
Abstract
Glioblastoma multiforme (GBM) develops on glial cells and is the most common as well as the deadliest form of brain cancer. As in other cancers, distinct combinations of genetic alterations in GBM subtypes induce a diversity of metabolic phenotypes, which explains the variability of GBM sensitivity to current therapies targeting its reprogrammed metabolism. Therefore, it is becoming imperative for cancer researchers to account for the temporal and spatial heterogeneity within this cancer type before making generalized conclusions about a particular treatment’s efficacy. Standard therapies for GBM have shown little success as the disease is almost always lethal; however, researchers are making progress and learning how to combine therapeutic strategies most effectively. GBMs can be classified initially into two subsets consisting of primary and secondary GBMs, and this categorization stems from cancer development. GBM is the highest grade of gliomas, which includes glioma I (low proliferative potential), glioma II (low proliferative potential with some capacity for infiltration and recurrence), glioma III (evidence of malignancy), and glioma IV (GBM) (malignant with features of necrosis and microvascular proliferation). Secondary GBM develops from a low-grade glioma to an advanced-stage cancer, while primary GBM provides no signs of progression and is identified as an advanced-stage glioma from the onset. The differences in prognosis and histology correlated with each classification are generally negligible, but the demographics of individuals affected and the accompanying genetic/metabolic properties show distinct differentiation [3].
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Glioblastoma
- Metabolic profile
- Glutamine metabolism
- IDH1 mutation
- mTOR signaling
- Liquid biopsy
- Glucose metabolism
-
Glioblastoma (GBM) can be categorized into different subtypes based on diverse metabolic profiles.
-
Characteristic genomic alterations lead to transformed metabolism.
-
Synergistic therapies are beneficial to combat dynamic adaptations of glioblastoma metabolism.
-
Advanced-grade brain tumors exhibit distinct metabolic profiles compared to lower grade tumors.
1 Introduction
Glioblastoma multiforme (GBM) develops on glial cells and is the most common as well as the deadliest form of brain cancer [1]. As in other cancers, distinct combinations of genetic alterations in GBM subtypes induce a diversity of metabolic phenotypes, which explains the variability of GBM sensitivity to current therapies targeting its reprogrammed metabolism. Therefore, it is becoming imperative for cancer researchers to account for the temporal and spatial heterogeneity within this cancer type before making generalized conclusions about a particular treatment’s efficacy. Standard therapies for GBM have shown little success as the disease is almost always lethal; however, researchers are making progress and learning how to combine therapeutic strategies most effectively. GBMs can be classified initially into two subsets consisting of primary and secondary GBMs, and this categorization stems from cancer development. GBM is the highest grade of gliomas, which includes glioma I (low proliferative potential), glioma II (low proliferative potential with some capacity for infiltration and recurrence), glioma III (evidence of malignancy), and glioma IV (GBM) (malignant with features of necrosis and microvascular proliferation) [2]. Secondary GBM develops from a low-grade glioma to an advanced-stage cancer, while primary GBM provides no signs of progression and is identified as an advanced-stage glioma from the onset. The differences in prognosis and histology correlated with each classification are generally negligible, but the demographics of individuals affected and the accompanying genetic/metabolic properties show distinct differentiation [3].
2 GBM Classifications and Intratumoral Heterogeneity
Previously, tumors had been classified based on histological and structural similarities without accounting for clinical disparities among them [4]. More recently, tumor classification has shifted toward a more molecular and genetic basis in combination with phenotypic information. This new-era classification allows practitioners to differentiate between biologically similar cases, allowing for more precise treatment and prognosis when encountering distinct mutant variants [4]. Phenotypic information must be used in accordance with genotypic data to determine tumor type and grade differentiation and to account for the rare occurrences when the phenotype differs from the usual criteria accompanying the defined genotype [5].
2.1 GBM Subtype Classification
A recent study identified four gene expression subtypes of GBM: mesenchymal, classical, proneural, and neural (Fig. 1).
-
The mesenchymal subtype is characterized by high mutation rates of the tumor-suppressor genes: neurofibromatosis type 1 (NF1), phosphatase and tensin homolog (PTEN), and phosphoprotein 53 (p53). Following aggressive treatment, mesenchymal subtypes frequently display substantial increases in length of survival [6].
The mesenchymal subtype was discovered to have a large association with both the tumor-promoting M2 macrophage gene and the deactivation of NF1 [7]. This suggests a pathway linking the loss of function of NF1 to promoted macrophage/microglia recruitment and invasion of the tumor microenvironment (TME), leading to a poorer prognosis for patients afflicted with mesenchymal subtype expression factors [7]. High-grade gliomas containing altered NF1 frequently have an associated deactivation of cyclin-dependent kinase inhibitor 2A (CDKN2A) that inhibits Ras-mediated growth signaling, suggesting NF1 as another tumor-suppressor gene in the central nervous system. As a consequence of losing the NF1 function, Ras activity stimulates Ras effectors (PI3K, PAK, RAF, ERK1/2), increasing the proliferation of astrocytes, contributing to GBM growth [8].
-
Classical GBM is defined by focal epidermal growth factor receptor (EGFR) mutation events in much larger frequencies than in the other three subtypes while containing zero mutations of the most altered gene in GBMs: p53. Similar to the mesenchymal subtype, classical subtypes tend to show the highest survival rates of all the subtypes when subjected to aggressive treatments [6].
-
The proneural subtype carries mutations of p53, platelet-derived growth factor receptor alpha (PDGFRA), and isocitrate dehydrogenase-1 (IDH1) [6]. IDH1 and PDGFRA mutations can result in irregular cell/tumor growth. Proneural patients are characteristically younger than other subtype patients and have more prolonged survival, but their survival remains constant whether they are exposed to aggressive treatment or not. Furthermore, studies have demonstrated that classical and mesenchymal subtypes have a better response to therapy and better prognosis compared to the proneural subtype [6].
-
The neural subtype is categorized based on the overexpression of neurofilament light (NEFL), gamma-aminobutyric acid type A receptor alpha-1 (GABRA1), synaptotagmin 1 (SYT1), and solute carrier family 12 member 5 (SLC12A5) neural markers. The gene expressions present within the neural subtype have been determined to be the most similar to normal brain tissue and are weakly characterized. Data suggests the average efficacy of treatment in the neural subtype, but it is not as effective as treatments of classical and mesenchymal subtypes [6]. These unique genetic alterations leading to subtype classifications result in different metabolic profiles of cancers depending on the specific genes altered.

