
Abstract
The goal of cancer immunotherapy is to induce specific and durable antitumor immunity. Adoptive T cell therapy (ACT) has garnered wide interest, particularly in regard to strategies to improve T cell efficacy in trials. There are many types of T cells (and subsets) which can be selected for use in ACT. CD4+ T cells are critical for the regulation, activation and aid of host defense mechanisms and, importantly, for enhancing the function of tumor-specific CD8+ T cells. To date, much research in cancer immunotherapy has focused on CD8+ T cells, in melanoma and other cancers. Both CD4+ T cells and CD8+ T cells have been evaluated as ACT in mice and humans, and both are effective at eliciting antitumor responses. IL-17 producing CD4+ T cells are a new subset of CD4+ T cells to be evaluated in ACT models. This review discusses the benefits of adoptive immunotherapy mediated by CD8+ and CD4+ cells. It also discusses the various type of T cells, source of T cells, and ex vivo cytokine growth factors for augmenting clinical efficacy of ACT.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The immune system is able to detect cancer, particularly in early stages of cancer, through a process referred to as immunosurveillance. Early tumors are eliminated by the immune system (“elimination” phase) and as cancers evolve, they continue to be held in check by the immune system (“equilibrium” phase) until their advanced growth becomes too challenging for the immune system to control and/or clear (immune “escape”) (Dunn et al., 2004a, b; Raval et al., 2014). Cancer immunotherapy attempts to harness the specificity and natural ability of the immune system to recognize and eliminate tumors, while overcoming immunosuppressive barriers present in the tumor microenvironment. For cancer immunotherapy trials to be effective the adaptive arm of the immune system, mediated in part by T cells, has to be activated.
Decades of evidence, initially shown with interleukin (IL)-2 treatment, have established that the immune system is capable of inducing complete and durable eradication, even of metastatic cancers (Klapper et al., 2008; McDermott, 2007). Today, there are a variety of approaches to elicit antitumor immunity. These approaches include therapeutic cancer vaccines (Cherryholmes et al., 2015; Guo et al., 2013; Knutson and Mittendorf, 2015), antitumor antibodies (Antonioli et al., 2016; Meng et al., 2016b), immune checkpoint blockade (Kakavand et al., 2016; Tang et al., 2016), and adoptive T cell therapy (ACT) (Phan-Lai et al., 2015; Restifo et al., 2012; Yee et al., 2002). In this review only ACT will be discussed. These platforms are often combined with other therapies, including radiation therapy, chemotherapy, gene therapy and immunomodulatory therapy (e.g. cytokines). This review will highlight the key properties and activation process of CD4+ and CD8+ T cells, their use in adoptive immunotherapy strategies for cancer, and some considerations (from source of T cells to ex vivo expansion) to achieve optimal efficacy of ACT.
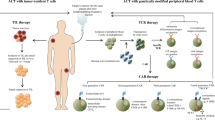
Adoptive t cell therapy
Cancer therapeutic strategies, which can be used alone or in combination, include: immunomodulation (via cytokines), antitumor antibodies, immune checkpoint blockade, cancer vaccines, dendritic cell (DC) therapy, and ACT. Unlike antitumor antibodies, which represent a form of passive immunotherapy, cancer vaccines and ACT are examples of active immunotherapy, i.e. they “actively” attempt to stimulate host endogenous immunity against the cancer. ACT harnesses the natural ability of T cells to specifically recognize and eliminate target tumor cells. Its main advantages are in generating long-lived antitumor immunity and overcoming tolerance, which is a major roadblock in cancer therapy. In principle, ACT involves the isolation of autologous antigen-specific T cells from patients for expansion ex vivo (with antigen restimulation and cytokine growth factors), followed by re-infusion into patients to treat the cancers. Sometimes, antigen is provided in vivo in the host (“vaccine priming”) to elicit more antigen-specific T cells in vivo prior to isolation. Due to excellent homing properties of T cells, ACT is beneficial for treating metastatic cancers, solid or non-solid cancers, and cancerous pockets in the body not easily excised with surgery. Furthermore, the antitumor action is tumor antigen-specific, minimizing adverse effects on normal cells.
Adaptive immunity
Innate and adaptive immunity protects the host from foreign pathogens. Both immune arms differentiate between “self” and “non-self” antigens. However, since innate and adaptive immunity are both generally tolerant towards host tissues and since cancer antigens are essential “self” antigens, cancer growth is often undetectable by the immune system. Unlike the innate immune arm, which recognizes pathogens in the context of non-specific molecular patterns, adaptive immunity is highly specific and is mediated by T cells and B cells. Similar to B cells, when T cells recognize antigen, via their antigen-specific T cell receptors (TCRs), they undergo activation and proliferation in a process known as clonal expansion; this leads to the generation of long-term memory T cells. Also, similar to B cells, the specificity and diversity of antigenspecific receptors result from V(D)J recombination (a random genetic combination of Variable, Diversity, and Joining gene segments). This recombination is what allows for such a vastly diverse and abundant repertoire of different highly specific receptors.
