
Abstract
A central pillar of the biotechnology and pharmaceutical industries continues to be the development of biological drug products manufactured from engineered mammalian cell lines. Since the hugely successful launch of human tissue plasminogen activator in 1987 and erythropoietin in 1988, the biopharmaceutical market has grown immensely. In 2014, biotherapeutics made up a significant portion of global drug sales as 7 of the top 10 and 21 of top 50 selling pharmaceuticals in the world were biologics with over US$100 billion in global sales (Table 1, [1]).
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others



Keywords
Introduction
A central pillar of the biotechnology and pharmaceutical industries continues to be the development of biological drug products manufactured from engineered mammalian cell lines. Since the hugely successful launch of human tissue plasminogen activator in 1987 and erythropoietin in 1988, the biopharmaceutical market has grown immensely. In 2014, biotherapeutics made up a significant portion of global drug sales as 7 of the top 10 and 21 of top 50 selling pharmaceuticals in the world were biologics with over US$100 billion in global sales (Table 1, [1]).
These marketed biopharmaceuticals will soon be joined by the numerous clinical candidates in development that promise to bring more and better treatments to patients. Compared to small molecule drugs, biotherapeutics show exquisite specificity with fewer off-target interactions and improved safety profiles. Protein engineering technologies have advanced to maximize the full potential of biologics by creating protein drugs with improved potency, specificity, stability, pharmacokinetics, and solubility. Strategies that have been employed to implement these changes include phage display, directed evolution methods, as well as rational design and structure-based computational approaches [2,3,4,5,6,7]. These advanced protein engineering technologies are creating novel drug designs and clever treatment strategies that are fuelling the biopharmaceutical market growth.
Antibodies are predominantly manufactured in recombinant mammalian cell systems and they continue their growth in the biotherapeutics industry since the early oncology successes of Rituxan (1997) and Herceptin (1998). As of the end of 2014, an additional 45 therapeutic antibodies have been approved in the US and Europe with $75 billion in sales and projections predicting ~70 approvals by 2020 with worldwide sales of $125 billion [8]. In 2014, 5 of the top 10 selling pharmaceuticals in the world were antibodies (Table 1, [1]).
The majority of all biotherapeutics are produced in cultivated mammalian cells with Chinese hamster ovary (CHO) systems manufacturing 60–70% of recombinant biologics on the market [9]. Mammalian systems are often preferred over other hosts, such as bacteria, plants, and yeast, because of their capability for proper protein folding, assembly and posttranslational modification. CHO systems have the added advantages that they can be grown in suspension at scales required to meet market demands; the cells have been genetically engineered to express high amounts of recombinant protein; the cells can be grown in serum-free and chemically defined media; and these rodent cells have less risks of propagating human viruses [10].
With the continuing expansion of the biotherapeutics market aided by the commercial successes of antibody therapeutics and emerging biosimilars, the demand for proteins derived from mammalian cells continues to grow and the biopharmaceutical industry is faced with the challenge of efficiently producing these proteins in large quantities. To keep up with demand while driving manufacturing costs down, mammalian cell production expectations are rising every year, with product titers reaching 10 g of protein/L of culture [11]. The high yields obtained today are the result of combined efforts made in improving host cells, expression vectors, screening methods, media optimization, and process development innovations. In this chapter, we outline the common methods applied to mammalian cell line development and describe the typical industrial processes currently used in cell culture and purification for the production of recombinant proteins. Recent advances in this field are also presented.
Cells Used for Industrial Production
This section focuses on the generation of engineered mammalian cell lines that stably produce therapeutic proteins. Hybridomas, transient expression systems, and insect and bacterial cell line development are outside the scope of this section. The commonly used cell lines, expression systems and vectors, as well as cell banking and stability are described.
Host Cell Lines
Mammalian cell lines have been developed as hosts to manufacture a variety of biotherapeutic proteins. CHO-derived cell lines are the preferred expression systems for biotherapeutic proteins although other systems have been used in the early days of the biologics era such as Sp2/0 and NS0 mouse myelomas, YB2/0 rat myeloma, and baby hamster kidney (BHK) cells. Recently, human-derived cell lines such as PER C6 and CAP were also developed for better posttranslation modification (PTM) and reduced immunogenicity of the biotherapeutic proteins. Numerous CHO-derived lines have been developed since the original Puck line of 1957 and the CHO-K1 cells which were derived over the course of the following 10 years from the same lab [12, 13] for an excellent history on CHO cell lines through the ages. In the 1970s and 1980s, Urlaub and Chasin used the Puck and Siminovitch lab cells to derive CHO-DXB11 and CHO-DG44 respectively. Figure 1 describes a lineage of the most popular CHO-derived cell lines. CHO-DXB11 and CHD-DG44 cell lines are engineered to have single or double knockouts respectively for the dihydrofolate reductase (DHFR) gene. Of note is the fact that human tissue plasminogen activator (TPA) was the first marketed biotherapeutic expressed in CHO cells and the CHO-DUXB11 line was used to produce hundreds of kilograms of protein.

A lineage of CHO-derived cell lines [12]
The CHO-K1 and the suspension culture variant, CHOK1SV, have risen in popularity due to their use in the Lonza, Inc. expression system that uses the glutamine synthetase selectable marker (GS system) originally designed for NS0 cells. As of 2013, five marketed biotherapeutics were developed with this system with numerous others in clinical development [12]. Lonza has recently developed another variant of the CHOK1SV cells with both alleles of the GS gene knocked out leading to increased selective pressure due to a requirement for exogenous glutamine for cell survival.
Although transient or stable transfected pools are frequently used for the production of research biologic material, stable cell lines are generated for clinical and commercial manufacturing by integrating the genes of interest into the genome of host cell lines. Selection of stable cell lines with high productivity after transfection requires proper selection system and most biopharmaceutical companies currently use either the methotrexate (MTX)-dihydrofolate reductase (DHFR) or methionine sulfoximine (MSX)-glutamine synthetase GS systems for clone screening. MTX inhibits DHFR in the MTX amplification system whereas MSX inhibits GS in the GS system. The productivity of the selected stable cell lines can be improved by further selection of the cell lines survived at increasing concentration of MTX or MSX, which can amplify the copy numbers of the genes of interest.
The CHO-Cumate system from the National Research Council (NRC), Canada has an engineered CHO-DUXB11 variant called CHOBRI with stably integrated Cumate operator transactivator-rcTA along with CymR repressor [14]. Stable cell lines are generated using the GS system without the gene of interest being expressed. In production mode, Cumate is added to the media which inactivates the repressor to allow production of the recombinant protein. The use of this inducible expression system has the benefit of separating cell culture growth and selection from production resulting in more efficient generation of stable pools and clones as well as being suited for the expression of recombinant proteins that are toxic to the CHO host cells.
The EuCODE™ system from Ambrx, Inc. uses an engineered CHO-K1 variant expressing a novel orthogonal set of amber codon tRNAs and tRNA synthetase pair [15]. This system allows for the production of recombinant proteins containing nonnatural amino acids. The nonnatural amino acid is introduced in the production cell culturing media and the orthogonal tRNA/tRNA synthetase incorporates it into the protein at specific sites designated by an amber codon. The nonnatural amino acids have reactive side chains that facilitate site-specific conjugation of toxins for making antibody drug conjugates (ADCs) or for attachment of other compounds such as polyethylene glycol (PEG) to extend the half-life of recombinant proteins.
In addition to being engineered for selection, host cells have also been genetically manipulated for cellular metabolism, anti-apoptosis [16], cell cycle interference, protein secretion, and glycosylation to generate better host cell lines [17]. With the sequencing of CHO genomes and the advent of targeted gene editing technologies such as the use of zinc-finger nuclease (ZFN), transcription activator-like effector nuclease (TALENs), and clustered regularly interspaced short palindromic repeat (CRISPR/Cas 9), designer CHO cell lines are in development. The current challenges center on choosing from hundreds of genes and dozens of relevant pathways to manipulate by knock in and knock out technologies. See [18] for an excellent review of these topics.
Expression Systems
The goal of cell line development is to engineer cells to express a large amount of a recombinant protein and to stably maintain this level of production for many cell doublings. The basic schema is:
-
1.
Generate plasmid expressing recombinant protein.
-
2.
Introduce plasmid into host cells.
-
3.
Identify clonal cell lines expressing high levels of recombinant protein.
-
4.
Select cell lines maintaining high levels of expression through scale-up and bioreactor processes.
Plasmids. The plasmid contains all genetic elements necessary for the expression of recombinant protein and for the selection of the cells generating the desired product.
The gene sequences of interest are frequently optimized for codon and tRNA usages, GC content, ribosome-binding sequences, and removal of unnecessary secondary structure, which result in the improvement of recombinant protein expression. For stable cell lines with high expression level of the gene of interest, the plasmids must integrate into transcriptionally active chromosomal regions. The vector promoter elements drive the expression of recombinant protein and are derived from viruses, mice, and human. The strong murine or human cytomegalo virus (CMV) promoter is in general use, although the weaker simian virus 40 (SV40) and rouse sarcoma virus (RSV) promoters are also used. In general, the strong promoters are used for expression of genes of interest whereas weak promoters are applied for expression of selection marker genes. Another strong promoter from the highly expressed endogenous housekeeping gene, CHO-EF1α (CHEF1) ([19]; ICOS, Bothell, Washington), has been generating CHO cell lines with high production levels. Also, the CR5 cumate inducible promoter has higher expression compared to CMV. The inclusion of a chimeric intron as an enhancer in the primary transcript leads to higher stable expression of recombinant protein through enhanced transport and processing of the mRNA from the nucleus into the cytoplasm where it is translated [20, 21].
To allow selection of cells expressing desired protein, the plasmid may also contain selectable markers such as the neo gene generating aminoglycoside 3′-phosphotransferase (APH 3′ II) for G418 geneticin resistance, the hph phosphotransferase gene for hygromycin resistance, the Sh ble gene for zeocin resistance, or puromycin N-acetyl-transferase encoded by the pac gene for puromycin resistance. The selection system most commonly used in industry is DHFR or GS, which is present in plasmids when using DHFR-negative or GS-negative host cell lines, such as CHO-DG44 and NS0-GS/CHOK1SV GS Knock out respectively. In the GS systems, a knock-out of endogenous GS is preferred but not necessary as the use of MSX in culture provides selective pressure by inhibiting GS. The use of an internal ribosomal entry site (IRES) element can facilitate the co-expression of selectable markers and protein product when integrated into the genome [22]. This system generates a single transcript accessible to ribosomes at two locations just prior to the start site of each gene. The fact that the selectable marker and the product gene are under the control of a single promoter, which generates one transcript, is likely to improve cell line stability. IRES elements can also be used for the co-expression of multicistronic peptides in a single transcript, which result in expression of multi-peptide proteins such as antibodies.
Elements such as scaffold or matrix attached regions (SARs or MARs) (Selexis, Geneva, Switzerland; [23, 24]) and ubiquitous chromatin opening elements (UCOEs) (Merck Millipore; [25]) can also be included in plasmids, as they are known to generate transcriptionally active genomic environments once integrated into the cell genome. Other systems direct site-specific integration of plasmid into highly transcriptionally active chromosomal regions using CHO host cells engineered with attB recombination sites and plasmids with attP sequences (Intrexon Inc.; [26]). The Cre/LoxP and Flp/FRT recombination systems utilize a similar approach [27]. The artificial chromosome expression (ACE) system consisting of a mammalian-based artificial chromosome known as Platform ACE, an ACE targeting vector (ATV), and a mutant λ integrase (ACE integrase) is also used for targeted recombination [28].
