
Abstract
Diabetes mellitus, commonly known as diabetes, and cancer are two of the most common diseases plaguing the world today. According to the Centers for Disease Control and Prevention (CDC), there are currently more than 20 million people with diabetes in the United States [1]. According to the International Agency for Research on Cancer (IARC), there were around 18 million people diagnosed with cancer, with approximately ten million deaths globally in 2018 [2]. Given the prevalence and deadliness of diabetes and cancer, these two diseases have long been the focus of many researchers with the goal of improving treatment outcomes. While diabetes and cancer may seem to be two very different diseases at first glance, they share several similarities, especially regarding their metabolic characteristics. This chapter discusses the similarities and relationships between the metabolism of diabetes, especially type 2 diabetes (T2D), and cancer, including their abnormal glucose and amino acid metabolism, the contribution of hyperglycemia to oncogenic mutation, and the contribution of hyperinsulinemia to cancer progression. Investigating the metabolic interplay between diabetes and cancer in an effort to exploit this connection for cancer treatment has the potential to significantly improve clinical efficacy.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Cancer
- Type 2 diabetes
- Glucose metabolism
- Hexosamine biosynthetic pathway
- KRAS mutation
- Hyperinsulinemia
- mTOR signaling
- Metformin
-
Diabetes is correlated with increased risk for several types of cancer.
-
Diabetes and cancer both exhibit abnormal glucose metabolism: hyperglycemia and the Warburg effect, respectively.
-
High glucose levels can lead to increased O-GlcNAcylation through upregulation of the hexosamine biosynthetic pathway (HBP), which contributes to insulin resistance in diabetes and de novo KRAS mutations which can result in pancreatic cancer.
-
Hyperinsulinemia, a condition commonly associated with diabetes, can promote cancer proliferation.
-
Obesity can result in insulin resistance and increased oxidative stress contributing to diabetes and cancer.
-
Dysregulation of leucine metabolism, which regulates insulin secretion and mTOR signaling, can promote type 2 diabetes (T2D) pathogenesis, insulin resistance, and cancer growth.
-
Glutamine regulates insulin secretion contributes to gluconeogenesis in T2D, and can promote cancer growth.
-
Different antidiabetic drugs have varied effects on cancer.
1 Introduction
Diabetes mellitus, commonly known as diabetes, and cancer are two of the most common diseases plaguing the world today. According to the Centers for Disease Control and Prevention (CDC), there are currently more than 20 million people with diabetes in the United States [1]. According to the International Agency for Research on Cancer (IARC), there were around 18 million people diagnosed with cancer, with approximately ten million deaths globally in 2018 [2]. Given the prevalence and deadliness of diabetes and cancer, these two diseases have long been the focus of many researchers with the goal of improving treatment outcomes. While diabetes and cancer may seem to be two very different diseases at first glance, they share several similarities, especially regarding their metabolic characteristics. This chapter discusses the similarities and relationships between the metabolism of diabetes, especially type 2 diabetes (T2D), and cancer, including their abnormal glucose and amino acid metabolism, the contribution of hyperglycemia to oncogenic mutations, and the contribution of hyperinsulinemia to cancer progression. Investigating the metabolic interplay between diabetes and cancer in an effort to exploit this connection for cancer treatment has the potential to significantly improve clinical efficacy.
2 The Epidemiological Association: Diabetes Correlates with Increased Risk for Many Types of Cancer
As two of the most common diseases in the world, diabetes and cancer have attracted the attention of a great number of investigators since the 1950s with the intention of discovering their epidemiological connections [3]. It was found through these studies that patients with diabetes have a higher risk of developing many different types of cancer, including liver, pancreatic, endometrium, breast, colon, and rectum cancers [3]. Specifically, it was found that the risk for colorectal cancer is 30% greater for patients with T2D, which is caused by insulin resistance, as compared to people who do not have diabetes [4, 5]. Among all types of cancers, pancreatic cancer has the most drastic association with diabetes. As Pannala et al. discovered, around 50% of the patients with pancreatic cancer have T2D [5, 6]. However, while the epidemiological connections between diabetes and cancer have long been known, the exact mechanisms behind the relationships are not well understood, and further studies are needed.
While it seems reasonable to speculate that high glucose levels in diabetes can promote cancer proliferation, some clinical data contradict this speculation. For example, it was found that there is a significantly decreased risk for prostate cancer in patients with T2D [7] and that the chances for developing lung [3] and ovarian cancers [7, 8] are not associated with T2D. These exceptions call for further investigations to fully understand the association between diabetes, especially T2D, and cancer, as well as the mechanisms behind it.