Subtypes of glioblastoma, including the major genes altered and effect on prognosis following treatment. NF1 neurofibromatosis type 1, PTEN phosphatase and tensin homolog, EGFR epidermal growth factor receptor, CDKN2A cyclin-dependent kinase inhibitor 2A, IDH1 isocitrate dehydrogenase 1, PDGFRA platelet-derived growth factor receptor alpha, NEFL neurofilament light, GABRA1 gamma-aminobutyric acid type A receptor alpha-1, SYT1 synaptotagmin 1
There have been speculations that GBM subtypes do not remain stagnant during disease progression, nor while being bombarded with varying treatments [7]. A study by Wang et al. investigated phenotypic plasticity and genetic drivers behind the evolution of proneural, classical, and mesenchymal subtypes. Samples were collected from varying gliomas at the time of diagnosis as well as at the first onset of GBM recurrence, and genetic profiles were obtained to establish their molecular subtypes for comparison. After analysis, 50 of 91 (55%) samples had their expression subtypes remain constant. After recurrence, the quantity of proneural and mesenchymal subtypes had increased, and the number of classical subtypes had decreased. There was no direct correlation observed between proneural and mesenchymal subtypes. The intratumoral heterogeneity of the initially collected samples was taken into account, and the samples with the lowest purity were typically the groups to undergo a transition of subtype.
GBMs are also divided into different groups based on the World Health Organization (WHO) classification system. Wild-type IDH accounts for 90% of diagnosed GBMs, which have a high correlation to primary GBM, especially in elderly patients. IDH-mutant GBM occurs in about 10% of patients and has a higher correlation to secondary GBM, which is GBM progressing from previous lower grade gliomas. The final classification is reserved for cases in which a complete IDH evaluation cannot be completed. IDHwt versus IDHmu has significant implications on GBM cells’ metabolism and needs to be investigated further. With all the heterogeneity that exists within glioblastomas, the classifications are continually changing, and the variants and patterns must frequently be updated to keep up with the evolving characteristics.
2.2 Intratumoral Heterogeneity
There are currently two proposed models that lead to tumor heterogeneity. The first model suggests that heterogeneity stems from clonal evolution, where there are changes in single cells that create survival advantages between clonal populations [9]. The second model relies on cancer stem cells to produce phenotypically diverse differentiated cells. These models are not mutually exclusive as the cancer stem cells have the opportunity to undergo clonal evolution for them to evolve into more aggressive, self-renewing stem cells [9].
The complexity of genotype and epigenetic states creates the intratumoral heterogeneity of metabolism [10,11,12,13,14,15]. In a study by Patel et al., single-cell RNA sequencing was used to create a profile for 430 cells harvested from 5 diverse glioblastomas. These cancer cells were categorized based on oncogenic signaling, proliferation, complement/immune response, and response to hypoxia. Variability between different tumors was evident, which led to different stages, gene and protein expression, and outcomes for therapeutic strategies [16].
2.2.1 Liquid Biopsy as a Method for Detecting Heterogeneity and Longitudinal Tracking
The existing spatial heterogeneity within individual gliomas has made therapy more difficult. Distinct cells contained within the same tumor can present with different mutations and phenotypic or epigenetic states, resulting in different subtypes being found in other compartments of the same tumor. These variances within the same tumor ultimately lead to the inefficiency of treatment and cancer recurrence. Thus, studies suggest that synergistic treatments will be the direction of new therapeutic strategies [16]. It is becoming more pressing to establish the spatial and temporal heterogeneity of any particular tumor in order to devise an effective treatment. As cancer develops, the longitudinal metabolic profile is not stagnant. Therefore, if we wish to inhibit metabolic targets, correct timing is essential. Surgical biopsies are the conventional strategy for gathering pathological information from GBM tumors. This fact is troublesome because a surgical biopsy will only provide a limited spatial and temporal snapshot of cancer, failing to reflect the intratumoral heterogeneity, not to mention there are significant risks associated with the procedure [17]. A newer approach is emerging that allows for assessment of the entire genetic landscape and longitudinal tracking, and is much less invasive: the liquid biopsy (Fig. 2).

Liquid biopsy and the analysis of biopsy: DNA, protein, and metabolites
Liquid biopsy has two potential sources, either the peripheral blood supply or the cerebrospinal fluid (CSF) via lumbar puncture. The liquid biopsy through the peripheral blood supply is best for measuring smaller metabolites that can cross the BBB easily and could be very important for longitudinal tracking of disease progression. Nguyen et al. demonstrated how concentrations of N-acetyl-aspartyl-glutamate (NAAG) within tumor tissue were directly correlated with the advancement of GBM and lower grade gliomas with very minimal concentrations of NAAG. NAAG concentration in plasma was also directly correlated with NAAG concentration in the tumor and glioma advancement [18].
Additionally, it has been shown that focused ultrasound (FUS) enhances the release of biomarkers into the bloodstream [19]. This technique, in concordance with the liquid biopsy, could be pivotal in establishing precise spatial heterogeneity within a tumor. Liquid biopsy via lumbar puncture is also a viable, less invasive option for patients where surgical resection is too dangerous. It has been shown that there are greatly enhanced detectable levels of cf-DNA from malignant brain tumors. Liquid biopsy is highly beneficial as it could provide the entire genome of a cancer and allow for personalized oncology to target specific biomarkers [20]. Liquid biopsy could provide a broader overall look at the spatial heterogeneity present within a particular tumor. However, there are still obstacles to be overcome with liquid biopsy of the CSF because the amount of tumor cf-DNA leakage into the CSF appears to be relevant to the proximity of the tumor to the CSF reservoir, as well as the progression of the tumor.
2.2.2 Glioblastoma Stem Cell Resistance and Recurrence Are Supported Through Mitochondrial Activity and Fatty Acid Oxidation
A recent discovery that has a substantial impact on the spatial and temporal pathogenesis is the existence of glioblastoma cancer stem-like cells (GSCs). GSCs are not traditional stem cells—they are tumor cells that develop an ultra-aggressive phenotype that facilitates resistance and survival following standard therapies [21]. The effect that GSCs have on spatial pathogenesis can be explained with the “Go or Grow hypothesis,” which postulates that infiltrative cells, such as GSCs, tend to diffuse and spread through tissues but have little proliferative potential. In contrast, tumor cells are proliferative and make up the bulk tumor mass (Fig. 3) [22]. The infiltration of these GSCs has the potential to seed different GBM cell subtype lineages, which contributes to the overall spatial heterogeneity.

Spatial representation of a GBM tumor exhibiting the proliferating mass of the bulk tumor compared with the diffuse infiltration of glioblastoma stem-like cells (GSCs) past the typical perimeter of surgical resection
The temporal pathogenesis of GBM is also affected by GSCs as they are the critical drivers for the recurrence of GBM, which is common following resection. Traits of GSCs that allow for recurrence are their diffuse infiltration, resistance, and initiation of growth following therapy. We have already discussed the diffuse infiltration of GSCs, but it is important to mention that slower proliferating GBM cells are better at initiating tumor growth and have increased resistance [23]. A recent discovery has determined that these infiltrating GSCs retain residual mitochondrial function, allowing them to perform oxidative phosphorylation along with fatty acid oxidation [23, 24]. This is the reason for resistance and recurrence: GSCs are able to withstand glucose deprivation and glycolytic disruption because they have other means to survive. These GSCs are incredibly adaptable to their environment as they are able to survive in four critical niches: hypoxic, perivascular, invasive, and acidic, allowing them to infiltrate various tissues without losing their stemness [21]. Recurrence typically occurs in resection-adjacent tissue, but even following a radical hemispherectomy, there is still contralateral recurrence. There is some evidence that injury, such as surgical resection, to the tumor may initiate proliferation and recurrence, but this process remains obscure [25]. Following the recurrence of GBM, when comparing the transcriptomes between primary and secondary tumors, the tumors have different metabolic signatures, with the recurrent tumor having similar metabolic activities to GSCs [23]. Experts are unsure as to the reason for the metabolic shifts, whether they are due to the changes in tumor microenvironment (TME) from first-line therapies or cell autonomous.
