
Abstract
The kappa opioid receptor (KOR) has emerged as a promising therapeutic target for pain and itch treatment. There is growing interest in biased agonists that preferentially activate select signaling pathways downstream of KOR activation on the cellular level due to their therapeutic promise in retaining the analgesic and antipruritic effects and eliminating the sedative and dysphoric effects of KOR signaling on the physiological level. The concept of ligand-selective signaling includes that biased ligands promote KOR to selectively recruit one transducer or regulator protein over another, introducing bias into the signaling cascade at the very receptor-proximal level. Measuring agonist effects directly at the receptor has remained challenging and previous studies have focused on inferring agonist-selective KOR engagement with G protein relative to β-arrestin based on downstream signaling readouts. Here we discuss novel strategies to directly assess ligand-selective effects on receptor activation using KOR-interacting biosensors. The conformation-specific cytoplasmic biosensors are disconnected from the endogenous signaling machinery and provide a direct receptor-proxy readout of ligand effects in living cells. Receptor–biosensor interaction is ligand concentration dependent and can be used to determine relative ligand potency and efficacy. In addition, the biosensors reveal the existence of two dimensions of agonist bias in the cellular context: Firstly, agonists can selectively produce discrete protein-engaged KOR states and secondly, agonists can differ in the precise subcellular location at which they activate KOR. We discuss the value and the limitations of using orthogonal receptor-interacting biosensors in the quest to understand functional selectivity amongst KOR agonists in the cellular context.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction: Ligand-Selective Effects at the Kappa Opioid Receptor
Agonists of the KOR comprise various endogenous and exogenous peptide and non-peptide ligands with diverse chemical scaffolds. The binding of agonists induces conformational changes in the receptor that allow binding of active state KOR to transducer and regulatory proteins on the cytosolic side, such as G proteins, GPCR kinases (GRKs), and β-arrestins (Bruchas and Chavkin 2010). Their coupling to KOR elicits transmembrane signal transduction. As a classical member of the GPCR family, KOR signals through allostery, which implies that signal transduction involves a reciprocal, cooperative coupling between the orthosteric agonist-binding site and the intracellular transducer-binding site. Orthosteric ligands can differ considerably in their potency and their efficacy to shift the equilibrium from inactive receptor to active receptor states and produce receptor–transducer coupling. It is also increasingly clear that chemically distinct agonists can introduce allosteric bias into the signaling cascade by promoting KOR to couple to specific downstream pathways (Dunn et al. 2018; Mores et al. 2019; Zhou et al. 2013). The ability of an agonist to activate a signal transduction pathway in vitro is a major determinant of its pharmacological activity and is important for predicting its effects (Smith et al. 2018). Current pharmacological approaches to determine the potency and efficacy of GPCR agonists are based on the measurement of G protein signaling, which can be sampled at multiple levels, e.g. at the level of GTP exchange (GTPγS), at the level of second messenger production (e.g. cAMP), or at the level of gene transcription (Mores et al. 2019). The functional assays of G protein coupling are based on an enzymatic process (nucleotide exchange) and involve different steps of signal amplification, rather than directly measuring protein–protein interaction at the receptor-proxy level. In parallel, pharmacological studies have determined the potency and efficacy of KOR agonists to drive KOR coupling to β-arrestin, another cellular transducer and regulatory protein. Arrestin coupling has been assayed by measuring β-arrestin recruitment to KOR tagged with reporter proteins, e.g. using bioluminescence resonance energy transfer (BRET), enzyme fragment complementation, or reporter gene expression readouts (Ho et al. 2018). While the assays are based on direct interaction between KOR and β-arrestin, it is now clear that β-arrestin recruitment to GPCRs involves multiple biochemical steps. In particular, full interaction with β-arrestin requires KOR to be phosphorylated by GPCR kinases (GRKs) in the cytoplasmic tail (Chen et al. 2016). Therefore, β-arrestin recruitment measured in such assays reflects a process that is more complex than allosteric coupling by the receptor (Eichel et al. 2018; Nuber et al. 2016). Due to the complexity in G protein- and β-arrestin-based readouts, it remains challenging to reliably determine the degree to which chemically distinct agonists can induce allosteric bias (Conibear and Kelly 2019; Gillis et al. 2020a; Kenakin 2019). An alternative approach to measuring agonist effects at the KOR has recently emerged, which relies on the use of conformation-specific biosensors. Recruitment of biosensors to KOR can serve as direct, unamplified readout for the relative efficacy and potency of distinct agonists. Furthermore, the differential recruitment of different biosensor probes in response to agonists provides new insight into allosteric bias at the receptor-proximal level. In this chapter, we describe the development and the characteristics of nanobody- and mini-G protein-based biosensors, discuss their value in determining ligand effects and ligand bias, and highlight the novel insights into agonist-selective effects at the KOR in living cells.