3 Abnormal Glucose Metabolism Serves as a Link Between Diabetes and Cancer
Abnormally high blood glucose levels, or hyperglycemia, is one of the defining characteristics of diabetes caused by either insulin secretion defects (T1D) or insulin resistance (T2D). Persistent hyperglycemia over a long period of time in diabetic patients eventually causes damage to important organs, including the eyes, heart, and kidneys, thus leading to the observed symptoms and detrimental effects of diabetes [9]. For many cancers, glucose metabolism is indispensable due to their high need for energy and materials to sustain growth and proliferation. The Warburg effect is one of the most well-known metabolic adaptations of cancers, where the cancer cells readily convert glucose to pyruvate and lactate even under normoxic conditions in order to satisfy their increased need for energy [10, 11]. Diabetes and cancer both have abnormal glucose metabolism, but with different mechanisms: one leads to deterioration of tissues while the other fuels uncontrolled cell growth.
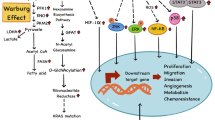
3.1 High Glucose Levels Lead to Hexosamine Biosynthetic Pathway (HBP) Upregulation, Contributing to Insulin Resistance in T2D and Oncogenic Mutations (Fig. 1)
As mentioned previously, it has been well established that patients with diabetes have a higher risk of developing many types of cancers. However, the mechanisms behind this association are less well known. Only recently was one possible mechanism discovered, where high glucose is linked to de novo mutation of the Kirsten rat sarcoma viral oncogene homolog (KRAS) in pancreatic cells, thus leading to a greater chance of tumorigenesis and an increased risk for cancer [12, 13].

Associations between type 2 diabetes, cancer, and obesity. High blood glucose levels in T2D lead to upregulation of the hexosamine biosynthetic pathway (HBP). The upregulation of HBP, in turn, results in high UDP-GlcNAc levels and increased protein O-glycosylation/O-GlcNAcylation. This can lead to insulin resistance in T2D and KRAS mutation in pancreatic cancer. Insulin resistance can contribute to hyperinsulinemia. Hyperinsulinemia can result in increased insulin receptor activation, which can further activate the MAPK and PI3K pathways that promote cell growth and proliferation. Obesity can result in increased protein tyrosine phosphatase expression, which can impair insulin signaling and contribute to insulin resistance. Obesity can also increase ROS levels which lead to DNA damages and mutations and contribute to cancer. Blue circles represent glucose molecules. Green squares represent insulin molecules
From the glycolysis pathway, glucose can be converted to glucose-6-phosphate, which then alternatively enters the hexosamine biosynthetic pathway (HBP). The HBP utilizes around 2–3% of the total glucose taken up by normal cells [14]. The final product of the HBP is uridine diphosphate N-acetylglucosamine (UDP-GlcNAc). UDP-GlcNAc is essential for the production of glycans, such as O-glycans and N-glycans, which are important for various cellular functions that are involved in cancer, such as signaling pathways and cell proliferation regulation [15]. UDP-GlcNAc is also the substrate for O-GlcNAc transferase (OGT), an enzyme responsible for the O-linked N-acetylglucosaminylation (O-GlcNAcylation) of many proteins in the cytoplasm, the nucleus, and the mitochondria [16]. O-GlcNAcylation is a posttranslational modification (PTM) where the O-linked GlcNAc moieties are attached to the serine or threonine residues of the proteins.
In diabetes, increased HBP activity is caused by high glucose levels and overexpression of glutamine: fructose-6-phosphate amidotransferase (GFA), the enzyme in HBP that converts fructose 6-phosphate to glucosamine 6-phosphate, which can lead to increased insulin resistance [17, 18]. This results in a feedback loop where hyperglycemia leads to an upregulation in HBP, which in turn leads to insulin resistance and worsens hyperglycemia. In the study by Hu et al., the upregulation of HBP and increased UDP-GlcNAc leads to increased O-GlcNAcylation of ribonucleotide reductase (RNR) [13]. Subsequently, this results in reduced activity of RNR, which coverts NDPs to the corresponding dNDPs, thus depleting the dNTP pool in the cells. Since dNTPs are indispensable for DNA synthesis and repair, the decreased dNTP pool impairs the DNA repair system leading to an increased mutation rate, including mutations in important oncogenes such as KRAS. High glucose levels thus led to upregulated HBP and increased O-GlcNAcylation, which eventually led to de novo KRAS mutation in pancreatic cells. Mutant KRAS is present in more than 95% of pancreatic ductal adenocarcinoma (PDAC) [19, 20]. It is responsible for regulating several key metabolic pathways of the cells, including glucose and glutamine metabolism, leading to PDAC’s aggressive growth and proliferation. KRAS mutations caused by increased O-GlcNAcylation thus lead to increased risk for developing pancreatic cancer. Besides contributing to KRAS mutation, O-GlcNAcylation is also involved in regulating key oncoproteins such as MYC and tumor suppressors such as p53 and is found to be upregulated in many cancers, such as colon and breast cancers, along with an increase in OGT level [21]. Furthermore, the inhibition of OGT has been shown to lead to breast cancer cell apoptosis [22].
