated intermediates (Masarone et al. 2018). As a consequence, free radicals, peroxides, and related products are generated and react with biological components such as proteins, DNA, but also lipids (Finkel and Holbrook 2000). Indeed, considering the increased amount of lipids present during NAFLD conditions, larger quantities of lipid peroxidation products are present in the liver of NAFLD patients (Sumida et al. 2013) and contribute to the transition toward more serious stages of NAFLD (Busch et al. 2017; Feldstein et al. 2010). In addition, ROS is known to mediate endoplasmic reticulum stress, thereby causing the formation of misfolded proteins, which is a critical factor in NAFLD (Ashraf and Sheikh 2015) as well as the progression of atherosclerosis (Hotamisligil 2010; Tabas 2010). Moreover, cholesterol oxidation products that are part of oxidized low-density lipoproteins (oxLDL) are majorly involved in inflammatory and fibrotic responses in the liver (also further discussed in next section) (Bieghs et al. 2013).
Considering the key role of mitochondria in cellular oxygen consumption and production of ROS, lipotoxic influences on mitochondria have the potential to further aggravate oxidative stress (Dominguez-Perez et al. 2019). Under physiological conditions, fatty acid transport into the mitochondria is mediated via carnitine palmitoyltransferase 1 (CPT-1) in order to stimulate beta-oxidation. Nevertheless, the expression of Cpt-1 was shown to be reduced in NAFLD (Kohjima et al. 2007), findings that were further supported by Francque et al., showing that peroxisome proliferator-activated receptor alpha (PPARα), an important nuclear receptor regulating CPT-1, inversely correlated with disease severity in patients with NASH (Francque et al. 2015). By using isolated mitochondria, it was also shown that short chain ceramides increase mitochondrial permeability due to the generation of ceramide channels and increased cytochrome C release (Colombini 2010), thereby mediating toxic effects. Moreover, mitochondrial cholesterol accumulation caused mitochondrial dysfunction (Balboa et al. 2017), and based on studies in the context of neurotoxicity (Barbero-Camps et al. 2014), mitochondrial cholesterol may play a role in endoplasmic reticulum stress and subsequent apoptosis.
2.1.3 Hepatic Inflammation and Fibrosis
An essential pathophysiological process during NAFLD that also unites the lipotoxic response with the generation of oxidative stress is the presence of hepatic inflammation which can progress into hepatic fibrosis. In contrast to the uptake of non-modified LDL, it has been established that the uptake of oxLDL contributes to cholesterol-induced foam cell formation and metabolic inflammation (Lara-Guzman et al. 2018) in NASH (Houben et al. 2017) but also in the context of atherosclerosis (Binder et al. 2003). Moreover, the accumulation of oxidized lipids into the lysosomal compartment of macrophages activates inflammatory cascades including inflammasome complexes and apoptosis (Bieghs et al. 2013; Grebe et al. 2018; Hendrikx et al. 2013; Jerome 2010). Indeed, recent studies show that specific inhibition of the NLRP3 inflammasome not only reverses hepatic inflammation and fibrosis (Mridha et al. 2017) but also reduces atherosclerotic lesion development (van der Heijden et al. 2017), pointing toward an important role for the inflammasome in chronic inflammatory diseases (Cai et al. 2017; Duewell et al. 2010; Pan et al. 2018). Furthermore, cholesterol-mediated activation of inflammasomes decreases cholesterol efflux, thereby disturbing the regulation of bile acid metabolism. Previously, it was indeed shown that mice lacking the bile acid receptor farnesoid X receptor (FXR) had pro-atherogenic lipoproteins (Mencarelli and Fiorucci 2010) and increased hepatic bile acid levels (Sinal et al. 2000), pointing toward a potential role for FXR in cholesterol-induced liver inflammation. Indeed, improving cholesterol efflux in hepatic macrophages by overexpressing Cyp27a1, an enzyme responsible for the conversion of cholesterol into bile acids, reduced hepatic inflammation and fibrosis in an experimental model (Hendrikx et al. 2015). Via accumulation of oxidized lipids into lysosomes, also disturbances in autophagy contribute to increased levels of inflammation both during NAFLD (Wu et al. 2018b) and atherosclerosis (Martinet and De Meyer 2009). Besides cholesterol, also other lipids such as phospholipids (Lee et al. 2012) and fatty acids (Reinaud et al. 1989), can (non)enzymatically interact with free radicals, triggering inflammation by a wide variety of underlying processes (Houben et al. 2017).
While macrophages play a key role in the inflammatory response, hepatic stellate cells are the main drivers of the fibrotic response (Friedman 1993). After a damaging insult, stellate cells are activated, thereby secreting collagens and related matrix proteins that lead to generation of scar tissue or fibrosis (Peters et al. 2018; Schneiderhan et al. 2001), a pathological process also described in atherosclerosis (Ostovaneh et al. 2018). Relevantly, Chu et al. recently demonstrated that exposing hepatic stellate cells to fatty acids resulted in an increased secretion of CCL20, resulting in a switch from a quiescent to an activated hepatic stellate cell. These findings were further confirmed in humans, showing increased circulating CCL20 protein levels in patients with NAFLD-related fibrosis (Chu et al. 2018). Further data based on an elegant co-culturing system using primary liver cells pointed toward CCL5 as an important hepatic stellate cell-derived chemokine capable of mediating steatosis and pro-inflammatory responses in initially healthy hepatocytes (Kim et al. 2018). Moreover, in vivo induction of CCL5 in response to high-fat diet was also shown to serve as an important regulator of vascular remodeling, revealing a role for CCL5 and its receptor in atherogenesis (Lin et al. 2018). Therefore, multiple reports indicate that lipids enable fibrotic responses by influencing hepatic stellate cells.
2.2 Metabolic Crosstalk in NAFLD
As previously mentioned, the capacity of adipose tissue to store lipids determines the quantity of free fatty acids to be released into the circulation under high lipid conditions. However, besides its storage capacity, adipose tissue is known as a “secretory” organ, releasing adipokines and adipocytokines that influence other organs (Ouchi et al. 2011). For this reason, lipid-induced adipose tissue function increases the release of adipocytokines such as TNFα, IL6, IL18, and ANGPTL, leading to inflammatory responses in other metabolic organs such as the liver (Ouchi et al. 2011; Reilly et al. 2015). Moreover, the release of these adipokines also influences circulating immune cells, contributing to a state of chronic inflammation (Bijnen et al. 2018; Mancuso 2016; Nakamura et al. 2014). Due to this systemic impact, it is not surprising that adipokines also influence atherosclerosis development. Indeed, adipose tissue-released TNFα directly influenced atherosclerosis development (Tanaka and Sata 2018). Besides modulating inflammation, the increased release of free fatty acids also hinders the anti-lipolytic role of insulin, aggravating insulin resistance (Engin 2017; Sears and Perry 2015).
Another extrahepatic organ that has been linked to NAFLD development is the thyroid. Being an endocrine organ, the thyroid secretes hormones that have a role in the regulation of energy homeostasis including the metabolism of cholesterol and fatty acids (Sinha et al. 2018). Specifically, hypothyroidism is characterized by increased serum LDL and HDL levels and decreased triglyceride levels (Duntas 2002). Besides indirectly influencing hepatic lipid metabolism by modulating circulating lipid levels, thyroid hormones also directly affect hepatic lipid metabolism mainly via the presence of hepatic thyroid hormone receptors (THR) (Sinha et al. 2018). THRs are nuclear hormone receptors that function as ligand-dependent transcription factors influencing downstream metabolic genes (Davis et al. 2016) but also disturb other metabolic transcription factors such as PPARy, LXR, and sterol regulatory element-binding protein 1 (SREBP1c) (Araki et al. 2009; Wang et al. 2015). For this regulatory role on hepatic lipid metabolism, THR agonists were also considered for the management of hepatic steatosis (Cable et al. 2009) but later observed adverse effects resulted in discontinuation of these clinical trials (Lammel Lindemann and Webb 2016). Nevertheless, thyroid hormones analogues (rather than THRs) are still considered as potential future NAFLD treatment (Perra et al. 2008).
Another organ that has gained attention in the context of NAFLD is the brain. At one hand, NAFLD-related inflammation has been demonstrated to influence microglia in the brain, leading to alterations in microvasculature of the brain (Ghareeb et al. 2011; Kim et al. 2016). Furthermore, NAFLD-associated endothelial dysfunction and the procoagulant state were linked to the same microvascular alterations, which may contribute to disturbances in brain circulation, damage, and cognitive impairment (Lombardi et al. 2019). Besides the link to the aforementioned cerebrovascular diseases (Airaghi et al. 2018), other brain-related associations have been established to NAFLD. Firstly, a recent report from Horwath et al. demonstrated that endoplasmic reticulum stress in the subfornical organ of the brain, a brain region previously linked to appetite (Matsuda et al. 2017), directly mediated hepatic steatosis, thereby directly linking the brain to the liver in the context of NAFLD. Moreover, Weinstein et al. recently linked NAFLD to lower cerebral brain volume hinting at a more profound role for the brain in NAFLD (Weinstein et al. 2018). Finally, as a regulation center for energy metabolism, brain regions such as the arcuate nucleus in the hypothalamus sense the metabolic status and govern food intake (Schwartz et al. 2000), making an obvious link to obesity-related NAFLD. An essential hormone involved with the homeostatic regulation of energy and acting via the hypothalamus is leptin (Kwon et al. 2016). Notably, variants of leptin receptors associated with increased NAFLD susceptibility, pointing toward a potential role for hypothalamic leptin sensitivity in NAFLD (Zain et al. 2013). Additional evidence linking hypothalamic inflammation to hepatic steatosis further substantiated the potential involvement of the hypothalamus in NAFLD (Valdearcos et al. 2015).
To end, multiple reports have indicated the involvement of the gastrointestinal tract to play a role in NAFLD development. Under physiological circumstances, the intestinal lining serves as a physical barrier that separates the host from contents in the gut. Disruption of this barrier leads to intestinal permeability (Winer et al. 2016), allowing for leakage of intestinal bacteria and other products into the circulation. Indeed, leakage of lipopolysaccharide (LPS) derived from intestinal bacteria into the circulation (Kitabatake et al. 2017) can activate KCs in the liver (Ye et al. 2012), thereby directly resulting in NASH development (Kitabatake et al. 2017; Wigg et al. 2001). Relevantly, gut-derived serum LPS was similarly associated with atherosclerosis development, reaffirming the tight link between NASH and atherosclerosis (Pastori et al. 2017). Though there are limited studies providing a causal role of the gut microbiome in NAFLD pathogenesis, the current amount of evidence suggests that the gut microbiota are at least involved with the development of NAFLD (Gregory et al. 2015; Kaden-Volynets et al. 2018; Martinez-Guryn et al. 2018; Turnbaugh et al. 2006) (Wang et al. 2018). Other well-known factors linking the gut to NAFLD are bile acid metabolism (Dumas et al. 2006; Tremaroli and Backhed 2012), bacterial-derived short-chain fatty acids (Canfora et al. 2019), and the toxic compounds dimethylamine and trimethylamine that were converted by bacteria from choline (Spencer et al. 2011; Wang et al. 2011). In line with our other descriptions, each of these compounds has also been associated with atherosclerosis development (Chambers et al. 2018; Charach et al. 2018; Tang et al. 2013).
Based on these evidences, it is clear that the development of NAFLD is linked to pathophysiological processes that arise in other (metabolic) organs. This information fuels a view of NAFLD being a complex, systemic disease influenced by a range of other organs. It is therefore likely that future management of NAFLD will require a systemic rather than a liver-specific approach.
2.3 Genetic Predisposition to NAFLD
NAFLD is considered a polygenic disease, implying the involvement of a variety of genetic factors in predisposing individuals to disease onset. While mutations in the patatin-like phospholipase domain-containing 3 (PNPLA3) gene were initially associated with hepatic steatosis (Romeo et al. 2008), other reports have also correlated the PNPLA3 variation to NASH progression (BasuRay et al. 2019; Valenti et al. 2010). Similarly, PNPLA3 genetic variants are also associated with carotid atherosclerosis in younger patients NAFLD (Petta et al. 2013). Though PNPLA3 variants were recently linked to the ubiquitylation processes (BasuRay et al. 2017), the exact underlying mechanism explaining the onset of NAFLD is still unclear.
Additionally, based on several population studies, it was recently described that mutations in the transmembrane 6 superfamily 2 (TM6SF2), a key regulator of very low-density lipoprotein (VLDL) export, correlated with NASH progression (Manne et al. 2018) and cardiovascular disease (Li et al. 2018), most likely via changes in plasma lipids. Indeed, plasma lipids appear to be one of the common denominators predicting severity of both NAFLD and coronary artery disease (Brouwers et al. 2019).