2 Nanobodies and Mini-G Proteins Act as Conformation-Specific KOR Binders
2.1 Active State Binding Nanobodies Nb39 and Nb33
Current biosensors for the KOR represent repurposed tools that originated from structural and biophysical studies into GPCR function. Over the past decade, tremendous progress in the field of GPCR structural biology has led to the determination of many high-resolution structures of inactive and active state GPCRs by X-ray crystallography or by cryoelectron microscopy (cryo-EM) techniques that reveal the molecular details and dynamics of GPCR signal transduction (Hilger et al. 2018; Mahoney and Sunahara 2016; Nygaard et al. 2013). Since GPCRs, even when bound by ligands, are highly dynamic and sample a continuum of conformational ensembles, researchers developed different tools to stabilize GPCRs in specific conformations. In particular nanobodies, recombinant camelid single-domain antibodies, have been instrumental in obtaining the active state structures of several GPCRs, including the KOR and the mu opioid receptor (MOR) (Che et al. 2018; Huang et al. 2015; Rasmussen et al. 2011a). The nanobodies bind to agonist-bound GPCRs on the cytosolic face and act as G protein mimetics, which means that the nanobodies recapitulate GPCR allostery by increasing the affinity of the agonist at the receptor, similar to G proteins. Pharmacological, spectroscopic, and structural studies show that conformation-selective nanobodies can stabilize active GPCRs in a confirmation that is strikingly similar to the G protein-coupled state, making them structural surrogates of cellular signaling partners. Nanobodies have unique properties that make them particularly well-suited for GPCR applications. They are stable and small (15 kDa), have a compact shape and three complementarity-determining regions (CDR) that can access small cavities which are inaccessible to conventional antibodies, and often bind conformational epitopes composed of discontinuous amino acid segments in the native protein target (Heukers et al. 2019; Manglik et al. 2017).
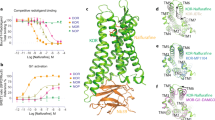
The currently available active state-selective nanobodies of KOR were generated by the Steyaert and Kobilka groups in an effort to generate G protein mimetics, originally for the MOR (Huang et al. 2015). For this purpose, lamas were immunized with purified agonist (DALDA)-bound MOR reconstituted into lipid vesicles. Lamas naturally produce heavy chain-only antibodies, which contain variable VHH domains that harbor the full antigen-binding capacity of the antibody and can be cloned and expressed as stable single-domain proteins, resulting in the so-called nanobodies. After immunization, the entire repertoire of variable VHH genes can be cloned and subjected to phage display to select for nanobodies of desired function (Pardon et al. 2014). For the MOR, a family of nanobodies, including Nb39 and Nb33 (Fig. 1a), was identified that binds MOR in an agonist-dependent manner at the intracellular surface and dramatically enhances the affinity of several peptide and non-peptide ligands at MOR (Huang et al. 2015). Subsequently, the crystal structure of active MOR bound to the agonist BU72 and Nb39 was solved, which indeed shows striking similarity to the structure of active MOR in complex with the agonist DAMGO and Gi (Koehl et al. 2018). The Nb39–MOR interface involves residues from ICL2, ICL3 and helix 8 of the MOR, all of which are conserved across the three canonical opioid receptors MOR, delta opioid receptor (DOR), and KOR. This motivated the Roth group to utilize Nb39 as crystallization chaperone for active state KOR (Che et al. 2018). Indeed, Nb39 also binds to active KOR in an agonist- and efficacy-dependent manner as shown by BRET between tagged nanobody and receptor proteins. Nb39 increases agonist affinity at KOR and attenuates the agonist dissociation rate. The crystal structure of active KOR bound to the agonist MP1104 and Nb39 was solved (Fig. 1d). The conformational rearrangements at the intracellular site of active KOR are stabilized by the KOR–Nb39 interactions and just like for MOR, Nb39 binds to a receptor cavity that overlaps with the Gi interaction surface (Che et al. 2018). Consistent with this notion, Gαi1 was found to inhibit the KOR–Nb39 interaction in a dose-dependent manner. Nb33 is another member of the originally detected nanobody family that binds to active state MOR in vitro. Nb33 differs from Nb39 in two amino acids located within CDR1 and CDR2 (Fig. 1a), while all six key residues in the nanobody that engage with active MOR and KOR are conserved. Nb33 has not been as extensively characterized as Nb39 in vitro. However, the sequence homology suggests that Nb33 bears highly similar biochemical and structural features.

Nanobodies and mini-G proteins act as conformation-specific KOR binders. (a) Sequence alignment of active state-specific nanobodies Nb39 and Nb33. Two amino acids highlighted in red differ between Nb39 and Nb33. The three CDRs of the nanobodies are highlighted and the blue arrows indicate key residues that mediate interactions with active state MOR and KOR (Che et al. 2018; Huang et al. 2015). (b) Sequence of inactive state-specific nanobody Nb6 with highlighted CDRs. Arrows point to key residues that interact with inactive state KOR. (c) Crystal structure of inactive state KOR (dark blue), bound to antagonist JDTic (gray) and Nb6 (orange), PDB: 6VI4. (d) Crystal structure of active state KOR (blue), bound to agonist MP1104 (yellow) and Nb39 (red), PDB: 6B73. (e) Crystal structure of active state A2AR (gray), bound to agonist NECA (yellow) and mini-Gs (dark red). PDB: 5G53. The C-terminal region, which differs between the mini-Gs and mini-Gsi probe, is highlighted in orange
Motivated by the fact that nanobodies can be functionally expressed in the cytoplasm of eukaryotic cells and building on the previous success of using an active state stabilizing nanobody of the β2-adrenergic receptor (β2AR) as sensor for receptor activation in intact cells (Irannejad et al. 2013), we and others have repurposed positive-allosteric Nb39 and Nb33 from crystallization chaperones into biosensors for KOR activation (Che et al. 2018; Stoeber et al. 2018). Of note, Nb39 and Nb33 can also act as biosensors for active state MOR and DOR (Stoeber et al. 2018).