Taken together, the HBP plays an important role in both diabetes and cancer. Given the role of O-GlcNAcylation in pancreatic cancer tumorigenesis, it is worth investigating its role in other cancer types. Downregulating the HBP by inhibiting enzymes such as GFA and/or inhibiting O-GlcNAcylation can be a potential target for cancer treatment. Indeed, several studies have shown that inhibiting GFA has tumor reduction effects in both in vitro and in vivo models and cancer patients [23]. In addition, with evidence supporting the association between high glucose levels and pancreatic cancer, more attention should be paid to mediating these high glucose levels early in diabetic patients not only to prevent symptoms associated with diabetes but also to prevent cancer development.
3.2 Hyperinsulinemia in Diabetes Promotes Cancer Growth Through Insulin Receptors and the Subsequent Signaling Pathways (Fig. 1)
Hyperinsulinemia is another common characteristic of T2D, where there are frequently high levels of insulin circulating in patients. It is usually caused by insulin resistance and subsequent treatments, such as the use of insulin secretagogues, which can lead to increased insulin secretion. Many studies have found that high levels of insulin can lead to cancer progression through increased activation of insulin receptors (IRs) and the subsequent cell signaling pathways, thus offering another possible explanation for the observed association between diabetes and cancer [3]. IRs are tyrosine kinases that are found in two isoforms: IR-A and IR-B. IR-A was found to activate the mitogen-activated protein kinase (MAPK) pathways [7]. These MAPK pathways are responsible for regulating many important cellular aspects, such as gene expression, metabolism, mitosis, and apoptosis through a series of phosphorylations [24]. IR-B, on the other hand, activates the phosphoinositide 3-kinase (PI3K) pathway [7], which promotes the synthesis of glycogens, lipids, and proteins [25]. Therefore, hyperinsulinemia can result in increased insulin binding to IRs, which can lead to increased activation of the MAPK pathways and the PI3K pathway. The activation of these pathways then promotes cell proliferation. Hence, diabetic treatments, which have the potential to promote hyperinsulinemia, should be considered more thoroughly before being administered to patients, especially those with cancer, in order to prevent cancer growth. Inhibiting IRs or decreasing insulin levels can also have potential as a cancer therapy.
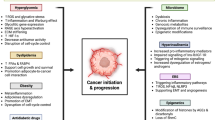
4 Obesity Leads to Insulin Resistance in Diabetes and Oxidative Stress, Which Can Lead to Cancer (Fig. 1)
Obesity, another common condition associated with dysregulated metabolism, serves as another link between type 2 diabetes (T2D) and cancer. Obesity has been shown to increase the expression and activity of protein tyrosine phosphatases (PTPs), enzymes that remove the phosphate group from protein tyrosine residues, which can lead to impaired insulin signaling and thus result in insulin resistance [26]. Insulin resistance can then result in T2D and hyperinsulinemia. Hyperinsulinemia, as discussed in Sect. 3.2, has the potential to promote cancer growth. Obesity can also increase reactive oxygen species (ROS) levels in the cells through adipokine production by adipose tissue leading to increased oxidative stress [27]. Elevated ROS levels can result in DNA damage and mutations. The accumulation of mutations in cells that escape apoptosis can ultimately lead to cancer [28]. It has been found that several types of cancer, including breast cancer [29], pancreatic cancer [20], and liver cancer, are associated with obesity [3]. Therefore, more effort should be devoted to raising awareness of the connections between obesity, diabetes, and cancer. Inhibiting PTPs and reducing adipokine production as well as ROS levels can potentially help prevent cancer in patients with obesity.
5 Amino Acid Metabolism Plays Important Roles in Both T2D and Cancer
Aside from glucose metabolism, amino acid metabolism is also deeply involved in both cancer and T2D. Amino acids are the building blocks for protein synthesis and are required for almost all cellular functions. Dysregulation of amino acid metabolism can thus contribute to the pathogenesis and progression of many diseases, including diabetes and cancer. Several key amino acids involved in diabetes and cancer include leucine, glutamine, methionine, and cysteine. Therefore, targeting dysregulated amino acid metabolism may serve as a promising strategy for cancer treatments [30, 31].