Types of T cells in act
Classical T cells (alpha-beta), unlike gamma-delta T cells (Silva-Santos et al., 2015) and B cells, cannot detect soluble antigen. Instead, they recognize antigen in the form of peptides bound to MHC molecules. The peptide/MHC complex (pMHC) is presented on the surface of antigen presenting cells (APCs), such as DCs (Palucka and Banchereau, 2013). The 2 main types of T cells are: CD8+ and CD4+ T cells. The CD8+ T cell subset binds to peptides (8-10 residues) bound to MHC class I molecules (MHC I), which are expressed by every nucleated cell, including APCs. Conversely, the CD4+ subset recognizes and binds to longer antigenic peptides presented in the context of MHC class II molecules (MHC II), which are mainly expressed by APCs.
Much emphasis in immunotherapy has been placed on CD8+ T cells because when activated, CD8+ T cells can induce potent cytotoxic effects on tumor cells, leading to tumor eradication; hence, they are also called cytotoxic T lymphocytes (CTLs). Tumor cytotoxicity is mediated by cytolytic molecules expressed by CTLs (e.g. granzyme and perforin). Upon encountering tumor-derived peptides presented on DCs, naive CD8+ T cells undergo cell expansion and differentiation, in a process mediated by co-stimulatory molecules (e.g. CD80, CD70 and 4-1BB) and DCderived cytokines (e.g. IL-12 and IL-15) (Zhang and Bevan, 2011). Following activation, most CD8+ T cells undergo programmed cell death to limit overactivation of the immune system. A minority of the activated and terminally differentiated CD8+ T cells (5-10%) become memory T cells (Zhang and Bevan, 2011). These memory antigen-specific T cells play an important role in preventing tumor relapse. Compared to naïve T cells, when the memory T cells encounter tumor antigen at a later time, they demonstrate enhanced recognition, robust antitumor responses and long-lasting memory (Raval et al., 2014). Therefore, induction of a memory response is a major goal of cancer immunotherapy strategies. Furthermore, CD8+ T cells express CD103 (i.e. integrin αE) which facilitates T cell adherence to E-cadherin, believed to play a role in tumor lysis (French et al., 2002).
CD8+ T cell differentiation is regulated by CD4+ T cells, also referred to as T helper (Th) cells (Pardoll and Topalian, 1998). Th cells influence the differentiation and expansion of tumor specific CTLs, enhance effector CTL priming and function, and induce memory T cell programming (Antony et al., 2005). Recapitulating CD4+ help in vivo is a major goal of immunotherapy. Recently, in a therapeutic cancer vaccine setting, CD4+ help was recapitulated by use of CD27 agonist antibodies plus immune checkpoint (PD-1) blockade (Ahrends et al., 2016; Pardoll, 2012). PD-1 expression is highly expressed on Tregs and is induced when T cells become activated. Blockade of PD-1 signaling enhances antitumor immunity by diminishing the number and activity of intratumoral regulatory T cells (Tregs), which are an immunosuppressive subtype of CD4+ cells.
CD4+ T cells are equally important as CD8+ cells. Upon engagement by an APC, a naïve CD4+ T cell differentiates into different kinds of effector cells, in a process mediated by cytokine signals in the microenvironment during activation. CD4+ T cells mainly differentiate into Th cells which, depending on the cytokine production, help activate other immune cells, including CTLs, B cells and NK cells. Th cells are further subdivided based on the type of immune response they generate, e.g. Th1, Th2 and Th17 (Majchrzak et al., 2016; Muranski and Restifo, 2009).
Th1 cells primarily produce interferon (IFN)-γ, tumor necrosis factor (TNF)-α, and interleukin (IL)-2; in contrast, Th2 cells produce IL-4, IL-5, IL-6, IL-10 and IL- 13. While Th1 cells tend to generate responses against intracellular parasites (e.g. viruses and bacteria), Th2 cells generate immune responses against extracellular parasites (e.g. helminths). Th2 cells can inhibit the generation of CTLs, through secretion of IL-4, which induces a CTL subpopulation called Tc2. Unlike Tc1, Tc2 cells have a low-killing capacity due to low expression of cytolytic molecules (Muranski and Restifo, 2009).