Further genetic screenings for NAFLD revealed that glucokinase regulator (GCKR) (Santoro et al. 2012) and lysophospholipid acyltransferase 7 (known as MBOAT7) (Mancina et al. 2016), key enzymes for glucose metabolism and reacetylation of phospholipids, respectively, as well as neurocan were associated with NAFLD development (Speliotes et al. 2011). Yet, a more recent study focusing on the aforementioned NAFLD-risk alleles (PNPLA3, TM6SF2, GCKR, and LYPLAL1) substantiated the heterogeneity of the NAFLD phenotype between patients, emphasizing the complexity of the disease (Sliz et al. 2018). As such, though genetic predisposition may influence disease onset, other pathophysiological factors that are independent of genetic predisposition are likely a stronger contributor to explain NAFLD development.
A line of research that has received increased attention is the influence of epigenetic changes on NAFLD development (Eslam et al. 2018). Epigenetic changes are induced by modifications in the regulators of DNA such as DNA methylation reactions, histone proteins, chromatin structure, and RNA-based mechanisms resulting in changes in genes expression (Eslam et al. 2018). These epigenetic modifications influence aging-related processes which contribute to NALFD (Horvath et al. 2014) but can also be transmitted to the progeny, thereby combining genetic and environmental factors involved in the development of disease. Mice that were rechallenged with a high-fat diet after being exposed to this diet during fetal life showed more several hepatic steatosis, inflammation, and fibrosis (Bruce et al. 2009). This influence of a detrimental fetal environment on NAFLD has been further substantiated by studies linking intrauterine growth retardation to increased risk of developing NAFLD (Nobili et al. 2007; Suomela et al. 2016; Valenti and Romeo 2016). Furthermore, methylation patterns of genes involved insulin signaling associated with the presence of NASH, which disappeared after bariatric surgery (Ahrens et al. 2013). As such, though being in its infancy, epigenetic modifications are expected to have an important role on NAFLD progression (Eslam et al. 2018).
3 Therapeutics
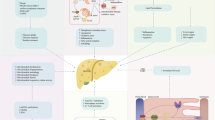
The involvement of different mechanisms in the pathogenesis of NAFLD also adds a level of complexity in finding appropriate therapeutic options to improve the different aspects of NAFLD. While therapies to reduce hepatic steatosis are known, a major problem is reversing the inflammatory component in the liver. Indeed, at present, no effective therapeutic approaches exist for reducing hepatic inflammation (Houben et al. 2017). From market size perspective, NASH-related therapeutics generated $1,179 million in 2017 and is estimated to reach $21,478 million by 2025 (Shinde 2018), pointing toward the huge demand for NASH treatments. Due to the magnitude of this health concern and its potential impact on healthcare, multiple treatments are currently being investigated with the aim to decrease inflammation and fibrosis (Oh et al. 2016). In this section, we provide a selection of currently investigated therapeutic approaches for NAFLD and demonstrate that these approaches are also investigated in the context of atherosclerosis (see Fig. 1). From this perspective, we further highlight the link between NAFLD and atherosclerosis.

Targets for NAFLD therapy. Besides exercise, changing the dietary pattern or surgical intervention and pharmacological intervention to improve NAFLD targets the pathological mechanisms of lipotoxicity, insulin/glucose metabolism, hepatic inflammation, fibrosis, as well as bile acid metabolism. HmG-CoA 3-hydroxy-3-methylglutarylcoenzyme A, PPAR peroxisome proliferator-activated receptor, GLP1R glucagon-like peptide-1 receptor, DPP4 dipeptidyl peptidase-4, SGLT2 sodium-glucose transport protein 2, FXR farnesoid X receptor
3.1 Dietary/Lifestyle Intervention and Bariatric Surgery
Dietary changes and lifestyle interventions resulting in weight reduction are currently the first-line therapy for NAFLD patients (Sumida and Yoneda 2018). Indeed, dietary restriction is the most effective way to reduce liver fat (Marchesini et al. 2016; Patel et al. 2015). Furthermore, it has been suggested that hepatic triglyceride content normalizes after a few weeks under a strictly hypocaloric diet (Patel et al. 2015), i.e., low fat and low carbohydrate, which has been proposed as the optimal composition of a diet for NAFLD patients (Asrih and Jornayvaz 2014). Apart from dietary changes, lifestyle modification is another way to lose weight, for instance, via physical activity instead of sedentariness (Fabricatore 2007). However, compared to dietary restriction, physical activity is less effective in losing weight due to reduced caloric consumption as compared with dietary restriction (Marchesini et al. 2016; Zou et al. 2018). While dietary change and lifestyle intervention are able to reduce body weight, many patients cannot adhere to these interventions. Therefore, bariatric surgery, and more recently termed metabolic surgery (Sasaki et al. 2014), typically results in massive weight loss and in concordant improvements in liver histology (Dixon et al. 2004). Indeed, Mummadi et al. reported that the resolution rates of steatosis, steatohepatitis, and fibrosis were 91.6%, 81.3%, and 65.5% in 15 studies using paired liver biopsies after bariatric surgery (Mummadi et al. 2008). Recently, a 1-year follow-up study by Nickel et al. also supported bariatric surgery as an effective treatment for NAFLD (Nickel et al. 2018). However, as not all NAFLD patients qualify for bariatric surgery, other interventions are necessary to combat NAFLD and related symptoms.
3.2 Targeting Lipotoxicity
As accumulation of lipids inside the liver comprises an essential component in the development of NAFLD, multiple therapeutic approaches have aimed to reduce hepatic lipids with the objective to concordantly reduce hepatic inflammation and fibrosis. The best known example of cholesterol-reducing agents are statins, which are drugs aimed at inhibiting 3-hydroxy-3-methylglutarylcoenzyme A (HmG-CoA) reductase, the rate-limiting enzyme in the cholesterol biosynthesis pathway (Stancu and Sima 2001). Showing beneficial results in the context of atherosclerosis (Bittencourt and Cerci 2015), statins were also investigated in NAFLD progression. Though some improvements were observed in hepatic damage and inflammation (Kargiotis et al. 2014), other reports declare only minor improvements or even increasing levels of inflammation and fibrosis when statins are administered over a longer period of time (Hyogo et al. 2008). Moreover, recent observations pointing toward the detrimental effects of statins on aging and associated processes (Cholesterol Treatment Trialists 2019; Izadpanah et al. 2015) raise drawbacks for using these drugs under certain conditions.
Finally, and potentially, the most promising pharmacological compound currently under investigation to regress NASH are agonists of PPAR. PPARs are nuclear receptor proteins exerting key regulatory functions as transcription factors on metabolism, among other physiological processes (Dubois et al. 2017). Currently, three types of PPARs (being PPARα, PPARβ/δ, and PPARγ) are known and used as targets to improve MetS-related symptoms. In the context of NAFLD, and specifically NASH, the PPARγ agonist class thiazolidinediones has been shown to improve hepatic inflammation and advanced fibrosis (Bril et al. 2018; Musso et al. 2017). Furthermore, a new agonist, named elafibranor (or GFT505) that targets PPARα and PPARδ, was recently shown to improve hepatic inflammation and fibrosis, along with improvements in systemic inflammation, lipid, and glucose metabolism (Ratziu et al. 2016; Staels et al. 2013).
Due to these positive results, both thiazolidinediones and elafibranor are currently under clinical investigation for the treatment of NASH (Connolly et al. 2018). With regard to their application in atherosclerosis, thiazolidinediones have also been proven to slow progression of atherosclerosis in patients (Saremi et al. 2013), while elafibranor was so far not tested in this context. However, preliminary results in an atherosclerotic mouse model suggest that this latter PPARα/δ dual agonist might also be beneficial in the context of atherosclerosis (Graham et al. 2005).
Besides pharmacological intervention, a more convenient manner of reducing lipids is by means of dietary intervention. Apart from following dietary regimens in which the composition of lipids, protein, and carbohydrates is modulated and caloric intake is minimized (Ratziu et al. 2015) in order to achieve improvements in energy metabolism (Kargulewicz et al. 2014; Perumpail et al. 2017), another approach is to increase the intake of food components named functional foods. Plant sterol and stanol esters are examples of such functional foods that have been proven to reduce serum total and LDL cholesterol (Lichtenstein and Deckelbaum 2001; Plat et al. 2019), leading to improvements in atherosclerosis (Kohler et al. 2017) and NAFLD (Plat et al. 2014). However, more studies are necessary to prove the potential benefit of plant sterol and stanol esters in NAFLD patients.
3.3 Targeting Insulin/Glucose Metabolism
As diabetes has been associated with several stages of NAFLD (Hazlehurst et al. 2016), researchers have investigated the impact of improving insulin and glucose metabolism in order to improve aspects of NAFLD. Firstly, glucagon-like peptide-1 receptor (GLP1R) agonists, which mimic the function of incretins, are currently investigated in NAFLD (Gastaldelli and Marchesini 2016). GLP is a peptide derived from the L cells of the lower gastrointestinal tract (the small intestine and proximal colon) and known to enhance insulin secretion from pancreatic β cells and inhibit glucagon release from pancreatic α cells (Campbell and Drucker 2013; Ratziu et al. 2015). Whereas the GLP1R agonist exenatide enhanced hepatic steatosis (Tanaka et al. 2014), hepatic oxidative stress, and hepatic inflammation (Shao et al. 2018) and improved adipose tissue lipolysis in different in vivo models, the application of dulaglutide, lixisenatide, liraglutide, and, recently, semaglutide also shows promising results in terms of improvements in hepatic fat, damage, inflammation, and fibrosis (Armstrong et al. 2016; Cusi et al. 2018; Ipsen et al. 2018; Koutsovasilis et al. 2018; Petit et al. 2017; Rakipovski et al. 2018). As such, liraglutide (Armstrong et al. 2016) and semaglutide were under extensive clinical investigation. While liraglutide will not be further evaluated in phase 3 development, Novo Nordisk has initiated a phase 2b trial (NCT02970942) evaluating semaglutide versus placebo in 372 participants with stage F2-F3 fibrosis and NAS ≥4 with a score of at least 1 for each of the components (steatosis, ballooning, and lobular inflammation) (Connolly et al. 2018). Relevantly, as diabetes has also been linked to formation and progression of the atherosclerotic plaque (Beckman et al. 2002; Chait and Bornfeldt 2009; Katakami 2018), several GLP1R agonists have also been shown to improve atherosclerosis including exenatide, liraglutide, and semaglutide (Li et al. 2017; Marso et al. 2013; Rakipovski et al. 2018; Yang et al. 2017).
Besides the GLRP1 agonists, another approach to improve the GLP1-related effects on insulin and glucose metabolism is by administration of dipeptidyl peptidase-4 (DPP4) inhibitors. DPP4 (also referred to as CD26) is an enzyme known to degrade GLP1. Hence, inhibition of DPP4 enhances the activity of GLP1. While administration of the DPP4 inhibitor sitagliptin has been successfully applied in diabetic patients (Derosa et al. 2015; Drucker and Nauck 2006), several reports demonstrated only minor to no beneficial effects on hepatic fat content or hepatic fibrosis (Cui et al. 2016; Joy et al. 2017). Relevantly, these negative results in the context of NAFLD were also confirmed in atherosclerosis, showing only minor effects on coronary artery plaque improvement (Katakami et al. 2018; Nozue et al. 2016).
Another class of pharmacological compounds that specifically improve glucose metabolism is inhibitors for sodium-glucose transport protein 2 (SGLT2), a transporter protein in the kidney responsible for the reabsorption of glucose (Hsia et al. 2017; Marshall 2018; van Baar et al. 2018). In contrast to the minor effects of the DPP4 inhibitors on NAFLD, the SGLT2 inhibitors canagliflozin, ipragliflozin, and luseogliflozin all show substantial improvements in hepatic steatosis, apoptosis, and fibrosis in in vivo models and NAFLD patients (Ito et al. 2017; Kabil and Mahmoud 2018; Shiba et al. 2018; Shibuya et al. 2018; Sumida et al. 2019). In line with the previously described similarities between NAFLD and atherosclerosis, SGLT2 inhibitors were shown to also positively impact atherosclerosis progression and development (Nakatsu et al. 2017; Nasiri-Ansari et al. 2018; Tanaka et al. 2016; Zelniker et al. 2019).
Together, several treatments aimed at improving insulin or glucose metabolism have positive effects on several aspects of NAFLD, substantiating the role of insulin and glucose metabolism in the progression of NAFLD. Moreover, therapeutic products that improve features of NAFLD also positively impact atherosclerosis, providing further evidence for the similarities between NAFLD and atherosclerosis.
3.4 Targeting Hepatic Inflammation and Fibrosis
Another therapeutic approach to ameliorate NAFLD is to directly target components of the inflammatory and/or fibrotic pathway, as these features are the main cause for hepatic symptoms in NAFLD patients and are also responsible for the development toward advanced liver diseases (Schuster et al. 2018). The caspase inhibitor, emricasan, is one of those investigated compounds targeting the inflammatory aspect of NAFLD. Specifically, caspases are enzymes involved with several physiological processes including inflammation, making them an attractive inflammatory drug target. Administration of emricasan to NAFLD patients showed improvements in hepatic damage (as evidenced by reductions in alanine transaminase (ALT) levels) (Shiffman et al. 2019). However, recent negative results with this compound have questioned its continuation for further clinical investigation (Garcia-Tsao et al. 2019). Another potential caspase-related target for inflammatory drugs is blocking the activation of inflammasomes (Schuster et al. 2018). Indeed, inhibition of the P2X7 receptor, which is known to activate the NLRP3 inflammasome (Amores-Iniesta et al. 2017), via SGM-1019 resulted in improvements in hepatic inflammation and fibrosis in mouse models and NASH patients (Dabbagh et al. 2018), substantiating its further clinical investigation in NAFLD.