5.1 Leucine’s Regulation of Insulin Secretion and mTOR Signaling Promotes T2D Pathogenesis, Insulin Resistance, and Cancer Growth
While it is well known that glucose uptake stimulates the secretion of insulin, the amino acid leucine can also lead to insulin secretion. Leucine allosterically activates glutamate dehydrogenase (GDH), an enzyme that converts glutamate to alpha-ketoglutarate (α-KG) to enter the tricarboxylic acid (TCA) cycle, which leads to the subsequent production of metabolic coupling factors which are needed for insulin secretion [32, 33]. Dysregulation of leucine metabolism can thus result in dysregulated insulin secretion contributing to diabetes. Specifically, it was found that constantly increased leucine levels for a long period of time can be detrimental to the function of β-cells [34], which are responsible for insulin synthesis and secretion. Leucine is also crucial for the activation of the mammalian target of rapamycin (mTOR) signaling pathway, specifically the mammalian target of rapamycin complex 1 (mTORC1) [35]. The activation of mTOCR1 can lead to the activation of S6 kinases (S6Ks), which results in the downregulation of insulin signaling by phosphorylating insulin receptor substrate 1 (IRS-1). This downregulation of insulin signaling then leads to insulin resistance and T2D [35]. Leucine and mTOR signaling pathway also play important roles in cancer. Studies have shown that mTORC1 is responsible for signaling cell cycle progression and cell survival through eIF4E-binding protein 1 (4E-BP1) and eukaryotic translation initiation factor 4E (eIF4E), thus promoting cancer growth [36]. In fact, various mTOR inhibitors are being tested in both preclinical and clinical studies for many cancer types with promising results [37]. Therefore, targeting leucine metabolism and inhibiting the mTOR signaling pathway are promising strategies for the treatment of cancer.
5.2 Glutamate Regulates Insulin Secretion, Contributes to Gluconeogenesis in T2D, and Promotes Cancer Growth
As mentioned above, GDH is regulated by leucine, which can lead to insulin secretion. Thus, glutamate, the substrate for GDH, is indirectly involved in regulating insulin secretion even though it cannot stimulate insulin secretion on its own [33]. Therefore, dysregulation in glutamine metabolism may also lead to dysregulated insulin secretion and diabetes. It has also been shown that mutations in GDH can result in hyperinsulinemia [32], which has a growth-promoting effect on cancer cells. Aside from its influence on insulin secretion, a study by Miller et al. has shown that glutamine’s contribution to gluconeogenesis is important in T2D with dysregulation of the glycogen signaling pathway, which can lead to hyperglycemia during the fasting state [38]. The role of glutamine in promoting cancer survival and proliferation as an important source of energy and building block materials is well known and also well described in the previous glutamine metabolism chapter [39, 40]. The important roles that glutamine and glutamate play in both diabetes and cancer thus serve as another link between the two diseases.
5.3 Increased Methionine and Cysteine Levels in T2D Can Promote Cancer Growth
Other amino acids are also implicated in T2D. Specifically, studies have found that the levels of methionine and cysteine are elevated in the blood of patients with T2D [41]. Methionine is involved in polyamine biosynthesis and one-carbon metabolism, which is important for redox homeostasis and nucleotide synthesis [42, 43]. Increased methionine levels can lead to increased nucleotide synthesis and contribute to cancer growth. Methionine metabolism also leads to S-adenosylmethionine (SAM) production, the substrate for methyltransferases, which are responsible for the methylation of secondary metabolites, lipids, nucleic acids, and proteins [44]. SAM is involved in histone methylation, which can control gene expression, the dysregulation of which can lead to cancer [43, 45]. In fact, for some cancers, methionine is indispensable, suggesting a condition called “methionine dependence” [46]. On the other hand, cysteine is an important component of glutathione (GSH), an antioxidant used by cancer cells to mediate oxidative stress [47]. Therefore, elevated cysteine levels can result in increased GSH production protecting cancer cells against oxidative stress. In short, increased methionine and cysteine levels in T2D can promote cancer growth, and inhibiting methionine and cysteine metabolism or decreasing their concentrations should be investigated as a potential therapy for cancer.