Th17 are a unique subset of Th cells that produce IL-17 (also called IL-17A) whose main role is to protect the host from extracellular pathogens, by facilitating recruitment of neutrophils and macrophages (Dong, 2008; Harrington et al., 2005; Ivanov et al., 2006). Th17 cells also play a role in the pathogenesis of autoimmune and pro-inflammatory disorders. Th17 cells express RAR-related orphan receptor gamma t (RORγt), a “master-regulator” transcription factor. RORγt deficiency diminishes Th17 activity (Dong, 2008). Recently, human Th1/Th17 cells could be induced by culture in IL-2 and IL-21 (an autocrine growth factor for Th17) (Korn et al., 2009; Phan-Lai et al., 2015). Moreover, gene expression of Th17 transcription factor, RORc, and IFN-γ regulatory factor, IRF-4, was significantly enhanced after IL-2/IL-21 culture. Although it is still unclear, IL-17 producing Th cells may represent a new type of effector T cells for ACT, with potentially greater proliferation and self-renewal potential in vivo due to reported stem-like properties (Muranski et al., 2011).
Unfortunately, CD4+ T cells can also differentiate into Tregs, when they recognize antigen in a cytokine milieu where transforming growth factor-β (TGF-β) and IL-10 signals are predominant (Roncarolo et al., 2001). Tregs are negative regulators of immunity; they are frequently found in the tumor microenvironment, where they inhibit adaptive and innate antitumor immunity. Tregs inhibit CTLs through secretion of IL-10 and/or TGF-β (Roncarolo et al., 2001). Through the constitutive expression of CD25 (i.e. IL-2Rα), Tregs also complete with CTLs (Kastenmuller et al., 2011). Many cancer immunotherapy strategies, therefore, target the inhibition of Treg function to promote tumor immunity.
As mentioned above, there are multiples types of T cells from which to select for use in ACT, including CD4+ (Th1, Th2, Th17) and CD8+ (Tc1, Tc2, Tc17) (Majchrzak et al., 2016). Other types of T cells that have been investigated for immunotherapy are natural killer T (NKT) cells and gamma delta T cells (γδ Tc) (Fujii et al., 2013; Silva-Santos et al., 2015). NKT cells are a heterogeneous group of T cells that have properties of both natural killer (NK) cells and T cells; they recognize CD1d molecule and constitute only approximately 0.1% of all peripheral blood T cells. Gamma delta (γδ) T cells are T cells which express a unique TCR comprised of a γ-chain and a δ-chain. Like NKT cells, gamma delta T cells are found in low abundance in the body.
ACT studies have mainly focused on autologous tumor- specific CD8+ T cells. Antigen-specific CD8+ T cells should theoretically recognize pMHCI complexes on tumor cells with high avidity, express high levels of cytolytic molecules and be able to penetrate the tumor bed, despite immunomodulatory mechanisms at the tumor microenvironment. In clinical trials, infusion of melanoma antigen-specific CTLs mediated rejection of even bulky metastatic tumors (Dudley et al., 2005; Yee et al., 2002). However, there are also a number of studies exploring CD4+ T cells (Disis et al., 2014; Knutson and Disis, 2004; Majchrzak et al., 2016; Phan-Lai et al., 2015). CD4+ T cells may have an advantage over CD8 T cells simply since they are capable of recognizing tumor antigens via both MHCI and MHCII processing pathways, not just MHCI (Muranski and Restifo, 2009). Moreover, In recent years, it has been demonstrated that CD4+ T cells can also contribute to the eradication of cancers, such as melanoma, by actively killing tumor cells and activating macrophages (Quezada et al., 2010).
T cell receptors
Via their TCRs, CTLs recognize short antigenic peptides presented by the major histocompatibility complex (pMHC) on the surface of professional APCs, such as DCs, or on the surface of infected or malignant cells (Hebeisen et al., 2015). T cell triggering or activation depends critically on TCR binding (avidity) to cognate pMHC. The avidity of the structural interaction impacts the efficiency of T cell-mediated immunity; strong TCR-pMHC interactions confer superior T cell activation, while weak interactions lead to suboptimal T cell activation. T cell activation requires 2 signals: binding of the TCR to the pMHC complex (signal 1) and simultaneous engagement of positive costimulatory molecules (signal 2; e.g. B7 on an APC and CD28 on T cells). After activation, T cells express coinhibitory receptors, e.g. CTLA-4 and PD-1 (Shi et al., 2016). The co-inhibitors turn down immune responses and are a mechanism used by tumors to suppress the host’s natural antitumor immunity. Understanding immune tolerance and immune inhibitory networks are important for ACT, as is knowing which immunemodulating agent(s) would augment ACT.