3 Genomic Alterations Lead to Distinct Metabolic Changes Allowing for Targeted Therapies
As previously mentioned, GBMs can be classified according to their genetic and metabolic profiles. Genetic mutations are the cause of deviation of metabolism from the status quo. As genes are overexpressed, inactivated, or mutated, it leads to downstream effects. These downstream metabolic effects can be identified and investigated for therapeutic strategies, which is the current goal of cancer metabolism research.
3.1 PTEN Mutations Lead to High Rates of Glycolysis, Facilitating Survival in Harsh Microenvironments
In a study by Wolf et al., GBMs with loss of PTEN activity had high expression of the glycolytic enzyme hexokinase 2 (HK2), the first enzyme of glycolysis, enabling those GBM cells to survive and proliferate in a harsh TME [26]. HK2 is expressed in basal levels in adipose and skeletal tissues, but it is not expressed in normal brain tissue, which typically expresses HK1. Inhibition of HK2, without interfering with HK1 function, by siRNA led to a reverse of the Warburg effect to oxidative glucose metabolism, which ultimately led to impaired tumor growth. Also, HK2 inhibition sensitized GBM cells to multiple treatments, including the following: (1) temozolomide, the current chemotherapeutic GBM treatment; (2) radiation; and (3) hypoxia-induced apoptosis. Also, high HK2 expression predicted lower overall survival [26]. These findings support the genetically evolved metabolic heterogeneity in cancer cells.
3.2 EGFR Mutations Shift Cancer Cells toward a Glycolytic Phenotype and Permit Survival under Glucose-Deprived Conditions
Mutations in EGFR provide an additional example of genetic alterations that lead to changes in cancer cell metabolism [27]. In their study, Babic et al. revealed an activating EGFRvIII mutation, which causes an intracellular increase in heterogeneous nuclear ribonucleoprotein (hnRNP) A1 splicing factor. This upregulation, in turn, promotes the splicing of MYC-associated factor X (MAX), a partner protein of MYC, which ultimately results in an upregulation of glycolysis, and shorter patient survival time [27]. There has been some success in utilizing the vulnerabilities created by the glycolytic phenotype of GBM as therapeutic strategies. A glucose analog, 2-deoxy-d-glucose (2DG), has been shown to inhibit glycolysis and was well tolerated in clinical trials for other advanced cancers [28].
Growth factor signaling pathways are responsible for cellular metabolism, proliferation, and environmental adaptation [29, 30]. The growth factor signaling pathways are heavily dependent upon regulation from receptor tyrosine kinases (RTKs), showing that genetic mutations in RTKs such as EGFR lead to variable progression and growth of tumors stemming from the changes in the signaling pathways [31]. Furnari et al. used mouse models in correlation with clinical samples wherein the mouse growth factor signaling pathways were genetically modified to match the clinical samples [32]. The corresponding GBMs were determined to be histologically identical, indicating the importance of RTK alterations in the progression of GBMs. In a study of 251 patient-derived GBMs comprised mainly of de novo GBMs (95%), there were alterations to RTKs in 66% of the samples, and the dominant alteration was to EGFR. This lesion was usually accompanied by activation of other PI3Ks, alteration, and deletion of CDKN2A. EGFRvIII+ cells had a higher proliferation rate with less cell death in xenograft models when using EGFR-targeted therapies and showed increased glycolysis to fulfill the energy demand [30, 31, 33, 34]. Further studies are required to evaluate the mechanisms utilized by GBMs to sustain growth based on their environment.
3.3 p53 Mutations Result in Activation of the Warburg Effect
Tumor-suppressor gene p53 has been identified as a gene commonly mutated in many cancers, including GBM [35]. p53 serves to initiate cell cycle arrest and apoptosis when the cell is subjected to stressors, including hypoxia, hyperproliferative signals, nutrient deprivation, and DNA damage [36, 37]. Mutant p53 genes typically lead to complete inactivation of p53, which is characterized by a higher malignancy of cancer through greater rates of metastasis, genetic instability, and cellular differentiation [38,39,40]. Novel functions of p53 have emerged, showing its potential to regulate cellular metabolism. A signature feature of cancer is its reliance on the Warburg effect [41]. p53 has been shown to counter this oncogenic hallmark by activating the synthesis of cytochrome oxidase 2 to promote oxidative phosphorylation and inhibit glycolysis by repressing glucose transporters (SLC2A1, SLC2A4). Thus, the Warburg effect is more profound when p53 is inactivated [42,43,44]. Recently, there have been contradictory studies showing that the impact of p53 as a tumor suppressor is not as compelling as initially suggested. These studies found evidence to support the claim that cell cycle arrest and apoptosis, as a result of DNA damage, are not large contributors to tumor suppression depending on the cancer tissue examined [35]. These findings have yet to be replicated when evaluating GBM development, but further examination is necessary.
3.4 GBM Exhibits Upregulated Glutamine Metabolism Allowing for Targeted Vulnerabilities Through GLS, GS, and mTOR
As is true for many other cancers, GBM also exhibits “glutamine addiction” [45]. Glutamine serves as the major contributor to cell growth and energy production after it is converted into glutamate via glutaminase (GLS), and then into α-ketoglutarate (aKG) via glutamate dehydrogenase (GDH) or several aminotransferases. For this reason, GLS inhibition has become a popular therapeutic strategy to treat cancer patients and has reached clinical trials (NCT03528642). Oizel et al. showed how metabolic phenotype leading to subtype classification could help with targeted therapies. In their study, Oizel showed that mesenchymal GBM cells had significantly more uptake of glutamine and that glutamate derived from glutamine was converted to aKG to generate ATP. This altered phenotype was facilitated by upregulated SLC1A5 (glutamine transporter), GLS, and mitochondrial and cytosolic aminotransferase. Consequently, mesenchymal GBM was shown to be more susceptible to GLS inhibition [46]. Glutamine is essential in normal brain tissue, but it appears to have even higher concentrations in GBM tissue [47]. The primary source of glutamine in the brain comes from glutamine synthase (GS) as it combines glutamate and ammonia to form glutamine. GS levels in GBM have been shown to correlate with patient survival time, as those with lower GS levels could have up to a twofold increase in survival time [48]. The impact of GS expression levels on GBM progression, resistance, and patient survival time needs further investigation, but GS appears to be a reliable prognostic biomarker and could potentially lead to another therapeutic strategy.