2.2 Inactive State Binding Nanobody Nb6
To generate KOR-specific nanobodies, the Roth group immunized llamas with purified agonist salvinorin A (SalA)-bound KOR and screened the nanobody library by phage display for KOR binders (Che et al. 2018). Selected nanobodies were further tested for agonist- or antagonist-dependency in their binding to KOR, which resulted in the discovery of Nb6, an inactive state-selective KOR nanobody (Fig. 1b, c). Nb6 binds to unliganded and antagonist-bound KOR and dissociates from the receptor upon activation by agonist. Radioligand binding and ligand dissociation studies showed that the presence of Nb6 attenuates agonist affinity and accelerates agonist dissociation, revealing the negative allosteric effect of Nb6 (Che et al. 2020). The crystal structure of inactive KOR bound to antagonist JDTic and Nb6 (Fig. 1c), which is overall strikingly similar to JDTic-only inactive KOR (Wu et al. 2012), revealed that Nb6 binds to KOR at an interface that is distinct from previously reported intracellular nanobody–GPCR interfaces, including the active state Nb39–KOR interface. Interestingly, the CDR3 loop of Nb6 inserts into a cavity between TM5 and TM6 of the KOR, which is different from the intracellular core region that mediates G protein interaction. The anchoring of TM5 and TM6 may prevent TM6 from moving outwards, thereby suppressing a hallmark event in the transition from an inactive to an active receptor conformation. The key residues of the KOR–Nb6 interface are conserved across the canonical opioid receptor family as well as the nociceptin/orphanin FQ peptide receptor. Accordingly, Nb6 binds all inactive state opioid receptors (Che et al. 2020). Since Nb6 and Nb39/Nb33 interact with discrete KOR conformations, Nb6 can serve as a complementary conformation-selective biosensor for the KOR.
2.3 Active State Binding Mini-G Protein Mini-Gsi
Another active state-selective KOR biosensor, which is unrelated to nanobodies in sequence and structure, is based on a minimal G protein (mini-G). The mini-G proteins represent an engineered domain of the G protein α-subunit that mimics heterotrimeric G proteins in inducing pharmacological and structural changes in GPCRs. They have been developed by the Tate group in order to facilitate structure determination of active conformations of GPCRs, similar to the nanobody-based approach (Carpenter and Tate 2016). The crystal structures of the β2AR bound to heterotrimeric Gs revealed that direct contacts between the G protein and the receptor are almost entirely mediated by the Ras-like GTPase domain of Gαs (Rasmussen et al. 2011b). Therefore, the domain was used as a basis to identify a minimum component of the Gαs subunit that could function as an effective mimetic of the heterotrimeric G protein, while being uncoupled from the βγ subunits (Gβγ), a lipid tether, and receptor binding-driven nucleotide exchange. The first engineered mini-G construct, mini-Gs (22 kDa), was shown to allosterically increase the agonist-binding affinity of Gs-coupled GPCRs to levels comparable to those elicited by heterotrimeric Gs. Furthermore, the crystal structure of the adenosine A2A receptor in complex with mini-Gs (Fig. 1e) revealed that the mini-G protein indeed recapitulated the native GPCR–G protein interface (Carpenter et al. 2016). Based on the design of mini-Gs, mini-G proteins for all major Gα families (Gαs, Gαi, Gαq, and Gα12) were developed and shown to retain the appropriate GPCR coupling specificity (Nehmé et al. 2017). Developing a mini-G probe based on Gαi1 proved challenging due to stability and coupling issues (Nehmé et al. 2017). However, a successful strategy in developing a mini-Gi probe was to create a chimera wherein specificity-determining residues at the distal C-terminus in mini-Gs were replaced with the corresponding residues from Gαi1. The C-terminal residues form a major determinant of G protein coupling specificity by folding into a helical structure (α5 helix) that occupies the agonist-activated GPCR core (Fig. 1e). The resulting mini-Gsi probe indeed gained coupling to Gi-coupled receptors and lost coupling to Gs-coupled receptors (Nehmé et al. 2017). In addition to their usefulness as surrogates for heterotrimeric G proteins in structural studies, mini-G proteins are currently used extensively as probes to report GPCR activation in living cells, similar to nanobodies (Wan et al. 2018). The mini-Gsi probe has proven to be a robust biosensor for KOR activation (Stoeber et al. 2020). Of note, it can bind to various active state Gi-coupled receptors, while Nb33, Nb39 and Nb6 couple selectively to members of the opioid receptor family.
3 Biosensors Robustly and Rapidly Report on KOR Activation and Deactivation
Measuring the dynamics of ligand-dependent GPCR signaling in living cells is essential for understanding how information is processed and transmitted in the complex cellular environment. In order to study GPCR signal transduction, different biosensors have been developed to monitor ligand binding and ligand-induced conformational changes in GPCRs, G proteins, and β-arrestins (Abreu and Levitz 2020; Haider et al. 2019). The usefulness of a biosensor relies on the recognition of the target under study and the subsequent conversion of recognition into a measurable signal. In addition, desired characteristics of a biosensor include specificity, reversibility, and the ability to report on GPCR signaling without interfering with it. The ability of Nb39, Nb33, Nb6, and mini-Gsi to act as optical biosensors for KOR activation in intact cells with high specificity, sensitivity, and high temporal and spatial resolution has recently been demonstrated (Che et al. 2020; Stoeber et al. 2020). As a straightforward approach, the different nanobodies and mini-G proteins have been fused with fluorescent proteins and ligand-dependent recruitment to fluorescently-labelled KOR has been determined by fluorescence microscopy. Alternatively, BRET measurements were performed in order to monitor close proximity (<10 nm) between the biosensor and KOR. Both assays rely on ligand-dependent biosensor relocalization from the cytosol (unbound) to the membrane (receptor-bound).