DNA Delivery. Transfection is a process that delivers foreign nucleic acids into cells and it can be mediated biologically, chemically, and physically. For the best outcome it should have high transfection efficiency, low cell toxicity, and minimal effects on normal physiology. The biological method uses the viral vectors for the nucleic acid delivery. Retroviral transduction has a long history for inserting DNA into cells, but use for manufacturing is only now emerging (GPEx® Gala Biotech, Middleton, WI). The chemical method involves various cationic reagents such as calcium phosphate, and lipids, and polymers. CaPO4 precipitation is the earliest method, but has been surpassed by the more convenient and consistent lipid-based reagents (Lipofectamine, Fugene, Transfectin). Polyethyleneimine (PEI) [29] is a low-cost alternative DNA delivery method. Direct injection by micro-needle [30], magnetofection by magnetic nanoparticles [31], and electroporation [32] are examples of physical methods. The electroporation becomes a method of choice for industrial manufacture cell lines for biotherapeutics because of convenience and no intellectual property protection. Transfection efficiencies can vary from 5 to 100% depending on cell line and DNA delivery method. CHO cells can achieve 5–40% with CaPO4, 20–60% with lipids, and ~100% with retroviral transduction.
Amplification Systems. The DHFR and GS amplification systems have successfully generated manufacturing cell lines with high protein titers (Lonza, Basel, Switzerland; [33, 34]). These systems employ a DHFR− or GS− cell line that is transfected with plasmid encoding product of interest along with DHFR or GS respectively. The continual adaptation of the recombinant cells to elevated concentrations of methotrexate and methionine sulfoximine results in chromosomal amplification events that increase the DHFR or GS gene copy number, respectively, to overcome the drug resistance. The gene encoding the product of interest is usually co-amplified with the DHFR or GS genes as they are inserted into the genome in the same locations. Ten-fold or greater improvements in expression can be achieved with this amplification system. Gene-amplified cell lines tend to be more unstable. The DHFR amplification system has the potential to experience the loss of transgene copy number [35,30,37]; consequently, stability studies are especially important to characterize cell lines derived from drug-induced genomic amplification approaches.
Identifying High-Expressing Clonal Cells
Identification of the cells with high productivity from polyclonal transfected pools is a critical process during cell line development. Effective screening methods are required to facilitate finding highly productive clones. Traditionally, selection begins with limiting dilution, a process where a polyclonal suspension of cells is diluted to very low cell density and the diluted cell suspension is then transferred to wells of microplates.
For secreted proteins, enzyme-linked immunosorbant assays (ELISA) on conditioned media can identify the cells expressing the highest protein levels. AlphaScreen™ (Perkin-Elmer, Boston, Massachusetts) is a homogeneous assay that is well suited for high-throughput quantification of protein production. The Guava easyCyte (EMD Millipore) microcapillary flow cytometer economically and conveniently generates fluorescence-activated cell sorting-like (FACS) expression profiles of cells with moderate throughput in 96-well microtiter plates. With this approach, clonal populations and cells with the highest average productivity can be identified. Systematic colony picking system from semisolid medium such as ClonePix was developed as an alternative high-throughput method. A critical element of generating stable cell lines is identifying clonal populations of expressing cells. Pools of expressing cells tend to express lower levels of desired protein; they can drift to lower expression levels, and are more difficult to adapt to serum-free suspension. Limited dilution methods have been used for years while FACS sorting of live cells has also proven successful. FACS can be used to simultaneously clone and enrich for the highest expressing cells [38]. Recent automated approaches for identifying clones include picking high-expressing colonies in semisolid media using ClonePix™ (Molecular Devices, New Milton, UK), Pickolo™ (Scirobotics, Kfar Saba Israel) and enriching for high-expressing cells by Laser-Enabled Analysis and Processing (LEAP™, Inrexon). Recently, assurance of monoclonality of the manufacture cell lines is drawn into regulatory attention but no regulatory guidance is available. The best way to prove the monoclonality of the cell lines is to image the cell lines during primary screening process in order to demonstrate that the selected cell lines are originated from a single progenitor. The limiting dilution and the FACS-sorting under low cell density plating conditions cannot guarantee the monoclonality of cells and high-resolution imaging of the cells in microplate wells may be required to support the monoclonality. If the ClonePix is applied for screening, at least two rounds of cloning are required for assurance of the monoclonality of the cells. Single cell-based genome sequencing may be used to examine the monoclonality. For example, investigation of the integration sites of genes of interest on genome which is unique to each single cell may be able to determine the monoclonality of the cells.
Cell Banking
A stock of cells must be preserved to ensure continuity for research, development, and manufacturing production programs. Once the lead candidate cell lines are identified, several research cell banks (RCB) of each cell line are prepared followed by the preparation of around 50 vials of development cell banks (DCS) from RCB. The DCB is used for the bioprocess development as well as for early material generation. For a small research program, only a small number of frozen vials may be needed. However, to continue to supply a cell line for the manufacture of therapeutic proteins, it is usually best to prepare two-tiered cell banks: a master cell bank (MCB) and working cell banks (WCB). A single cell line demonstrating suitable expression levels and stability is used to generate an MCB, and a WCB is derived from one vial of the MCB. Each MCB and WCB usually includes 100–300 vials. As a WCB is depleted during manufacturing runs, another frozen vial of MCB is used to generate a new WCB. Both MCB and WCB should be fully characterized for sterility, mycoplasma, and adventitious viral contamination before release.
Making cell banks involves the process of cryopreserving cells. During cryopreservation, cells can be damaged by the formation of intracellular ice crystals or by osmotic effects that occur with decreased water content. To minimize cell damage, the rate of freezing must be controlled and cryoprotectants must be used. Dimethyl sulfoxide (DMSO) at 7.5–10% is routinely used as a cryoprotectant. In some cases, a low percentage of fetal calf serum or serum albumin is added to the freezing medium. However, when freezing cell lines for the production of therapeutic proteins, it is generally preferable to eliminate any animal-derived material to minimize the risk of disease transmission from animal to human. To control the rate of freezing, Mr. Frosty™ (Thermo Fisher Scientific, Waltham, MA) and CoolCell® (BioCision, San Rafael, CA) freezing containers are routinely used in making small cell banks, while programmable controlled-rate cell freezers are needed for large cell banks.
Cell Stability
The properties of a cell line are likely to change during a long period of continuous passage. For example, cell lines can lose their expression and can generate undesired proteins clipped from the product. Therefore, it is critical to characterize the cells to ensure consistency for large-scale production and to guarantee that the properties of the protein derived from the cells are maintained. For a production cell line, an acceptable level of stability of the desired characteristics must be established and a maximum passage number must be defined so that comparison of the cells’ characteristics can be made after low and extensive passages. Tests such as peptide mapping, amino acid sequencing, DNA fingerprinting, and determination of gene copy number and phenotype markers must be conducted to ensure the cells’ genetic stability. In addition, productivity and product quality must be examined to assess the stability of the cell line. A good production cell line should be able to maintain its productivity and product quality through the many generations required to reach the end of large-scale production. In most instances, stability retained for 50 generations will satisfy the rigors of large-scale manufacturing [39]. Conventionally, the cell line stability is assessed by monitoring change of volumetric productivity over extended period of time in culture; if the cell lines maintain their volumetric productivity greater than 70% of the original values, they would be considered stable cell lines. The cell line stability assessment may take up to 2–3 months and is a bottleneck for the cell line development timeline. If we are able to predict the cell line stability at early cell line development stage, we can speed up the cell line development timeline. Dorai et al. [40] demonstrated that the unstable cell lines were potentially identified by examining the secondary population peak by flow cytometry after internal staining of the cell lines with fluorescence tagged anti-capture antibody. The causes of instability of production cell lines likely vary but the loss of gene copies and the silence of genes by methylation are two of primary causes well studied.
Media
Mammalian cell culture is the most important source of therapeutic proteins. Just as mammalian cells are more complicated than microorganisms, the media required for their growth is also more complex. The medium must provide the same nutrients and growth factors that mammalian cells are exposed to in vivo in order for them to survive and proliferate. Serum contains many important components that support the growth of mammalian cells including growth factors, hormones, transport and binding proteins, attachment factors, protease inhibitors, and lipids. Serum was, therefore, a common media supplement in early mammalian cell culture and for large-scale production of therapeutic proteins and monoclonal antibodies in the 1980s. However, the use of serum in mammalian cell culture has many disadvantages: (1) it is a potential source of bacterial, mycoplasmal, and viral contamination; (2) it is the most expensive single additive to cell culture media; (3) it has a high degree of batch variability, making production consistency difficult; (4) it contains a high concentration of proteins that can interfere with product recovery. In the early 1990s, these drawbacks, especially the serious concern about the risk of transferring diseases such as Bovine Spongiform Encephalopathy from animals to humans, led to an important regulatory-driven trend to eliminate serum and animal-derived components from mammalian cell growth media. This trend sparked an industry-wide interest in developing serum-free media.
Commercial Serum-Free Media
Many new media companies formed in the early 1990s. Today, more than two decades later, the media development industry has matured. Currently, a variety of serum-free (SF) media are available commercially. Table 2 lists some SF media produced in the U.S. Many of these media are also made in powder form to facilitate use in large-scale production. As listed in Table 2, there are different types of SF media, which can be categorized into protein-free (PF), chemical-defined (CD), and animal-component-free (ACF) media. The relationships among these different types of media are illustrated in Fig. 2.

Relationships among serum-free, protein-free, chemical-defined and animal-component-free media
SF, PF, ACF, and CD media tend to be highly specific to one cell type and sometimes even to one particular cell line. It is not uncommon that a different optimal medium is required for a particular cell line. Development of SF, PF, ACF, or CD media requires considerable experience and expertise and can be very time-consuming. One approach is to start with a commercially available SF medium and add necessary nutrients to optimize growth and production for a particular cell type. This approach can shorten the timeline needed for in-house medium development. However, using commercial media has two major drawbacks: (1) commercial SF media are expensive, which can lead to a high cost of goods for large-scale production; (2) the composition of a commercial SF medium is proprietary to the medium company and the buyer will not know its formulation. This makes it difficult to gain full knowledge about how logical changes to it will affect the growth and production characteristics of a cell line. To fully understand the metabolism and characteristics of a cell line, there is no substitute for developing a media de novo.
Approaches for Serum-Free Medium Development
Medium development is part of the process of cell culture optimization. It can be very complicated and time-consuming, and will require expertise and resources. In this section, we outline the common as well as novel methods used for SF medium development. Details of each strategy can be found in the referenced literature.
In the early 1980s, efforts were initiated to eliminate serum and animal-derived components from the culture media used to produce human therapeutics. At that time, two different strategies were commonly utilized for the development of SF media:
-
1.
Limiting factor method [41]: Starting with an existing formulation, the serum concentration is lowered until cell growth becomes limited; then the concentration of each component of the medium is optimized until cell growth recovers.
-
2.
Synthetic method [42]: A variety of growth factors are added to the existing basal media to replace the serum’s functions.
The last decade saw the development of many new genomic and automated screening tools. These advancements, as well as an improved understanding of mammalian cell culture, allowed novel concepts and approaches to be applied to the development of SF and CD media. Three representative strategies are briefly summarized below:
-
1.