Another way to reduce inflammation and fibrosis is by blocking the effect of cytokines and chemokines that propagate the inflammatory reaction. With this regard, the C-C chemokine receptor type 2/C-C chemokine receptor 5 (CCR2/CCR5) inhibitor cenicriviroc has been successfully created. Specifically, cenicriviroc reduced hepatic fibrosis, inflammation, as well as systemic inflammatory parameters in NAFLD patients and animal models (Friedman et al. 2018; Lefebvre et al. 2016; Tacke 2018). Currently, cenicriviroc is being evaluated in phase 3 trials, targeting patients with F2-F3 fibrosis and having an anticipated enrollment of 2,000 participants (Connolly et al. 2018). Additionally, inhibition of galectin-3, a protein belonging to the lectin family and previously linked to NASH severity, has provided promising first results (Harrison et al. 2016), which need to be further validated. Finally, inhibition of apoptosis signal-regulating kinase 1 (ASK1) has also been investigated as drug target to reduce hepatic inflammation and fibrosis. ASK1 is part of the mitogen-activated protein kinase family and has been shown an essential role in NASH development in patients and mouse models (Wang et al. 2017; Xiang et al. 2016; Zhang et al. 2018). In line with this observation, inhibition of ASK1 using selonsertib has shown impressive improvements in hepatic inflammation and fibrosis (Loomba et al. 2017; Younossi et al. 2018b). Selonsertib is currently under evaluation in two phase 3 clinical trials (STELLAR-3 [NCT03053050] and STELLAR-4 [NCT03053063]) for the treatment of NASH (Connolly et al. 2018).
Similar to the dual therapeutic effects of approaches targeting insulin and glucose metabolism, therapeutic approaches targeting inflammation and fibrosis also show dual positive influences in atherosclerosis and NASH. Galectin-3 has, for example, been linked atherosclerotic plaque progression (Papaspyridonos et al. 2008), and its inhibition results in reductions of atherosclerotic lesion size in vivo (MacKinnon et al. 2013). However, compared to drugs targeting insulin and glucose metabolism, targeting inflammation and fibrosis pathways is less investigated in the context of atherosclerosis, as exemplified by no described clinical studies for selonsertib, emricasan, ASK1 inhibitors, or cenicriviroc.
3.5 Targeting Bile Acid Metabolism
Hepatic components that have been extensively linked to different aspects of NAFLD include bile acids. Bile acids have regulatory functions on lipid and glucose metabolism, impact gut microbiota composition, and influence hepatic inflammation and damage (Schuster et al. 2018), explaining why modulation of bile acid metabolism has been an attractive therapeutic target for NAFLD. Firstly, the hepatoprotective natural bile acid ursodeoxycholic acid (UDCA) has been shown to exert beneficial effects on immune function, has anti-apoptotic and insulin-sensitizing effects, and reduces harmful effects of reactive oxygen species (Kars et al. 2010; Ljubuncic et al. 1996; Rodrigues et al. 1998), all aspects present during NAFLD. Indeed, besides improvements in hepatic steatosis, inflammation, and damage in NASH animal models, two randomized controlled trials showed improvements in lobular inflammation and hepatic fibrosis along with reductions in ALT levels upon UDCA administration (Leuschner et al. 2010; Ratziu et al. 2011). However, other studies showed no effect of UDCA administration in NASH patients (Liechti and Dufour 2012), emphasizing the need for further investigation. In addition, agonists of FXR, a nuclear receptor that has been linked to NAFLD (Zhang et al. 2009), have also been tested in NAFLD. Obeticholic acid (OCA), a semisynthetic variant of chenodeoxycholic acid, showed reductions in steatosis and fibrosis (Fiorucci et al. 2005; Goto et al. 2018), and recently, the first promising results were published from the FLINT study, investigated OCA in NAFLD patients (Neuschwander-Tetri et al. 2015). Currently, OCA is being evaluated in the phase 3 study REGENERATE (NCT02548351) for the treatment of NASH (Connolly et al. 2018).
Strikingly, though bile acids are produced by the liver, multiple evidences have pointed toward their systemic effects on inflammation, cell death, and apoptosis (Chiang 2013). As such, recent evidences have also shown beneficial effects of UDCA (Bode et al. 2016) and OCA (Hageman et al. 2010; Moris et al. 2017) in atherosclerotic models.
4 Conclusion
As the hepatic component of the MetS, NAFLD comprises one of the largest global health threats of the twenty-first century. Though the exact etiology of why NAFLD patients progress from hepatic steatosis to hepatic inflammation and fibrosis is unclear, several studies have established key pathophysiological processes contributing to hepatic inflammation. Considering this large amount of processes involved with NAFLD (which have intra- and extrahepatic origins), it is clear that NAFLD is a complex, systemic disease with high interindividual variation, pointing toward combination therapies or personalized medicine as potential future directions for NAFLD. Moreover, due to this systemic nature, it is clear that NAFLD and atherosclerosis are very closely linked (Bieghs et al. 2012), implying the liver as a potential target to manage atherosclerosis.
Abbreviations
- ALT:
-
Alanine transaminase
- ASK1:
-
Apoptosis signal-regulating kinase 1
- CCR2/CCR5:
-
C-C chemokine receptor type 2/C-C chemokine receptor 5
- CKD:
-
Chronic kidney disease
- CPT-1:
-
Carnitine palmitoyltransferase 1
- DPP4:
-
Dipeptidyl peptidase-4
- ESLD:
-
End-stage of liver disease
- FXR:
-
Farnesoid X receptor
- GCKR:
-
Glucokinase regulator
- GLP1R:
-
Glucagon-like peptide-1 receptor
- HCC:
-
Hepatocellular carcinoma
- HDL:
-
High-density lipoproteins
- HmG-CoA:
-
3-Hydroxy-3-methylglutarylcoenzyme A
- HR-QOL:
-
Health-related quality of life
- KCs:
-
Kupffer cells
- LDL:
-
Low-density lipoproteins
- LPS:
-
Lipopolysaccharide
- LSECs:
-
Liver sinusoidal endothelial cells
- LXR:
-
Liver X receptor
- MetS:
-
Metabolic syndrome
- NAFLD:
-
Nonalcoholic fatty liver disease
- NASH:
-
Nonalcoholic steatohepatitis
- OCA:
-
Obeticholic acid
- oxLDL:
-
Oxidized low-density lipoproteins
- PBPLA3:
-
Patatin-like phospholipase domain-containing 3
- PCOS:
-
Polycystic ovary syndrome
- PPARα:
-
Peroxisome proliferator-activated receptor alpha
- ROS:
-
Reactive oxygen species
- SGLT2:
-
Sodium-glucose transport protein 2
- SREBP1c:
-
Sterol regulatory element-binding protein 1
- T2DM:
-
Type 2 diabetes mellitus
- THR:
-
Thyroid hormone receptors
- TM6SF2:
-
Transmembrane 6 superfamily 2
- UDCA:
-
Ursodeoxycholic acid
- VLDL:
-
Very low-density lipoprotein
References
Abdelmalek MF (2016) NAFLD: the clinical and economic burden of NAFLD: time to turn the tide. Nat Rev Gastroenterol Hepatol 13:685–686
Ackerman Z, Oron-Herman M, Grozovski M, Rosenthal T, Pappo O, Link G, Sela BA (2005) Fructose-induced fatty liver disease: hepatic effects of blood pressure and plasma triglyceride reduction. Hypertension 45:1012–1018
Afendy A, Kallman JB, Stepanova M, Younoszai Z, Aquino RD, Bianchi G, Marchesini G, Younossi ZM (2009) Predictors of health-related quality of life in patients with chronic liver disease. Aliment Pharmacol Ther 30:469–476
Ahn SB, Jang K, Jun DW, Lee BH, Shin KJ (2014) Expression of liver X receptor correlates with intrahepatic inflammation and fibrosis in patients with nonalcoholic fatty liver disease. Dig Dis Sci 59:2975–2982
Ahrens M, Ammerpohl O, von Schonfels W, Kolarova J, Bens S, Itzel T, Teufel A, Herrmann A, Brosch M, Hinrichsen H, Erhart W, Egberts J, Sipos B, Schreiber S, Hasler R, Stickel F, Becker T, Krawczak M, Rocken C, Siebert R, Schafmayer C, Hampe J (2013) DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab 18:296–302
Airaghi L, Rango M, Maira D, Barbieri V, Valenti L, Lombardi R, Biondetti P, Fargion S, Fracanzani AL (2018) Subclinical cerebrovascular disease in NAFLD without overt risk factors for atherosclerosis. Atherosclerosis 268:27–31
Albillos A, Lario M, Alvarez-Mon M (2014) Cirrhosis-associated immune dysfunction: distinctive features and clinical relevance. J Hepatol 61:1385–1396
Amores-Iniesta J, Barbera-Cremades M, Martinez CM, Pons JA, Revilla-Nuin B, Martinez-Alarcon L, di Virgilio F, Parrilla P, Baroja-Mazo A, Pelegrin P (2017) Extracellular ATP activates the NLRP3 inflammasome and is an early danger signal of skin allograft rejection. Cell Rep 21:3414–3426
Angulo P (2002) Nonalcoholic fatty liver disease. N Engl J Med 346:1221–1231
Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC, Lafferty HD, Stahler A, Haflidadottir S, Bendtsen F (2015) Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 149:389–97.e10
Anstee QM, Day CP (2013) The genetics of NAFLD. Nat Rev Gastroenterol Hepatol 10:645–655
Araki O, Ying H, Zhu XG, Willingham MC, Cheng SY (2009) Distinct dysregulation of lipid metabolism by unliganded thyroid hormone receptor isoforms. Mol Endocrinol 23:308–315
Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, Hazlehurst JM, Guo K, Team LT, Abouda G, Aldersley MA, Stocken D, Gough SC, Tomlinson JW, Brown RM, Hubscher SG, Newsome PN (2016) Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 387:679–690
Artwohl M, Lindenmair A, Sexl V, Maier C, Rainer G, Freudenthaler A, Huttary N, Wolzt M, Nowotny P, Luger A, Baumgartner-Parzer SM (2008) Different mechanisms of saturated versus polyunsaturated FFA-induced apoptosis in human endothelial cells. J Lipid Res 49:2627–2640
Ashraf NU, Sheikh TA (2015) Endoplasmic reticulum stress and oxidative stress in the pathogenesis of non-alcoholic fatty liver disease. Free Radic Res 49:1405–1418
Asrih M, Jornayvaz FR (2014) Diets and nonalcoholic fatty liver disease: the good and the bad. Clin Nutr 33:186–190
Balboa E, Castro J, Pinochet MJ, Cancino GI, Matias N, Saez PJ, Martinez A, Alvarez AR, Garcia-Ruiz C, Fernandez-Checa JC, Zanlungo S (2017) MLN64 induces mitochondrial dysfunction associated with increased mitochondrial cholesterol content. Redox Biol 12:274–284
Barber MC, Price NT, Travers MT (2005) Structure and regulation of acetyl-CoA carboxylase genes of metazoa. Biochim Biophys Acta 1733:1–28
Barbero-Camps E, Fernandez A, Baulies A, Martinez L, Fernandez-Checa JC, Colell A (2014) Endoplasmic reticulum stress mediates amyloid beta neurotoxicity via mitochondrial cholesterol trafficking. Am J Pathol 184:2066–2081
Basaranoglu M, Basaranoglu G, Bugianesi E (2015) Carbohydrate intake and nonalcoholic fatty liver disease: fructose as a weapon of mass destruction. Hepatobiliary Surg Nutr 4:109–116
BasuRay S, Smagris E, Cohen JC, Hobbs HH (2017) The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 66:1111–1124
BasuRay S, Wang Y, Smagris E, Cohen JC, Hobbs HH (2019) Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc Natl Acad Sci U S A 116:9521–9526
Baumeister SE, Volzke H, Marschall P, John U, Schmidt CO, Flessa S, Alte D (2008) Impact of fatty liver disease on health care utilization and costs in a general population: a 5-year observation. Gastroenterology 134:85–94
Beckman JA, Creager MA, Libby P (2002) Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA 287:2570–2581
Bellentani S, Marino M (2009) Epidemiology and natural history of non-alcoholic fatty liver disease (NAFLD). Ann Hepatol 8(Suppl 1):S4–S8
Bellentani S, Saccoccio G, Masutti F, Croce LS, Brandi G, Sasso F, Cristanini G, Tiribelli C (2000) Prevalence of and risk factors for hepatic steatosis in northern Italy. Ann Intern Med 132:112–117