The mechanistic target of rapamycin (mTOR) is a protein kinase that promotes oncogenic signaling through the phosphoinositide 3-kinase (PI3K) pathway, which in turn promotes cancer growth [49]. This has also made mTOR a popular target for cancers that use PI3K as a major pathway [50]. mTOR has been identified as a primary factor in downstream signaling for EGFR-mutant GBM, which is resistant to kinase inhibitors [51]. In a study by Tanaka et al., they found that mTOR-targeted treatments affected glutamine catabolism, increasing GLS expression, which is already highly expressed in GBM patients. mTOR-targeted therapies (by rapamycin or PP242) limited cell proliferation, glucose usage, and lactate production [50]. However, they were ineffective in promoting cell death. Following these results, Tanaka et al. performed an experiment in which the U87 and EGFRvIII GBM cells were subjected to glutamine deprivation through compound 968 (GLS inhibitor) and then treated with PP242, which was seen as more effective than mTOR-targeted treatment alone. Results showed that when used in combination, the GLS- and mTOR-targeted therapies yielded a synergistic effect triggering enhanced tumor cell death compared to when either treatment was applied individually [50]. This combined treatment was then tested on normal human astroglial cells, and the results revealed that the treatment did not cause any normal cell death to occur. This synergistic treatment was then tested on GBM xenograft models of U87 and EGFRvIII GBM samples. The treatment resulted in 80% shrinkage of tumors and a sixfold increase in cell death from mTOR-targeted treatment alone. To determine the effects of the drug on the whole body and motor function, the same treatment was applied to normal mice. There were no changes in body weight, motor function, or cell morphology, indicating that the combination of GLS and mTOR inhibition has the potential to develop into an effective treatment for mTOR-targeted resistant GBM cancers [50].
3.5 Lipid Metabolism Dysregulation Following BRAF Mutations and EGFR Signaling Provides Clues for New GBM Therapeutic Strategies
Lipid synthesis is a limiting factor for cellular proliferation. GBMs must synthesize their own lipid components for proliferation leading to a potential vulnerability. Indeed, certain lipid levels—specifically free fatty acid levels—in malignant tissue are elevated when compared to normal brain tissue [52]. To synthesize fatty acids, cells must first generate cytosolic acetyl-CoA, and both tumor-suppressor genes and oncogenes regulate this crucial step in GBM. When there is a BRAF mutation concomitant with TP53 and PTEN deletion, it results in an increased expression of acetyl-CoA synthetase enzyme 2 (ACSS2), the enzyme responsible for converting acetate to acetyl-CoA in the cytosol [53, 54]. Another study discovered that EGFR-PI3K-Akt signaling, which results in sterol regulatory element-binding protein 1 (SREBP-1) cleavage and activation of acetyl coenzyme A (CoA) carboxylase (ACC) and fatty acid synthase (FAS), plays a role in the upregulation of lipogenesis in malignant gliomas. The reaction catalyzed by ACC has been determined as the rate-limiting step for de novo lipogenesis. For cells with upregulated EGFR signaling, targeting the downstream effectors of the pathway (SREBP-1, ACC, and FAS) results in GBM cell death but does not affect normal cells [55]. GSCs also contain elevated levels of lipid metabolites, which contribute to their resistance to glycolytic inhibition or glucose deprivation because when facing glycolytic inhibition or glucose deprivation, they have the ability to rely on fatty acid oxidation for energy. To combat this, Hoang-Minh et al. experimented with pharmacologic inhibition and CRISPR intervention of the fatty acid transporter fatty acid-binding protein 7 (FABP7) to prevent fatty acid uptake, which led to an increase in the overall survival of mice carrying GBM in vivo model [23].
3.6 GBMs Rely on the TCA Cycle and Its Reductants
Alterations of tumor-suppressor genes and oncogenes lead to dysregulations of the TCA cycle in GBM, which creates aberrant metabolism. IDH enzymes play a significant role in the TCA cycle for normal cells by catalyzing the oxidative decarboxylation of isocitrate to aKG using NADP+ or NAD+ as cofactors. IDHwt expression levels are elevated fourfold in GBM tumors when compared to normal brain tissue making IDH the most significant NADPH-producing enzyme and thus a promising therapeutic target. NADPH is necessary for GBM tumors as a reductant for antioxidant biomolecules that help to mitigate oxidative stress. The knockdown of IDH1 depletes stores of NADPH and sensitizes GBM cells to radiation, increasing cellular senescence [56].
In SF188 glioblastoma, high amplification of MYC [57, 58] activates glutamate dehydrogenase (GDH), enabling cancer cell survival under glucose deprivation [59]. GDH, an enzyme necessary for the conversion of glutamate to aKG for incorporation into the TCA cycle, is upregulated in the absence of glucose. This upregulation allows glioblastoma cells to maximize the use of glutamine and thus contributes to the growth and proliferation of neoplastic cells in the absence of glucose [59].
As seen with the previous metabolic pathways, dysregulation of the TCA cycle results in aggressive GBM phenotypes resulting in greater proliferation and growth.
3.7 IDH1 Mutations Lead to Oncometabolite Production and Glutamine Addiction and Act as a Prognostic Marker
A study by Dang et al. demonstrated that mutations in IDH1 give rise to a novel function of this enzyme, which produces the oncometabolite (R)-2-hydroxyglutarate (2HG) from alpha-ketoglutarate (aKG) [60]. Struys et al. reported that “2HG,” in fact, does not exist [61]. The compound has an asymmetric carbon atom that leads to L-2HG and D-2HG, which are both stable endogenous metabolites in all bodily fluids. Routine analytical methods measure the sum of these two metabolites, which creates problems because IDH1 mutations solely result in increased levels of D-2HG. Therefore, an increase in L-2HG could yield false positives, and a small rise in D-2HG may be missed. This is why analytical methods able to distinguish L-2HG from D-2HG must be used [61, 62]. D-2HG not only is found in glioblastoma but has also been found to be sufficient in promoting several other types of cancers, such as leukemia, through mutations in a homolog to IDH1 and IDH2 [63, 64]. D-2HG retains a structure similar to that of aKG, thus inhibiting enzymes from binding to aKG.