There are mechanisms that prevent TCRs from identifying and reacting with host tissues. Immunologic self-tolerance can occur when a T cell receptor recognizes its cognate antigen (signal 1 only), in the absence of appropriate co-stimulation. An example occurs when CD8+ T cells interact with tumor cells, which do not express appropriate costimulatory molecules, leading to T cell anergy. Furthermore, when activated T cells receive excessive stimulation (via signal 1 with or without signal 2), this leads to T cell exhaustion; T cells become incapable of further activation despite exposure to antigen in a pro-stimulatory environment. Moreover, since ‘self’ tumor antigen-reactive T cells are mostly eliminated during central and peripheral deletion mechanisms, there is a rarity of high avidity TCRs to tumor antigens which, in essence, are ‘self’ antigens. Therefore, the identification of TCRs of optimal avidity for tumor antigens is an important component for ACT to potentially predict which T cells may be more effective in the clinic. To identify the most functionally relevant T cells for ACT, strategies and technologies have been developed to assess TCRpMHC interaction kinetics, and are reviewed in (Hebeisen et al., 2015).
Moreover, for effective cytolysis of tumor cells, effector T cells must interact with target antigens on tumor cells. However, the antigens to be targeted for optimal antitumor immunity still need to be investigated. Advancement in technology, structural proteomics and information from sequencing data have enabled more efficient screening and validation of tumor-associated antigens (TAAs) that may be targeted by T cells (Battaglia and Muhitch, 2016). For instance, the understanding of the structure of tumor antigen p53, in ovarian cancer, led to the design of an effective p53 peptide inhibitor (Soragni et al., 2016). Structural understanding of TAAs recognized by effector T cells and design of therapies based on the TCR-pMHC interaction could be useful in combination with ACT. Molecular identification of human TAAs and their surface interaction with T cells should benefit targeted antigen- specific immunotherapy for cancer (Battaglia and Muhitch, 2016).
Sources of T cells in act
For ACT, multiple kinds of T cells have been evaluated. These include tumor-infiltrating T cells (TILs) that have been expanded and activated ex vivo after isolation from tumor (Dudley et al., 2005; Meng et al., 2016a), T cell receptor (TCR)-modified T cells that are modified ex vivo to express a particular TCR for more specific tumor recognition (Spear et al., 2016), and chimeric antigen receptor (CAR)-engineered T cells (where the receptor is engineered by fusing an antibody (Ab) with the TCR to improve tumor recognition and interaction) (Brown et al., 2015). The above manipulations are part of an effort to improve the clinical efficacy of ACT. Of the list of approaches above, the two most common and least labor-intensive, and which will be discussed in this review, are: 1) TILs extracted from tumor, and 2) peripheral blood monocytes (PBMC) isolated from patients.
For TIL-based ACT, TILs are extracted from the patient’s tumor, expanded in vivo with IL-2 and reinfused in the patients (Dudley et al., 2005). While TILs represent a suitable option for T cells in ACT, their major disadvantages include: adverse side effects (including pneumonia and other respiratory complications), limited quantity that can be extracted even from bulky tumors, and the limitation of the approach to solid tumors (from which the TILs are harvested). In melanoma disease, use of autologous TILs demonstrated a 50% response rate (Dudley et al., 2005). Therefore, another approach is to harvest endogenous peripheral tumor-specific T cells (via several apheresis). The PBMC develop into tumor-specific T cells during ex vivo expansion, in a process which is much more labor intensive- from start to finish- than that for TIL expansion. The endogenous T cells in peripheral blood are sometimes primed in vivo first by several rounds of vaccination with tumor antigen before the PBMC isolation process (Dang et al., 2007). The primed T cells are then isolated from PBMC, expanded during ex vivo culture, and re-infused into the patient.
Act trials: CD8+ versus CD4+
ACT has been studied in many cancers, including melanoma, ovarian cancer and renal cell carcinoma (TILs), breast cancer (PBMCs), as well as B-cell lymphoma, glioblastoma and hematological malignancies (CAR-Tc); overall, ACT strategy (via CD8+ TILs) has mostly been effective for melanoma (Yang and Rosenberg, 2016). One of the first immunotherapy trials, conducted in the 1990s, investigated the efficacy of infusing high dose IL-2 (a prototypic T cell growth factor) to stimulate endogenous T cells against solid tumors, e.g. renal cell carcinoma (McDermott, 2007). Not only did high dose IL-2 induce complete responses in renal cell carcinoma patients (up to 10%) but it was also effective in eliciting responses in metastatic melanoma patients (Atkins et al., 1999; Klapper et al., 2008).
Infused T cells require maintenance of homeostatic proliferation. For this, patients are typically pretreated with lymphodepleting agents (e.g. cyclophosphamide and fludarabine) and may also be treated with total body irradiation. Patients on this regimen continue to receive IL-2 as part of their maintenance therapy. Lymphodepletion enhances ACT by activating innate immunity via microbes released in the gut from injury induced by radiation. Lipopolysaccharide (LPS) is a microbial compound and toll-like receptor (TLR) agonist, which mimics the responses induced by total body irradiation (TBI). LPS has been evaluated as a strategic use in ACT to boost T cell responses.