Real-time KOR activation and deactivation using Nb39 and Nb6 as biosensors in living cells was first assessed by Che and colleagues (Che et al. 2018, 2020). Ligand-dependent recruitment of the biosensors to KOR was detected using BRET in HEK293 cells co-expressing KOR C-terminally fused to Renilla Luciferase (RLuc, donor) and Nb39 or Nb6 fused to yellow fluorescent protein (YFP, acceptor). Adding the agonist SalA to cells drove recruitment of Nb39 to active KOR, which was detectable as a pronounced increase in the BRET signal due to close proximity of Rluc and YFP. The signal increase occurred within seconds of agonist addition. Subsequent addition of the antagonist JDTic could rapidly reverse the BRET signal back to baseline, indicative of Nb39 dissociation from inactive KOR. Conversely, adding SalA to cells expressing Nb6-YFP and KOR-Rluc drove dissociation of Nb6 from the receptor, detectable as a strong decrease in the BRET signal. This effect was reversed upon adding the antagonist JDTic reflecting the reassociation of Nb6 with the inactive receptor. The experiments established the usefulness of active state-selective Nb39 and inactive state-selective Nb6 as ligand-dependent and reversible conformational biosensors in living cells. The BRET assay, set up in a 96-well format and based on luminescence- and fluorescence-measurements, further provided an approach to determine ligand concentration-response curves and detect differences in ligand potency and efficacy at the cell population-level. Importantly, the ligand effects on KOR determined by nanobody recruitment corresponded to the known pharmacology of full and partial KOR ligands previously established by classical G protein signaling measurements.
In a parallel approach, we described a total internal reflection fluorescence (TIRF) microscopy-based assay to determine KOR activation and deactivation using Nb33 and mini-Gsi (Stoeber et al. 2020). In TIRF microscopy, the laser illuminates the sample above a critical angle that results in total reflection of the laser beam at the glass-specimen interface, which creates an evanescent excitation field. The evanescent field decays exponentially from the interface and penetrates into the sample medium only to a depth of approximately 100 nm. Thus, TIRF microscopy enables to selectively visualize fluorophores at the plasma membrane and the cytoplasmic zone immediately beneath it (Fig. 2a). We used this approach to follow the ligand-induced relocalization of biosensors from the cytosol to KOR in the plasma membrane in transfected HEK293 cells. To simultaneously image biosensors and receptors in living cells, we fused the biosensors with fluorescent proteins and labelled FLAG-tagged KOR in the cell surface with a fluorescent monoclonal antibody. Binding of mini-Gsi and Nb33 to KOR was monitored upon adding agonists, such as Dynorphin A (DynA), by bath application or perfusion and dissociation measured upon adding the competitive antagonist 5’GNTI in excess. Quantification of the fluorescence intensity throughout time lapse movies acquired at 5 s intervals provided real-time biosensor association traces that showed robust, rapid, and reversible recruitment of Nb33 and mini-Gsi to KOR in response to the full agonists DynA, U50,488, and U69,593. KOR surface levels did not significantly change during the 5–6 min movies, showing that biosensor recruitment could be reliably measured without possible complications of later KOR trafficking (Fig. 2b). Confirming the biosensor specificity, the mini-Gs probe, differing in only nine residues at the distal C-terminus from mini-Gsi, was not recruited in response to KOR activation. The TIRF setup also allowed recording single cell concentration-response curves by following biosensor intensity while increasing agonist concentration in a stepwise manner (Fig. 2c, d). Each dose response was internally normalized to the maximal biosensor intensity measured after adding the reference agonist DynA in excess at the end of each time series. Both Nb33 and mini-Gsi were robustly recruited to KOR in a concentration-dependent manner by chemically diverse full agonists (Stoeber et al. 2020).

Biosensors directly detect KOR activation and ligand pharmacology in living cells. (a) TIRF assay for measuring biosensor (Nb39, Nb33, or mini-Gsi) recruitment to KOR in the plasma membrane. Biosensor fused to a fluorescent protein (FP, red) re-localizes from the cytosol to active state KOR (blue) in the plasma membrane upon agonist addition. The total internal reflection fluorescence (TIRF) microscopy light beam is indicated. (b) Schematic of the fluorescence changes of an active state-specific biosensor (red) and of KOR (blue) detected in the TIRF assay. Agonist and antagonist additions are depicted. (c) Schematic of the stepwise increase in biosensor fluorescence intensity during concentration response measurements of single cells using TIRF. The maximum signal of a given ligand is normalized to the intensity generated by an internal reference ligand, which is applied in excess at the beginning (and then washed out) or end of the time series. (d) Concentration-dependent recruitment of biosensors to KOR measured by TIRF reveals ligand differences in efficacy and potency
Taken together, several KOR-interacting proteins, known to recognize different structural features of the receptor without requiring or engaging other known cellular proteins, can be used as rapid and robust biosensors for KOR activation in living cells. The current assays are based on receptor-proximal biosensor recruitment and present a linear system well-suited to detect differences in relative agonist efficiency and potency.
4 Biosensors Reveal Ligand-Selective Effects at KOR
4.1 Ligand-Selective Recruitment of Distinct Biosensors to KOR
Ligand selectivity or ligand bias is the process whereby chemically distinct agonists can produce different receptor-based effects and represents a well-established pharmacological concept. It is based on the hypothesis that different agonists can drive GPCRs to recruit different cytoplasmic proteins in living cells, introducing allosteric bias into the signaling system at the very receptor-proximal level. To date, most studies on functional selectivity have focused on classifying ligands based on their ability to drive G protein relative to β-arrestin-based pathways and the indirect downstream readouts have required subsequent calculation of bias by operational analysis. We tested whether the GPCR-interacting biosensors could offer a more simple and direct approach for assessing ligand-selective protein recruitment to receptors. While the active state biosensors of KOR have been engineered with the goal to sense and stabilize activation-associated conformational changes in the receptor, it is clear that nanobodies and mini-G proteins present distinct protein folds and that each probe recognizes different structural features of the activated receptor (Fig. 1). Therefore, we reasoned that nanobody-bound KOR and mini-Gsi-bound KOR may represent discrete protein-engaged receptor states that might be selectively produced by diverse agonists (Stoeber et al. 2020). Using mini-Gsi and Nb33 comparatively as KOR-interacting probes, we first noticed that the concentration-response curve for mini-Gsi relative to Nb33 recruitment was consistently left-shifted for all agonists (Fig. 3a, b). The potency shift indicates a difference in allosteric communication of the distinct ligand–KOR–biosensor complexes and further highlights that mini-Gsi and Nb33 interactions with active KOR are not identical. Of note, a similar left shift for mini-Gsi relative to Nb33 was also detected for various agonists at MOR, as determined in BRET and TIRF assays (Gillis et al. 2020b; Stoeber et al. 2020).