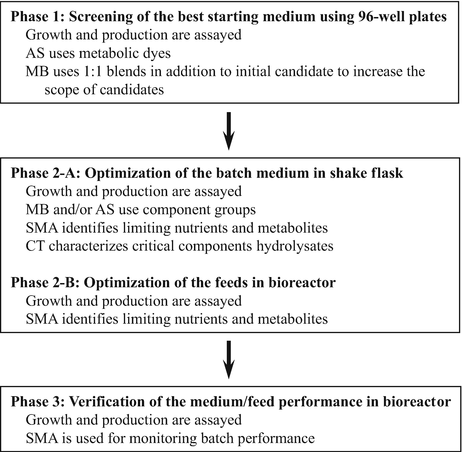
Rational design [43]: This method uses four complementary methods, including component titration (CT), media blending (MB), spent media analysis (SMA), and automated screening (AS), to achieve the best SF culture medium in the shortest timeline. An example of how to use this strategy is outlined below.
-
2.
Factorial design [44,45,46]: This method identifies key growth factors in a lean basal medium by performing experiments using a full factorial matrix. It allows calculating the maximum number of interaction effects and gives insights into growth factor biology.
-
3.
Genomic tools [47]: This method takes advantage of select genomic (microarray- or PCR-based approaches) and proteomic (antibody array analysis) tools to identify the receptors for growth factors, hormones, cytokines, and other components of cell signaling pathways expressed by a culture of interest.

Chemically Defined Media Development
There are several advantages of utilizing a chemically defined (CD) media, specifically CD media has less lot-to-lot variability compared to media with undefined components, such as hydrolysates [48]. Also, CD media allows the scientist to have more insight into the metabolic state of the cells. For example, the metabolism of short peptides by mammalian cells is not well understood, thus media containing them is not desirable [48]. One of the primary drawbacks of CD media is that most CD media formulations typically do not support cell growth as well as media formulations with undefined components [49]. However, using similar methods to those detailed in the previous section, Ma et al. [48] and Huang et al. [50] were able to overcome this drawback. Huang et al. [50] utilized a rapid screening methodology to test several media components during the latter half of several fed-batch cultures. These experiments were followed by additional rounds of CD optimization experiments using trace metals and vitamins. Huang et al. [50] were able to more than double the specific productivity of the culture and final titer using this optimized formulation. Ma et al. [48] implemented a top-down CD formulation development; starting with an over-rich formulation, which contained over 90 components. Ma et al [48]. systematically removed components or groups of components that did not improve the performance of the culture. The concentrations of amino acids in this simplified formulation were further optimized through the use of spent media analysis. This final optimized formulation improved the culture longevity and decreased lactate accumulation while simultaneously doubling the final titer [48]. The top-down approach implemented by Ma et al. [48] led to a well-defined, robust process however this approach required a substantial investment in both time and resources. A cell culture scientist may not have the wherewithal to attain the optimal cell culture media and feed formulation using a top-down approach and instead will have to implement a more empirical methodology.
Bioreactor Systems
Many therapeutic proteins are produced using genetically modified mammalian cells, as described in the preceding sections. This section describes the basic design and function of bioreactors used for suspended mammalian cell culture. Bioreactors should provide a sterile environment, adequate mixing, ease of operation, and control of temperature, pH, and dissolved oxygen. Traditionally, these requirements were met using glass or stainless-steel stirred tank systems. At production scale, therapeutic proteins are primarily produced in stainless-steel stirred tank bioreactors. However, single use, disposable systems such as the Thermo SUB™ and Sartorius Cultibag STR™ bioreactors are gaining acceptance for certain applications.
Stirred Tank Systems
Stirred tank bioreactors are generally glass or stainless-steel tanks with an impeller to provide mixing. Air or oxygen is usually bubbled through the media to supply oxygen to the cells. An example of a stirred tank bioreactor is depicted in Fig. 3. Sterility is obviously an important issue in mammalian cell culture, and therefore traditional glass or stainless-steel stirred tank systems require extensive cleaning and sterilization. A significant portion of the labor involved with operating these systems is related to cleaning and sterilizing.

Standard bench-scale, water-jacketed stirred tank bioreactor (permission of Sartorius BBI Systems Inc.)
Mixing in Stirred Tank Bioreactors. Adequate mixing is essential to suspend the cells and to facilitate heat and mass transfer. Historically, however, due to concerns regarding the sensitivity of mammalian cells to hydrodynamic stress, most stirred tank bioreactors were agitated just enough to keep the cells in suspension [51]. This low level of mixing can result in large concentration gradients of pH, oxygen, and other nutrients. Ozturk [52] demonstrated this experimentally by adding base to a poorly mixed bioreactor. The base was added to the top of the bioreactor, and because of poor mixing, a high pH region was created at the top. The cells in this region ruptured, forming a “snow ball” of cellular debris in the vicinity of the base inlet.
Proper impeller selection and sizing will improve mixing. Generally, the diameter of the impeller should be approximately one third to one half of the tank diameter. The lowest impeller should produce a radial flow pattern to aid in gas dispersion, and the upper impeller(s) should produce axial flow to eliminate “zones of mixing.” Paddle or Rushton impellers produce radial flow, and hydrofoils and pitched blade impellers produce axial flow. Retrofitting existing bioreactors with different impellers, however, may prove difficult because the motor driving the impellers may not produce enough torque to turn them. Adequate baffling will also improve mixing; baffles prevent solid body rotation and vortex formation [53].
Aeration. Oxygen can be introduced to the culture in many different ways. Membrane aeration provides efficient oxygen transfer with minimum shear damage to the mammalian cells and minimal foaming. However, due to the design complexity and the difficulty involved in cleaning and sterilizing membrane reactors, membrane aeration has limited utility in large-scale bioreactor systems. Sparger aeration offers high oxygen transfer rates and is widely used in both bench and production-scale bioreactors. Since oxygen is only sparingly soluble in water, a large surface area is needed to maximize diffusion of oxygen into the cell culture media. Frit spargers with micro-pores provide a large surface area for diffusion, but this type of sparger can cause foaming problems at large scale, and frit spargers are not effective removing dissolved carbon dioxide [54, 55]. Traditional large-hole-ring spargers tend to have fewer issues with foaming, remove dissolved carbon dioxide more efficiently, and are easier to clean. Therefore, traditional spargers are often used in production-scale stirred tank bioreactors [54, 56].
Biosensors. Sensors are required to adequately monitor bioreactor performance. Ideally, one would want online sensors to minimize the number of samples to be taken from the bioreactor and to automate the bioreactor process. Most bioreactors have autoclavable pH and dissolved oxygen (D.O.) electrodes as online sensors, and use off-line detectors, such as the Nova Flex or YSI 2950D analyzers, to measure other critical parameters such as lactate, ammonium, glucose and glutamine concentration, cell density, and carbon dioxide partial pressure (pCO2). An online pCO2 probe is commercially available from Mettler-Toledo. Probes are also commercially available that determine viable cell density by measuring the capacitance of a cell suspension. Data from perfusion and batch cultures indicate that these probes are reasonably accurate at cell concentrations greater than 0.5 × 106 cells/mL [57, 58]. The accuracy of the capacitance biomass probes can be improved by using dielectric spectroscopy data [59].
Disposable Bioreactors
One of the major drawbacks of the stainless-steel stirred tank bioreactors is the difficulty of cleaning and sterilizing the vessels. Disposable bioreactors are now commercially available that are based on the use of presterilized plastic bags. There are two primary designs of disposable bioreactors, rocker motion bags and stirred tanks. For the rocker motion bioreactors, a sterile bag is partially filled with cell culture media and is then pressurized with a mixture of carbon dioxide and air. The bag is placed on a platform that rocks back and forth creating waves inside the bag. The wave motion provides sufficient mixing and gas transfer to easily support cell growth of over 20 × 106 cells/mL [60]. Currently, rocker bags are available in sizes up to 500 L culture volume and have been proven for the Good Manufacturing Practice (GMP) production of human therapeutics. Rocker bioreactors have been primarily used as batch culture for inoculum preparation [61] and transient production; however, internal perfusion filters can be applied, allowing the option of use for high-density perfusion culture. Stirred tank single-use bioreactors are the other major class of disposable bioreactors, and are the most popular type of disposable production bioreactor [62]. Disposable stirred tank bioreactors usually consist of a cylindrical presterilized bag, supported by a stainless-steel shell, with either mechanically or magnetically coupled drive shafts turning axial flow impellers. The bag also contains either a drilled hole or a frit sparger, depending on the vendor, and an apparatus to insert autoclaved pH and DO probes or presterilized optical pH and DO patches. These patches contain a luminescent dye that changes color in proportion to changes in pH or dissolved oxygen. The color shifts of these dyes are detected using an external fluorometer [63, 64]. Disposable bioreactors are available in sizes up to 2000 L and have been shown to give comparable cell culture performance, titer, and product quality as stainless-steel bioreactors [65].
Modes of Bioreactor Operation
Industrial mammalian cell culture can be divided into three primary modes of operation: batch, fed-batch, and perfusion. Each mode of operation has its attributes and drawbacks, which are described in detail below.
Batch. Batch culture is the simplest of the three modes to operate. Cells are inoculated into media, and with the exception of agitation, temperature, pH, and D.O. control, no additional nutrients or media are added. Batch processes are easy to operate and require the least optimization effort of the three modes. However, cell densities and protein titer are significantly lower for batch processes, typically less than 5 × 106 cells/mL and 0.5 g/L of product, respectively [66]. Also, some protein products are degraded in the media during the batch process.
Fed-Batch. Fed-batch processes start out as batch cultures; after a few days of growth – when a crucial nutrient is depleted – a concentrated solution of nutrients is added to the media. Fed-batch cultures persist for one to two weeks and may produce high cell density and product titers, typically greater than 10 × 106 cells/mL and 1.5 g/L, respectively [66]. However, one needs to optimize the contents of the feed solution as well as the feeding strategy. With optimization, peak viable cell density, and harvest titers one can attain over 25 × 106 cells/mL and 10 g/L, respectively, in fed-batch cultures [50]. Similar to batch cultures, fragile proteins may be degraded during the course of the culture.
Perfusion. Perfusion cultures can be considered continuous cultures, since conditioned media is continually removed from the bioreactor. The cells are separated from the medium using a cell retention device and are returned to the bioreactor. The cell-free medium is collected for later protein purification. There are a variety of cell retention devices available, including spin filters, alternating tangential flow (ATF) filtration, acoustic separators, continuous centrifuges, and gravity settlers. Spin filters and, to a lesser extent, acoustic settlers are the two most commonly used cell retention devices for large-scale perfusion cultures [67], and recently many new perfusion-based large-scale processes are using ATF filtration [68]. Perfusion cultures usually last several weeks, but require a longer time for process optimization, and more effort in bioreactor operation than either batch or fed-batch cultures. The increased effort is primarily due to continuous feeding/harvesting and fouling of the cell retention devices. The cell densities attained in perfusion cultures are usually on the order of 50–100 × 106 cells/mL [69], and the product titer of the medium, in most cases, is lower than that from fed-batch culture [70]. However, the volumetric productivity is typically ten times greater than that of fed-batch cultures [70]. Since conditioned media is continually removed from the bioreactor, labile proteins can be separated immediately from cell proteases and other components that can cause degradation of the product. Some companies have explored the use of perfusion bioreactors in conjunction with novel purification schemes to create a continuous process, whereby the product is purified immediately after it is produced in the bioreactor [71].
Cell Culture Process and Control
Mammalian cell culture processes must be tightly controlled to attain acceptable cell density, maximize product titer, and maintain acceptable product quality. Slight deviations in pH, temperature, nutrient, or catabolite concentrations can cause irreversible damage to the cells. This section covers the effects of pH, shear stress, catabolite, and carbon dioxide accumulation on cell growth and product formation, and discusses the importance of controlling glucose and glutamine concentrations in fed-batch and perfusion cultures. A brief discussion of scale-up heuristics in mammalian cell culture is also included.