6 Exploiting the Similarities and Relationships Between Diabetes and Cancer Metabolism for Cancer Treatment (Fig. 2)
6.1 Metformin, a Drug Developed for Diabetes, Can Inhibit Cancer Growth and Proliferation
As new research provides more insights into these similarities and relationships, it opens a new avenue of repurposing therapeutic strategies used for one disease for the treatment of another. One prominent example is the use of metformin, which was developed decades ago and is a commonly used drug for the treatment of diabetes [48]. It has the ability to reduce the production of glucose in the liver, activate adenosine monophosphate-activated protein kinase (AMPK), and improve insulin sensitivity [48]. Metformin administration leads to reduced pancreatic and breast cancer risk and prolonged survival for cancer patients even though the specific mechanisms behind its effects are not yet well understood [49]. Metformin has yielded positive results in in vivo studies, including in lung cancer xenografts [50], gastric cancer xenografts [51], and prostate cancer xenografts [52]. It is also currently under clinical trials (e.g., NCT01101438, NCT02122185, NCT01750567) for cancer treatment [53]. A study by Elgogary et al. demonstrated that combination therapy using metformin with the glutaminase inhibitor bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES) encapsulated in nanoparticles resulted in a more pronounced reduction in pancreatic tumor growth in vivo than either treatment alone since both glucose metabolism and glutamine metabolism of the cancer cells were inhibited [54]. Metformin thus serves as a positive example for which a drug developed for one disease, in this case, diabetes, can be repurposed for the treatment of cancer because of the metabolic similarities between the two diseases. Therefore, the strategy of using drugs developed for diabetes for cancer treatment seems promising and is worth investigating further.

Antidiabetic drugs and their effects on cancer. Metformin can activate AMPK, increase insulin sensitivity, and decrease hepatic glucose production in T2D while it also leads to reduced pancreatic and breast cancer risk, increased survival, and reduced tumor growth together with glutamine metabolism inhibition by BPTES. Polyphenols such as flavonoids regulate glucose metabolism, increase β-cell function, and inhibit mTOR signaling in T2D while they impair signaling pathways such as PI3K/AKT/mTOR pathways, decrease cell proliferation, and increase apoptosis in cancer. Thiazolidinediones such as pioglitazone and troglitazone can activate PPARγ, leading to increased insulin sensitivity in T2D. However, in cancer, while they can lead to decreased cell growth and increased apoptosis in vitro, pioglitazone is associated with increased bladder cancer risk, and troglitazone showed no promising results in patients. Insulin secretagogues can promote insulin secretion in T2D which can potentially help to explain their observed association with increased liver and pancreatic cancer risk through the effects of hyperinsulinemia
6.2 Polyphenols Prevent Diabetes and Reduce Cancer Growth
Polyphenols, such as flavonoids, are naturally occurring compounds found in plants that are being investigated for the treatment of several diseases, including diabetes and cancer [55]. A study by Rienks et al. found that polyphenols, especially flavonoids, can help reduce the risk for T2D [56]. Polyphenols’ antihyperglycemic effects are associated with their ability to regulate glucose metabolism and enhance β-cell function as well as their inhibition of mTOR signaling [35, 57]. In cancers, flavonoids have been shown to modulate several signaling pathways leading to reduced cell proliferation and increased apoptosis [58]. Specifically, Zhang et al. showed that treatment with flavonoids resulted in reduced levels of phospho-PI3K, phospho-AKT, phospho-mTOR, phospho-p70S6K, and phospho-ULK, thereby impairing the PI3K/AKT/mTOR/p70S6K/ULK signaling pathways, contributing to their observed effects [59]. Polyphenols and flavonoids thus should be investigated further as potential drugs for cancer treatment.
6.3 Thiazolidinediones and Their Varied Effects on Cancer
Thiazolidinediones are antidiabetic drugs that can help improve insulin sensitivity by activating peroxisome proliferator-activated receptor γ (PPARγ) [3, 49]. While PPARγ agonists have been shown to inhibit cell growth and increase apoptosis in vitro, in vivo studies have shown that they can actually lead to tumorigenesis, which is potentially caused by varied conditions within the model used that can lead to varied peroxisome proliferator-responsive element (PPRE) activation by PPARγ agonists [3, 60]. In fact, several cohort studies with patients have indicated an increased risk for bladder cancer associated with the long-term and high-dose exposure to pioglitazone, a thiazolidinedione [49]. Troglitazone, another thiazolidinedione, has been tested in phase II clinical trial for the treatment of refractory metastatic breast cancer but did not exhibit promising results [61]. Therefore, although thiazolidinediones should theoretically have anticancer effects as PPARγ agonists, more research is required before applying them to cancer treatment in patients because of their observed association with increased cancer risk.