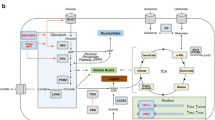
Both L- and D-2HG inhibit aKG-dependent histone demethylases and D-2HG occupies the active site of CeKDM7A, which is where aKG usually binds. Both L- and D-2HG also interferes with 10–11 translocation (TET) family interactions and regulates HIF-1a, which promotes hyper-methylation, triggering cancer proliferation, and preservation [65, 66]. The effect of IDH1mu on HIF-1a is currently being debated. HIF-1a-induced overexpression of target enzymes GLUT1, vascular endothelial growth factor (VEGF), and pyruvate dehydrogenase kinase 1 (PDK1) leads to increased tumor growth, invasion, and angiogenesis [67]. These mutations render cancer cells addicted to glutamine. Thus, glioblastoma cells with IDH1 mutations are more sensitive to GLS inhibition by bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES), a small-molecule selective inhibitor of GLS, as compared to their wild-type counterparts [68] (Fig. 4). Metabolic dependence on aKG was confirmed through rescue experiments showing that the supplement of aKG reduced the impact of BPTES on proliferation hindrance. However, wild-type IDH1 and IDH1 mutants were equally sensitive to glutamine deprivation, suggesting that there are different downstream effectors active when considering inhibition of glutamine metabolism versus inhibition of glutamine uptake. Metabolomics analysis [69] provided interesting findings that 2HG levels remained constant between the IDH1-mutant BPTES-treated group and the IDH1-mutant non-treated group. However, the glycolytic intermediate levels increased, and the TCA cycle intermediate levels decreased in the group treated with BPTES when compared to the non-treated group. The increase of glycolytic intermediates and the decrease of TCA cycle intermediates can be attributed to compensatory mechanisms attempting to maintain aKG levels by reallocating glutamine carbon from the TCA cycle to glycolysis [68]. The message from this specific example is that the mutations of IDH1 form a subset of glioblastomas that exhibit a shift toward glutamine-dependent energy pathways. These pathways allow tumor cells to best utilize glutamine and its products in order to produce ATP as a fuel source for biosynthetic pathways. This ability is particularly advantageous when glucose is scarce. This signifies that these IDHmu tumors exhibit glutamine addiction, and therapies to target glutamine metabolism should prove to be particularly useful here. GLS inhibition could serve as a potential therapeutic target, and several BPTES analogs have been developed [70]. Still, they will most likely need to be used in conjunction with other treatment regiments to combat the dynamic properties of cellular metabolism [68].

The paradoxical nature of downstream effects of 2HG. 2HG inhibition of aKG-dependent histone demethylases, 2HG regulation of HIF-1a which induces overexpression of target enzymes GLUT1, VEGF, and PDK1 leading to increased tumor growth, proliferation, and glutamine addiction. However, that glutamine addiction creates a vulnerability within those GBM cells to targeted glutamine inhibition. 2HG also functions to inhibit ATP synthase and interferes with mTOR signaling causing decreased cancer cell growth and viability
In a study by Nobusawa et al., individuals with secondary glioblastoma who possessed IDH1 mutations had a more favorable prognosis than those with IDH1 wild-type [71]. Histologically, primary and secondary glioblastomas are identical; thus, clinical data is relied upon to determine their subdivision because they occur in patients of different ages and develop through different pathways [72, 73]. Through statistical analyses, it was determined that there was a positive correlation between IDH1 mutations accompanied by p53 mutations, diagnosis of secondary glioblastoma, more prolonged median survival, and younger patient age [74]. IDH1wt genes correlated with older patients, shorter median survival, and higher EGFR amplification. Secondary glioblastomas make up a smaller fraction of the disease compared to primary glioblastomas, but of the secondary cases sampled, an IDH1mu affected the majority, while a minority of primary glioblastomas were affected by the IDH1 mutation. For the patients initially diagnosed as secondary GBM, which did not harbor an IDH1 mutation, and for the cases initially diagnosed as primary, which contained an IDH1 mutation, further analysis showed that these cases were likely misdiagnosed. The “primary” diagnosed patients with IDH1mu had every characteristic of secondary glioblastoma, while the opposite was true for “secondary” glioblastomas with IDH1wt. These discrepancies suggest incorrect diagnoses for gliomas that may have started at a low grade and then progressed quickly, making them look like a primary or higher grade glioma with some progression disguising it as secondary. Nobusawa et al. accurately identified IDH1 as a reliable signature marker for secondary glioblastoma with a more favorable outcome [71].
Furthermore, another study by Labussiere et al. showed that individuals with IDH1mu tumors lived longer than those who had IDH1wt tumors, despite all tumors being of the same grade [75]. Another classical function of IDH1 is to support oxidative decarboxylation of isocitrate to aKG coupled with the reduction of NADPH, allowing NADPH to promote the further reduction of glutathione (GSH), a crucial antioxidant [76, 77]. When glioblastomas have IDH1mu, the loss of normal enzymatic function lowers the production of aKG and NADPH, subsequently lowering GSH, while increasing 2HG [60]. The surge of D-2HG increases oxidative stress present in cancer cells and the accumulation of reactive oxygen species (ROS), which encourages tumor cell growth [78]. These characteristics seemingly create a paradox wherein the accumulation of ROS could serve to further tumor development due to the increase of genetic instability, but on the other hand, the decrease of GSH levels leaves the tumor cells vulnerable for oxidative damage [75]. Both L- and D-2HG have also been shown to function as an ATP synthase inhibitor and interferes with mTOR signaling, which leads to a decrease in tumor growth and cell viability [79] (Fig. 4). There appears to be a greater response to radiation in patients with IDHmu tumors, which may be due to the effect D-2HG has as a radiosensitizing agent [80]. Both enantiomers of 2HG inhibit homologous recombination allowing for the greater effect of alkylating agents [81]. Controversy exists over the therapeutic potential of targeting 2HG because there appear to be both oncometabolite and tumor-suppressing capabilities of 2HG. These conclusions suggest the need for further analyses on the mechanistic links between metabolic phenotype and clinical outcome. There appears to be a limited therapeutic window with IDHmu tumors where inhibition of IDH1/2 or 2HG could be beneficial earlier in pathogenesis to prevent invasion and progression, but further in development, these metabolic aberrations could hold the key to increased longevity due to the vulnerabilities created. This conflict illustrates the need for a deeper dive into the temporal pathogenesis of GBM with IDH mutations.
4 Benefits of Combined Therapy
Among the many struggles in treating cancer, tackling its inherent metabolic heterogeneity is a significant obstacle [69]. Metabolic pathways relevant to GBM have been established, but those pathways are dynamic, and cancer cells alter their metabolism as their environments change [82]. When a pathway is hit and deactivated, the ability of a cancer cell to work around it contributes to the complexity of treatments. After multiple pathways are inhibited, a cancer cell’s metabolism will eventually be cornered with nowhere to turn (Fig. 5). Tanaka et al. successfully combined therapies inhibiting mTOR and GLS to limit cancer cell proliferation [50]. As mTOR-targeted treatments inactivate the PI3K, GBM switches to higher expressions of GLS to rescue it from apoptosis [50]. Combining the mTOR-targeted treatment with GLS inhibition essentially traps specific GBM cells so that their metabolism cannot shift pathways to encourage cell survival. Heterogeneity creates problems when determining treatment because different metabolic profiles result in differences as to how cancer metabolism will change in response to treatment. These synergistic treatments are beneficial because they can work together when attempting to target different pathways. Still, the challenge remains to affect only the cancerous cells while not having a detrimental impact on benign, healthy cells.