Process Parameters
General Parameter Values. The optimal pH range for mammalian cell growth is 6.8–7.4, which is typically maintained using carbon dioxide and sodium bicarbonate. The optimal osmolality of the media is between 280 and 320 mOsm/kg. In general, mammalian cells achieve high cell densities most quickly with the temperature set to 37 °C; however, a lower temperature may be advantageous in some cases for extending cell life, thereby increasing product titer. The oxygen demand for mammalian cells is 0.5–4 mM/hr. [50] and is one of the more critical control parameters in cell culture. The optimal ranges of common control parameters in cell culture are summarized in Table 3.
Mitigating Effects of Physical and Chemical Stress
Shear Stress. Because mammalian cells lack a cell wall and are larger than bacteria, they are more susceptible to hydrodynamic forces, or shear stress. Several studies have investigated the effects of shear stress on mammalian cells [72,63,64,75]. Many indicate that the action of the impeller alone does not decrease the viability of suspension-adapted mammalian cells [72, 75, 76]. Some bioprocess engineers in industry have observed a few cell lines that appear to be less robust, and anecdotally might have been damaged by the impeller. However, bubble rupture does cause sufficient hydrodynamic force to kill all the cells attached to the bubble [75]. The effects of bubble rupture can be greatly reduced by the addition of surfactants, such as Pluronic F-68. Pluronic F-68 renders cell to bubble adhesion thermodynamically unfavorable, so the cells do not adhere to bubbles [77]. However, Pluronic F-68 offers very little shear protection; cells rupture at the same level of hydrodynamic force regardless of the Pluronic F-68 concentration [78]. Cells attached to microcarriers, however, are very susceptible to shear stress and can quite easily be removed and killed by the action of the impeller [74].
pH Perturbations. As mentioned in Sect. 4, significant pH gradients within the bioreactor are common, due to inadequate mixing. Cell lysis occurs at extreme pH; however, even moderate deviations from the optimal pH may be detrimental. Osman et al. [79] found that pH values greater than 8.0 or less than 6.8 cause a considerable decrease in cell viability and a reduced culture time. Antibody titers increased when the pH setpoint was reduced from 7.2–7.3 to 7.0 [79, 80]. The best way to eliminate transient deviations from optimal pH is to improve mixing (reduce the mixing time); this can be achieved by increasing the agitation or aeration rate, adding baffles, or optimizing the impeller design or placement.
Catabolite Accumulation. Several byproducts of cellular metabolism accumulate during the course of a bioreactor run. Many of these catabolites, such as lactate, ammonia, and carbon dioxide, are detrimental to cell growth and protein production. Lactate is a product of glycolysis and lactate accumulation greater than 2 g/L tends to inhibit cell growth and increase glucose and glutamine consumption [81, 82]. Lao and Toth [81] pointed out the difficulty in completely decoupling the effects of lactate accumulation from the effects of increased osmolality (osmolality increases with increasing lactate concentration). For some cell lines, the effects of lactate accumulation can be mitigated by keeping glucose levels low, which can be achieved by optimizing media composition and feeding strategies ([48, 83], respectively). Other nutrient feeding strategies may limit lactate accumulation without limiting the glucose concentration in the culture [84].
In cell culture, ammonia is produced as a cellular metabolite and is converted from glutamine in the media through deamination. An ammonia concentration of 20 mM, and possibly less, can inhibit cell growth, induce apoptosis, and alter glycosylation in certain cell lines [85,86,87]. The primary method of reducing ammonia concentrations in fed-batch and perfusion cultures is to optimize feeding strategies. Genzel et al. [85] showed that substituting glutamine with pyruvate can greatly reduce ammonia production.
Carbon dioxide is a product of cellular respiration. In mammalian cell culture, carbon dioxide and sodium bicarbonate are normally used to control bioreactor pH. Elevated partial pressure of carbon dioxide (pCO2) hinders cell growth and protein production [88,79,90]. As with lactate accumulation, the effect of elevated pCO2 is difficult to completely decouple from the effect of elevated osmolality [88, 90]. Generally, one can reduce pCO2 by increasing the volumetric mass-transfer coefficient (kLa), typically by increasing the air sparge rate and/or the agitation rate [91]. Mostafa and Gu [92] were able to reduce pCO2 in a 1000 L culture and nearly double the titer by increasing the sparge rate and using an open pipe instead of a sparger. Table 4 summarizes the effects of shear force, pH deviation, and accumulation of lactate, ammonia, and carbon dioxide on cell culture and lists methods to minimize these effects.
Temperature Shifts. Culture temperature is one of the primary control parameters in mammalian cell culture. As stated previously, most mammalian cells grow optimally at 37 °C. However, reducing the temperature set point slows cellular growth rate and metabolism, extends the period of high cell viability, arrests cells in the G0/G1 phase of the cycle, and possibly reduces intracellular protease activity [93,94,95,96]. By decreasing the temperature to 31–33 °C, the period with high cell viability is extended for several days, leading to higher product titers. Fox et al. [96] and Bollati-Fogolin et al. [93] reaped the benefits of a cooler temperature without excessively extending culture time by shifting from 37 °C to 32 °C or 33 °C, respectively. The temperature shift occurred toward the end of the exponential growth phase of the cultures, approximately 3–4 days after inoculation in a batch culture. Different cell lines and culture conditions may have different optimal time points for the temperature shift. However, finding the optimal time point is important and worth investing the time required. Operating cell cultures at a reduced temperature often increases the production of the target protein, but to avoid substantially increasing the culture time, the culture is generally started at 37 °C, then decreased to 31–33 °C at a predetermined time point based on cell growth.
Fed-Batch Process Control and Optimization
In general, fed-batch cultures are initially operated in batch mode until a key nutrient(s) is exhausted. Then, a solution containing the nutrient(s) is added to the media. As mentioned in Sect. 4, the feed solution and feeding strategy should be optimized. A common approach is to use partial concentrates (i.e., 10×) of the basal media as the feed solution; typically, most of the salts found in the basal media are not included in the feed solution. The bioreactor is sampled at regular intervals and when one or more of the key nutrients (usually glucose and/or glutamine) are below a certain concentration, a precise amount of feed solution is added to raise the nutrient concentration to its setpoint. A useful rule for choosing the nutrient setpoint concentrations is that it should provide enough nutrients to support cell growth and product production while avoiding formation of toxic levels of catabolites due to excess feeding. Huang et al. [50] followed this heuristic and fed a concentrated amino acid solution based on the projected uptake rates of the amino acids from their platform processes. Another method is to determine which media components are depleted during the culture and add those particular nutrients to the bioreactor independently [97, 98]. Wong et al. [99] selected glutamine and glucose as key control nutrients, and used two feed solutions (glucose and glutamine solutions) to maintain them at specified concentrations. Using this strategy, they maximized cell viability and density while decreasing accumulation of lactate and other catabolites. Recent advances in proteomics and metabolomics have given researchers additional insight into the internal machinery of CHO cells, and could potentially lead to more targeted approaches to media and feed optimization [100]. Another common approach to nutrient feed optimization relies on the use of non-animal-derived hydrolysates, such as soy, wheat gluten, and yeast [49]. Hydrolysates are undefined, and variability between lots of the same type of hydrolysate is a concern [49]. The use of size exclusion filtration can reduce the lot-to-lot variability of hydrolysates [101]. After a series of optimization experiments, based on statistical design of experiments, Kim and Lee (2009) were able to increase titer by approximately two- to three-fold using a mixture of soy, wheat, and yeast hydrolysates.
Perfusion Process Control and Optimization
In perfusion bioreactors, culture is removed from the bioreactor at certain times. The cells are separated from the conditioned media, the conditioned media is collected, and the cells are returned to the bioreactor. Perfusion bioreactors can be operated in a variety of modes. The simplest mode is to consistently remove a certain amount of conditioned media each day (i.e., one bioreactor volume/day) and replace with fresh media. This mode is relatively easy to control. However, as the cell density increases, the required nutrient level may not be met. Also, the protein product tends to become diluted in the collected supernatant [52, 87]. Another mode of perfusion operation is to remove relatively small volumes of media at a time and replace the volume with a concentrated solution of nutrients. This mode is similar to fed-batch bioreactors. One might think of this as a perfusion/fed-batch hybrid, and like fed-batch bioreactors, considerable effort goes into determining the optimal nutrient setpoint concentrations and the feeding strategy. Sophisticated analysis and control schemes have been developed for this type of perfusion bioreactor [102, 103]. A third mode of perfusion bioreactor operation attempts to maintain a pseudo-steady state cell density, after an initial growth period. Dowd et al. [57] employed an online cell density meter and was able to maintain a relatively constant cell density by altering the perfusion rate – the amount of media removed from the bioreactor. This mode of perfusion reduces the frequency of sampling and analysis required to maintain a set nutrient concentration, but requires a well-characterized online cell density probe and a well-calibrated pump control scheme. Warikoo et al. [71] also employed an online, capacitance-based biomass probe to maintain a pseudo-steady state cell density, but achieved the steady-state condition by keeping the perfusion rate constant while implementing an automated cell bleed. In other words, Warikoo et al. [71] would automatically discard a small amount of cell culture to maintain a predefined cell concentration.
Scale-Up of Mammalian Cell Bioreactors
When scaling up a process to large scale, it is important to maintain the same physical and chemical conditions as in small scale. The chemical conditions include pH, oxygen level, concentration of medium components, and concentrations of toxic metabolites. These must be monitored and controlled to keep the cells in the proper physiological environment. The physical conditions include the bioreactor configuration and the power provided to the bioreactor. In scale-up, it is advisable to preserve a similar geometrical configuration of the bioreactor in order to facilitate duplication of mixing patterns.
Impellers are an important physical component in a stirred bioreactor; they convert mechanical energy to hydrodynamic motion and generate the turbulence required to keep the cells in suspension and achieve sufficient mass transfer. At large scale, efficient oxygen delivery and carbon dioxide stripping become increasingly difficult due to suboptimal mixing and relatively low air sparge rates [54, 55]. At the same time, the energy generated at the tip of the impeller blades must be limited, as certain cell lines can be damaged by the elevated shear force. Therefore, to achieve good mixing and minimize possible cell damage from high shear force, one must determine the proper impeller shape, ratio of impeller to vessel diameter, and impeller tip speed [54].
The mixing time as well as the oxygen and carbon dioxide mass transfer rates can be correlated to the power per unit volume of the reactor, also known as the average or overall energy dissipation rate [55, 104]. Maintaining constant power per unit volume is a commonly used scale-up strategy because of its simplicity. Other strategies include keeping the average shear force experienced by the cells constant, or keeping the maximal shear force constant (the shear force experienced by the cells when passing the impeller tip).
In addition to scale-up models, scale-down models are widely used to establish the operating ranges of critical large-scale process variables [105, 106]. Conducting the many experiments required to define a validated range for each parameter is not feasible at large scale. A scale-down model based on an existing large-scale process therefore becomes an efficient and economical tool in reaching this goal, and is an expectation of most regulatory agencies [91]. It might be necessary to operate the small-scale bioreactor in a nonideal manner to adequately mimic the large-scale bioreactor. For example, Li et al. [91] completely eliminated the air sparge in their small-scale bioreactors in order to reduce the carbon dioxide stripping capacity of the small-scale bioreactors and make the dissolved carbon dioxide profile more comparable between the small- and large-scale bioreactors.
Purification Process
Biopharmaceuticals have a vast scope of different products, including monoclonal antibodies (mAbs) and mAb fragments, vaccines, cytokines, hormones, enzymes, gene therapies, and blood products. The scope of this section is primarily focused on recombinant protein purification.