Bieghs V, Rensen PC, Hofker MH, Shiri-Sverdlov R (2012) NASH and atherosclerosis are two aspects of a shared disease: central role for macrophages. Atherosclerosis 220:287–293
Bieghs V, Walenbergh SM, Hendrikx T, van Gorp PJ, Verheyen F, Olde Damink SW, Masclee AA, Koek GH, Hofker MH, Binder CJ, Shiri-Sverdlov R (2013) Trapping of oxidized LDL in lysosomes of Kupffer cells is a trigger for hepatic inflammation. Liver Int 33:1056–1061
Bijnen M, Josefs T, Cuijpers I, Maalsen CJ, van de Gaar J, Vroomen M, Wijnands E, Rensen SS, Greve JWM, Hofker MH, Biessen EAL, Stehouwer CDA, Schalkwijk CG, Wouters K (2018) Adipose tissue macrophages induce hepatic neutrophil recruitment and macrophage accumulation in mice. Gut 67:1317–1327
Binder CJ, Horkko S, Dewan A, Chang MK, Kieu EP, Goodyear CS, Shaw PX, Palinski W, Witztum JL, Silverman GJ (2003) Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med 9:736–743
Bittencourt MS, Cerci RJ (2015) Statin effects on atherosclerotic plaques: regression or healing? BMC Med 13:260
Bode N, Grebe A, Kerksiek A, Lutjohann D, Werner N, Nickenig G, Latz E, Zimmer S (2016) Ursodeoxycholic acid impairs atherogenesis and promotes plaque regression by cholesterol crystal dissolution in mice. Biochem Biophys Res Commun 478:356–362
Boursier J, Fabron C, Lafuma A, Bureau I (2018) NASH/NAFLD patients with end stage liver disease experienced high inpatient hospitalization costs and substantial disease progression: results of a French national database on hospital care analysis. J Hepatol 68:S238–S239
Bril F, Kalavalapalli S, Clark VC, Lomonaco R, Soldevila-Pico C, Liu IC, Orsak B, Tio F, Cusi K (2018) Response to pioglitazone in patients with nonalcoholic steatohepatitis with vs without type 2 diabetes. Clin Gastroenterol Hepatol 16:558–566.e2
Brouwers M, Simons N, Stehouwer CDA, Koek GH, Schaper NC, Isaacs A (2019) Relationship between nonalcoholic fatty liver disease susceptibility genes and coronary artery disease. Hepatol Commun 3:587–596
Bruce KD, Cagampang FR, Argenton M, Zhang J, Ethirajan PL, Burdge GC, Bateman AC, Clough GF, Poston L, Hanson MA, McConnell JM, Byrne CD (2009) Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 50:1796–1808
Busch CJ, Hendrikx T, Weismann D, Jackel S, Walenbergh SM, Rendeiro AF, Weisser J, Puhm F, Hladik A, Goderle L, Papac-Milicevic N, Haas G, Millischer V, Subramaniam S, Knapp S, Bennett KL, Bock C, Reinhardt C, Shiri-Sverdlov R, Binder CJ (2017) Malondialdehyde epitopes are sterile mediators of hepatic inflammation in hypercholesterolemic mice. Hepatology 65:1181–1195
Caballero F, Fernandez A, de Lacy AM, Fernandez-Checa JC, Caballeria J, Garcia-Ruiz C (2009) Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J Hepatol 50:789–796
Cable EE, Finn PD, Stebbins JW, Hou J, Ito BR, van Poelje PD, Linemeyer DL, Erion MD (2009) Reduction of hepatic steatosis in rats and mice after treatment with a liver-targeted thyroid hormone receptor agonist. Hepatology 49:407–417
Cai C, Zhu X, Li P, Li J, Gong J, Shen W, He K (2017) NLRP3 deletion inhibits the non-alcoholic steatohepatitis development and inflammation in Kupffer cells induced by palmitic acid. Inflammation 40:1875–1883
Calzadilla Bertot L, Adams LA (2016) The natural course of non-alcoholic fatty liver disease. Int J Mol Sci 17:774
Campbell JE, Drucker DJ (2013) Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab 17:819–837
Canfora EE, Meex RCR, Venema K, Blaak EE (2019) Gut microbial metabolites in obesity, NAFLD and T2DM. Nat Rev Endocrinol 15:261–273
Chait A, Bornfeldt KE (2009) Diabetes and atherosclerosis: is there a role for hyperglycemia? J Lipid Res 50(Suppl):S335–S339
Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ (2012) The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 55:2005–2023
Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, Harrison SA, Brunt EM, Sanyal AJ (2018) The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67:328–357
Chambers ES, Preston T, Frost G, Morrison DJ (2018) Role of gut microbiota-generated short-chain fatty acids in metabolic and cardiovascular health. Curr Nutr Rep 7:198–206
Charach G, Argov O, Geiger K, Charach L, Rogowski O, Grosskopf I (2018) Diminished bile acids excretion is a risk factor for coronary artery disease: 20-year follow up and long-term outcome. Ther Adv Gastroenterol 11. https://doi.org/10.1177/1756283X17743420
Chiang JY (2013) Bile acid metabolism and signaling. Compr Physiol 3:1191–1212
Chinen I, Shimabukuro M, Yamakawa K, Higa N, Matsuzaki T, Noguchi K, Ueda S, Sakanashi M, Takasu N (2007) Vascular lipotoxicity: endothelial dysfunction via fatty-acid-induced reactive oxygen species overproduction in obese Zucker diabetic fatty rats. Endocrinology 148:160–165
Cholesterol Treatment Trialists (2019) Efficacy and safety of statin therapy in older people: a meta-analysis of individual participant data from 28 randomised controlled trials. Lancet 393:407–415
Chu X, Jin Q, Chen H, Wood GC, Petrick A, Strodel W, Gabrielsen J, Benotti P, Mirshahi T, Carey DJ, Still CD, DiStefano JK, Gerhard GS (2018) CCL20 is up-regulated in non-alcoholic fatty liver disease fibrosis and is produced by hepatic stellate cells in response to fatty acid loading. J Transl Med 16:108
Colombini M (2010) Ceramide channels and their role in mitochondria-mediated apoptosis. Biochim Biophys Acta 1797:1239–1244
Connolly JJ, Ooka K, Lim JK (2018) Future pharmacotherapy for non-alcoholic steatohepatitis (NASH): review of phase 2 and 3 trials. J Clin Transl Hepatol 6:264–275
Cui J, Philo L, Nguyen P, Hofflich H, Hernandez C, Bettencourt R, Richards L, Salotti J, Bhatt A, Hooker J, Haufe W, Hooker C, Brenner DA, Sirlin CB, Loomba R (2016) Sitagliptin vs. placebo for non-alcoholic fatty liver disease: a randomized controlled trial. J Hepatol 65:369–376
Cusi K, Sattar N, Garcia-Perez LE, Pavo I, Yu M, Robertson KE, Karanikas CA, Haupt A (2018) Dulaglutide decreases plasma aminotransferases in people with type 2 diabetes in a pattern consistent with liver fat reduction: a post hoc analysis of the AWARD programme. Diabet Med 35:1434–1439
Dabbagh K, Dodson GS, Yamamoto L, Baeza-Raja B, Goodyear AW (2018) Preclinical and first-in human development of SGM-1019, a first-in-class novel small molecule modulator of inflammasome activity for the treatment of nonalcoholic steatohepatitis (NASH). J Hepatol 68:S60–S60
Dan AA, Kallman JB, Wheeler A, Younoszai Z, Collantes R, Bondini S, Gerber L, Younossi ZM (2007) Health-related quality of life in patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther 26:815–820
Davis PJ, Goglia F, Leonard JL (2016) Nongenomic actions of thyroid hormone. Nat Rev Endocrinol 12:111–121
Derosa G, D'Angelo A, Maffioli P (2015) Sitagliptin in type 2 diabetes mellitus: efficacy after five years of therapy. Pharmacol Res 100:127–134
Dixon JB, Bhathal PS, Hughes NR, O'Brien PE (2004) Nonalcoholic fatty liver disease: improvement in liver histological analysis with weight loss. Hepatology 39:1647–1654
Dominguez-Perez M, Simoni-Nieves A, Rosales P, Nuno-Lambarri N, Rosas-Lemus M, Souza V, Miranda RU, Bucio L, Uribe Carvajal S, Marquardt JU, Seo D, Gomez-Quiroz LE, Gutierrez-Ruiz MC (2019) Cholesterol burden in the liver induces mitochondrial dynamic changes and resistance to apoptosis. J Cell Physiol 234:7213–7223
Drosatos K, Schulze PC (2013) Cardiac lipotoxicity: molecular pathways and therapeutic implications. Curr Heart Fail Rep 10:109–121
Drucker DJ, Nauck MA (2006) The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 368:1696–1705
Dubois V, Eeckhoute J, Lefebvre P, Staels B (2017) Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J Clin Invest 127:1202–1214
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464:1357–1361
Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, Fearnside J, Tatoud R, Blanc V, Lindon JC, Mitchell SC, Holmes E, McCarthy MI, Scott J, Gauguier D, Nicholson JK (2006) Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci U S A 103:12511–12516
Duntas LH (2002) Thyroid disease and lipids. Thyroid 12:287–293
Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S, Hultcrantz R (2015) Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 61:1547–1554
El-Serag HB, Rudolph KL (2007) Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 132:2557–2576
Elwing JE, Lustman PJ, Wang HL, Clouse RE (2006) Depression, anxiety, and nonalcoholic steatohepatitis. Psychosom Med 68:563–569
Engin AB (2017) What is lipotoxicity? Adv Exp Med Biol 960:197–220
Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, Fazio S, Wiest MM, Watkins SM, Linton MF, Hotamisligil GS (2009) Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med 15:1383–1391
Eslam M, Valenti L, Romeo S (2018) Genetics and epigenetics of NAFLD and NASH: clinical impact. J Hepatol 68:268–279
Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ (2018) Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 67:123–133
Fabricatore AN (2007) Behavior therapy and cognitive-behavioral therapy of obesity: is there a difference? J Am Diet Assoc 107:92–99
Fassio E, Alvarez E, Dominguez N, Landeira G, Longo C (2004) Natural history of nonalcoholic steatohepatitis: a longitudinal study of repeat liver biopsies. Hepatology 40:820–826
Fazel Y, Koenig AB, Sayiner M, Goodman ZD, Younossi ZM (2016) Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism 65:1017–1025
Feldstein AE, Lopez R, Tamimi TA, Yerian L, Chung YM, Berk M, Zhang R, McIntyre TM, Hazen SL (2010) Mass spectrometric profiling of oxidized lipid products in human nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. J Lipid Res 51:3046–3054
Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, Hegele RA, Krauss RM, Raal FJ, Schunkert H, Watts GF, Boren J, Fazio S, Horton JD, Masana L, Nicholls SJ, Nordestgaard BG, van de Sluis B, Taskinen MR, Tokgozoglu L, Landmesser U, Laufs U, Wiklund O, Stock JK, Chapman MJ, Catapano AL (2017) Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 38:2459–2472
Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408:239–247
Fiorucci S, Rizzo G, Antonelli E, Renga B, Mencarelli A, Riccardi L, Morelli A, Pruzanski M, Pellicciari R (2005) Cross-talk between farnesoid-X-receptor (FXR) and peroxisome proliferator-activated receptor gamma contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. J Pharmacol Exp Ther 315:58–68
Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B, Lefebvre P, Taskinen MR, van Hul W, Mertens I, Hubens G, van Marck E, Michielsen P, van Gaal L, Staels B (2015) PPARalpha gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J Hepatol 63:164–173
Friedman SL (1993) Seminars in medicine of the Beth Israel Hospital, Boston. The cellular basis of hepatic fibrosis. Mechanisms and treatment strategies. N Engl J Med 328:1828–1835
Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, Francque S, Farrell G, Kowdley KV, Craxi A, Simon K, Fischer L, Melchor-Khan L, Vest J, Wiens BL, Vig P, Seyedkazemi S, Goodman Z, Wong VW, Loomba R, Tacke F, Sanyal A, Lefebvre E (2018) A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 67:1754–1767
Garcia-Tsao G, Bosch J, Kayali Z, Harrison S, Abdelmalek M, Lawitz E, Satapathy S, Ghabril M, Shiffman M, Younes ZH, Thuluvath PJ, Berzigotti A, Albillos A, Robinson J, Chan JL, Hagerty D, Sanyal A (2019) Multicenter, double-blind, placebo-controlled, randomized trial of emricasan in subjects with NASH cirrhosis and severe portal hypertension. J Hepatol 70:E127–E127
Gastaldelli A, Marchesini G (2016) Time for glucagon like peptide-1 receptor agonists treatment for patients with NAFLD? J Hepatol 64:262–264