Metabolic targets for GBM therapy (indicated by multicolored panels) accompanied by the specific metabolic pathways in which they participate. These targeted therapies can be used to supplement the standard therapy for treating GBM possibly resulting in synergistic effects. HK2 hexokinase 2, mTOR mechanistic target of rapamycin or mammalian target of rapamycin, GLS glutaminase, GS glutamine synthase, IDH isocitrate dehydrogenase, GDH glutamate dehydrogenase, SREBP-1 sterol regulatory element-binding protein 1, FABP7 fatty acid-binding protein 7
The current treatment protocol for GBM is surgical resection, radiation, and chemotherapy, typically through temozolomide (TMZ), which is an alkylating agent that causes DNA damage to tumor cells to trigger apoptosis. Even with aggressive treatment, this disease is universally lethal. Interfering with GBM’s metabolic pathways through metabolic targeting could serve as a way to enhance the effectiveness of standard therapies. Glycolytic phenotypes are typically accompanied by radiation resistance. Therefore if glycolytic inhibition is accompanied by radiation, it could improve the effectiveness of treatment. Indeed, these are the results obtained by Wolf et al. in their in vitro studies. As they depleted HK2 (a glycolytic enzyme) and combined it with radiation therapy, they observed reduced proliferation and increased apoptosis [26]. As discussed earlier, NADPH is a powerful reductant to help mediate oxidative stress created by radiation. Decreasing NADPH levels is also linked to high rates of cellular senescence in the presence of radiation [56]. As IDHwt is the driver for increased NADPH concentrations, IDHwt inhibition appears to be another potential synergistic strategy. Combination therapy with high pharmacological doses of ascorbate, which becomes a powerful prooxidant to inflict oxidative damage, and radiation is also a viable strategy to combat radiation resistance. Specifically, Herst et al. used in vitro studies to show that high doses of ascorbate combined with 1 Gy of radiation (which is lower than the typical amount of 2 Gy) resulted in greater cell death than either singular strategy and had less harmful effects on normal astrocytes [83]. Oxidative stress can be mediated by altering glutamine metabolism. Glutamine is converted to glutamate via GLS, and glutamate is an amino acid incorporated into GSH. Therefore, if GLS inhibition indirectly inhibits the formation of GSH, there are fewer antioxidants to provide radioprotection. CB-839, a potent GLS inhibitor, is currently undergoing a clinical trial (NCT03528642) in combination with TMZ and radiation.
Currently, it is not well understood how glycolytic metabolites contribute to chemotherapy resistance. However, as seen with radiation, the depletion of HK2 also appears to sensitize GBM to TMZ [26]. Tumor protein 73 antisense RNA 1 (TP73-AS1) has been found to have connections with GSCs that confers TMZ resistance. Mazor et al. demonstrated that silencing TP73-AS1 led to an increased sensitivity of GBM to TMZ therapy. The mechanism behind this effect is currently being investigated. TP73-AS1 is known to encode aldehyde dehydrogenase 1 family member A1 (ALDH1A1), which is an established marker for cancer stem cells and promotes resistance. ALDH1A1 inhibition has a very similar sensitizing effect on GBM cells to TMZ [84].
5 Advanced Brain Tumors (GBM) Display Distinct Metabolic Profiles Compared to Lower Grade Tumors
While IDH1 mutations can distinguish between primary and secondary GBMs, another metabolic pathway involving cysteine catabolism is not highly activated in lower grade tumorigenesis [85]. The GSH synthesis pathway involving cysteine begins with a simultaneous efflux of glutamate and influx of cystine, which is then reduced to cysteine and converted to GSH when combined with glutamate and glycine. GSH functions as a central nervous system antioxidant increasing cancer cell survival when subjected to redox stress and hypoxia [86]. Prabhu et al. investigated another pathway involving cysteine, which resulted in the accumulation of cysteine sulfinic acid (CSA) through the regulatory enzyme cysteine dioxygenase-1 (CDO1). When comparing with grade 2 gliomas, there was a 23-fold increase in the relative accumulation of CSA in GBM, ranking it as the highest change in concentration of any metabolite. This increase of metabolite concentration correlated with a higher expression of the CDO1 enzyme in GBM when evaluated using Western blot analysis. The buildup of CSA is associated with inhibited cellular respiration and decreased both oxidative phosphorylation and ATP production. CSA modulates mitochondrial function through inhibition of pyruvate dehydrogenase (PDH). PDH functions as a channel enzyme controlling the rate at which glycolysis occurs, and this enzyme was inhibited in a dose-dependent manner when treated with CSA using an established GBM cell line (U251). Further investigation is needed to determine how this alternative pathway of cysteine catabolism contributes to GBM tumorigenesis [85]. An analysis was conducted to uncover the impacts PDH modulation had on tumorigenesis in GBM and showed that PDH phosphatase expression regulated PDH activity as a result of Ras-mediated signaling. When the impairment of PDH was reversed, it inhibited tumor growth, making this pathway a possible therapeutic target to treat GBM in the future [87].
Nguyen et al. discovered a metabolite, NAAG, that serves as a glutamate reservoir for cancer cells in a glutamine-deprived environment, as mentioned earlier. Additionally, NAAG was found to be significantly elevated in GBM when compared to grade II–III gliomas and meningiomas [18]. As more of these biomarkers emerge, they could provide the key for tracking longitudinal pathogenesis and discovering new metabolic targets.
6 Conclusion
Glioblastoma is the most common and deadliest form of brain cancer in humans. Its poor prognosis and unreliable diagnosis are the results of its intricate heterogeneity and evolutionary characteristics. Experts have made substantial progress in characterizing this cancer with the use of improved technologies; most recently, there has been a spotlight on the use of metabolomics to discover its underlying molecular mechanisms. As more data and results are obtained to determine how different glioblastomas function and why they exhibit certain metabolic phenotypes, more therapeutic strategies can be developed to treat patients individually with respect to their genotypic and phenotypic profiles.
Abbreviations
- 2DG:
-
2-Deoxy-d-glucose
- 2HG:
-
2-Hydroxyglutarate
- ACC:
-
Acetyl coenzyme A (CoA) carboxylase
- ACSS2:
-
Acetyl-CoA synthetase enzyme 2
- aKG:
-
Alpha-ketoglutarate
- ALDH1A1:
-
Aldehyde dehydrogenase 1 family member A1
- BBB:
-
Blood-brain barrier
- BPTES:
-
Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide
- CDKN2A:
-
Cyclin-dependent kinase inhibitor 2A
- CDO:
-
Cysteine dioxygenase
- CSA:
-
Cysteine sulfinic acid
- EGFR:
-
Epidermal growth factor receptor
- FABP7:
-
Fatty acid-binding protein 7
- FAS:
-
Fatty acid synthase
- FUS:
-
Focused ultrasound
- GABRA1:
-
Gamma-aminobutyric acid type A receptor alpha-1
- GBM:
-
Glioblastoma
- GDH:
-
Glutamate dehydrogenase
- GLS:
-
Glutaminase
- GSC:
-
Glioblastoma stem-like cell
- GSH:
-
Glutathione
- HK:
-
Hexokinase
- IDH:
-
Isocitrate dehydrogenase
- M2:
-
Macrophage 2
- MAX:
-
Myc-associated factor X
- mTOR:
-
Mechanistic target of rapamycin or mammalian target of rapamycin
- NAAG:
-
N-acetyl-aspartyl-glutamate
- NEFL:
-
Neurofilament light
- NF1:
-
Neurofibromatosis type 1
- p53:
-
Phosphoprotein 53
- PDGFRA:
-
Platelet-derived growth factor receptor alpha
- PDH:
-
Pyruvate dehydrogenase
- PDK1:
-
Pyruvate dehydrogenase kinase 1
- PI3K:
-
Phosphoinositide 3-kinase
- PTEN:
-
Phosphatase and tensin homolog
- ROS:
-
Reactive oxygen species
- RTK:
-
Receptor tyrosine kinase
- SLC12A5:
-
Solute carrier family 12 member 5
- SREBP-1:
-
Sterol regulatory element-binding protein 1
- SYT1:
-
Synaptotagmin 1
- TCA:
-
Tricarboxylic acid
- TET:
-
Ten-eleven translocation
- TME:
-
Tumor microenvironment
- TMZ:
-
Temozolomide
- TP73-AS1:
-
Tumor protein 73 antisense RNA 1
- VEGF:
-
Vascular endothelial growth factor
References
Purow, B. W., & Schiff, D. (2010). Glioblastoma genetics: In rapid flux. Discovery Medicine, 9(45), 125–131.