6.4 Insulin Secretagogues Can Lead to Increased Cancer Risks
Despite the positive effects of certain diabetes drugs on cancer, there are some drugs that can actually promote cancer, especially the drugs that induce insulin secretion in diabetic patients. For example, insulin secretagogues, such as sulfonylureas and meglitinides, which stimulate insulin secretion, have been found to be associated with increased risks for liver and pancreatic cancers [49]. This is likely the result of increased insulin binding to insulin receptors, which, as discussed in Sect. 4, can promote cancer growth through subsequent signaling pathways. Therefore, caution must be exercised, and the drug’s mechanism of action and effects must be thoroughly investigated in preclinical models before repurposing it for the treatment of another disease.
7 Conclusion
Diabetes and cancer are two of the most common diseases around the world that have many similarities and associations regarding their metabolic characteristics. Diabetes is correlated with an increased risk for many types of cancer. Certain features of diabetes metabolism, such as upregulated HBP and hyperinsulinemia, can actually contribute to cancer pathogenesis and growth. Understanding these associations with the use of metabolomics technologies [62] and exploiting them for cancer treatment, as demonstrated by the repurposing of metformin for cancer treatment, can potentially improve current clinical outcomes. There should also be heightened awareness about the connections between diabetes and cancer so that more efforts can be directed to prevent cancer in diabetic patients, given the metabolic similarities and associations between these two diseases.
Abbreviations
- 4E-BP1:
-
eIF4E-binding protein 1
- AMPK:
-
Adenosine monophosphate-activated protein kinase
- BPTES:
-
Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide
- CDC:
-
Centers for Disease Control and Prevention
- eIF4E:
-
Eukaryotic translation initiation factor 4E
- GDH :
-
Glutamate dehydrogenase
- GFA:
-
Glutamine:fructose-6-phosphate amidotransferase
- GLS:
-
Glutaminase
- GSH:
-
Glutathione
- HBP:
-
Hexosamine biosynthetic pathway
- IARC:
-
International Agency for Research on Cancer
- IR:
-
Insulin receptor
- IRS-1:
-
Insulin receptor substrate 1
- KRAS:
-
Kirsten rat sarcoma viral oncogene homolog
- MAPK:
-
Mitogen-activated protein kinase
- mTOR:
-
Mammalian target of rapamycin
- mTORC1:
-
Mammalian target of rapamycin complex 1
- O-GlcNAcylation:
-
O-linked N-acetylglucosaminylation
- OGT:
-
O-GlcNAc transferase
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PI3K:
-
Phosphoinositide 3-kinase
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- PPRE:
-
Peroxisome proliferator-responsive element
- PTM:
-
Posttranslational modification
- PTP:
-
Protein tyrosine phosphatase
- RNR:
-
Ribonucleotide reductase
- ROS:
-
Reactive oxygen species
- S6K:
-
S6 kinase
- SAM:
-
S-adenosylmethionine
- T1D:
-
Type 1 diabetes
- T2D:
-
Type 2 diabetes
- TCA:
-
Tricarboxylic acid
- UDP-GlcNAc:
-
Uridine diphosphate N-acetylglucosamine
- α-KG:
-
Alpha-ketoglutarate
References
Prevention, C.f.D.C.a. (2020). In U.S.D.o.H.a.H. Services (Ed.), National diabetes statistics report, 2020. GA: Atlanta.
Bray, F., et al. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 68(6), 394–424.
Giovannucci, E., et al. (2010). Diabetes and cancer: A consensus report. Diabetes Care, 33(7), 1674–1685.
Sacerdote, C., & Ricceri, F. (2018). Epidemiological dimensions of the association between type 2 diabetes and cancer: A review of observational studies. Diabetes Research and Clinical Practice, 143, 369–377.
Le, A., Udupa, S., & Zhang, C. (2019). The Metabolic Interplay between Cancer and Other Diseases. Trends Cancer, 5(12), 809–821.
Pannala, R., et al. (2008). Prevalence and clinical profile of pancreatic cancer-associated diabetes mellitus. Gastroenterology, 134(4), 981–987.
Orgel, E., & Mittelman, S. D. (2013). The links between insulin resistance, diabetes, and cancer. Current Diabetes Reports, 13(2), 213–222.
Gapstur, S. M., et al. (2012). Type II diabetes mellitus and the incidence of epithelial ovarian cancer in the cancer prevention study-II nutrition cohort. Cancer Epidemiology, Biomarkers & Prevention, 21(11), 2000–2005.
American Diabetes, A. (2010). Diagnosis and classification of diabetes mellitus. Diabetes Care, 33(Suppl 1), S62–S69.