The process of purification, also called “downstream processing,” depends on the product and the degree of purification required [107]. Conventional strategies used for purification of therapeutic proteins generally involve four steps: harvest and clarification, product capture, intermediate purification (removal of bulk impurities), and polishing (removal of trace impurities). When affinity chromatography is used for product capture, the intermediate purification step can often be eliminated.
Microbial cells such as E. coli and yeast are limited in their ability to make glycoproteins or correctly structured glycoproteins. Therefore, therapeutic proteins including mAbs that require glycosylation for their biological activity are often produced via culture of mammalian cells. Compared to bacterial fermentation, mammalian cell culture generates complex broths with a variety of impurities. Typical impurities found in culture broths include:
-
1.
Host cells and cell debris
-
2.
Host cell protein (HCP) and DNA released by the cells
-
3.
Aggregated proteins or cleaved proteins produced by the cells
-
4.
Medium additives (such as nutrients and protein used to support cell growth)
These impurities pose risks for the safety of therapeutic proteins and must be removed to a final concentration below their target limit. In addition, the product stream contacts materials such as filters and resins. Extractables, such as leachates from protein A resins, can pose an immunogenic risk to the patient and must be eliminated [108]. Finally, adventitious agents such as viruses and bacterial pathogens or related contaminants such as endotoxins can lead to serious problems with the safety of the protein preparation and therefore must be minimized. Table 5 lists concentrations for the above impurities that are generally considered acceptable in a final protein product [109, 110].
This section describes some general processes used for recombinant protein purification, particularly mAb purification, including methods and tools currently employed by the biopharmaceutical industry to achieve clarification, capture, and removal of impurities.
Platform Purification Processes
It is impossible to have one purification process to fit a vast scope of different recombinant therapeutic proteins. However, increasing availability of affinity resins can greatly simplify the purification process of any proteins, since over 90% product purity can often be achieved by one affinity chromatography step.
mAbs can bind to Protein A affinity resin through their Fc region. Over the last decade, a common purification platform has been developed using Protein A affinity resin as the capture step (Fig. 4, [111]). Some process development-related attributes with respect to production of mAbs include: (1) significant increase in titer levels over the past two decades (from 0.5 g/L to 5–10 g/L); (2) high product requirement (as much as tons) due to higher doses of typical mAb products; (3) larger manufacturing facilities (from 1–5 kL to 10–20 kL).

Flow diagram of a typical mAb platform purification process. The polishing chromatography can be ion exchange column or membrane, or mixed mode column
The platform approach begins with a boilerplate purification process that is adjusted and optimized for each new product candidate. This usually entails verifying process fit, establishing dynamic binding capacity and loading parameters for the chromatography steps, defining the pH and conductivity of the buffers, and verifying impurity removal and product quality. The key advantages of a platform purification process include: (1) Shortening of process development timelines using minimal resources; (2) Ease of process scale-up, validation, and technology transfer; (3) Reduced capital expense when bringing in a new product to the manufacturing facility; (4) Reduction of raw material inventory through the use of common components; (5) Streamlined documentation through the use of templates. Since the Fc-fusion proteins also bind to Protein A affinity resin, the mAb purification platform can also be applied to purification of therapeutic Fc-fusion proteins.
Antibody fragments (e.g., Fab, scFv, dAb, minibody, diabody, etc.) are becoming the next important class of protein-based biotherapeutics after mAbs [112]. One of the advantages is that due to their structure and smaller sizes, antibody fragments possess unique properties (e.g., easier tissue penetration and shorter half-life) that suit a range of diagnostic and therapeutic applications. With the introduction of Capto L, the first industrial platform for the purification of antibody fragments is now available (Fig. 5). With its recombinant Protein L ligand, Capto L is a chromatography medium with a broad range affinity for antibody fragments of different sizes containing kappa light chains. Table 6 lists the binding characteristics of Protein L, Protein A, and Protein G for immunoglobulins of different species.

Flow diagram of a typical platform purification process for mAb fragments. The polishing chromatography can be ion exchange column or membrane, or mixed mode column
Selection of the purification strategy will depend on the specific properties of the sample and the required level of purification. Due to the widely differing properties of proteins, a final purification strategy that is most suitable for one protein may be unsuitable for another. A logical combination of chromatographic steps can usually achieve the final goal of protein purification.
Cell Culture Harvest and Clarification
For proteins secreted into cell culture broth, the first unit operation in the downstream process is to remove cells, cell debris, and large particles, to clarify the supernatant by removing small and submicron particles, and to remove any potential microbial contamination. The primary harvest techniques commonly used in the industry are centrifugation, microfiltration, and depth filtration, depending on the production scale, cell density, and overall cost effectiveness. Flocculants can be added prior to the cell culture harvest to facilitate the harvest and clarification [111]. The final step prior to capture chromatography is usually the sterile filtration of the clarified supernatant.
Depth filtration refers to the use of a porous medium capable of retaining particles from the mobile phase throughout its matrix rather than just on its surface. It is often used for the whole cell culture harvest at or below 2000-L bioreactor scales due to its operational convenience and disposable design, and also commonly used following a primary harvest technique such as centrifugation because there is a practical lower limit to the particle size that can be removed by centrifugation technique. Depth filters have different pore sizes (0.5–10 μm) that can be used for primary depth filtration (using large pores) and secondary depth filtration (using smaller pores). In addition to removing particles by size exclusion, some positively charged depth filters can potentially remove negatively charged impurities such as endotoxins, DNA, and viruses by absorption. The major depth filter suppliers for biopharmaceutical industry include 3 M/CUNO, Millipore, Pall, and Sartorius.
Microfiltration is used in tangential flow mode by which cell culture fluid flows tangential to the micro-porous membrane and pressure-driven filtrate flow separates the soluble product from large particles. It requires thorough process monitoring of pressure, feed flow, and flux, and therefore is more delicate to use than normal flow filtration such as depth filtration and sterile filtration. The advantage of the tangential flow microfiltration is its ability to reduce the cell culture supernatant volume by concentration and perform buffer exchanges by diafiltration. Tangential flow microfiltration is best suited for cell culture solid load less than 3% packed cell volume due to the pressure limitation. At higher solid load, centrifugation is preferred for large-scale manufacturing.
Centrifugation separates soluble product from insoluble particles in the cell culture broth by their density difference and can be performed in a batch mode or continuous mode. In the batch mode, an intermediate tank is advisable to hold the centrate before secondary clarification because the centrate flow is discontinuous due to intermittent solid discharge phases. Most industrial applications utilize disk-stack centrifuges to remove cells and cell debris continuously, rendering their throughput consistent with the desire to limit the time for harvest operation. However, cells can be disrupted during the process and many particles of submicron size cannot be removed in the centrifuge, thereby increasing the burden on the subsequent depth filtration.
High cell density cell culture, which produces increased cell debris and colloids in the cell culture fluid, presents a burden to the harvest and clarification. This can result in a high depth filter surface area requirement that can exceed the existing filtration train equipment capacity. To improve the efficiency and capacity of primary clarification techniques, various feed pretreatment methods have been developed in recent years [113]. Pretreatment options include use of acidic or cationic polyelectrolytes, such as acetic acid, calcium chloride, potassium phosphate, and cationic polymers such as chitosan. These pretreatment methods generate larger-sized particulates that can be more easily removed by centrifugation or depth filtration.
Figure 6 shows the common harvest and clarification schemes for large-scale mammalian cell culture process.

Common harvest and clarification schemes for large-scale mammalian cell culture process
Capture Chromatography
After cell culture harvest and clarification, the next step in a protein purification process is to concentrate and isolate a protein product by capture chromatography. This step is best accomplished with affinity chromatography due to its high selectivity. Protein A chromatography is the most commonly used unit operation for antibody capturing. The degree of the product purity is consistently higher than 95%, with HCP reduction over 1000 times. It also removes other process-related impurities such as DNA, media components, and virus particles. Over the last decade, the dynamic-binding capacity of Protein A resins has been significantly improved (over 80 g mAb/L) and alkaline-resistant Protein A resins are also available thereby relieving the previous concern of the column cleaning issue.
In recent years, alternative affinity media for mAb purification have become available including CaptureSelect affinity resins from ThermoFisher. For example, CaptureSelect™ FcXL affinity matrix can be used to purify human IgG, Fc fusion proteins, and certain antibody fragments such as minibodies from cell culture medium and human plasma. The affinity matrix possesses a ligand that is a 13 kDa llama heavy chain antibody fragment specific for the CH3 domain of human IgG (all four subclasses including IgG1, IgG2, IgG3, and IgG4). The major advantage of the CaptureSelect™ FcXL affinity matrix versus Protein A resin lies in its requirement of only gentle pH condition for elution, thereby retaining better biological activity and minimizing aggregation of IgG and Fc fusion proteins caused by low pH exposure. It has been observed that the salts in the low pH elution buffer are inhibitory of some hydrophobic minibody elution from CaptureSelect™ FcXL column but can be counteracted by adding a small percentage of propylene glycol in the elution buffer [114]. Other CaptureSelect affinity resins provide specific purification of human growth hormone, follicles-stimulating hormone, human albumin, gonadotropin, transferrin, fibrinogen, antithrombin III, IgM, and IgA.
Another alternative of Protein A chromatography are synthetic mimetic affinity resins provided by ProMetic. ProMetic’s MAbsorbent affinity resins can be used to purify all IgG-type monoclonal antibodies, antibody fragments, and antibody-fusion proteins with the advantage of alkaline-resistant property. Additionally, ProMetic also provides other types of affinity resins for purification of plasma proteins.
Cation exchange chromatography (CEX) represents another type of capture chromatography. The charge characteristics of a protein can be altered by changing the pH of the separation. Based on the charge differences of a targeted protein and other impurities, conditions at the capture step are selected to maximally bind the targeted protein and minimally bind the impurities to achieve isolation of the product. CEX offers many advantages such as high capacity (as high as over 100 g/L), long resin lifetime, and low cost. However, it requires a pH shift of the feed and a decrease of the conductivity before loading onto the column in order to optimize the dynamic binding capacity. CEX can be used for capturing antibodies and other proteins with a basic isoelectric point. Antibody variants such as charge variants and aggregates can be removed as well as most negatively charged impurities. The drawback of CEX lies in its relatively low selectivity, therefore lower product purity in comparison to affinity chromatography. To reduce the host cell protein burden before CEX, selective precipitation in combination with adsorptive depth filtration needs to be used for CHO cell culture clarification [115].
Removal of Impurities
Although the capture step, particularly the affinity chromatography step, can dramatically enrich a targeted protein and remove majority of impurities, some product-related impurities such as aggregates and undesirable charge variants and degradation products, and process-related impurities such as HCP, DNA, endotoxins, virus particles, and leached affinity ligand, can remain in the eluted pool. Additional polishing steps are needed to eliminate these residual impurities [111, 116].
CEX can be used not only as capture chromatography for proteins with high isoelectric point (>7.5), but also is more commonly used as a polishing chromatography after the affinity chromatography. In the latter case, elution conditions of the affinity column are preferably adjusted to fit the CEX loading condition without further conditioning. Since most process-related impurities have lower isoelectric points, CEX is very effective for the removal of HCP, endotoxins, DNA, and virus particles. With properly optimized elution condition, CEX is also effective to remove aggregates, undesirable charge variants, and degradation products. Based on the author’s experience, the efficiency of impurity removal by CEX is negatively related to the resin load; the higher the resin load, the less removal of the impurities.