Ghareeb DA, Hafez HS, Hussien HM, Kabapy NF (2011) Non-alcoholic fatty liver induces insulin resistance and metabolic disorders with development of brain damage and dysfunction. Metab Brain Dis 26:253–267
Golabi P, Otgonsuren M, Cable R, Felix S, Koenig A, Sayiner M, Younossi ZM (2016) Non-alcoholic fatty liver disease (NAFLD) is associated with impairment of health related quality of life (HRQOL). Health Qual Life Outcomes 14:18
Goto T, Itoh M, Suganami T, Kanai S, Shirakawa I, Sakai T, Asakawa M, Yoneyama T, Kai T, Ogawa Y (2018) Obeticholic acid protects against hepatocyte death and liver fibrosis in a murine model of nonalcoholic steatohepatitis. Sci Rep 8:8157
Graham TL, Mookherjee C, Suckling KE, Palmer CN, Patel L (2005) The PPARdelta agonist GW0742X reduces atherosclerosis in LDLR(−/−) mice. Atherosclerosis 181:29–37
Grebe A, Hoss F, Latz E (2018) NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res 122:1722–1740
Gregory JC, Buffa JA, Org E, Wang Z, Levison BS, Zhu W, Wagner MA, Bennett BJ, Li L, DiDonato JA, Lusis AJ, Hazen SL (2015) Transmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem 290:5647–5660
Guo Y, Yuan W, Yu B, Kuai R, Hu W, Morin EE, Garcia-Barrio MT, Zhang J, Moon JJ, Schwendeman A, Eugene Chen Y (2018) Synthetic high-density lipoprotein-mediated targeted delivery of liver x receptors agonist promotes atherosclerosis regression. EBioMedicine 28:225–233
Hageman J, Herrema H, Groen AK, Kuipers F (2010) A role of the bile salt receptor FXR in atherosclerosis. Arterioscler Thromb Vasc Biol 30:1519–1528
Han MS, Park SY, Shinzawa K, Kim S, Chung KW, Lee JH, Kwon CH, Lee KW, Lee JH, Park CK, Chung WJ, Hwang JS, Yan JJ, Song DK, Tsujimoto Y, Lee MS (2008) Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J Lipid Res 49:84–97
Harrison SA, Marri SR, Chalasani N, Kohli R, Aronstein W, Thompson GA, Irish W, Miles MV, Xanthakos SA, Lawitz E, Noureddin M, Schiano TD, Siddiqui M, Sanyal A, Neuschwander-Tetri BA, Traber PG (2016) Randomised clinical study: GR-MD-02, a galectin-3 inhibitor, vs. placebo in patients having non-alcoholic steatohepatitis with advanced fibrosis. Aliment Pharmacol Ther 44:1183–1198
Hazlehurst JM, Woods C, Marjot T, Cobbold JF, Tomlinson JW (2016) Non-alcoholic fatty liver disease and diabetes. Metabolism 65:1096–1108
Hendrikx T, Bieghs V, Walenbergh SM, van Gorp PJ, Verheyen F, Jeurissen ML, Steinbusch MM, Vaes N, Binder CJ, Koek GH, Stienstra R, Netea MG, Hofker MH, Shiri-Sverdlov R (2013) Macrophage specific caspase-1/11 deficiency protects against cholesterol crystallization and hepatic inflammation in hyperlipidemic mice. PLoS One 8:e78792
Hendrikx T, Jeurissen ML, Bieghs V, Walenbergh SM, van Gorp PJ, Verheyen F, Houben T, Guichot YD, Gijbels MJ, Leitersdorf E, Hofker MH, Lutjohann D, Shiri-Sverdlov R (2015) Hematopoietic overexpression of Cyp27a1 reduces hepatic inflammation independently of 27-hydroxycholesterol levels in Ldlr(−/−) mice. J Hepatol 62:430–436
Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schonfels W, Ahrens M, Heits N, Bell JT, Tsai PC, Spector TD, Deloukas P, Siebert R, Sipos B, Becker T, Rocken C, Schafmayer C, Hampe J (2014) Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci U S A 111:15538–15543
Hotamisligil GS (2010) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140:900–917
Houben T, Brandsma E, Walenbergh SMA, Hofker MH, Shiri-Sverdlov R (2017) Oxidized LDL at the crossroads of immunity in non-alcoholic steatohepatitis. Biochim Biophys Acta Mol Cell Biol Lipids 1862:416–429
Hsia DS, Grove O, Cefalu WT (2017) An update on sodium-glucose co-transporter-2 inhibitors for the treatment of diabetes mellitus. Curr Opin Endocrinol Diabetes Obes 24:73–79
Hyogo H, Tazuma S, Arihiro K, Iwamoto K, Nabeshima Y, Inoue M, Ishitobi T, Nonaka M, Chayama K (2008) Efficacy of atorvastatin for the treatment of nonalcoholic steatohepatitis with dyslipidemia. Metabolism 57:1711–1718
Ibrahim SH, Kohli R, Gores GJ (2011) Mechanisms of lipotoxicity in NAFLD and clinical implications. J Pediatr Gastroenterol Nutr 53:131–140
Ipsen DH, Rolin B, Rakipovski G, Skovsted GF, Madsen A, Kolstrup S, Schou-Pedersen AM, Skat-Rordam J, Lykkesfeldt J, Tveden-Nyborg P (2018) Liraglutide decreases hepatic inflammation and injury in advanced lean non-alcoholic steatohepatitis. Basic Clin Pharmacol Toxicol 123:704–713
Ito D, Shimizu S, Inoue K, Saito D, Yanagisawa M, Inukai K, Akiyama Y, Morimoto Y, Noda M, Shimada A (2017) Comparison of ipragliflozin and pioglitazone effects on nonalcoholic fatty liver disease in patients with type 2 diabetes: a randomized, 24-week, open-label, active-controlled trial. Diabetes Care 40:1364–1372
Izadpanah R, Schachtele DJ, Pfnur AB, Lin D, Slakey DP, Kadowitz PJ, Alt EU (2015) The impact of statins on biological characteristics of stem cells provides a novel explanation for their pleiotropic beneficial and adverse clinical effects. Am J Physiol Cell Physiol 309:C522–C531
Jerome WG (2010) Lysosomes, cholesterol and atherosclerosis. Clin Lipidol 5:853–865
Jou J, Choi SS, Diehl AM (2008) Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin Liver Dis 28:370–379
Joy TR, McKenzie CA, Tirona RG, Summers K, Seney S, Chakrabarti S, Malhotra N, Beaton MD (2017) Sitagliptin in patients with non-alcoholic steatohepatitis: a randomized, placebo-controlled trial. World J Gastroenterol 23:141–150
Kabil SL, Mahmoud NM (2018) Canagliflozin protects against non-alcoholic steatohepatitis in type-2 diabetic rats through zinc alpha-2 glycoprotein up-regulation. Eur J Pharmacol 828:135–145
Kadayifci A, Tan V, Ursell PC, Merriman RB, Bass NM (2008) Clinical and pathologic risk factors for atherosclerosis in cirrhosis: a comparison between NASH-related cirrhosis and cirrhosis due to other aetiologies. J Hepatol 49:595–599
Kaden-Volynets V, Basic M, Neumann U, Pretz D, Rings A, Bleich A, Bischoff SC (2018) Lack of liver steatosis in germ-free mice following hypercaloric diets. Eur J Nutr 58:1933–1945
Kakisaka K, Cazanave SC, Fingas CD, Guicciardi ME, Bronk SF, Werneburg NW, Mott JL, Gores GJ (2012) Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. Am J Physiol Gastrointest Liver Physiol 302:G77–G84
Kalaany NY, Mangelsdorf DJ (2006) LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol 68:159–191
Kargiotis K, Katsiki N, Athyros VG, Giouleme O, Patsiaoura K, Katsiki E, Mikhailidis DP, Karagiannis A (2014) Effect of rosuvastatin on non-alcoholic steatohepatitis in patients with metabolic syndrome and hypercholesterolaemia: a preliminary report. Curr Vasc Pharmacol 12:505–511
Kargulewicz A, Stankowiak-Kulpa H, Grzymislawski M (2014) Dietary recommendations for patients with nonalcoholic fatty liver disease. Przeglad Gastroenterol 9:18–23
Kars M, Yang L, Gregor MF, Mohammed BS, Pietka TA, Finck BN, Patterson BW, Horton JD, Mittendorfer B, Hotamisligil GS, Klein S (2010) Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 59:1899–1905
Katakami N (2018) Mechanism of development of atherosclerosis and cardiovascular disease in diabetes mellitus. J Atheroscler Thromb 25:27–39
Katakami N, Mita T, Irie Y, Takahara M, Matsuoka TA, Gosho M, Watada H, Shimomura I, Sitagliptin Preventive study of Intima-media thickness Evaluation C (2018) Effect of sitagliptin on tissue characteristics of the carotid wall in patients with type 2 diabetes: a post hoc sub-analysis of the sitagliptin preventive study of intima-media thickness evaluation (SPIKE). Cardiovasc Diabetol 17:24
Kim DG, Krenz A, Toussaint LE, Maurer KJ, Robinson SA, Yan A, Torres L, Bynoe MS (2016) Non-alcoholic fatty liver disease induces signs of Alzheimer’s disease (AD) in wild-type mice and accelerates pathological signs of AD in an AD model. J Neuroinflammation 13:1
Kim BM, Abdelfattah AM, Vasan R, Fuchs BC, Choi MY (2018) Hepatic stellate cells secrete Ccl5 to induce hepatocyte steatosis. Sci Rep 8:7499
Kitabatake H, Tanaka N, Fujimori N, Komatsu M, Okubo A, Kakegawa K, Kimura T, Sugiura A, Yamazaki T, Shibata S, Ichikawa Y, Joshita S, Umemura T, Matsumoto A, Koinuma M, Sano K, Aoyama T, Tanaka E (2017) Association between endotoxemia and histological features of nonalcoholic fatty liver disease. World J Gastroenterol 23:712–722
Kohjima M, Enjoji M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, Fujino T, Yada M, Yada R, Harada N, Takayanagi R, Nakamuta M (2007) Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med 20:351–358
Kohler J, Teupser D, Elsasser A, Weingartner O (2017) Plant sterol enriched functional food and atherosclerosis. Br J Pharmacol 174:1281–1289
Koruk M, Savas MC, Yilmaz O, Taysi S, Karakok M, Gundogdu C, Yilmaz A (2003) Serum lipids, lipoproteins and apolipoproteins levels in patients with nonalcoholic steatohepatitis. J Clin Gastroenterol 37:177–182
Koutsovasilis A, Sotiropoulos A, Papadaki D, Bletsa E, Kokotos G, Kounelakis I, Bousboulas S, Peppas T (2018) Qualitative and quantitative effect of IDegLira compared with the nonfixed administration of degludec and liraglutide. Diabetes 67. https://doi.org/10.2337/db18-1104-P
Kwon O, Kim KW, Kim MS (2016) Leptin signalling pathways in hypothalamic neurons. Cell Mol Life Sci 73:1457–1477
Lam C, Bandsma R, Ling S, Mouzaki M (2016) More frequent clinic visits are associated with improved outcomes for children with NAFLD. Can J Gastroenterol Hepatol 2016:8205494
Lammel Lindemann J, Webb P (2016) Sobetirome: the past, present and questions about the future. Expert Opin Ther Targets 20:145–149
Lara-Guzman OJ, Gil-Izquierdo A, Medina S, Osorio E, Alvarez-Quintero R, Zuluaga N, Oger C, Galano JM, Durand T, Munoz-Durango K (2018) Oxidized LDL triggers changes in oxidative stress and inflammatory biomarkers in human macrophages. Redox Biol 15:1–11
Lee S, Birukov KG, Romanoski CE, Springstead JR, Lusis AJ, Berliner JA (2012) Role of phospholipid oxidation products in atherosclerosis. Circ Res 111:778–799
Lefebvre E, Moyle G, Reshef R, Richman LP, Thompson M, Hong F, Chou HI, Hashiguchi T, Plato C, Poulin D, Richards T, Yoneyama H, Jenkins H, Wolfgang G, Friedman SL (2016) Antifibrotic effects of the dual CCR2/CCR5 antagonist cenicriviroc in animal models of liver and kidney fibrosis. PLoS One 11(6):e0158156
Leuschner UF, Lindenthal B, Herrmann G, Arnold JC, Rossle M, Cordes HJ, Zeuzem S, Hein J, Berg T, Group NS (2010) High-dose ursodeoxycholic acid therapy for nonalcoholic steatohepatitis: a double-blind, randomized, placebo-controlled trial. Hepatology 52:472–479
Levey AS, Eckardt KU, Tsukamoto Y, Levin A, Coresh J, Rossert J, de Zeeuw D, Hostetter TH, Lameire N, Eknoyan G (2005) Definition and classification of chronic kidney disease: a position statement from kidney disease: improving global outcomes (KDIGO). Kidney Int 67:2089–2100
Li J, Liu X, Fang Q, Ding M, Li C (2017) Liraglutide attenuates atherosclerosis via inhibiting ER-induced macrophage derived microvesicles production in T2DM rats. Diabetol Metab Syndr 9:94
Li B, Zhang C, Zhan YT (2018) Nonalcoholic fatty liver disease cirrhosis: a review of its epidemiology, risk factors, clinical presentation, diagnosis, management, and prognosis. Can J Gastroenterol 2018:2784537
Li J, Zou B, Yeo YH, Feng Y, Xie X, Lee DH, Fujii H, Wu Y, Kam LY, Ji F, Li X, Chien N, Wei M, Ogawa E, Zhao C, Wu X, Stave CD, Henry L, Barnett S, Takahashi H, Furusyo N, Eguchi Y, Hsu YC, Lee TY, Ren W, Qin C, Jun DW, Toyoda H, Wong VW, Cheung R, Zhu Q, Nguyen MH (2019) Prevalence, incidence, and outcome of non-alcoholic fatty liver disease in Asia, 1999-2019: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol 4:389–398