Recently, it was shown that host preconditioning may not be as critical as thought. Toll-like receptor agonist therapy, e.g. LPS therapy, was sufficient to augment reactivity of tumor-specific CTLs in ACT in a melanoma mouse model (Nelson et al., 2016). Exogenous administration of LPS enhanced the ability of adoptively transferred CD8+ T cells to destroy tumor. The timing of administration was critical; administering LPS during or one day prior to ACT therapy did not augment T cell function whereas administering LPS after ACT augmented antitumor efficacy of ACT, indicating expansion of adoptively transferred tumorspecific CD8+ T cells. Other TLR agonists, including TLR4 agonist, monophosphoryl lipid A (MPL), and TLR9 agonist, CpG Oligodeoxynucleotide (CpG ODN), also augmented infused CD8+ T cell activity. In mice depleted of host CD4+ T cells and treated with CD8+ ACT and LPS, TBI treatment was not needed for antitumor response. Therefore, the use of TLR agonists, and appropriate timing, may circumvent preconditioning. In designing next generation ACT for cancer, lymphodepletion may not be an absolute requirement and thus, side effects associated with lymphodepletion could be minimized for the patient.
It has become evident that the barriers to ACT efficacy, including peripheral tolerance and tumor-associated immunosuppression at the tumor microenvironment, limit the effectiveness of ex vivo generated antigenspecific CTLs (i.e. CD8+ T cells). While CD8+ T cells have been the main focus of ACT trials, for their robust tumor cell killing ability (Restifo et al., 2012; Yee et al., 2002), it is also important to evaluate Th cells as a strategic alternative since they play a pivotal role in augmenting CTL responsiveness. Next-generation cancer vaccines, including Th epitopes, have been used to prime Th cells in vivo prior to extraction for ex vivo expansion. The identification of appropriate Th epitopes of tumor antigens has become an important alternative strategy for targeted immune induction and help. Other barriers to the success of ACT include tumor stroma associated immunosuppression (Phan and Disis, 2008). Cellular components in tumor stroma, including cancer-associated fibroblasts (CAFs), myeloid-derived suppressor cells (MDSC), and tumorassociated macrophages (TAMs) inhibit T cell function (Arina and Bronte, 2015; Arina et al., 2014; Phan- Lai et al., 2013). However, reports have shown the ability of adoptively transferred T cells to eradicate well-established tumors in mice, despite the abundant presence of MDSC and TAMs, which exert strong immunosuppression in the tumor stroma (Arina and Bronte, 2015).
Improving potency of T cells via ex vivo expansion
During ex vivo culture of antitumor T cells, one strategy to optimize the potency of tumor-specific T cells is to expand T cells in appropriate cytokine growth factors which optimally stimulate antitumor T cell immunity. Studies have already shown that tumor antigen- specific T cells, e.g. HER2-specific T cells, derived from HER2+ breast cancer patients after expansion with IL-2/IL-12, can induce partial tumor regression (Knutson and Disis, 2004). IL-2/IL-15 have both been beneficial and tested in vaccine and ACT design (Dang et al., 2007; Knutson and Disis, 2004; Waldmann, 2006). Comparative investigations on the role of cytokine growth factors (used in combination with IL-2) to induce optimal T cell immunity are limited and unclear. Recent data suggest that IL2/IL21 may be optimal cytokines to mediate robust T cell immunity and elicit tumor regression (Phan-Lai et al., 2015). In a TgMMTV-neu murine mammary tumor model, infusion of neu p98-specific T cell lines derived from IL2/IL21 culture led to a different cytokine secretion pattern, including significantly higher production of TNF-α and IL17 (p<0.5). Following infusion of IL2/IL21 cultured T cells, tumor-infiltrating CD8+ cells were significantly increased (Phan-Lai et al., 2015). Moreover, results with IL2/IL21 cultured human antigen- specific T cells were similar to those of murine T cells. Thus, cytokine growth factors used in ex vivo expansion of vaccine-primed CD4+ T cells can significantly improve antitumor immune responses. The choice of cytokine growth factor, therefore, is critical (Meng et al., 2016a; Phan-Lai et al., 2015). Specifically, IL2/IL21 (perhaps also in combination with other cytokines) can give rise to Th1/Th17 polyfunctional T cells and induce durable responses in mice and patients (Phan-Lai et al., 2015). Enhancement of CD8+ T cell infiltration was associated with use of those cytokines.