Anion exchange chromatography (AEX) is another commonly used polishing method. In contrast to CEX, most impurities bind to AEX column more strongly than antibodies do. Therefore AEX is usually operated in a flow-through mode. With optimized loading pH and conductivity, the target protein flows through the column while leaving impurities on the column. The main advantages of AEX in the flow-through mode include high resin load (as high as >1000 g/L), simple operation, and low cost. From the author’s own experience, AEX operated in the flow-through mode is more effective to remove the process-related impurities than the product-related impurities. For the target proteins with relatively low isoelectric points (< 7), use of the bind-elute mode is more effective to remove impurities than using the flow-through mode.
Ceramic hydroxyapatite chromatography is increasingly used as polishing step for antibody purification. Hydroxyapatite (Ca5(PO4) 3OH)2 is a form of calcium phosphate that can be used for purification of different subclasses of antibodies, antibodies that differ in light chain composition, antibody fragments, isozymes, supercoiled DNA from linear duplexes, and single-stranded from double-stranded DNA. CHT ceramic hydroxyapatite is a spherical, macro-porous form of hydroxyapatite (BioRad Laboratories). Unlike other chromatography adsorbents, CHT is both the ligand and the support matrix. Hydroxyapatite has unique selectivity and resolution due to its multimodal interactions with proteins. It can theoretically retain solutes by anion exchange with positively charged calcium, by metal affinity with calcium, by cation exchange with phosphate groups, and by hydrogen bonding with crystal hydroxyl groups. Based on the author’s own experience, CHT is extremely effective in removing product aggregates and can reduce aggregates from the initial over 30% to below 1% [114]. It is also effective in removing HCP, DNA, endotoxins, and virus particles [117]. The main drawbacks of CHT resin include relatively low protein-binding capacity and shorter resin life time. Other resins with multimodal ligands such as Capto MMC and Capto Adhere (GE Healthcare) can be used as polishing steps for removal of impurities.
Over the last decade, membrane chromatography or membrane adsorber has been gaining popularity as a polishing method for antibody purification due to its speed, simplicity, and disposable design. It functions similarly to packed columns, but in the format of conventional filtration modules, with symmetric microfiltration membranes functionalized with specific ligands attached to the convective pores. Diffusive pores are eliminated, mass transfer of biomolecules depends on convection, and the binding capacity is largely independent of flow rates. The most widely used membrane adsorber is the anion exchange membrane adsorber, which is usually used in the flow-through mode, and has essentially replaced the AEX column as a polishing method in antibody purification. The loading conditions for anion exchange membrane adsorbers are similar to AEX column. Impurities such as DNA, HCP, endotoxins, and virus particles bind to the membrane at neutral or slightly basic pH and low conductivity while the product flows through. The major brands of membrane adsorbers include Sartobind from Sartorius, Mustang from Pall, and ChromoSorp from Millipore. Similar to anion exchange membrane adsorbers, adsorptive depth filtration containing diatomaceous earth has been shown to be able to remove most process-related impurities [118].
Thanks to the availability of ion exchange membrane adsorbers, continuous antibody purification processes have become possible by performing both cationic and anionic exchange chromatography in a flow-through mode [119, 120]. The continuous processes can significantly reduce the buffer and equipment usage and shorten the length of the whole purification process.
Virus Filtration
Mammalian cell culture used in manufacturing mAbs and other therapeutic recombinant proteins produce endogenous retroviruses and may occasionally be infected with adventitious viruses during processing. It is generally accepted that mammalian cell-derived products should be controlled to contain less than one retrovirus particle per million doses. To meet the virus safety requirement by the regulatory agencies, the purification processes need to demonstrate sufficient built-in virus clearance capacity with orthogonal clearance mechanisms. Three complementary virus clearance mechanisms are generally designed into the purification processes: (1) virus inactivation by low-pH or detergent exposure; (2) virus removal through differential binding in chromatography; (3) virus removal through size-exclusion by nanofiltration. Since most viruses are bigger than 20 nm in size, a nanofilter with 20 nm cutoff is usually used for viral filtration. The common brands of nanofilters include Planova (Asahi-Kasei), Viresolve (Millipore), Ultipor (Pall), and Virosart (Sartorious).
During virus filtration, a not uncommon problem is nanofilter fouling. Fouling is often caused by the presence of high molecular weight species such as protein aggregates and can be significantly reduced using a 0.1–0.2 μm prefilter before the nanofilter. For some hydrophobic mAbs, fouling can be caused by inappropriate pH and salt concentrations [121]. It has been observed that some hydrophobic mAbs cannot pass through 20 nm viral filter at pH above 5 or conductivity above 15 mS/cm. Light scattering analysis has confirmed that these mAbs had increased hydrodynamic radius close to the pore size of the viral filter at neutral pH and above physical saline concentration. The underlining mechanism is reversible self-association, since these mAbs can pass through the same size of the viral filter after lowering the load pH to 4 and the salt concentration to below 50 mM. Lower pH renders the mAbs more positively charged thereby suppressing self-association. On the other hand, higher salt concentration can enhance hydrophobic interaction thereby promoting self-association.
Product Formulation by Ultrafiltration, Diafiltration, and Sterile filtration
The last unit operation in therapeutic protein purification is product formulation. This is achieved by product concentration through ultrafiltration, buffer exchange by diafiltration, and final sterile filtration to remove bioburden.
Ultrafiltration is a pressure-driven membrane process that is widely used for protein concentration and buffer exchange. Ultrafiltration is a size-based separation, where the product is retained and buffer components pass through freely. Buffer exchange is achieved using a diafiltration mode in which the buffer of the final desired composition is added to the retentate system at the same rate in which filtrate is removed thereby maintaining a constant retentate volume. Ultrafiltration is normally carried out in tangential flow filtration mode, in which fluid passes across the filter (cross-flow), tangential to the plane of the filter surface, resulting in higher throughput than normal flow filtration mode. Ultrafiltration and diafiltration processes are typically controlled using constant retentate pressure, constant trans-membrane pressure, or constant filtrate flux.
Proteins, particularly hydrophobic proteins, tend to aggregate and precipitate at higher concentrations during ultrafiltration. Based on the author’s experience, the addition of arginine at final 50–200 mM concentration prior to the product concentration can significantly suppress the protein aggregation and precipitation [114]. The spiked arginine can be removed by subsequent diafiltration if it is not part of the formulation components.
Strategies for Scaling Up Purification Process
The great demand for high quality and quantity of therapeutic proteins, particularly monoclonal antibodies, requires robust and economical manufacturing processes, both upstream and downstream. When bioreactors are scaled up from bench to pilot to production scale (2000–20,000 L), downstream processes must be scaled up accordingly. Equally complicated as upstream processes, scale-up of downstream processes requires more than simply increasing the size and volume of the laboratory equipment. The use of larger piping dimensions, larger filters, different types of pumps, and larger dead volumes can introduce variation to the large-scale process. We’ve limited our description here to common strategies used by the industry to scale up chromatography columns.
Chromatography process development is often conducted at lab scales using columns with a bed volume around of 10–20 mL at manufacturing-relevant bed heights, whereas the column bed volumes used in a manufacturing process can be 10,000-fold greater. The scale-up of chromatography operations is usually achieved by increasing the column diameter but maintaining the resin bed height and linear flow rate. This ensures that the residence time is kept the same at all scales of operations. However, linear scale-up of the chromatography column to the manufacturing scale is not always practical for the existing manufacturing facility capability and can often be cost prohibitive. Therefore, to reduce the column resin cost and prevent other issues associated with larger-sized columns such as potential column pressure increase and poor column packing homogeneity, multiple-cycle runs with appropriately sized smaller columns for each column chromatography step are often performed at manufacturing scales. However, the number of cycles for each unit operation is limited by the total downstream process time, which should be the same or shorter than the upstream cell culture process time.
Successful process scale-up depends on determining, measuring, and monitoring critical scale-independent process parameters, then designing and operating equipment appropriately to deliver those same parameters at larger scales. A scale-up process evaluation from the bench scale to the manufacturing scale can be divided into several stages [122].
-
1.
Initial calculation. It is risky to directly apply the process conditions developed at lab scale to the manufacturing scale that is often 10,000-fold greater. Therefore, a multiple-step scale-up evaluation is required. The manufacturing column sizes are chosen with consideration of the cost of the resins, resin dynamic binding capacity, flow rate limitation of the purification skid, and number of the cycles for each unit operation. Once the manufacturing column size is determined, a column size at 10 to 50-fold smaller scale can be chosen for the pilot-scale evaluation. A further 50 to 100-fold scale-down column can be used for lab scale confirmation of the process initially developed using 10–20 mL columns.
-
2.
Process confirmation and further optimization in lab-scale scale-up runs. Key parameters for optimization include maximum resin load and flow rate, process time, buffer volumes for each step, pooling criteria, and in-process pool stability (hold time) compatible with manufacturing operation. Shortening process time is critical for improving throughput, preventing product deterioration in unformulated condition and growth of potential adventitious agents.
-
3.
One or more pilot scale runs for process confirmation and necessary adjustment.
-
4.
One or more at-scale engineering runs to test the equipment compatibility with the developed process and the documentation accuracy of process parameters and description.
The elution volume and product concentration from each column step should be monitored during scale-up since they are associated with the actual elution buffer conditions. High product concentration in the elution pool can lead to product precipitation and aggregation. In addition, pooling criteria can be modified during scale-up to strike a balance between the product yield, purity, and impurity levels.