Ligand-selective recruitment of distinct biosensors to KOR. (a and b): Left-shifted concentration response curve of mini-Gsi relative to Nb33. (a) mini-Gsi (mGsi) and Nb33 intensity during TIRF microscopy time-lapse series of a HEK293 cell, co-expressing KOR and Venus-mGsi or EGFP-Nb33, adding increasing concentrations of U69,593 (U69, 1 nM–10 μM) followed by reference compound dynorphin A (10 μM). (b) Concentration-dependent recruitment of mini-Gsi and Nb33 probes to KOR upon U69 addition. (c and d): Selective KOR recruitment of mini-Gsi relative to Nb33 observed after receptor activation by etorphine (ET). (c) mini-Gsi and Nb33 intensity during TIRF microscopy time-lapse series as in (a), adding increasing concentrations of ET (1 nM–10 μM), followed by reference compound DynA (10 μM). (d) Concentration-dependent recruitment of mGsi and Nb33 probes to KOR upon ET addition. All data as in Stoeber et al. (2020), published under Creative Commons Attribution Licence CC-BY 4.0
We subsequently investigated the effect of the alkaloid agonist etorphine on mini-Gsi and Nb33 recruitment by KOR. Etorphine is an opiate alkaloid drug that efficaciously promotes G protein signaling but drives β-arrestin-mediated KOR internalization poorly and is therefore classified as a G protein-biased agonist by operational criteria (DiMattio et al. 2015; Jordan et al. 2000). In the TIRF-based biosensor recruitment assay, etorphine behaved as a potent but partial agonist for mini-Gsi recruitment, and remarkably, etorphine drove no recruitment of Nb33 even at very high concentrations (Fig. 3c, d). The differential biosensor recruitment indicates that mini-Gsi and Nb33 probes can distinguish receptor-proximal agonist effects in intact cells. In other words, the results provided direct experimental evidence that ligands can impose selectivity on protein recruitment by GPCRs. Differential recruitment of mini-Gsi and Nb33 was not unique to etorphine at KOR but was observed for a range of chemically diverse MOR agonists, including morphine, PZM21, and mitragynine pseudoindoxyl (Gillis et al. 2020b; Stoeber et al. 2020).
While the different biosensors allow to directly measure receptor-proximal protein recruitment in living cells, they present engineered probes that do not directly relate to receptor function. Moving beyond biosensors, we found that agonist-selective protein recruitment to KOR also applies to the physiological relevant GPCR-interacting kinase GRK2. GRK2 drives ligand-dependent KOR phosphorylation in the cytoplasmic tail and it is clear that KOR agonists differ in their ability to stimulate receptor phosphorylation (Chen et al. 2016; Chiu et al. 2017). Using an adapted TIRF protocol that allows to differentiate plasma membrane recruitment from receptor recruitment, we detected that etorphine did not promote GRK2 recruitment to KOR, which was in striking contrast to DynA that strongly drove GRK2–KOR engagement (Stoeber et al. 2020). Ligand-selective GRK2 binding may explain why KOR exhibits agonists-selective phosphorylation and subsequent receptor internalization (Chu et al. 1997; Li et al. 2003). It also suggests that the Nb33 probe can report allosteric effects relevant to GRK engagement.
The findings that conformational biosensors, such as mini-Gsi and Nb33, can be differentially recruited to KOR render them particularly interesting as straightforward tools to probe for ligand bias at KOR. The differential recruitment provides evidence for the hypothesis that agonist bias can manifest in discrete receptor-proximal molecular selection events in the cell. To date, the underlying biophysical differences between the Nb33- and miniG-engaged KOR states remain unresolved. It is possible that the distinct complexes present unique active receptor conformations that are selectively stabilized by agonists. It is also possible that even more diversity in ligand-activated receptor states exists that is not detected by the currently available biosensor probes. Measuring mini-Gsi and Nb33 recruitment comparatively for a larger panel of KOR agonists with diverse pharmacological profiles will deepen our understanding of both agonist bias and the newly available tools to directly assess it.
4.2 Agonist-Selective Activation of KOR at Distinct Cellular Locations
In the past decade, it has become increasingly clear that in addition to receptors at the cell surface, GPCRs in intracellular organelles, such as endosomes, the Golgi apparatus, or the nuclear envelope, can be ligand-activated and mediate physiologically important signaling (Eichel and von Zastrow 2018; Jong et al. 2018). It was also uncovered that ligands differ strikingly in their ability to access GPCRs at different locations in the cell. For example, in order to activate internal β1-adrenergic receptors, agonists must be able to pass membranes either by diffusion or by the help of membrane-embedded transporters (Irannejad et al. 2017). Accumulating evidence shows that the location of GPCR signaling can affect both the specificity and the timing of downstream events, demonstrating that cellular location bias in ligand action represents an important new dimension of ligand selectivity (Godbole et al. 2017; Nash et al. 2019; Stoeber et al. 2018; Tsvetanova and von Zastrow 2014).