Lichtenstein AH, Deckelbaum RJ (2001) AHA science advisory. Stanol/sterol ester-containing foods and blood cholesterol levels. A statement for healthcare professionals from the Nutrition Committee of the Council on nutrition, physical activity, and metabolism of the American Heart Association. Circulation 103:1177–1179
Liechti F, Dufour JF (2012) Treatment of NASH with ursodeoxycholic acid: cons. Clin Res Hepatol Gastroenterol 36(Suppl 1):S46–S52
Lin CS, Hsieh PS, Hwang LL, Lee YH, Tsai SH, Tu YC, Hung YW, Liu CC, Chuang YP, Liao MT, Chien S, Tsai MC (2018) The CCL5/CCR5 axis promotes vascular smooth muscle cell proliferation and atherogenic phenotype switching. Cell Physiol Biochem 47:707–720
Lindenmeyer CC, McCullough AJ (2018) The natural history of nonalcoholic fatty liver disease-an evolving view. Clin Liver Dis 22:11–21
Ljubuncic P, Fuhrman B, Oiknine J, Aviram M, Bomzon A (1996) Effect of deoxycholic acid and ursodeoxycholic acid on lipid peroxidation in cultured macrophages. Gut 39:475–478
Lombardi R, Fargion S, Fracanzani AL (2019) Brain involvement in non-alcoholic fatty liver disease (NAFLD): a systematic review. Dig Liver Dis 51(9):1214–1222
Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, Diehl AM, Djedjos CS, Han L, Myers RP, Subramanian GM, McHutchison JG, Goodman ZD, Afdhal NH, Charlton MR, Investigators G.U (2017) The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: a randomized, phase 2 trial. Hepatology 67:549–559
Loria A, Escheik C, Gerber NL, Younossi ZM (2013) Quality of life in cirrhosis. Curr Gastroenterol Rep 15:301
Macavei B, Baban A, Dumitrascu DL (2016) Psychological factors associated with NAFLD/NASH: a systematic review. Eur Rev Med Pharmacol Sci 20:5081–5097
MacKinnon AC, Liu X, Hadoke PW, Miller MR, Newby DE, Sethi T (2013) Inhibition of galectin-3 reduces atherosclerosis in apolipoprotein E-deficient mice. Glycobiology 23:654–663
Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, Boren J, Montalcini T, Pujia A, Wiklund O, Hindy G, Spagnuolo R, Motta BM, Pipitone RM, Craxi A, Fargion S, Nobili V, Kakela P, Karja V, Mannisto V, Pihlajamaki J, Reilly DF, Castro-Perez J, Kozlitina J, Valenti L, Romeo S (2016) The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 150(1219–1230):e6
Mancuso P (2016) The role of adipokines in chronic inflammation. Immunotargets Ther 5:47–56
Manne V, Handa P, Kowdley KV (2018) Pathophysiology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Clin Liver Dis 22:23–37
Marchesini G, Petta S, Dalle Grave R (2016) Diet, weight loss, and liver health in nonalcoholic fatty liver disease: pathophysiology, evidence, and practice. Hepatology 63:2032–2043
Marcuccilli M, Chonchol M (2016) NAFLD and chronic kidney disease. Int J Mol Sci 17:562
Marshall SM (2018) The bark giving diabetes therapy some bite: the SGLT inhibitors. Diabetologia 61:2075–2078
Marso SP, Poulter NR, Nissen SE, Nauck MA, Zinman B, Daniels GH, Pocock S, Steinberg WM, Bergenstal RM, Mann JF, Ravn LS, Frandsen KB, Moses AC, Buse JB (2013) Design of the liraglutide effect and action in diabetes: evaluation of cardiovascular outcome results (LEADER) trial. Am Heart J 166:823–30.e5
Martinet W, de Meyer GR (2009) Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res 104:304–317
Martinez-Guryn K, Hubert N, Frazier K, Urlass S, Musch MW, Ojeda P, Pierre JF, Miyoshi J, Sontag TJ, Cham CM, Reardon CA, Leone V, Chang EB (2018) Small intestine microbiota regulate host digestive and absorptive adaptive responses to dietary lipids. Cell Host Microbe 23:458–469.e5
Masarone M, Rosato V, Dallio M, Gravina AG, Aglitti A, Loguercio C, Federico A, Persico M (2018) Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxidative Med Cell Longev 2018:9547613
Maslak E, Buczek E, Szumny A, Szczepnski W, Franczyk-Zarow M, Kopec A, Chlopicki S, Leszczynska T, Kostogrys RB (2015) Individual CLA isomers, c9t11 and t10c12, prevent excess liver glycogen storage and inhibit lipogenic genes expression induced by high-fructose diet in rats. Biomed Res Int 2015:535982
Matsuda T, Hiyama TY, Niimura F, Matsusaka T, Fukamizu A, Kobayashi K, Kobayashi K, Noda M (2017) Distinct neural mechanisms for the control of thirst and salt appetite in the subfornical organ. Nat Neurosci 20:230–241
Matsumoto M, Zhang J, Zhang X, Liu J, Jiang JX, Yamaguchi K, Taruno A, Katsuyama M, Iwata K, Ibi M, Cui W, Matsuno K, Marunaka Y, Itoh Y, Torok NJ, Yabe-Nishimura C (2018) The NOX1 isoform of NADPH oxidase is involved in dysfunction of liver sinusoids in nonalcoholic fatty liver disease. Free Radic Biol Med 115:412–420
Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H, Yokoyama M, Honda M, Zen Y, Nakanuma Y, Miyamoto K, Kaneko S (2007) Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 46:1392–1403
Mencarelli A, Fiorucci S (2010) FXR an emerging therapeutic target for the treatment of atherosclerosis. J Cell Mol Med 14:79–92
Moris D, Giaginis C, Tsourouflis G, Theocharis S (2017) Farnesoid-X receptor (FXR) as a promising pharmaceutical target in atherosclerosis. Curr Med Chem 24:1147–1157
Mota M, Banini BA, Cazanave SC, Sanyal AJ (2016) Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 65:1049–1061
Mozaffarian D, Rimm EB, Herrington DM (2004) Dietary fats, carbohydrate, and progression of coronary atherosclerosis in postmenopausal women. Am J Clin Nutr 80:1175–1184
Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, van Rooyen DM, Haczeyni F, Teoh NC, Savard C, Ioannou GN, Masters SL, Schroder K, Cooper MA, Feldstein AE, Farrell GC (2017) NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol 66:1037–1046
Muhidin SO, Magan AA, Osman KA, Syed S, Ahmed MH (2012) The relationship between nonalcoholic fatty liver disease and colorectal cancer: the future challenges and outcomes of the metabolic syndrome. J Obes 2012:637538
Mullerova H, Meeraus WH, Galkin DV, Albers FC, Landis SH (2019) Clinical burden of illness among patients with severe eosinophilic COPD. Int J Chron Obstruct Pulmon Dis 14:741–755
Mummadi RR, Kasturi KS, Chennareddygari S, Sood GK (2008) Effect of bariatric surgery on nonalcoholic fatty liver disease: systematic review and meta-analysis. Clin Gastroenterol Hepatol 6:1396–1402
Muro E, Atilla-Gokcumen GE, Eggert US (2014) Lipids in cell biology: how can we understand them better? Mol Biol Cell 25:1819–1823
Musso G, Gambino R, de Michieli F, Cassader M, Rizzetto M, Durazzo M, Faga E, Silli B, Pagano G (2003) Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology 37:909–916
Musso G, Gambino R, Tabibian JH, Ekstedt M, Kechagias S, Hamaguchi M, Hultcrantz R, Hagstrom H, Yoon SK, Charatcharoenwitthaya P, George J, Barrera F, Hafliethadottir S, Bjornsson ES, Armstrong MJ, Hopkins LJ, Gao X, Francque S, Verrijken A, Yilmaz Y, Lindor KD, Charlton M, Haring R, Lerch MM, Rettig R, Volzke H, Ryu S, Li G, Wong LL, Machado M, Cortez-Pinto H, Yasui K, Cassader M (2014) Association of non-alcoholic fatty liver disease with chronic kidney disease: a systematic review and meta-analysis. PLoS Med 11:e1001680
Musso G, Cassader M, Cohney S, de Michieli F, Pinach S, Saba F, Gambino R (2016) Fatty liver and chronic kidney disease: novel mechanistic insights and therapeutic opportunities. Diabetes Care 39:1830–1845
Musso G, Cassader M, Paschetta E, Gambino R (2017) Thiazolidinediones and advanced liver fibrosis in nonalcoholic steatohepatitis: a meta-analysis. JAMA Intern Med 177:633–640
Nagaya T, Tanaka N, Suzuki T, Sano K, Horiuchi A, Komatsu M, Nakajima T, Nishizawa T, Joshita S, Umemura T, Ichijo T, Matsumoto A, Yoshizawa K, Nakayama J, Tanaka E, Aoyama T (2010) Down-regulation of SREBP-1c is associated with the development of burned-out NASH. J Hepatol 53:724–731
Nakamura K, Fuster JJ, Walsh K (2014) Adipokines: a link between obesity and cardiovascular disease. J Cardiol 63:250–259
Nakatsu Y, Kokubo H, Bumdelger B, Yoshizumi M, Yamamotoya T, Matsunaga Y, Ueda K, Inoue Y, Inoue MK, Fujishiro M, Kushiyama A, Ono H, Sakoda H, Asano T (2017) The SGLT2 inhibitor luseogliflozin rapidly normalizes aortic mRNA levels of inflammation-related but not lipid-metabolism-related genes and suppresses atherosclerosis in diabetic ApoE KO mice. Int J Mol Sci 18:1704
Nasiri-Ansari N, Dimitriadis GK, Agrogiannis G, Perrea D, Kostakis ID, Kaltsas G, Papavassiliou AG, Randeva HS, Kassi E (2018) Canagliflozin attenuates the progression of atherosclerosis and inflammation process in APOE knockout mice. Cardiovasc Diabetol 17:106
Neuschwander-Tetri BA, Ford DA, Acharya S, Gilkey G, Basaranoglu M, Tetri LH, Brunt EM (2012) Dietary trans-fatty acid induced NASH is normalized following loss of trans-fatty acids from hepatic lipid pools. Lipids 47:941–950
Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B, Kowdley KV, McCullough A, Terrault N, Clark JM, Tonascia J, Brunt EM, Kleiner DE, Doo E, Network NCR (2015) Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 385:956–965
Newton JL, Jones DE, Henderson E, Kane L, Wilton K, Burt AD, Day CP (2008) Fatigue in non-alcoholic fatty liver disease (NAFLD) is significant and associates with inactivity and excessive daytime sleepiness but not with liver disease severity or insulin resistance. Gut 57:807–813
Ni Y, Li JM, Liu MK, Zhang TT, Wang DP, Zhou WH, Hu LZ, Lv WL (2017) Pathological process of liver sinusoidal endothelial cells in liver diseases. World J Gastroenterol 23:7666–7677
Nickel F, Tapking C, Benner L, Sollors J, Billeter AT, Kenngott HG, Bokhary L, Schmid M, von Frankenberg M, Fischer L, Mueller S, Muller-Stich BP (2018) Bariatric surgery as an efficient treatment for non-alcoholic fatty liver disease in a prospective study with 1-year follow-up: BariScan study. Obes Surg 28:1342–1350
Nobili V, Marcellini M, Marchesini G, Vanni E, Manco M, Villani A, Bugianesi E (2007) Intrauterine growth retardation, insulin resistance, and nonalcoholic fatty liver disease in children. Diabetes Care 30:2638–2640
Noureddin M, Vipani A, Bresee C, Todo T, Kim IK, Alkhouri N, Setiawan VW, Tran T, Ayoub WS, Lu SC, Klein AS, Sundaram V, Nissen NN (2018) NASH leading cause of liver transplant in women: updated analysis of indications for liver transplant and ethnic and gender variances. Am J Gastroenterol 113:1649–1659
Nowak WN, Deng J, Ruan XZ, Xu Q (2017) Reactive oxygen species generation and atherosclerosis. Arterioscler Thromb Vasc Biol 37:e41–e52
Nozue T, Fukui K, Koyama Y, Fujii H, Kunishima T, Hikita H, Hibi K, Miyazawa A, Michishita I, Investigators FT (2016) Effects of sitagliptin on coronary atherosclerosis in patients with type 2 diabetes-A serial integrated backscatter-intravascular ultrasound study. Am J Cardiovasc Dis 6:153–162
Oh H, Jun DW, Saeed WK, Nguyen MH (2016) Non-alcoholic fatty liver diseases: update on the challenge of diagnosis and treatment. Clin Mol Hepatol 22:327–335
Ostovaneh MR, Ambale-Venkatesh B, Fuji T, Bakhshi H, Shah R, Murthy VL, Tracy RP, Guallar E, Wu CO, Bluemke DA, Lima JAC (2018) Association of liver fibrosis with cardiovascular diseases in the general population: the multi-ethnic study of atherosclerosis (MESA). Circ Cardiovasc Imaging 11:e007241
Ouchi N, Parker JL, Lugus JJ, Walsh K (2011) Adipokines in inflammation and metabolic disease. Nat Rev Immunol 11:85–97
Pan J, Ou Z, Cai C, Li P, Gong J, Ruan XZ, He K (2018) Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell Immunol 332:111–120
Paoletti R, Bolego C, Poli A, Cignarella A (2006) Metabolic syndrome, inflammation and atherosclerosis. Vasc Health Risk Manag 2:145–152