Conclusions
Cancer immunotherapy attempts to harness the specificity and natural ability of the immune system to recognize and eliminate tumors. Robust, specific and durable antitumor immunity depends on T cells. Adoptive T cell therapy using autologous tumor antigen specific T cells have been studied in a variety of cancers, although the best tumor responses have been observed in metastatic melanoma. Adoptive immunotherapy, via CD8+ or CD4+ T cells, has shown antitumor responses. CD4+ T cells represent an ideal cell choice for ACT since they are capable of differentiating into Th1, Th2, or Th17, and providing cytokine “help” to CD8+ cells and macrophages in the tumor. IL-17 producing CD4+ T cells are a new subset of CD4+ T cells to be evaluated in ACT models. The activation and magnitude of the T cell response depend on the cytokine milieu during antigen recognition. Therefore, important considerations for ACT include: 1) the type of T cells, 2) the source of T cells, 3) the strength of the TCR/pMHC interaction, and 4) the ex vivo cytokine culture conditions for T cells.
References
Ahrends, T., Babala, N., Xiao, Y., Yagita, H., van Eenennaam, H., and Borst, J. (2016). CD27 agonism plus PD-1 blockade recapitulates CD4 + T cell help in therapeutic anti-cancer vaccination. Cancer research.
Antonioli, L., Yegutkin, G.G., Pacher, P., Blandizzi, C., and Hasko, G. (2016). Anti-CD73 in cancer immunotherapy: awakening new opportunities. Trends in cancer 2, 95–109.
Antony, P.A., Piccirillo, C.A., Akpinarli, A., Finkelstein, S.E., Speiss, P.J., Surman, D.R., Palmer, D.C., Chan, C.C., Klebanoff, C.A., Overwijk, W.W., et al. (2005). CD8 + T cell immunity against a tumor/self-antigen is augmented by CD4 + T helper cells and hindered by naturally occurring T regulatory cells. J Immunol 174, 2591–2601.
Arina, A., and Bronte, V. (2015). Myeloid-derived suppressor cell impact on endogenous and adoptively transferred T cells. Current pinion in immunology 33, 120–125.
Arina, A., Schreiber, K., Binder, D.C., Karrison, T.G., Liu, R.B., and Schreiber, H. (2014). Adoptively transferred immune T cells eradicate established tumors despite cancer-induced immune suppression. J Immunol 192, 1286–1293.
Atkins, M.B., Lotze, M.T., Dutcher, J.P., Fisher, R.I., Weiss, G., Margolin, K., Abrams, J., Sznol, M., Parkinson, D., Hawkins, M., et al. (1999). High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 17, 2105–2116.
Battaglia, S., and Muhitch, J.B. (2016). Unmasking targets of antitumor immunity via high-throughput antigen profiling. Curr Opin Biotechnol 42, 92–97.
Brown, C.E., Badie, B., Barish, M.E., Weng, L., Ostberg, J.R., Chang, W.C., Naranjo, A., Starr, R., Wagner, J., Wright, C., et al. (2015). Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8 + T Cells in Patients with Recurrent Glioblastoma. Clinical cancer research: an official journal of the American Association for Cancer Research 21, 4062–4072.
Cherryholmes, G.A., Stanton, S.E., and Disis, M.L. (2015). Current methods of epitope identification for cancer vaccine design. Vaccine 33, 7408–7414.
Dang, Y., Knutson, K.L., Goodell, V., dela Rosa, C., Salazar, L.G., Higgins, D., Childs, J., and Disis, M.L. (2007). Tumor antigen-specific T-cell expansion is greatly facilitated by in vivo priming. Clinical cancer research: an official journal of the American Association for Cancer Research 13, 1883–1891.
Disis, M.L., Dang, Y., Coveler, A.L., Marzbani, E., Kou, Z.C., Childs, J.S., Fintak, P., Higgins, D.M., Reichow, J., Waisman, J., et al. (2014). HER-2/neu vaccine-primed autologous T-cell infusions for the treatment of advanced stage HER-2/neu expressing cancers. Cancer immunology, immunotherapy: CII 63, 101–109.
Dong, C. (2008). TH17 cells in development: an updated view of their molecular identity and genetic programming. Nature reviews Immunology 8, 337–348.
Dudley, M.E., Wunderlich, J.R., Yang, J.C., Sherry, R.M., Topalian, S.L., Restifo, N.P., Royal, R.E., Kammula, U., White, D.E., Mavroukakis, S.A., et al. (2005). Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 23, 2346–2357.
Dunn, G.P., Old, L.J., and Schreiber, R.D. (2004a). The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21, 137–148.
Dunn, G.P., Old, L.J., and Schreiber, R.D. (2004b). The three Es of cancer immunoediting. Annual review of immunology 22, 329–360.
French, J.J., Cresswell, J., Wong, W.K., Seymour, K., Charnley, R.M., and Kirby, J.A. (2002). T cell adhesion and cytolysis of pancreatic cancer cells: a role for E-cadherin in immunotherapy? British journal of cancer 87, 1034–1041.