The use of novel conformation-specific GPCR binders as biosensors in living cells has been instrumental in advancing our understanding of the subcellular organization of GPCR signaling. Recently, we have delineated the spatiotemporal pattern of MOR and DOR activation in living neurons using Nb33 as a biosensor (Fig. 4) (Stoeber et al. 2018). Opioid receptor ligands comprise structurally diverse peptide and non-peptide agonists that differ substantially in their physicochemical properties, which affect membrane permeability. Focusing first on peptide agonists, we found that the endogenous neuropeptides met-enkephalin and β-endorphin drive two spatially and temporally resolved “waves” of MOR and DOR activation, first in the plasma membrane and then in endosomes following agonist-induced internalization of ORs. Extending our studies to clinically relevant opioid drugs, we then found that non-peptide opioid ligands, such as the prototypic alkaloid morphine, drive a discrete and additional wave of MOR and DOR activation in the Golgi apparatus. Golgi-localized OR activation does not require receptor trafficking and is specific to opioid drugs relative to opioid neuropeptides, since drugs can access this internal location due to their ability to diffuse freely across membranes (Fig. 4). Together with studies on other GPCRs, the findings provide a novel cellular framework for understanding how drugs may exert their specific effects (or side-effects) on the cellular level. First inroads into probing the functional significance of internal OR activation show that endosomal ORs contribute a sustained component of adenylyl cyclase inhibition and a subset of ERK and PKC signals on the cellular level and sustained inhibitory actions in sensory neurons on the physiological level (Jimenez-Vargas et al. 2020; Stoeber et al. 2018). The physiological importance of Golgi-localized OR signaling remains to be determined.

Location bias of ligand effects: OR activation occurs at distinct subcellular membrane compartments. Peptide ligands (orange circles) first bind and activate ORs at the plasma membrane and initiate a second wave of receptor activation in endosomes following receptor internalization. In addition, membrane-permeant ligands (green-circles) can access receptors that localize inside the cell at steady state and drive OR activation in the Golgi apparatus. Nanobodies allow detecting the spatiotemporal activation profile of distinct agonists
Like for the other GPCRs, KOR activation and signaling has generally been assumed to be restricted to the plasma membrane. However, first evidence that KOR ligands strikingly differ in the subcellular location at which they produce receptor activation comes from a recent study that used the biosensor Nb39. Che et al. followed KOR activation in real-time by confocal microscopy and detected a striking difference between the endogenous peptide ligand DynA and the non-peptide hallucinogen SalA (Che et al. 2020). Within seconds, SalA activated the Golgi-localized pool of KOR as detected by recruitment of Nb39 to an internal organelle containing KOR and co-labelling with the Golgi marker GalT. In contrast, DynA drove KOR activation only in the plasma membrane. It is likely that the difference in cellular activation patterns lies in the differential access of ligands to Golgi-localized receptors. SalA may freely diffuse across the membrane, while the peptide DynA penetrates membranes inefficiently. Given the large and increasing number of chemically diverse peptide and non-peptide ligands for KOR, it will be exciting to probe ligand-specific subcellular patterns of KOR activation more broadly and study the contribution of location-specific activation to the physiological effects of drugs. The KOR-binding nanobodies provide ideal and validated tools for gaining insights into the spatiotemporal organization of KOR signaling and ligand selectivity at the cellular level.
5 Conclusions and Outlook
It is increasingly clear that GPCRs can relay significantly more information about the local chemical environment than the mere binding of an agonist. GPCRs can convey what agonist is binding more specifically by triggering agonist-selective cellular responses. How agonist-selective effects are encoded and transmitted by GPCRs has been studied on many levels and remains an area of intense investigation (Smith et al. 2018). The allosteric complex of ligand, receptor, and transducer mediates communication across the cellular membrane and defines the downstream signaling response. High resolution structural and biophysical studies reveal that distinct agonists can impose bias on the conformational landscape of individual receptors and suggest that unique receptor conformations can couple to distinct cytosolic transducers, leading to a biased cellular response (Weis and Kobilka 2018). The development of orthogonal conformation-specific GPCR binders, including here described nanobodies and mini-G probes for KOR, has opened up new approaches to directly test the hypothesis of ligand-dependent “allosteric processing” at GPCRs.
As G protein mimetics, biosensors can serve as tools in pharmacological assays that measure ligand potency and efficacy. Some advantages over classical G protein assays exist, which include the linear assay system, the receptor-proxy readout, the lack of ceiling effect if sensors are in excess, and their usefulness in in vitro and cellular assays. Moreover, the distinct protein probes differ in their interactions with receptors and can reveal agonist-selective effects directly at the receptor. The differential recruitment of mini-Gsi and Nb33 to KOR provides direct evidence that different agonists stabilize receptors in distinct conformational ensembles that can interact with selective proteins. Therefore, the probes are promising orthogonal and straightforward screening tools in exploring diversity and selectivity between KOR ligands. Kinetic differences in agonist action at GPCRs can be another contributor to bias and future time-dependent analysis of biosensor recruitment to KOR could provide additional insights into selectivity among agonists (Klein Herenbrink et al. 2016; Livingston et al. 2018). As novel tools, it still remains to be determined what the biophysical basis for the observed selectivity of protein recruitment is and also, if and how the distinct allosteric complexes relate to receptor function.