References
Paul D (2014) World preview 2014, outlook to 2020. Evaluate pharma. http://info.evaluategroup.com/rs/evaluatepharmaltd/images/EP240614.pdf
Brekke OH, Loset GA (2003) New technologies in therapeutic antibody development. Curr Opin Pharmacol 3(5):544–550
Graddis TJ, Remmele RL, McGrew JT (2002) Designing proteins that work using recombinant technologies. Curr Pharm Biotechnol 3(4):285–297
Kumar S, Singh (eds) (2015) Developability of biotherapeutics: computational approaches. CRC Press
Lazar GA, Marshall SA, Plecs JJ, Mayo SL, Desjarlais JR (2003) Designing proteins for therapeutic applications. Curr Opin Struct Biol 4:513–518
Marshall SA, Lazar GA, Chirino AJ, Desjarlais JR (2003) Rational design and engineering of therapeutic proteins. Drug Discov Today 8:212–221
Vasserot AP, Dickinson CD, Tang Y, Huse WD, Manchester KS, Watkins JD (2003) Optimization of protein therapeutics by directed evolution. Drug Discov Today 8:118–126
Ecker DM, Jones SD, Levine H (2015) The therapeutic monoclonal antibody market. MAbs 7(1):9–14
McAtee AG, Templeton N, Young JD (2014) Role of Chinese hamster ovary central carbon metabolism in controlling the quality of secreted biotherapeutic proteins. Pharm Bioprocess 2(1):63–74
Lai T, Yang Y, Ng SK (2013) Advances in mammalian cell line development technologies for recombinant protein production. Pharmaceuticals 6:570–603
Kim JY, Kim YG, Lee GM (2012) CHO cells in biotechnology for production of recombinant proteins: current state and further potential. Appl Microbiol Biotechnol 93:917–930
Wurm FM (2013) CHO quasispecies—implications for manufacturing processes. Processes 1:296–311
Jayapal KP, Wlaschin KF, Yap MGS, Hu WS (2007) Recombinant protein therapeutics from CHO cells—20 years and counting. Chem Eng Prog 103:40–47
Gaillet B, Gilbert R, Broussau S, Pilotte A, Malenfant F, Mullick A, Garnier A, Massie B (2010) High-level recombinant protein production in CHO cells using lentiviral vectors and the cumate gene-switch. Biotechnol Bioeng 106(2):203–215
Tian F, Lu Y, Manibusan A, Sellers A, Tran H, Sun Y, Phuong T, Barnet R, Hehli B, Song F, DeGuzman MJ, Ensari S, Pinkstaff JK, Sullivan LM, Biroc SL, Cho H, Schultz PG, DiJoseph J, Dougher M, Ma D, Dushin R, Leal M, Tchistiakova L, Feyfant E, Gerber HP, Sapra P (2014) A general approach to site-specific antibody drug conjugates. Proc Natl Acad Sci U S A 111(5):1766–1771
Dorai H, Kyung YS, Ellis D, Kinney C, Lin C, Jan D, Moore G, Betenbaugh MJ (2009) Expression of anti-apoptosis genes alters lactate metabolism of Chinese Hamster Ovary cells in culture. Biotechnol Bioeng 103(3):592–608
Le H, Vishwanathan N, Jacob NM, Gadgil M, Hu WS (2015) Cell line development for biomanufacturing processes: recent advances and an outlook. Biotechnol Lett 37(8):1553–1564
Fischer S, Handrick R, Otte K (2015) The art of CHO cell engineering: a comprehensive retrospect and future perspectives. Biotechnol Adv 33:1878–1879
Running Deer J, Allison DS (2004) High-level expression of proteins in mammalian cells using transcription regulatory sequences from the Chinese hamster EF-1alpha gene. Biotechnol Prog 20(3):880–889
Brinster RL, Allen JM, Behringer RR, Gelinas RE, Palmiter RD (1988) Introns increase transcriptional efficiency in transgenic mice. Proc Natl Acad Sci U S A 85(3):836–840
Palmiter RD, Sandgren EP, Avarbock MR, Allen DD, Brinster RL (1991) Heterologous introns can enhance expression of transgenes in mice. Proc Natl Acad Sci U S A 88(2):478–482
Pestova TV, Kolupaeva VG, Lomakin IB, Pilipenko EV, Shatsky IN, Agol VI, Hellen CU (2001) Molecular mechanisms of translation initiation in eukaryotes. Proc Natl Acad Sci U S A 98(13):7029–7036
Kim JM, Kim JS, Park DH, Kang HS, Yoon J, Baek K, Yoon Y (2004) Improved recombinant gene expression in CHO cells using matrix attachment regions. J Biotechnol 107(2):95–105
Zahn-Zabal M, Kobr M, Girod PA, Imhof M, Chatellard P, de Jesus M, Wurm F, Mermod N (2001) Development of stable cell lines for production or regulated expression using matrix attachment regions. J Biotechnol 87(1):29–42
Harland L, Crombie R, Anson S, deBoer J, Ioannou PA, Antoniou M (2002) Transcriptional regulation of the human TATA binding protein gene. Genomics 79(4):479–482
Campbell M, Corisdeo S, McGee C, Kraichely D (2010) Utilization of site-specific recombination for generating therapeutic protein producing cell lines. Mol Biotechnol 45(3):199–202
Crawford Y, Zhou M, Hu Z, Joly J, Snedecor B, Shen A, Gao A (2013) Fast identification of reliable host for targeted cell line develoment from a limited-genome screening using combined φC31 integrase and CRE-Lox technologies. Biotechnol Prog 29(5):1307–1315
Kennard ML, Goosney DL, Monteith D, Zhang L, Moffat M, Fischer D, Mott J (2009) The generation of stable, high MAb expressing CHO cell lines based on the artificial chromosome expression (ACE) technology. Biotechnol Bioeng 104:540–553
Kichler A (2004) Gene transfer with modified polyethylenimines. J Gene Med 6(Suppl 1):S3–S10
Mehier-Humbert S, Guy RH (2005) Physical methods for gene transfer: improving the kinetics of gene delivery into cells. Adv Drug Deliv Rev 57(5):733–753
Scherer F, Anton M, Schillinger U, Henke J, Bergemann C et al (2002) Magnetofection: enhancing and targeting gene delivery by magnetic force in vitro and in vivo. Gene Ther 9(2):102–109
Wong TK, Neumann E (1982) Electric field mediated gene transfer. Biochem Biophys Res Commun 107(2):584–587
Cockett MI, Bebbington CR, Yarranton GT (1990) High level expression of tissue inhibitor of metalloproteinases in Chinese hamster ovary cells using glutamine synthetase gene amplification. Biotechnology 8(7):662–667
Lucas BK, Giere LM, DeMarco RA, Shen A, Chisholm V, Crowley CW (1996) High-level production of recombinant proteins in CHO cells using a dicistronic DHFR intron expression vector. Nucleic Acids Res 24(9):1774–1779
Fann CH, Guirgis F, Chen G, Lao MS, Piret JM (2000) Limitations to the amplification and stability of human tissue-type plasminogen activator expression by Chinese hamster ovary cells. Biotechnol Bioeng 69(2):204–212
Kim SJ, Lee GM (1999) Cytogenetic analysis of chimeric antibody-producing CHO cells in the course of dihydrofolate reductase-mediated gene amplification and their stability in the absence of selective pressure. Biotechnol Bioeng 64(6):741–749
Yoshikawa T, Nakanishi F, Ogura Y, Oi D, Omasa T, Katakura Y, Kishimoto M, Suga K (2000) Amplified gene location in chromosomal DNA affected recombinant protein production and stability of amplified genes. Biotechnol Prog 16(5):710–715
Brezinsky SC, Chiang GG, Szilvasi A, Mohan S, Shapiro RI, MacLean A, Sisk W, Thill G (2003) A simple method for enriching populations of transfected CHO cells for cells of higher specific productivity. J Immunol Methods 277:141–155
Bailey LA, Hatton D, Field R, Dickerson AJ (2012) Determination of Chinese hamster ovary cell line stability and recombinant antibody expression during long-term culture. Biotechnol Bioeng 109(8):2093–2103
Dorai H, Corisdeo S, Ellis D, Kinney C, Chomo M, Hawley-Nelson P, Moore G, Betenbaugh MJ, Ganguly S (2012) Early prediction of instability of Chinese hamster ovary cell lines expressing recombinant antibodies and antibody-fusion proteins. Biotechnol Bioeng 109(4):1016–1030
Ham RG (1981) Tissue growth factors. In: Baserga R (ed) Handbook of experimental pharmacology, vol 57. Springer, New York, p 13
Sato GH, Pardee A, Sirbasku DA (eds) (1982) Growth of cells in hormonally defined media. Cold Spring Harbor Conference on Cell Proliferation, vol 9. Cold Spring Harbor Press, Cold Spring Harbor, New York
Fletcher T (2005) Designing culture media for recombinant protein production: a rational approach. BioProcess Int 3(1):30–36
Chun C, Heineken K, Szeto D, Ryll T, Chamow S, Chung JD (2003) Application of factorial design to accelerate identification of CHO growth factor requirements. Biotechnol Prog 19(1):52–57
Lee GM, Kim EJ, Kim NS, Yoon SK, Ahn YH, Song JY (1999) Development of a serum-free medium for the production of erythropoietin by suspension culture of recombinant Chinese hamster ovary cells using a statistical design. J Biotechnol 69(2,3):85–93
Liu CH, Chu IM, Hwang SM (2001) Factorial designs combined with the steepest ascent method to optimize serum-free media for CHO cells. Enzyme Microb Technol 28(4–5):314–321
Allison DW, Aboytes KA, Fong DK, Leugers SL, Johnson TK, Loke HN, Donahue LM (2005) Development and optimization of cell culture media: genomic and proteomic approaches. BioProcess Int 3(1):38–45
Ma N, Ellet J, Okediadi C, Hermes P, McCormick E, Casnocha S (2009) A single nutrient feed supports both chemically defined NS0 and CHO fed-batch processes: improved productivity and lactate metabolism. Biotechnol Prog 25(5):1353–1363
Butler M, Meneses-Acosta A (2012) Recent advances in technology supporting biopharmaceutical production from mammalian cells. Appl Microbiol Biotechnol 96:885–894
Huang Y, Hu W, Rustandi E, Chang K, Yusuf-Makagiansar H, Ryll T (2010) Maximizing productivity of CHO cell-based fed-batch culture using chemically defined media conditions and typical manufacturing equipment. Biotechnol Prog 26(5):1400–1410
Varley J, Birch J (1999) Reactor design for large-scale suspension animal cell culture. Cytotechnology 29:177–205
Ozturk S (1996) Engineering challenges in high density cell culture systems. Cytotechnology 22:3–16
Myers KJ, Reeder MF, Fasano JB (2002) Optimize mixing by using the proper baffles. Chem Eng Prog 98(2):42–47
Marks D (2003) Equipment design considerations for large scale cell culture. Cytotechnology 42:21–33
Xing Z, Kenty B, Li Z, Lee S (2009) Scale-up analysis for a CHO cell culture process in large-scale bioreactors. Biotechnol Bioeng 103(4):733–746
Christi Y (1993) Animal cell culture in stirred bioreactors: observations on scale up. Bioprocess Eng 9:191–196
Dowd JE, Jubb A, Kwok K, Ezra P, James M (2003) Optimization and control of perfusion cultures using a viable cell probe and cell specific perfusion rates. Cytotechnology 42(1):35–45
Noll T, Biselli M (1998) Dielectric spectroscopy in the cultivation of suspended and immobilized hybridoma cells. J Biotechnol 63(3):187–198
Downey B, Graham L, Breit J, Glutting N (2014) A novel approach for using dielectric spectroscopy to predict viable cell volume (VCV) in early process development. Biotechnol Prog 30(2):479–487
Singh V (1999) Disposable bioreactor for cell culture using wave-induced agitation. Cytotechnology 30(1–3):149–158
Shukla A, Gottschalk U (2013) Single-use disposable technologies for biopharmaceutical manufacturing. Trends Biotechnol 31(3):147–154
Eibl R, Kaiser S, Lombriser R, Eibl D (2010) Disposable bioreactors: the current state-of-the-art and recommended applications in biotechnology. Appl Microbiol Biotechnol 86:41–49
Gupta A, Rao G (2003) A study of oxygen transfer in shake flasks using a non-invasive oxygen sensor. Biotechnol Bioeng 84(3):351–358
Ge X, Kostov Y, Rao G (2003) High-stability non-invasive autoclavable naked optical CO2 sensor. Biosens Bioelectron 18(7):857–865
Smelko J, Wiltberger K, Hickman E, Morris B, Blackburn T, Ryll T (2011) Performance of high intensity fed-batch mammalian cell cultures in disposable bioreactor systems. Biotechnol Prog 27(5):1358–1364