Fujii, S., Shimizu, K., Okamoto, Y., Kunii, N., Nakayama, T., Motohashi, S., and Taniguchi, M. (2013). NKT cells as an ideal antitumor immunotherapeutic. Frontiers in immunology 4, 409.
Guo, C., Manjili, M.H., Subjeck, J.R., Sarkar, D., Fisher, P.B., and Wang, X.Y. (2013). Therapeutic cancer vaccines: past, present, and future. Advances in cancer research 119, 421–475.
Harrington, L.E., Hatton, R.D., Mangan, P.R., Turner, H., Murphy, T.L., Murphy, K.M., and Weaver, C.T. (2005). Interleukin 17- producing CD4 + effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nature immunology 6, 1123–1132.
Hebeisen, M., Allard, M., Gannon, P.O., Schmidt, J., Speiser, D.E., and Rufer, N. (2015). Identifying Individual T Cell Receptors of Optimal Avidity for Tumor Antigens. Frontiers in immunology 6, 582.
Ivanov, II., McKenzie, B.S., Zhou, L., Tadokoro, C.E., Lepelley, A., Lafaille, J.J., Cua, D.J., and Littman, D.R. (2006). The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17 + T helper cells. Cell 126, 1121–1133.
Kakavand, H., Wilmott, J.S., Long, G.V., and Scolyer, R.A. (2016). Targeted therapies and immune checkpoint inhibitors in the treatment of metastatic melanoma patients: a guide and update for pathologists. Pathology 48, 194–202.
Kastenmuller, W., Gasteiger, G., Subramanian, N., Sparwasser, T., Busch, D.H., Belkaid, Y., Drexler, I., and Germain, R.N. (2011). Regulatory T cells selectively control CD8 + T cell effector pool size via IL-2 restriction. J Immunol 187, 3186–3197.
Klapper, J.A., Downey, S.G., Smith, F.O., Yang, J.C., Hughes, M.S., Kammula, U.S., Sherry, R.M., Royal, R.E., Steinberg, S.M., and Rosenberg, S. (2008). High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma: a retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer 113, 293–301.
Knutson, K.L., and Disis, M.L. (2004). IL-12 enhances the generation of tumor antigen-specific Th1 CD4 T cells during ex vivo expansion. Clin Exp Immunol 135, 322–329.
Knutson, K.L., and Mittendorf, E.A. (2015). Cancer vaccines in the new era of cancer immunotherapy. Vaccine 33, 7376.
Korn, T., Bettelli, E., Oukka, M., and Kuchroo, V.K. (2009). IL-17 and Th17 Cells. Annual review of immunology 27, 485–517.
Majchrzak, K., Nelson, M.H., Bailey, S.R., Bowers, J.S., Yu, X.Z., Rubinstein, M.P., Himes, R.A., and Paulos, C.M. (2016). Exploiting IL- 17-producing CD4(+) and CD8(+) T cells to improve cancer immunotherapy in the clinic. Cancer immunology, immunotherapy: CII 65, 247–259.
McDermott, D.F. (2007). Update on the application of interleukin-2 in the treatment of renal cell carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research 13, 716 s-720 s.
Meng, Q., Liu, Z., Rangelova, E., Poiret, T., Ambati, A., Rane, L., Xie, S., Verbeke, C., Dodoo, E., Del Chiaro, M., et al. (2016a). Expansion of Tumor-reactive T Cells From Patients With Pancreatic Cancer. J Immunother 39, 81–89.
Meng, Y., Zheng, L., Yang, Y., Wang, H., Dong, J., Wang, C., Zhang, Y., Yu, X., Wang, L., Xia, T., et al. (2016b). A monoclonal antibody targeting ErbB2 domain III inhibits ErbB2 signaling and suppresses the growth of ErbB2-overexpressing breast tumors. Oncogenesis 5, e211.
Muranski, P., Borman, Z.A., Kerkar, S.P., Klebanoff, C.A., Ji, Y., Sanchez-Perez, L., Sukumar, M., Reger, R.N., Yu, Z., Kern, S.J., et al. (2011). Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity 35, 972–985.
Muranski, P., and Restifo, N.P. (2009). Adoptive immunotherapy of cancer using CD4(+) T cells. Current opinion in immunology 21, 200–208.
Nelson, M.H., Bowers, J.S., Bailey, S.R., Diven, M.A., Fugle, C.W., Kaiser, A.D., Wrzesinski, C., Liu, B., Restifo, N.P., and Paulos, C.M. (2016). Toll-like receptor agonist therapy can profoundly augment the antitumor activity of adoptively transferred CD8(+) T cells without host preconditioning. J Immunother Cancer 4, 6.