Warburg, O. (1924). Über den stoffwechsel der carcinomzelle. Naturwissenschaften, 1924, 1131–1137.
Bose, S., Zhang, C., & Le, A. (2021). Glucose metabolism in cancer: The Warburg effect and beyond. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_1.
Waters, A. M., & Der, C. J. (2018). KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harbor Perspectives in Medicine, 8, 9.
Hu, C. M., et al. (2019). High glucose triggers nucleotide imbalance through O-GlcNAcylation of key enzymes and induces KRAS mutation in pancreatic cells. Cell Metabolism, 29(6), 1334–1349. e10.
Marshall, S., Bacote, V., & Traxinger, R. R. (1991). Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. The Journal of Biological Chemistry, 266(8), 4706–4712.
Stowell, S. R., Ju, T., & Cummings, R. D. (2015). Protein glycosylation in cancer. Annual Review of Pathology, 10, 473–510.
Yang, X., & Qian, K. (2017). Protein O-GlcNAcylation: Emerging mechanisms and functions. Nature Reviews. Molecular Cell Biology, 18(7), 452–465.
Cooksey, R. C., & McClain, D. A. (2011). Increased hexosamine pathway flux and high fat feeding are not additive in inducing insulin resistance: Evidence for a shared pathway. Amino Acids, 40(3), 841–846.
Buse, M. G. (2006). Hexosamines, insulin resistance, and the complications of diabetes: Current status. American Journal of Physiology. Endocrinology and Metabolism, 290(1), E1–E8.
Bryant, K. L., et al. (2014). KRAS: Feeding pancreatic cancer proliferation. Trends in Biochemical Sciences, 39(2), 91–100.
Camelo, F., & Le, A. (2021). The intricate metabolism of pancreatic cancers. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_5.
Fardini, Y., et al. (2013). O-GlcNAcylation: A new cancer hallmark? Frontiers in Endocrinology (Lausanne), 4, 99.
Barkovskaya, A., et al. (2019). O-GlcNAc transferase inhibition differentially affects breast cancer subtypes. Scientific Reports, 9(1), 5670.
Yang, C., et al. (2016). High expression of GFAT1 predicts poor prognosis in patients with pancreatic cancer. Scientific Reports, 6, 39044.
Cargnello, M., & Roux, P. P. (2011). Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiology and Molecular Biology Reviews, 75(1), 50–83.
Wilcox, G. (2005). Insulin and insulin resistance. Clinical Biochemist Reviews, 26(2), 19–39.
Kahn, B. B., & Flier, J. S. (2000). Obesity and insulin resistance. The Journal of Clinical Investigation, 106(4), 473–481.
Fernandez-Sanchez, A., et al. (2011). Inflammation, oxidative stress, and obesity. International Journal of Molecular Sciences, 12(5), 3117–3132.
Liou, G. Y., & Storz, P. (2010). Reactive oxygen species in cancer. Free Radical Research, 44(5), 479–496.
Tan, J., & Le, A. (2021). The heterogeneity of breast cancer metabolism. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_6.
Dang, C. V., et al. (2011). Therapeutic targeting of cancer cell metabolism. Journal of Molecular Medicine (Berlin), 89(3), 205–212.
Hirschey, M. D., et al. (2015). Dysregulated metabolism contributes to oncogenesis. Seminars in Cancer Biology, 35(Suppl), S129–S150.
Li, C., et al. (2003). Regulation of leucine-stimulated insulin secretion and glutamine metabolism in isolated rat islets. The Journal of Biological Chemistry, 278(5), 2853–2858.
Newsholme, P., et al. (2005). New insights into amino acid metabolism, beta-cell function and diabetes. Clinical Science (London, England), 108(3), 185–194.
Liu, Z., et al. (2012). Chronic exposure to leucine in vitro induces beta-cell dysfunction in INS-1E cells and mouse islets. The Journal of Endocrinology, 215(1), 79–88.
Melnik, B. C. (2012). Leucine signaling in the pathogenesis of type 2 diabetes and obesity. World Journal of Diabetes, 3(3), 38–53.
Zoncu, R., Efeyan, A., & Sabatini, D. M. (2011). mTOR: From growth signal integration to cancer, diabetes and ageing. Nature Reviews. Molecular Cell Biology, 12(1), 21–35.
Tian, T., Li, X., & Zhang, J. (2019). mTOR signaling in cancer and mTOR inhibitors in solid tumor targeting therapy. International Journal of Molecular Sciences, 20, 3.
Miller, R. A., et al. (2018). Targeting hepatic glutaminase activity to ameliorate hyperglycemia. Nature Medicine, 24(4), 518–524.