Ozturk S (2005) Batch versus perfusion: a real case comparison of highly developed cell culture processes for the production of monoclonal antibodies. 229th National Meeting American Chemical Society, San Diego, CA, USA, 13–17 March
Voisard D, Meuwly F, Ruffieux PA, Baer G, Kadouri A (2003) Potential of cell retention techniques for large-scale high-density perfusion culture of suspended mammalian cells. Biotechnol Bioeng 82(7):751–765
Pollock J, Ho S, Farid S (2013) Fed-batch and perfusion culture processes: economic, environmental, and operational feasibility under uncertainty. Biotechnol Bioeng 110(1):206–219
Clincke M, Mӧlleryd C, Zhang Y, Lindskog E, Walsh K, Chotteau V (2013) Very high density of CHO cells in perfusion by ATF or TFF in WAVE BioreactorTM. Part I. Effect of the cell density on the process. Biotechnol Prog 29(3):754–767
Bonham-Carter J, Shevitz J (2011) A brief history of perfusion biomanufacturing. BioProcess Int 9(9):24–30
Warikoo V, Godawat R, Brower K, Jain S, Cummings D, Simons E, Johnson T, Walther J, Yu M, Wright B, McLarty J, Karey K, Hwang C, Zhou W, Riske F, Konstantinov K (2012) Integrated continuous production of recombinant therapeutic proteins. Biotechnol Bioeng 109(12):3018–3029
Al-Rubeai M, Singh RP, Goldman MH, Emery AN (1995b) Death mechanisms of animal cells in conditions of intensive agitation. Biotechnol Bioeng 45(6):463–472
Al-Rubeai M, Singh R, Emery A, Zhang Z (1995a) Cell cycle and cell size dependence of susceptibility to hydrodynamic forces. Biotechnol Bioeng 46(1):88–92
Gregoriades N, Clay J, Ma N, Koelling K, Chalmers JJ (2000) Cell damage of microcarrier culture as a function of local energy dissipation created by a rapid extensional flow. Biotechnol Bioeng 69:171–182
Ma N, Koelling K, Chalmers JJ (2002) The fabrication and use of a transient contraction flow device to quantify the sensitivity of mammalian and insect cells to hydrodynamic forces. Biotechnol Bioeng 80:428–437
Kunus KT, Papoutsakis ET (1990) Damage mechanisms of suspended animal cells in agitated bioreactors with and without bubble entrainment. Biotechnol Bioeng 36:476–483
Chattopadhyay D, Rathman J, Chalmers J (1995) Thermodynamic approach to explain cell adhesion to air-medium interfaces. Biotechnol Bioeng 48(6):649–658
Ma N, Chalmers J, Aunins J, Zhou W, Xie L (2004) Quantitative studies of cell-bubble interactions and cell damage at different puronic F-68 and cell concentrations. Biotechnol Prog 20:1183–1191
Osman JJ, Birch J, Varley J (2001) The response of GS-NS0 myeloma cells to pH shifts and pH perturbations. Biotechnol Bioeng 75(1):63–73
Sauer PW, Burky JE, Wesson MC, Sternard HD, Qu L (2000) A high-yielding, generic fed-batch cell culture process for production of recombinant antibodies. Biotechnol Bioeng 67(5):585–597
Lao SM, Toth D (1997) Effects of ammonium and lactate on growth and metabolism of a recombinant Chinese hamster ovary cell culture. Biotechnol Prog 13(5):688–691
Ozturk S, Thrift J, Blackie J, Naveh D (1997) Real-time monitoring and control of glucose and lactate concentrations in a mammalian cell perfusion reactor. Biotechnol Bioeng 53(4):372–378
Gagnon M, Hiller G, Luan YT, Kittredge A, DeFelice J, Drapeau D (2011) High-end pH-controlled delivery of glucose effectively suppresses lactate accumulation in CHO fed-batch cultures. Biotechnol Bioeng 108(6):1328–1337
Li J, Wong C, Vijayasankaran N, Hudson T, Amanullah A (2012) Feeding lactate for CHO cell culture processes: impact on culture metabolism and performance. Biotechnol Bioeng 109(5):1173–1186
Genzel Y, Ritter JB, Koenig S, Alt R, Reichl U (2005) Substitution of glutamine by pyruvate to reduce ammonia formation and growth inhibition of mammalian cells. Biotechnol Prog 21(1):58–69
Martinelle K, Westlund A, Haggstrom L (1996) Ammonium ion transport – a cause of cell death. Cytotechnology 22:251–254
Yang M, Butler M (2000) Effect of ammonia on the glycosylation of human recombinant erythropoietin in culture. Biotechnol Prog 16(5):751–759
DeZengotita VM, Schmelzer AE, Miller WM (2002) Characterization of hybridoma cell responses to elevated pCO2 and osmolality: intracellular pH, cell size, apoptosis, and metabolism. Biotechnol Bioeng 77(4):369–380
Gray DR, Chen S, Howarth W, Inlow D, Maiorella B (1996) CO2 in large-scale and high-density CHO cell perfusion culture. Cytotechnology 22:65–78
Zhu MM, Goyal AR, Rank DL, Gupta SK, Vanden Boom T, Lee SS (2005) Effects of elevated pCO2 and osmolality on growth of CHO Cells and production of antibody-fusion protein B1: a case study. Biotechnol Prog 21(1):70–77
Li F, Hashimura Y, Pendleton R, Harms J, Collins E, Lee B (2006) A systematic approach for scale-down model development and characterization of commercial cell culture processes. Biotechnol Prog 22:696–703
Mostafa SS, Gu XS (2003) Strategies for improved dCO2 removal in large-scale fed-batch cultures. Biotechnol Prog 19(1):45–51
Bollati-Fogolin M, Forno G, Nimtz M, Conradt HS, Etcheverrigaray M, Kratje R (2005) Temperature reduction in cultures of hgm-CSF-expressing CHO cells: effect on productivity and product quality. Biotechnol Prog 21(1):17–21
Chen ZL, Wu BC, Liu H, Huang PT (2004) Temperature shift as a process optimization step for the production of pro-urokinase by a recombinant Chinese hamster ovary cell line in high-density perfusion culture. J Biosci Bioeng 97(4):239–243
Clark KJR, Chaplin FWR, Harcum SW (2004) Temperature effects on product-quality-related enzymes in batch CHO cell cultures producing recombinant tPA. Biotechnol Prog 20(6):1888–1892
Fox SR, Patel UA, Yap MGS, Wang DIC (2004) Maximizing interferon-gamma production by Chinese hamster ovary cells through temperature shift optimization: experimental and modeling. Biotechnol Bioeng 85(2):177–184
Dempsey J, Ruddock S, Osborne M, Ridley A, Sturt S, Field R (2003) Improved fermentation processes for NS0 cell lines expressing human antibodies and glutamine synthetase. Biotechnol Prog 19(1):175–178
Yu M, Hu Z, Pacis E, Vijayasankaran N, Shen A, Li F (2011) Understanding the intracellular effect of enhanced nutrient feeding toward high titer antibody production process. Biotechnol Bioeng 108(5):1078–1088
Wong DCF, Wong KTK, Goh LT, Heng CK, Yap MGS (2005) Impact of dynamic online fed-batch strategies on metabolism, productivity and N-glycosylation quality in CHO cell cultures. Biotechnol Bioeng 89(2):164–177
Chong W, Goh L, Reddy S, Yusufi F, Lee D, Wong N, Heng C, Yap M, Ho Y (2009) Metabolomics profiling of extracellular metabolites in recombinant Chinese Hamster Ovary fed-batch culture. Rapid Commun Mass Spectrom 23:3763–3771
Franek F, Hohenwarter O, Katinger H (2000) Plant protein hydrolysates: preparation of defined peptide fractions promoting growth and production in animal cells cultures. Biotechnol Prog 16(5):688–692
Dowd JE, Kwok K, Ezra P, James M (2000) Increased t-PA yields using ultrafiltration of an inhibitory product from CHO fed-batch culture. Biotechnol Prog 16(5):786–794
Dowd JE, Kwok K, Ezra P, James M (2001) Predictive modeling and loose-loop control for perfusion bioreactors. Biochem Eng J 9(1):1–9
Nienow AW, Langheinrich C, Stevenson NC, Emery AN, Clayton TM, Slater NK (1996) Homogenisation and oxygen transfer rates in large agitated and sparged animal cell bioreactors: some implications for growth and production. Cytotechnology 22(6):87–94
Gardner AR, Smith TM (2000) Identification and establishment of operating ranges of critical process variables. In: Sofer G, Zabriskie DW (eds) Biopharmaceutical process validation. Marcel Dekker, Inc., New York, pp 61–76
Moran EB, McGowan ST, McGuire JM, Frankland JE, Oyebade IA, Waller W, Archer LC, Morris LO, Pandya J, Nathan SR, Smith L, Cadette ML, Michalowski JT (2000) A systematic approach to the validation of process control parameters for monoclonal antibody production in fed-batch culture of a Murine Myeloma. Biotechnol Bioeng 69(3):242–255
Lyddiatt A (1991) Downstream processing: protein recovery. Mamm Cell Biotechnol 187–206
The Biopharmaceutical Process Extractables Core Team (2002) Evaluation of extractables from product-contact surfaces. BioPharm 15(12):22–34
Kemp G, O’Neil P (2004) Large-scale production of therapeutic antibodies: considerations for optimizing product capture and purification. In: Subramanian G (ed) Antibodies, volume 1: production and purification, Kluwer Academic Press, p 101–131
Wang F, Richardson D, Shameem M (2015) Host cell protein measurement and control. Biopharm Int 28(6):32–38
Liu FH, Ma J, Winter C, Bayer R (2010) Recovery and purification process development for monoclonal antibody production. MAbs 2(5):480–499
Rodrigo G, Gruvegard M, Van Alstine JM (2015) Antibody fragments and their purification by Protein L affinity chromatography. Antibodies 4:259–277
LeMerdy S: Evolving clarification strategies to meet new challenges. BioProcess Int (2014) 12 (9): insert 10–12.
Zhang GG (2016) A simple and robust two-column purification platform for antibody fragment manufacturing. Presentation at CHI’s 15th Annual PepTalk, Protein Purification and Recovery, San Diego, USA, 18–22 January
Arunakumari A, Wang J, Ferreira G (2009) Advances in non-Protein A purification processes for human monoclonal antibodies. BioPharm Int Suppl:22–26
Gronemeyer P, Ditz R, Strube J (2014) Trends in upstream and downstream process development for antibody manufacturing. Bioengineering 1:188–212
Gagnon P, Ng P, Zhen J, Aberin C, He J, Mekosh H, Cummings L, Zaidi S, Richieri R (2006) A ceramic hydroxyapatite- based purification platform. BioProcess Int 4(2):50–60
Schreffler J, Bailley M, Klimek T, Agneta P, Wiltsie WE, Felo M, Maisey P, Zuo X, Routhier E (2015) Charaterization of postcapture impurity removal across an adsorptive depth filter. BioProcess Int 13(3):36–45
Xenopoulos A (2013) A new, integrated, continuous purification process template for monoclonal antibodies. Presentation at Integrated Continuous Biomanufacturing, Castelldefels, Spain, 20–24 October
Zhang GG, O’Brien C, Rheuban J, Zhu MM (2011) Development of a simple and semi-continuous monoclonal antibody purification process. Presentation at BioProcess International Conference & Exhibition: Recovery and Purification, Long Beach, USA, October 31–November 4
Zhang GG, O’Brien C, Rheuban J, Zhu MM (2012) Identification and characterization of the critical factors for enhancing the viral filtration performance of nanofilters for hydrophobic monoclonal antibodies. Presentation at IBC’s 9th Annual Viral Safety for Biologicals, San Diego, USA, February 29–March 2
Aldington S, Bonnerjea J (2007) Scale-up of monoclonal antibody purification processes. J. Chromatogr B 848:64–78
Acknowledgments
The authors would like thank all the colleagues who helped in many invaluable ways in the production of this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Zhu, M.M., Mollet, M., Hubert, R.S., Kyung, Y.S., Zhang, G.G. (2017). Industrial Production of Therapeutic Proteins: Cell Lines, Cell Culture, and Purification. In: Kent, J., Bommaraju, T., Barnicki, S. (eds) Handbook of Industrial Chemistry and Biotechnology. Springer, Cham. https://doi.org/10.1007/978-3-319-52287-6_29
Download citation
DOI: https://doi.org/10.1007/978-3-319-52287-6_29
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-52285-2
Online ISBN: 978-3-319-52287-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)