Papaspyridonos M, McNeill E, de Bono JP, Smith A, Burnand KG, Channon KM, Greaves DR (2008) Galectin-3 is an amplifier of inflammation in atherosclerotic plaque progression through macrophage activation and monocyte chemoattraction. Arterioscler Thromb Vasc Biol 28:433–440
Pastori D, Carnevale R, Nocella C, Novo M, Santulli M, Cammisotto V, Menichelli D, Pignatelli P, Violi F (2017) Gut-derived serum lipopolysaccharide is associated with enhanced risk of major adverse cardiovascular events in atrial fibrillation: effect of adherence to Mediterranean diet. J Am Heart Assoc 6:e005784
Patel NS, Doycheva I, Peterson MR, Hooker J, Kisselva T, Schnabl B, Seki E, Sirlin CB, Loomba R (2015) Effect of weight loss on magnetic resonance imaging estimation of liver fat and volume in patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol 13:561–568.e1
Patil R, Sood GK (2017) Non-alcoholic fatty liver disease and cardiovascular risk. World J Gastrointest Pathophysiol 8:51–58
Perra A, Simbula G, Simbula M, Pibiri M, Kowalik MA, Sulas P, Cocco MT, Ledda-Columbano GM, Columbano A (2008) Thyroid hormone (T3) and TRbeta agonist GC-1 inhibit/reverse nonalcoholic fatty liver in rats. FASEB J 22:2981–2989
Perumpail BJ, Cholankeril R, Yoo ER, Kim D, Ahmed A (2017) An overview of dietary interventions and strategies to optimize the management of non-alcoholic fatty liver disease. Diseases 5:23
Peters KM, Wilson RB, Borradaile NM (2018) Non-parenchymal hepatic cell lipotoxicity and the coordinated progression of non-alcoholic fatty liver disease and atherosclerosis. Curr Opin Lipidol 29:417–422
Petit JM, Cercueil JP, Loffroy R, Denimal D, Bouillet B, Fourmont C, Chevallier O, Duvillard L, Verges B (2017) Effect of liraglutide therapy on liver fat content in patients with inadequately controlled type 2 diabetes: the Lira-NAFLD study. J Clin Endocrinol Metab 102:407–415
Petta S, Valenti L, Marchesini G, di Marco V, Licata A, Camma C, Barcellona MR, Cabibi D, Donati B, Fracanzani A, Grimaudo S, Parrinello G, Pipitone RM, Torres D, Fargion S, Licata G, Craxi A (2013) PNPLA3 GG genotype and carotid atherosclerosis in patients with non-alcoholic fatty liver disease. PLoS One 8:e74089
Pfaffenbach KT, Gentile CL, Nivala AM, Wang D, Wei Y, Pagliassotti MJ (2010) Linking endoplasmic reticulum stress to cell death in hepatocytes: roles of C/EBP homologous protein and chemical chaperones in palmitate-mediated cell death. Am J Physiol Endocrinol Metab 298:E1027–E1035
Piscaglia F, Svegliati-Baroni G, Barchetti A, Pecorelli A, Marinelli S, Tiribelli C, Bellentani S, Group H-NIS (2016) Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: a multicenter prospective study. Hepatology 63:827–838
Plat J, Hendrikx T, Bieghs V, Jeurissen ML, Walenbergh SM, van Gorp PJ, de Smet E, Konings M, Vreugdenhil AC, Guichot YD, Rensen SS, Buurman WA, Greve JW, Lutjohann D, Mensink RP, Shiri-Sverdlov R (2014) Protective role of plant sterol and stanol esters in liver inflammation: insights from mice and humans. PLoS One 9:e110758
Plat J, Baumgartner S, Vanmierlo T, Lutjohann D, Calkins KL, Burrin DG, Guthrie G, Thijs C, Te Velde AA, Vreugdenhil ACE, Sverdlov R, Garssen J, Wouters K, Trautwein EA, Wolfs TG, van Gorp C, Mulder MT, Riksen NP, Groen AK, Mensink RP (2019) Plant-based sterols and stanols in health & disease: “consequences of human development in a plant-based environment?”. Prog Lipid Res 74:87–102
Polyzos SA, Kountouras J, Mantzoros CS (2017) Adipose tissue, obesity and non-alcoholic fatty liver disease. Minerva Endocrinol 42:92–108
Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, Sanyal AJ (2007) A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 46:1081–1090
Rakipovski G, Rolin B, Nohr J, Klewe I, Frederiksen KS, Augustin R, Hecksher-Sorensen J, Ingvorsen C, Polex-Wolf J, Knudsen LB (2018) The GLP-1 analogs liraglutide and semaglutide reduce atherosclerosis in ApoE(−/−) and LDLr(−/−) mice by a mechanism that includes inflammatory pathways. JACC Basic Transl Sci 3:844–857
Ratziu V, de Ledinghen V, Oberti F, Mathurin P, Wartelle-Bladou C, Renou C, Sogni P, Maynard M, Larrey D, Serfaty L, Bonnefont-Rousselot D, Bastard JP, Riviere M, Spenard J, FRESGUN (2011) A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. J Hepatol 54:1011–1019
Ratziu V, Goodman Z, Sanyal A (2015) Current efforts and trends in the treatment of NASH. J Hepatol 62:S65–S75
Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, Romero-Gomez M, Boursier J, Abdelmalek M, Caldwell S, Drenth J, Anstee QM, Hum D, Hanf R, Roudot A, Megnien S, Staels B, Sanyal A, Group G-IS (2016) Elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and –delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 150:1147–1159.e5
Reilly SM, Ahmadian M, Zamarron BF, Chang L, Uhm M, Poirier B, Peng X, Krause DM, Korytnaya E, Neidert A, Liddle C, Yu RT, Lumeng CN, Oral EA, Downes M, Evans RM, Saltiel AR (2015) A subcutaneous adipose tissue-liver signalling axis controls hepatic gluconeogenesis. Nat Commun 6:6047
Reinaud O, Delaforge M, Boucher JL, Rocchiccioli F, Mansuy D (1989) Oxidative metabolism of linoleic acid by human leukocytes. Biochem Biophys Res Commun 161:883–891
Rodrigues CM, Fan G, Ma X, Kren BT, Steer CJ (1998) A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J Clin Invest 101:2790–2799
Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH (2008) Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 40:1461–1465
Santoro N, Zhang CK, Zhao H, Pakstis AJ, Kim G, Kursawe R, Dykas DJ, Bale AE, Giannini C, Pierpont B, Shaw MM, Groop L, Caprio S (2012) Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology 55:781–789
Saremi A, Schwenke DC, Buchanan TA, Hodis HN, Mack WJ, Banerji M, Bray GA, Clement SC, Henry RR, Kitabchi AE, Mudaliar S, Ratner RE, Stentz FB, Musi N, Tripathy D, DeFronzo RA, Reaven PD (2013) Pioglitazone slows progression of atherosclerosis in prediabetes independent of changes in cardiovascular risk factors. Arterioscler Thromb Vasc Biol 33:393–399
Sasaki A, Nitta H, Otsuka K, Umemura A, Baba S, Obuchi T, Wakabayashi G (2014) Bariatric surgery and non-alcoholic fatty liver disease: current and potential future treatments. Front Endocrinol (Lausanne) 5:164
Sayiner M, Koenig A, Henry L, Younossi ZM (2016) Epidemiology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in the United States and the rest of the world. Clin Liver Dis 20:205–214
Schaffer JE (2016) Lipotoxicity: many roads to cell dysfunction and cell death: introduction to a thematic review series. J Lipid Res 57:1327–1328
Schneiderhan W, Schmid-Kotsas A, Zhao J, Grunert A, Nussler A, Weidenbach H, Menke A, Schmid RM, Adler G, Bachem MG (2001) Oxidized low-density lipoproteins bind to the scavenger receptor, CD36, of hepatic stellate cells and stimulate extracellular matrix synthesis. Hepatology 34:729–737
Schuster S, Cabrera D, Arrese M, Feldstein AE (2018) Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol 15:349–364
Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG (2000) Central nervous system control of food intake. Nature 404:661–671
Sears B, Perry M (2015) The role of fatty acids in insulin resistance. Lipids Health Dis 14:121
Shao N, Yu XY, Ma XF, Lin WJ, Hao M, Kuang HY (2018) Exenatide delays the progression of nonalcoholic fatty liver disease in C57BL/6 mice, which may involve inhibition of the NLRP3 inflammasome through the mitophagy pathway. Gastroenterol Res Pract 2018:1864307
Shiba K, Tsuchiya K, Komiya C, Miyachi Y, Mori K, Shimazu N, Yamaguchi S, Ogasawara N, Katoh M, Itoh M, Suganami T, Ogawa Y (2018) Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH. Sci Rep 8:2362
Shibuya T, Fushimi N, Kawai M, Yoshida Y, Hachiya H, Ito S, Kawai H, Ohashi N, Mori A (2018) Luseogliflozin improves liver fat deposition compared to metformin in type 2 diabetes patients with non-alcoholic fatty liver disease: a prospective randomized controlled pilot study. Diabetes Obes Metab 20:438–442
Shiffman M, Freilich B, Vuppalanchi R, Watt K, Chan JL, Spada A, Hagerty DT, Schiff E (2019) Randomised clinical trial: emricasan versus placebo significantly decreases ALT and caspase 3/7 activation in subjects with non-alcoholic fatty liver disease. Aliment Pharmacol Ther 49:64–73
Shinde PJS (2018) Non-alcoholic steatohepatitis (NASH) market by drug type (vitamin E & pioglitazone, ocaliva, elafibranor, and selonsertib & cenicriviroc), and sales channel (Hospital pharmacy, online provider, and retail pharmacy) – global opportunity analysis and industry forecast, 2021–2025, p 155
Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ (2000) Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102:731–744
Singal AK, Salameh H, Kamath PS (2014) Prevalence and in-hospital mortality trends of infections among patients with cirrhosis: a nationwide study of hospitalised patients in the United States. Aliment Pharmacol Ther 40:105–112
Sinha RA, Singh BK, Yen PM (2018) Direct effects of thyroid hormones on hepatic lipid metabolism. Nat Rev Endocrinol 14:259–269
Sliz E, Sebert S, Wurtz P, Kangas AJ, Soininen P, Lehtimaki T, Kahonen M, Viikari J, Mannikko M, Ala-Korpela M, Raitakari OT, Kettunen J (2018) NAFLD risk alleles in PNPLA3, TM6SF2, GCKR and LYPLAL1 show divergent metabolic effects. Hum Mol Genet 27:2214–2223
Sookoian S, Pirola CJ (2008) Non-alcoholic fatty liver disease is strongly associated with carotid atherosclerosis: a systematic review. J Hepatol 49:600–607
Sookoian S, Pirola CJ (2018) Systematic review with meta-analysis: the significance of histological disease severity in lean patients with nonalcoholic fatty liver disease. Aliment Pharmacol Ther 47:16–25
Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, Gudnason V, Eiriksdottir G, Garcia ME, Launer LJ, Nalls MA, Clark JM, Mitchell BD, Shuldiner AR, Butler JL, Tomas M, Hoffmann U, Hwang SJ, Massaro JM, O'Donnell CJ, Sahani DV, Salomaa V, Schadt EE, Schwartz SM, Siscovick DS, Nash CRN, Consortium G, Investigators M, Voight BF, Carr JJ, Feitosa MF, Harris TB, Fox CS, Smith AV, Kao WH, Hirschhorn JN, Borecki IB, Consortium G (2011) Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 7:e1001324
Spencer MD, Hamp TJ, Reid RW, Fischer LM, Zeisel SH, Fodor AA (2011) Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 140:976–986
Staels B, Rubenstrunk A, Noel B, Rigou G, Delataille P, Millatt LJ, Baron M, Lucas A, Tailleux A, Hum DW, Ratziu V, Cariou B, Hanf R (2013) Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 58:1941–1952
Stancu C, Sima A (2001) Statins: mechanism of action and effects. J Cell Mol Med 5:378–387
Stewart KE, Levenson JL (2012) Psychological and psychiatric aspects of treatment of obesity and nonalcoholic fatty liver disease. Clin Liver Dis 16:615–629
Stewart KE, Haller DL, Sargeant C, Levenson JL, Puri P, Sanyal AJ (2015) Readiness for behaviour change in non-alcoholic fatty liver disease: implications for multidisciplinary care models. Liver Int 35:936–943
Sumida Y, Yoneda M (2018) Current and future pharmacological therapies for NAFLD/NASH. J Gastroenterol 53:362–376
Sumida Y, Niki E, Naito Y, Yoshikawa T (2013) Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic Res 47:869–880
Sumida Y, Murotani K, Saito M, Tamasawa A, Osonoi Y, Yoneda M, Osonoi T (2019) Effect of luseogliflozin on hepatic fat content in type 2 diabetes patients with non-alcoholic fatty liver disease: a prospective, single-arm trial (LEAD trial). Hepatol Res 49:64–71