Palucka, K., and Banchereau, J. (2013). Cancer immunotherapy via dendritic cells. Nature reviews Cancer 12, 265–277.
Pardoll, D.M. (2012). The blockade of immune checkpoints in cancer immunotherapy. Nature reviews Cancer 12, 252–264.
Pardoll, D.M., and Topalian, S.L. (1998). The role of CD4 + T cell responses in antitumor immunity. Current opinion in immunology 10, 588–594.
Phan, V., and Disis, M.L. (2008). Tumor stromal barriers to the success of adoptive T cell therapy. Cancer immunology, munotherapy: CII 57, 281–283.
Phan-Lai, V., Dang, Y., Gad, E., Childs, J., and Disis, M.L. (2015). The Antitumor Efficacy of IL2/IL21-Cultured Polyfunctional Neu-Specific T Cells Is TNFalpha/IL17 Dependent. Clinical cancer research: an official journal of the American Association for Cancer Research.
Phan-Lai, V., Florczyk, S.J., Kievit, F.M., Wang, K., Gad, E., Disis, M.L., and Zhang, M. (2013). Three-dimensional scaffolds to evaluate tumor associated fibroblast-mediated suppression of breast tumor specific T cells. Biomacromolecules 14, 1330–1337.
Quezada, S.A., Simpson, T.R., Peggs, K.S., Merghoub, T., Vider, J., Fan, X., Blasberg, R., Yagita, H., Muranski, P., Antony, P.A., et al. (2010). Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. The Journal of experimental medicine 207, 637–650.
Raval, R.R., Sharabi, A.B., Walker, A.J., Drake, C.G., and Sharma, P. (2014). Tumor immunology and cancer immunotherapy: summary of the 2013 SITC primer. J Immunother Cancer 2, 14.
Restifo, N.P., Dudley, M.E., and Rosenberg, S.A. (2012). Adoptive immunotherapy for cancer: harnessing the T cell response. Nature reviews Immunology 12, 269–281.
Roncarolo, M.G., Bacchetta, R., Bordignon, C., Narula, S., and Levings, M.K. (2001). Type 1 T regulatory cells. Immunological reviews 182, 68–79.
Shi, L., Chen, L., Wu, C., Zhu, Y., Xu, B., Zheng, X., Sun, M., Wen, W., Dai, X., Yang, M., et al. (2016). PD-1 Blockade Boosts Radiofrequency Ablation-Elicited Adaptive Immune Responses against Tumor. Clinical cancer research: an official journal of the American Association for Cancer Research 22, 1173–1184.
Silva-Santos, B., Serre, K., and Norell, H. (2015). gammadelta T cells in cancer. Nature reviews Immunology 15, 683–691.
Soragni, A., Janzen, D.M., Johnson, L.M., Lindgren, A.G., Thai-Quynh Nguyen, A., Tiourin, E., Soriaga, A.B., Lu, J., Jiang, L., Faull, K.F., et al. (2016). A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer cell 29, 90–103.
Spear, T.T., Callender, G.G., Roszkowski, J.J., Moxley, K.M., Simms, P.E., Foley, K.C., Murray, D.C., Scurti, G.M., Li, M., Thomas, J.T., et al. (2016). TCR gene-modified T cells can efficiently treat established hepatitis C-associated hepatocellular carcinoma tumors. Cancer immunology, immunotherapy: CII 65, 293–304.
Tang, H., Wang, Y., Chlewicki, L.K., Zhang, Y., Guo, J., Liang, W., Wang, J., Wang, X., and Fu, Y.X. (2016). Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer cell 29, 285–296.
Waldmann, T.A. (2006). The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nature reviews Immunology 6, 595–601.
Yang, J.C., and Rosenberg, S.A. (2016). Adoptive T-Cell Therapy for Cancer. Advances in immunology 130, 279–294.
Yee, C., Thompson, J.A., Byrd, D., Riddell, S.R., Roche, P., Celis, E., and Greenberg, P.D. (2002). Adoptive T cell therapy using antigenspecific CD8 + T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proceedings of the National Academy of Sciences of the United States of America 99, 16168–16173.
Zhang, N., and Bevan, M.J. (2011). CD8(+) T cells: foot soldiers of the immune system. Immunity 35, 161–168.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare they have no competing interests.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License (CC-BY 4.0) which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Cite this article as:
Phan-Lai, V. (2016). Adoptive immunotherapy via CD4+ versus CD8+ T cells. Biomedical Research And Therapy, 3(4), 588-595.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Phan-Lai, V. Adoptive immunotherapy via CD4+ versus CD8+ T cells. Biomed Res Ther 3, 16 (2016). https://doi.org/10.7603/s40730-016-0016-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.7603/s40730-016-0016-6