Le, A., et al. (2012). Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metabolism, 15(1), 110–121.
Li, T., Copeland, C., & Le, A. (2021). Glutamine metabolism in cancer. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_2.
Adams, S. H. (2011). Emerging perspectives on essential amino acid metabolism in obesity and the insulin-resistant state. Advances in Nutrition, 2(6), 445–456.
Gao, X., et al. (2019). Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature, 572(7769), 397–401.
Gao, X., et al. (2017). Metabolic interactions with cancer epigenetics. Molecular Aspects of Medicine, 54, 50–57.
Yang, M., & Vousden, K. H. (2016). Serine and one-carbon metabolism in cancer. Nature Reviews. Cancer, 16(10), 650–662.
Song, Y., Wu, F., & Wu, J. (2016). Targeting histone methylation for cancer therapy: Enzymes, inhibitors, biological activity and perspectives. Journal of Hematology & Oncology, 9(1), 49.
Cavuoto, P., & Fenech, M. F. (2012). A review of methionine dependency and the role of methionine restriction in cancer growth control and life-span extension. Cancer Treatment Reviews, 38(6), 726–736.
Traverso, N., et al. (2013). Role of glutathione in cancer progression and chemoresistance. Oxidative Medicine and Cellular Longevity, 2013, 972913.
Rena, G., Hardie, D. G., & Pearson, E. R. (2017). The mechanisms of action of metformin. Diabetologia, 60(9), 1577–1585.
Nguyen, Q. T., et al. (2012). Diabetes medications and cancer risk: Review of the literature. American Health & Drug Benefits, 5(4), 221–229.
Yousef, M., & Tsiani, E. (2017). Metformin in lung cancer: Review of in vitro and in vivo animal studies. Cancers (Basel), 9, 5.
Kato, K., et al. (2012). The antidiabetic drug metformin inhibits gastric cancer cell proliferation in vitro and in vivo. Molecular Cancer Therapeutics, 11(3), 549–560.
Ben Sahra, I., et al. (2008). The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene, 27(25), 3576–3586.
Chae, Y. K., et al. (2016). Repurposing metformin for cancer treatment: Current clinical studies. Oncotarget, 7(26), 40767–40780.
Elgogary, A., et al. (2016). Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proceedings of the National Academy of Sciences of the United States of America, 113(36), E5328–E5336.
Pandey, K. B., & Rizvi, S. I. (2009). Plant polyphenols as dietary antioxidants in human health and disease. Oxidative Medicine and Cellular Longevity, 2(5), 270–278.
Rienks, J., et al. (2018). Polyphenol exposure and risk of type 2 diabetes: Dose-response meta-analyses and systematic review of prospective cohort studies. The American Journal of Clinical Nutrition, 108(1), 49–61.
Bahadoran, Z., Mirmiran, P., & Azizi, F. (2013). Dietary polyphenols as potential nutraceuticals in management of diabetes: A review. Journal of Diabetes and Metabolic Disorders, 12(1), 43.
Abotaleb, M., et al. (2018). Flavonoids in cancer and apoptosis. Cancers (Basel), 11, 1.
Zhang, H. W., et al. (2018). Flavonoids inhibit cell proliferation and induce apoptosis and autophagy through downregulation of PI3Kgamma mediated PI3K/AKT/mTOR/p70S6K/ULK signaling pathway in human breast cancer cells. Scientific Reports, 8(1), 11255.
Clay, C. E., et al. (2001). Magnitude of peroxisome proliferator-activated receptor-gamma activation is associated with important and seemingly opposite biological responses in breast cancer cells. Journal of Investigative Medicine, 49(5), 413–420.
Burstein, H. J., et al. (2003). Use of the peroxisome proliferator-activated receptor (PPAR) gamma ligand troglitazone as treatment for refractory breast cancer: A phase II study. Breast Cancer Research and Treatment, 79(3), 391–397.
Hoang, G., Udupa, S., & Le, A. (2019). Application of metabolomics technologies toward cancer prognosis and therapy. International Review of Cell and Molecular Biology, 347, 191–223.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Zhang, C., Le, A. (2021). Diabetes and Cancer: The Epidemiological and Metabolic Associations. In: Le, A. (eds) The Heterogeneity of Cancer Metabolism. Advances in Experimental Medicine and Biology, vol 1311. Springer, Cham. https://doi.org/10.1007/978-3-030-65768-0_16
Download citation
DOI: https://doi.org/10.1007/978-3-030-65768-0_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-65767-3
Online ISBN: 978-3-030-65768-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)