Suomela E, Oikonen M, Pitkanen N, Ahola-Olli A, Virtanen J, Parkkola R, Jokinen E, Laitinen T, Hutri-Kahonen N, Kahonen M, Lehtimaki T, Taittonen L, Tossavainen P, Jula A, Loo BM, Mikkila V, Telama R, Viikari JSA, Juonala M, Raitakari OT (2016) Childhood predictors of adult fatty liver. The cardiovascular risk in Young Finns study. J Hepatol 65:784–790
Surdea-Blaga T, Dumitrascu DL (2011) Depression and anxiety in nonalcoholic steatohepatitis: is there any association? Rom J Intern Med 49:273–280
Tabas I (2010) Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol 10:36–46
Tacke F (2018) Cenicriviroc for the treatment of non-alcoholic steatohepatitis and liver fibrosis. Expert Opin Investig Drugs 27:301–311
Tanaka K, Sata M (2018) Roles of perivascular adipose tissue in the pathogenesis of atherosclerosis. Front Physiol 9:3
Tanaka K, Masaki Y, Tanaka M, Miyazaki M, Enjoji M, Nakamuta M, Kato M, Nomura M, Inoguchi T, Kotoh K, Takayanagi R (2014) Exenatide improves hepatic steatosis by enhancing lipid use in adipose tissue in nondiabetic rats. World J Gastroenterol 20:2653–2663
Tanaka A, Murohara T, Taguchi I, Eguchi K, Suzuki M, Kitakaze M, Sato Y, Ishizu T, Higashi Y, Yamada H, Nanasato M, Shimabukuro M, Teragawa H, Ueda S, Kodera S, Matsuhisa M, Kadokami T, Kario K, Nishio Y, Inoue T, Maemura K, Oyama J, Ohishi M, Sata M, Tomiyama H, Node K, Investigators PS (2016) Rationale and design of a multicenter randomized controlled study to evaluate the preventive effect of ipragliflozin on carotid atherosclerosis: the PROTECT study. Cardiovasc Diabetol 15:133
Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL (2013) Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 368:1575–1584
Targher G, Chonchol M, Miele L, Zoppini G, Pichiri I, Muggeo M (2009) Nonalcoholic fatty liver disease as a contributor to hypercoagulation and thrombophilia in the metabolic syndrome. Semin Thromb Hemost 35:277–287
Targher G, Day CP, Bonora E (2010) Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med 363:1341–1350
Targher G, Byrne CD, Lonardo A, Zoppini G, Barbui C (2016) Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: a meta-analysis. J Hepatol 65:589–600
Trauner M, Arrese M, Wagner M (2010) Fatty liver and lipotoxicity. Biochim Biophys Acta 1801:299–310
Tremaroli V, Backhed F (2012) Functional interactions between the gut microbiota and host metabolism. Nature 489:242–249
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031
Valdearcos M, Xu AW, Koliwad SK (2015) Hypothalamic inflammation in the control of metabolic function. Annu Rev Physiol 77:131–160
Valenti L, Romeo S (2016) Destined to develop NAFLD? The predictors of fatty liver from birth to adulthood. J Hepatol 65:668–670
Valenti L, Al-Serri A, Daly AK, Galmozzi E, Rametta R, Dongiovanni P, Nobili V, Mozzi E, Roviaro G, Vanni E, Bugianesi E, Maggioni M, Fracanzani AL, Fargion S, Day CP (2010) Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 51:1209–1217
van Baar MJB, van Ruiten CC, Muskiet MHA, van Bloemendaal L, IJzerman RG, van Raalte DH (2018) SGLT2 inhibitors in combination therapy: from mechanisms to clinical considerations in type 2 diabetes management. Diabetes Care 41:1543–1556
van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slutter B, Foks AC, Bot I, Kuiper J (2017) NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler Thromb Vasc Biol 37:1457–1461
van Herpen NA, Schrauwen-Hinderling VB (2008) Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol Behav 94:231–241
van Rooyen DM, Larter CZ, Haigh WG, Yeh MM, Ioannou G, Kuver R, Lee SP, Teoh NC, Farrell GC (2011) Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 141:1393–1403.e1-5
Vassilatou E (2014) Nonalcoholic fatty liver disease and polycystic ovary syndrome. World J Gastroenterol 20:8351–8363
Villanova N, Moscatiello S, Ramilli S, Bugianesi E, Magalotti D, Vanni E, Zoli M, Marchesini G (2005) Endothelial dysfunction and cardiovascular risk profile in nonalcoholic fatty liver disease. Hepatology 42:473–480
Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL (2011) Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472:57–63
Wang Y, Viscarra J, Kim SJ, Sul HS (2015) Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol 16:678–689
Wang PX, Ji YX, Zhang XJ, Zhao LP, Yan ZZ, Zhang P, Shen LJ, Yang X, Fang J, Tian S, Zhu XY, Gong J, Zhang X, Wei QF, Wang Y, Li J, Wan L, Xie Q, She ZG, Wang Z, Huang Z, Li H (2017) Targeting CASP8 and FADD-like apoptosis regulator ameliorates nonalcoholic steatohepatitis in mice and nonhuman primates. Nat Med 23:439–449
Wang R, Li H, Yang X, Xue X, Deng L, Shen J, Zhang M, Zhao L, Zhang C (2018) Genetically obese human gut microbiota induces liver steatosis in germ-free mice fed on normal diet. Front Microbiol 9:1602
Weinstein AA, Kallman Price J, Stepanova M, Poms LW, Fang Y, Moon J, Nader F, Younossi ZM (2011) Depression in patients with nonalcoholic fatty liver disease and chronic viral hepatitis B and C. Psychosomatics 52:127–132
Weinstein G, Zelber-Sagi S, Preis SR, Beiser AS, DeCarli C, Speliotes EK, Satizabal CL, Vasan RS, Seshadri S (2018) Association of nonalcoholic fatty liver disease with lower brain volume in healthy middle-aged adults in the Framingham study. JAMA Neurol 75:97–104
Welte T, Torres A, Nathwani D (2012) Clinical and economic burden of community-acquired pneumonia among adults in Europe. Thorax 67:71–79
Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG (2001) The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 48:206–211
Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, Landt CL, Harrison SA (2011) Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology 140:124–131
Winer DA, Luck H, Tsai S, Winer S (2016) The intestinal immune system in obesity and insulin resistance. Cell Metab 23:413–426
Wojcik-Cichy K, Koslinska-Berkan E, Piekarska A (2018) The influence of NAFLD on the risk of atherosclerosis and cardiovascular diseases. Clin Exp Hepatol 4:1–6
Wong VW, Wong GL, Tsang SW, Fan T, Chu WC, Woo J, Chan AW, Choi PC, Chim AM, Lau JY, Chan FK, Sung JJ, Chan HL (2011) High prevalence of colorectal neoplasm in patients with non-alcoholic steatohepatitis. Gut 60:829–836
Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, Lutjohann D, Kerksiek A, van Kruchten R, Maeda N, Staels B, van Bilsen M, Shiri-Sverdlov R, Hofker MH (2008) Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology 48:474–486
Wu J, Yao XY, Shi RX, Liu SF, Wang XY (2018a) A potential link between polycystic ovary syndrome and non-alcoholic fatty liver disease: an update meta-analysis. Reprod Health 15:77
Wu WKK, Zhang L, Chan MTV (2018b) Autophagy, NAFLD and NAFLD-related HCC. Adv Exp Med Biol 1061:127–138
Xiang M, Wang PX, Wang AB, Zhang XJ, Zhang Y, Zhang P, Mei FH, Chen MH, Li H (2016) Targeting hepatic TRAF1-ASK1 signaling to improve inflammation, insulin resistance, and hepatic steatosis. J Hepatol 64:1365–1377
Yang G, Lei Y, Inoue A, Piao L, Hu L, Jiang H, Sasaki T, Wu H, Xu W, Yu C, Zhao G, Ogasawara S, Okumura K, Kuzuya M, Cheng XW (2017) Exenatide mitigated diet-induced vascular aging and atherosclerotic plaque growth in ApoE-deficient mice under chronic stress. Atherosclerosis 264:1–10
Ye D, Li FY, Lam KS, Li H, Jia W, Wang Y, Man K, Lo CM, Li X, Xu A (2012) Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of X-box binding protein-1 in mice. Gut 61:1058–1067
Younossi ZM, Henry L (2015) Economic and quality-of-life implications of non-alcoholic fatty liver disease. PharmacoEconomics 33:1245–1253
Younossi Z, Henry L (2016) Contribution of alcoholic and nonalcoholic fatty liver disease to the burden of liver-related morbidity and mortality. Gastroenterology 150:1778–1785
Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, Srishord M (2011a) Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol 9:524–530.e1; quiz e60
Younossi ZM, Stepanova M, Rafiq N, Makhlouf H, Younoszai Z, Agrawal R, Goodman Z (2011b) Pathologic criteria for nonalcoholic steatohepatitis: interprotocol agreement and ability to predict liver-related mortality. Hepatology 53:1874–1882
Younossi ZM, Zheng L, Stepanova M, Venkatesan C, Mishra A (2014) Clinical outcomes and resource utilisation in medicare patients with chronic liver disease: a historical cohort study. BMJ Open 4:e004318
Younossi ZM, Otgonsuren M, Henry L, Venkatesan C, Mishra A, Erario M, Hunt S (2015a) Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 62:1723–1730
Younossi ZM, Zheng L, Stepanova M, Henry L, Venkatesan C, Mishra A (2015b) Trends in outpatient resource utilizations and outcomes for medicare beneficiaries with nonalcoholic fatty liver disease. J Clin Gastroenterol 49:222–227
Younossi ZM, Blissett D, Blissett R, Henry L, Stepanova M, Younossi Y, Racila A, Hunt S, Beckerman R (2016a) The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 64:1577–1586
Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M (2016b) Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64:73–84
Younossi Z, Tacke F, Arrese M, Sharma BC, Mostafa I, Bugianesi E, Wong VW, Yilmaz Y, George J, Fan J, Vos MB (2018a) Global perspectives on non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Hepatology 69:2672–2682
Younossi ZM, Stepanova M, Lawitz E, Charlton M, Loomba R, Myers RP, Subramanian M, McHutchison JG, Goodman Z (2018b) Improvement of hepatic fibrosis and patient-reported outcomes in non-alcoholic steatohepatitis treated with selonsertib. Liver Int 38:1849–1859
Younossi ZM, Tampi R, Priyadarshini M, Nader F, Younossi IM, Racila A (2019) Burden of illness and economic model for patients with nonalcoholic steatohepatitis in the United States. Hepatology 69:564–572
Zain SM, Mohamed Z, Mahadeva S, Cheah PL, Rampal S, Chin KF, Mahfudz AS, Basu RC, Tan HL, Mohamed R (2013) Impact of leptin receptor gene variants on risk of non-alcoholic fatty liver disease and its interaction with adiponutrin gene. J Gastroenterol Hepatol 28:873–879
Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Furtado RHM, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Sabatine MS (2019) SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet 393:31–39
Zhang S, Wang J, Liu Q, Harnish DC (2009) Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol 51:380–388
Zhang Q, Liu J, Liu J, Huang W, Tian L, Quan J, Wang Y, Niu R (2014) oxLDL induces injury and defenestration of human liver sinusoidal endothelial cells via LOX1. J Mol Endocrinol 53:281–293
Zhang P, Wang PX, Zhao LP, Zhang X, Ji YX, Zhang XJ, Fang C, Lu YX, Yang X, Gao MM, Zhang Y, Tian S, Zhu XY, Gong J, Ma XL, Li F, Wang Z, Huang Z, She ZG, Li H (2018) The deubiquitinating enzyme TNFAIP3 mediates inactivation of hepatic ASK1 and ameliorates nonalcoholic steatohepatitis. Nat Med 24:84–94
Zou L, Li X, Wu N, Jia P, Liu C, Jia D (2017) Palmitate induces myocardial lipotoxic injury via the endoplasmic reticulum stressmediated apoptosis pathway. Mol Med Rep 16:6934–6939
Zou TT, Zhang C, Zhou YF, Han YJ, Xiong JJ, Wu XX, Chen YP, Zheng MH (2018) Lifestyle interventions for patients with nonalcoholic fatty liver disease: a network meta-analysis. Eur J Gastroenterol Hepatol 30:747–755
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this chapter

Cite this chapter
Ding, L., Oligschlaeger, Y., Shiri-Sverdlov, R., Houben, T. (2020). Nonalcoholic Fatty Liver Disease. In: von Eckardstein, A., Binder, C.J. (eds) Prevention and Treatment of Atherosclerosis . Handbook of Experimental Pharmacology, vol 270. Springer, Cham. https://doi.org/10.1007/164_2020_352
Download citation
DOI: https://doi.org/10.1007/164_2020_352
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-86075-2
Online ISBN: 978-3-030-86076-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)