Delgado-Martin, B., & Medina, M. A. (2020). Advances in the knowledge of the molecular biology of glioblastoma and its impact in patient diagnosis, stratification, and treatment. Advanced Science (Weinh), 7(9), 1902971.
Kleihues, P., & Ohgaki, H. (1999). Primary and secondary glioblastomas: From concept to clinical diagnosis. Neuro-Oncology, 1(1), 44–51.
Louis, D. N., et al. (2016). The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathologica, 131(6), 803–820.
Hainfellner, J., et al. (2014). Letter in response to David N. Louis et al, International Society Of Neuropathology-Haarlem Consensus Guidelines for Nervous System Tumor Classification and Grading, Brain Pathology, doi: 10.1111/bpa.12171. Brain Pathology, 24(6), 671–672.
Verhaak, R. G., et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell, 17(1), 98–110.
Wang, Q., et al. (2017). Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell, 32(1), 42–56. e6.
Rubin, J. B., & Gutmann, D. H. (2005). Neurofibromatosis type 1—a model for nervous system tumour formation? Nature Reviews. Cancer, 5(7), 557–564.
Visvader, J. E., & Lindeman, G. J. (2008). Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nature Reviews. Cancer, 8(10), 755–768.
Navin, N., et al. (2011). Tumour evolution inferred by single-cell sequencing. Nature, 472(7341), 90–94.
Gerlinger, M., et al. (2012). Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England Journal of Medicine, 366(10), 883–892.
Driessens, G., et al. (2012). Defining the mode of tumour growth by clonal analysis. Nature, 488(7412), 527–530.
Schepers, A. G., et al. (2012). Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science, 337(6095), 730–735.
Bedard, P. L., et al. (2013). Tumour heterogeneity in the clinic. Nature, 501(7467), 355–364.
Nabi, K., & Le, A. (2021). The intratumoral heterogeneity of cancer metabolism. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_11
Patel, A. P., et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science, 344(6190), 1396–1401.
Simonelli, M., et al. (2020). Cerebrospinal fluid tumor DNA for liquid biopsy in glioma patients’ management: Close to the clinic? Critical Reviews in Oncology/Hematology, 146, 102879.
Nguyen, T., et al. (2019). Uncovering the role of N-acetyl-aspartyl-glutamate as a glutamate reservoir in cancer. Cell Reports, 27(2), 491–501. e6.
Zhu, L., et al. (2018). Focused ultrasound-enabled brain tumor liquid biopsy. Scientific Reports, 8(1), 6553.
Pan, W., et al. (2015). Brain tumor mutations detected in cerebral spinal fluid. Clinical Chemistry, 61(3), 514–522.
Badr, C. E., et al. (2020). Metabolic heterogeneity and adaptability in brain tumors. Cellular and Molecular Life Sciences, 77(24), 5101–5119.
Xie, Q., Mittal, S., & Berens, M. E. (2014). Targeting adaptive glioblastoma: An overview of proliferation and invasion. Neuro-Oncology, 16(12), 1575–1584.
Hoang-Minh, L. B., et al. (2018). Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. The EMBO Journal, 37, 23.
Lin, H., et al. (2017). Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro-Oncology, 19(1), 43–54.
Osswald, M., et al. (2015). Brain tumour cells interconnect to a functional and resistant network. Nature, 528(7580), 93–98.
Wolf, A., et al. (2011). Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. The Journal of Experimental Medicine, 208(2), 313–326.
Babic, I., et al. (2013). EGFR mutation-induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell Metabolism, 17(6), 1000–1008.
Dwarakanath, B. S., et al. (2009). Clinical studies for improving radiotherapy with 2-deoxy-D-glucose: Present status and future prospects. Journal of Cancer Research and Therapeutics, 5(Suppl 1), S21–S26.
Gschwind, A., Fischer, O. M., & Ullrich, A. (2004). The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nature Reviews. Cancer, 4(5), 361–370.
Lemmon, M. A., Schlessinger, J., & Ferguson, K. M. (2014). The EGFR family: Not so prototypical receptor tyrosine kinases. Cold Spring Harbor Perspectives in Biology, 6(4), a020768.
Brennan, C. W., et al. (2013). The somatic genomic landscape of glioblastoma. Cell, 155(2), 462–477.
Furnari, F. B., et al. (2015). Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nature Reviews. Cancer, 15(5), 302–310.
Nathanson, D. A., et al. (2014). Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science, 343(6166), 72–76.
Dunn, G. P., et al. (2012). Emerging insights into the molecular and cellular basis of glioblastoma. Genes & Development, 26(8), 756–784.
Bieging, K. T., Mello, S. S., & Attardi, L. D. (2014). Unravelling mechanisms of p53-mediated tumour suppression. Nature Reviews. Cancer, 14(5), 359–370.
Giaccia, A. J., & Kastan, M. B. (1998). The complexity of p53 modulation: Emerging patterns from divergent signals. Genes & Development, 12(19), 2973–2983.
Hu, W., Feng, Z., & Levine, A. J. (2012). The regulation of multiple p53 stress responses is mediated through MDM2. Genes & Cancer, 3(3-4), 199–208.
Song, H., Hollstein, M., & Xu, Y. (2007). p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nature Cell Biology, 9(5), 573–580.
Olivier, M., & Taniere, P. (2011). Somatic mutations in cancer prognosis and prediction: Lessons from TP53 and EGFR genes. Current Opinion in Oncology, 23(1), 88–92.
Rivlin, N., et al. (2011). Mutations in the p53 tumor suppressor gene: Important milestones at the various steps of tumorigenesis. Genes & Cancer, 2(4), 466–474.
Bose, S., Zhang, C., & Le, A. (2021). Glucose metabolism in cancer: The Warburg effect and beyond. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_1
Maddocks, O. D., & Vousden, K. H. (2011). Metabolic regulation by p53. Journal of Molecular Medicine (Berlin), 89(3), 237–245.