An emerging additional mechanism that differentiates agonists is the specific cellular location at which they drive GPCR signaling. GPCRs have been recognized for many years to be present at internal membrane locations as well as at the plasma membrane; however, internal receptors have been considered a reserve pool (secretory pathway) or a desensitized pool (endosomal pathway) with no contribution to acute signaling. Assays with high spatiotemporal resolution have revealed that many GPCRs are subject to ligand-dependent activation at internal membrane locations and that ligands differ significantly in their ability to access intracellular GPCR pools. Biosensors provide straightforward inroads into analyzing the ligand-selective subcellular activation pattern of receptor activation. A future challenge is to adapt the biosensor methodology to probe receptor activation in neurons with endogenous receptor expression and to possibly turn the tools into real-time optical sensors for in vivo applications.
Opioid receptors are clinically important targets that provide a striking example for biased agonism. The finding that agonists with different pharmacological profiles can cause different outcomes on the physiological level is the driving force behind the quest to develop novel analgesics with a reduced side-effects profile. However, understanding and quantifying bias in the cellular context remains challenging in part due to the lack in direct comparative transducer-specific assays. The novel orthogonal biosensors for opioid receptor activation can report on various aspects of ligand-selective signaling and are likely to gain traction as valuable tools in cellular pharmacology.
References
Abreu N, Levitz J (2020) Optogenetic techniques for manipulating and sensing G protein-coupled receptor signaling. Methods Mol Biol 2173:21–51
Bruchas MR, Chavkin C (2010) Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology 210:137–147
Carpenter B, Tate CG (2016) Engineering a minimal G protein to facilitate crystallisation of G protein-coupled receptors in their active conformation. Protein Eng Des Sel 29:583–594
Carpenter B, Nehmé R, Warne T, Leslie AGW, Tate CG (2016) Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 536:104–107
Che T, Majumdar S, Zaidi SA, Ondachi P, McCorvy JD, Wang S, Mosier PD, Uprety R, Vardy E, Krumm BE et al (2018) Structure of the Nanobody-stabilized active state of the kappa opioid receptor. Cell 172:55–67.e15
Che T, English J, Krumm BE, Kim K, Pardon E, Olsen RHJ, Wang S, Zhang S, Diberto JF, Sciaky N et al (2020) Nanobody-enabled monitoring of kappa opioid receptor states. Nat Commun 11:1145
Chen C, Chiu Y-T, Wu W, Huang P, Mann A, Schulz S, Liu-Chen L-Y (2016) Determination of sites of U50,488H-promoted phosphorylation of the mouse κ opioid receptor (KOPR): disconnect between KOPR phosphorylation and internalization. Biochem J 473:497–508
Chiu Y-T, Chen C, Yu D, Schulz S, Liu-Chen L-Y (2017) Agonist-dependent and -independent κ opioid receptor phosphorylation: distinct phosphorylation patterns and different cellular outcomes. Mol Pharmacol 92:588–600
Chu P, Murray S, Lissin D, von Zastrow M (1997) Delta and kappa opioid receptors are differentially regulated by dynamin-dependent endocytosis when activated by the same alkaloid agonist. J Biol Chem 272:27124–27130
Conibear AE, Kelly E (2019) A biased view of μ-opioid receptors? Mol Pharmacol 96:542–549
DiMattio KM, Ehlert FJ, Liu-Chen L-Y (2015) Intrinsic relative activities of κ opioid agonists in activating Gα proteins and internalizing receptor: differences between human and mouse receptors. Eur J Pharmacol 761:235–244
Dunn AD, Reed B, Guariglia C, Dunn AM, Hillman JM, Kreek MJ (2018) Structurally related kappa opioid receptor agonists with substantial differential signaling bias: neuroendocrine and behavioral effects in C57BL6 mice. Int J Neuropsychopharmacol 21:847–857
Eichel K, von Zastrow M (2018) Subcellular organization of GPCR signaling. Trends Pharmacol Sci 39:200–208
Eichel K, Jullié D, Barsi-Rhyne B, Latorraca NR, Masureel M, Sibarita J-B, Dror RO, von Zastrow M (2018) Catalytic activation of β-arrestin by GPCRs. Nature 557:381–386
Gillis A, Sreenivasan V, Christie MJ (2020a) Intrinsic efficacy of opioid ligands and its importance for apparent bias, operational analysis and therapeutic window. Mol Pharmacol 98:410
Gillis A, Gondin AB, Kliewer A, Sanchez J, Lim HD, Alamein C, Manandhar P, Santiago M, Fritzwanker S, Schmiedel F et al (2020b) Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci Signal 13:eaaz3140
Godbole A, Lyga S, Lohse MJ, Calebiro D (2017) Internalized TSH receptors en route to the TGN induce local Gs-protein signaling and gene transcription. Nat Commun 8:443
Haider RS, Godbole A, Hoffmann C (2019) To sense or not to sense-new insights from GPCR-based and arrestin-based biosensors. Curr Opin Cell Biol 57:16–24
Heukers R, De Groof TWM, Smit MJ (2019) Nanobodies detecting and modulating GPCRs outside in and inside out. Curr Opin Cell Biol 57:115–122
Hilger D, Masureel M, Kobilka BK (2018) Structure and dynamics of GPCR signaling complexes. Nat Struct Mol Biol 25:4–12
Ho J-H, Stahl EL, Schmid CL, Scarry SM, Aubé J, Bohn LM (2018) G protein signaling-biased agonism at the κ-opioid receptor is maintained in striatal neurons. Sci Signal 11:eaar4309
Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC et al (2015) Structural insights into μ-opioid receptor activation. Nature 524:315–321
Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SGF, Sunahara RK, El-Samad H, Huang B et al (2013) Conformational biosensors reveal GPCR signalling from endosomes. Nature 495:534–538
Irannejad R, Pessino V, Mika D, Huang B, Wedegaertner PB, Conti M, von Zastrow M (2017) Functional selectivity of GPCR-directed drug action through location bias. Nat Chem Biol 13:799–806
Jimenez-Vargas NN, Gong J, Wisdom MJ, Jensen DD, Latorre R, Hegron A, Teng S, DiCello JJ, Rajasekhar P, Veldhuis NA et al (2020) Endosomal signaling of delta opioid receptors is an endogenous mechanism and therapeutic target for relief from inflammatory pain. Proc Natl Acad Sci U S A 117:15281–15292
Jong Y-JI, Harmon SK, O’Malley KL (2018) GPCR signalling from within the cell. Br J Pharmacol 175:4026–4035
Jordan BA, Cvejic S, Devi LA (2000) Kappa opioid receptor endocytosis by dynorphin peptides. DNA Cell Biol 19:19–27
Kenakin T (2019) Biased receptor signaling in drug discovery. Pharmacol Rev 71:267–315
Klein Herenbrink C, Sykes DA, Donthamsetti P, Canals M, Coudrat T, Shonberg J, Scammells PJ, Capuano B, Sexton PM, Charlton SJ et al (2016) The role of kinetic context in apparent biased agonism at GPCRs. Nat Commun 7:10842
Koehl A, Hu H, Maeda S, Zhang Y, Qu Q, Paggi JM, Latorraca NR, Hilger D, Dawson R, Matile H et al (2018) Structure of the μ-opioid receptor-Gi protein complex. Nature 558:547–552
Li J-G et al (2003) Differential regulation of the human κ opioid receptor by agonists: Etorphine and Levorphanol reduced Dynorphin A- and U50,488H-induced internalization and phosphorylation. J Pharmacol Exp Ther 305(2):531–540
Livingston KE, Mahoney JP, Manglik A, Sunahara RK, Traynor JR (2018) Measuring ligand efficacy at the mu-opioid receptor using a conformational biosensor. eLife 7
Mahoney JP, Sunahara RK (2016) Mechanistic insights into GPCR-G protein interactions. Curr Opin Struct Biol 41:247–254
Manglik A, Kobilka BK, Steyaert J (2017) Nanobodies to study G protein-coupled receptor structure and function. Annu Rev Pharmacol Toxicol 57:19–37
Mores KL, Cummins BR, Cassell RJ, van Rijn RM (2019) A review of the therapeutic potential of recently developed G protein-biased kappa agonists. Front Pharmacol 10:407
Nash CA, Wei W, Irannejad R, Smrcka AV (2019) Golgi localized β1-adrenergic receptors stimulate Golgi PI4P hydrolysis by PLCε to regulate cardiac hypertrophy. eLife 8:e48167
Nehmé R, Carpenter B, Singhal A, Strege A, Edwards PC, White CF, Du H, Grisshammer R, Tate CG (2017) Mini-G proteins: novel tools for studying GPCRs in their active conformation. PLoS One 12:e0175642
Nuber S, Zabel U, Lorenz K, Nuber A, Milligan G, Tobin AB, Lohse MJ, Hoffmann C (2016) β-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 531:661–664
Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP et al (2013) The dynamic process of β(2)-adrenergic receptor activation. Cell 152:532–542
Pardon E, Laeremans T, Triest S, Rasmussen SGF, Wohlkönig A, Ruf A, Muyldermans S, Hol WGJ, Kobilka BK, Steyaert J (2014) A general protocol for the generation of Nanobodies for structural biology. Nat Protoc 9:674–693
Rasmussen SGF, Choi H-J, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS et al (2011a) Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature 469:175–180
Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D et al (2011b) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477:549–555
Smith JS, Lefkowitz RJ, Rajagopal S (2018) Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov 17:243–260
Stoeber M, Jullié D, Lobingier BT, Laeremans T, Steyaert J, Schiller PW, Manglik A, von Zastrow M (2018) A genetically encoded biosensor reveals location Bias of opioid drug action. Neuron 98:963–976.e5
Stoeber, M., Jullié, D., Li, J., Chakraborty, S., Majumdar, S., Lambert, N.A., Manglik, A., and von Zastrow, M. (2020). Agonist-selective recruitment of engineered protein probes and of GRK2 by opioid receptors in living cells. eLife 9, e54208.
Tsvetanova NG, von Zastrow M (2014) Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol 10:1061–1065
Wan Q, Okashah N, Inoue A, Nehmé R, Carpenter B, Tate CG, Lambert NA (2018) Mini G protein probes for active G protein–coupled receptors (GPCRs) in live cells. J Biol Chem 293:7466–7473
Weis WI, Kobilka BK (2018) The molecular basis of G protein-coupled receptor activation. Annu Rev Biochem 87:897–919
Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang X-P, Carroll FI et al (2012) Structure of the human κ-opioid receptor in complex with JDTic. Nature 485:327–332
Zhou L, Lovell KM, Frankowski KJ, Slauson SR, Phillips AM, Streicher JM, Stahl E, Schmid CL, Hodder P, Madoux F et al (2013) Development of functionally selective, small molecule agonists at kappa opioid receptors. J Biol Chem 288:36703–36716
Acknowledgments
We thank Zoé Valbret, Damien Jullié, and Mark von Zastrow for critical reading of the manuscript and helpful suggestions. Work in the authors’ lab is supported by an Eccellenza Professorial Fellowship to M.S. (PCEFP3_181282) from the Swiss National Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Oberhauser, L., Stoeber, M. (2021). Biosensors Monitor Ligand-Selective Effects at Kappa Opioid Receptors. In: Liu-Chen, LY., Inan, S. (eds) The Kappa Opioid Receptor. Handbook of Experimental Pharmacology, vol 271. Springer, Cham. https://doi.org/10.1007/164_2020_427
Download citation
DOI: https://doi.org/10.1007/164_2020_427
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-89073-5
Online ISBN: 978-3-030-89074-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)