Kawauchi, K., et al. (2008). p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nature Cell Biology, 10(5), 611–618.
Schwartzenberg-Bar-Yoseph, F., Armoni, M., & Karnieli, E. (2004). The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Research, 64(7), 2627–2633.
Li, T., Copeland, C., & Le, A. (2021). Glutamine metabolism in cancer. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_2
Oizel, K., et al. (2017). Efficient mitochondrial glutamine targeting prevails over glioblastoma metabolic plasticity. Clinical Cancer Research, 23(20), 6292–6304.
Marin-Valencia, I., et al. (2012). Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metabolism, 15(6), 827–837.
Rosati, A., et al. (2013). Glutamine synthetase expression as a valuable marker of epilepsy and longer survival in newly diagnosed glioblastoma multiforme. Neuro-Oncology, 15(5), 618–625.
Cancer Genome Atlas Research N. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature, 455(7216), 1061–1068.
Tanaka, K., et al. (2015). Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. The Journal of Clinical Investigation, 125(4), 1591–1602.
Duvel, K., et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular Cell, 39(2), 171–183.
Guo, D., Bell, E. H., & Chakravarti, A. (2013). Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncolology, 2(3), 289–299.
Mashimo, T., et al. (2014). Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell, 159(7), 1603–1614.
Park, J. K., et al. (2021). The heterogeneity of lipid metabolism in cancer. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_3
Guo, D., et al. (2009). EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Science Signaling, 2(101), ra82.
Wahl, D. R., et al. (2017). Glioblastoma therapy can be augmented by targeting IDH1-mediated NADPH biosynthesis. Cancer Research, 77(4), 960–970.
Dang, C. V., Le, A., & Gao, P. (2009). MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clinical Cancer Research, 15(21), 6479–6483.
Le, A., & Dang, C. V. (2013). Studying Myc’s role in metabolism regulation. Methods in Molecular Biology, 1012, 213–219.
Yang, C., et al. (2009). Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Research, 69(20), 7986–7993.
Dang, L., et al. (2010). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature, 465(7300), 966.
Struys, E. A. (2013). 2-Hydroxyglutarate is not a metabolite; D-2-hydroxyglutarate and L-2-hydroxyglutarate are! Proceedings of the National Academy of Sciences of the United States of America, 110(51), E4939.
Karlstaedt, A., et al. (2016). Oncometabolite d-2-hydroxyglutarate impairs alpha-ketoglutarate dehydrogenase and contractile function in rodent heart. Proceedings of the National Academy of Sciences of the United States of America, 113(37), 10436–10441.
Losman, J. A., et al. (2013). (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science, 339(6127), 1621–1625.
Gross, S., et al. (2010). Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. The Journal of Experimental Medicine, 207(2), 339–344.
Xu, W., et al. (2011). Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell, 19(1), 17–30.
Zhao, S., et al. (2009). Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science, 324(5924), 261–265.
Fu, Y., et al. (2012). Glioma derived isocitrate dehydrogenase-2 mutations induced up-regulation of HIF-1alpha and beta-catenin signaling: Possible impact on glioma cell metastasis and chemo-resistance. The International Journal of Biochemistry & Cell Biology, 44(5), 770–775.
Seltzer, M. J., et al. (2010). Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Research, 70(22), 8981–8987.
Hoang, G., Udupa, S., & Le, A. (2019). Application of metabolomics technologies toward cancer prognosis and therapy. International Review of Cell and Molecular Biology, 347, 191–223.
Zimmermann, S. C., et al. (2016). Allosteric glutaminase inhibitors based on a 1,4-di(5-amino-1,3,4-thiadiazol-2-yl)butane scaffold. ACS Medicinal Chemistry Letters, 7(5), 520–524.
Nobusawa, S., et al. (2009). IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clinical Cancer Research, 15(19), 6002–6007.
Ohgaki, H., et al. (2004). Genetic pathways to glioblastoma: A population-based study. Cancer Research, 64(19), 6892–6899.
Ohgaki, H., & Kleihues, P. (2007). Genetic pathways to primary and secondary glioblastoma. The American Journal of Pathology, 170(5), 1445–1453.
Parsons, D. W., et al. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science, 321(5897), 1807–1812.
Labussiere, M., et al. (2010). IDH1 gene mutations: A new paradigm in glioma prognosis and therapy? The Oncologist, 15(2), 196–199.
Thompson, C. B. (2009). Metabolic enzymes as oncogenes or tumor suppressors. The New England Journal of Medicine, 360(8), 813–815.
Lee, S. M., et al. (2002). Cytosolic NADP(+)-dependent isocitrate dehydrogenase status modulates oxidative damage to cells. Free Radical Biology & Medicine, 32(11), 1185–1196.
Latini, A., et al. (2003). D-2-hydroxyglutaric acid induces oxidative stress in cerebral cortex of young rats. The European Journal of Neuroscience, 17(10), 2017–2022.
Fu, X., et al. (2015). 2-Hydroxyglutarate inhibits ATP synthase and mTOR signaling. Cell Metabolism, 22(3), 508–515.
Molenaar, R. J., et al. (2015). Radioprotection of IDH1-mutated cancer cells by the IDH1-mutant inhibitor AGI-5198. Cancer Research, 75(22), 4790–4802.
Sulkowski, P. L., et al. (2017). 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Science Translational Medicine, 9, 375.
Antonio, M. J., Zhang, C., & Le, A. (2021). Different tumor microenvironments lead to different metabolic phenotypes. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_10.
Herst, P. M., et al. (2012). Pharmacological concentrations of ascorbate radiosensitize glioblastoma multiforme primary cells by increasing oxidative DNA damage and inhibiting G2/M arrest. Free Radical Biology & Medicine, 52(8), 1486–1493.
Mazor, G., et al. (2019). The lncRNA TP73-AS1 is linked to aggressiveness in glioblastoma and promotes temozolomide resistance in glioblastoma cancer stem cells. Cell Death & Disease, 10(3), 246.
Prabhu, A., et al. (2014). Cysteine catabolism: A novel metabolic pathway contributing to glioblastoma growth. Cancer Research, 74(3), 787–796.
Chung, W. J., et al. (2005). Inhibition of cystine uptake disrupts the growth of primary brain tumors. The Journal of Neuroscience, 25(31), 7101–7110.
Prabhu, A., et al. (2015). Ras-mediated modulation of pyruvate dehydrogenase activity regulates mitochondrial reserve capacity and contributes to glioblastoma tumorigenesis. Neuro-Oncology, 17(9), 1220–1230.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Quinones, A., Le, A. (2021). The Multifaceted Glioblastoma: From Genomic Alterations to Metabolic Adaptations. In: Le, A. (eds) The Heterogeneity of Cancer Metabolism. Advances in Experimental Medicine and Biology, vol 1311. Springer, Cham. https://doi.org/10.1007/978-3-030-65768-0_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-65768-0_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-65767-3
Online ISBN: 978-3-030-65768-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)