
Abstract
Background
Multiple hereditary exostoses (MHE) is characterized by multiple benign projections of bone capped by cartilage, most numerous in metaphyses of long bones. HME are usually inherited in autosomal dominant mode, chief genes EXT1 and EXT2.
Methods
Two MHE patients were identified from clinic and enrolled in genetic study, complete coding regions of EXT1 and EXT2, including intron/exon boundaries, sequenced via DNA samples drawn from participants.
Results
DNA sequencing revealed mutant EXT1 gene in both cases, within which frame-shift mutation c.447delC (p.Ser149fsX156) in exon1 and nonsense mutation c.2034T>G (p.Tyr678X) in exon10, emerged. Neither mutation was detected in control group.
Conclusions
Our results extended the spectrum of EXT1 mutations, revealing similar incidence of EXT1 and EXT2 in Taiwanese MHE patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1. Introduction
Multiple hereditary exostoses (MHE; also known as multiple osteochondromas, MIM#133700, 133701), most frequent human benign bone tumors, are characterized by multiple outgrowth of bone capped by cartilage, mostly in the metaphyses but also occurring on diaphyses of long bones. Flat bones, vertebrae, and ribs are also affected, skull rarely involved [1]. Onset is variable, from early childhood to puberty, stopping increase until closure of growth plate [2]; prevalence is estimated at 1/50,000 among the European population [1].
MHE may be asymptomatic, but generally clinical presentation is heterogeneous. Because exostoses come from the growth plate, they may consist of deformities and various levels of functional limitation (sensory or motor deficits). Complications, such as compression of nerves and blood vessels, pain caused by pressure on neighboring tissue, and short stature, are also common [3]. Most severe secondary complication is malignant transformation into secondary peripheral chondrosarcoma (in 0.5-5.0% of cases) [4]. Patients with milder forms require no active therapy; physical therapy, pain management, and surgery are common practice in MHE cases, clinical outcome less than beneficial at times [5].
MHE, autosomal dominant disease, links with exostosin 1 (EXT1) and 2 (EXT2) genes. EXT1, assigned to chromosome 8q24.11-q24.13, comprises 11 exons spanning less than 350kb and encoding a polypeptide of 746 amino acids [6, 7]. EXT2 maps to chromosome 11p11-p11.2, consists of 16 exons, and spans almost 108 kb. Alternative splicing allows three transcript variants of mRNA produced, a major one (transcript variant 2) encoding protein of 718 amino acids [8-10]. Both encode ubiquitously expressed Type-II transmembrane glycoproteins [6, 8, 9] that catalyze elongation of the heparan sulphate- glycosaminoglycan chain of matrix proteoglycans [11].
According to records from the Multiple Osteochondromas Mutation Database (MOdb) (http://medgen.ua.ac.be/LOVDv.2.0/), as well as recent publications, over 440 mutations in EXT1 and 230 in EXT2 have been described [12-16]. Depending on race, for all MHE cases, about 56-78% mutation is detected in EXT1 versus 21-44% for EXT2 [17]. We sequenced DNA of two Taiwanese patients to determine mutations in EXT1 or EXT2 genes, also collecting published genetic analysis of Taiwanese MHE patients, then briefly summarized a mutation profile of the Taiwanese population.
2. Patients and Methods
2.1. Patients
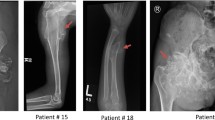
Patient #1, six-year-old girl, firstborn of nonconsanguineous healthy parents, was brought to our genetic outpatient clinic because of progressive deformation over both forearms for a period of time. Clinical examination revealed shortened forearms with bony projection over distal radial and ulnar sides of right forearm (Figure 1A). Radiologic image disclosed bowing of radius and ulnar bones of both forearms with bony exostosis over distal ulnar bones (Figure 1B).


Photograph and radiograph of Patient #1. (A) Both forearms and (B) X-ray image. Note bowing of radius and ulna with bony exostosis over distal ulna.
Patient #2, 23-year-old female, presented typical manifestations of multiple long bones exostosis as index case and was referred by Department of Genetics of National Taiwan University Hospital (Taipei, Taiwan) for further genetic test. Importantly, the propositus’ mother and younger sister and brother had similar clinical phenotypes.
Prior to genetic analysis, informed consent (as per national law) was obtained from adult patients or parents of each study subject.
2.2. DNA preparation and sequencing
Genomic DNA extraction from each participant's peripheral blood leukocytes used MagNA Pure LC DNA Isolation Kit (Roche, Mannheim, Germany). Complete EXT1 and EXT2 coding regions were amplified according to the protocol published by Plilippe et al. (1997) and Wuyts et al. (1998) [18,19]. In addition, some of the primers were redesigned in this study (Table 1). PCR products were purified from the agarose gel using QIAEX II (Qiagen, Hilden, Germany) and then used for direct sequencing to detect gene mutations. The direct sequencing process was performed using BigDye 3.1 Terminator cycle sequencing kit (Applied Biosystems, Forest City, CA) with ABI 3100 Genetic Analyzer (Applied Biosystems, Forest City, CA).
To determine carrier-rate of novel mutations detected in Taiwanese population, EXT1 gene profile of 100 matched controls was analyzed by procedure mentioned above, reference sequence and base-pair numbers of EXT1 and EXT2 obtained from GenBank by accession numbers NM_000127 and NM_207122, respectively.
3. Results
To identify possible exonic mutations in EXT1 and EXT2 causing MHE, entire coding sequence on 26 DNA fragments, each covering an exon and its flanking regions, was amplified. Analysis of EXT1 and EXT2 in both patients identified one frame-shift and one nonsense mutation in EXT1. To our knowledge, neither has been described in prior article (Figure 2).
In Patient #1, one base C at nucleotide 447 was deleted (c.447delC) in exon1, causing protein translation frame-shift after codon 149, and early terminated at codon 156 (p.Ser149fsX156). This mutation did not appear in her parents’ EXT1 gene and should be spontaneous mutation. Patient #2 showed nonsense mutation c.2034T>G (p.Tyr678X) located in exon10, inherited from her MHE mother. We detected this mutation site in other family members, confirming this mutation as cosegregated with disease phenotype in those afflicted with MHE. Neither mutation mentioned above was detected in 100 healthy volunteers enrolled in normal control group.
This study found another single polymorphism in EXT1, located at nucleotide 1761 (p.Glu587Glu), substitution of G to A. This site was described previously in another population [18].


Sequences of novel mutations observed from patients’ EXT1 gene. Arrows indicate mutation sites. (A) A one-base C deleted at nucleotide 447 (c.447delC), (B) G to A substitution at nucleotide 2034 (c.2034T>G) resulting in p.Tyr678X
4. Discussion
EXT1 and EXT2 are ubiquitously expressed tumor suppressors of the EXT family, which includes three EXT-like genes (EXTL1, EXTL2, and EXTL3) [20-22]. To date, only mutant EXT1 and EXT2 are involved with MHE, no MHE cases have been identified with mutant EXTL1-3. All members of the EXT gene family share homologous domains: [1] exostosin located in N-terminal region and [2] glycosyltransferase in C-terminal region. The latter is involved in the biosynthesis of heparin sulfate at heparin sulfate proteoglycans [23]. These heparin sulfate proteoglycans play major roles in cell growth/differentiation signal pathways and interact with diffusion of signaling molecules like Indian Hedgehog, an important regulator of chondrocyte proliferation and differentiation in growth plate [24]. These results reflect cartilage growth regulatory function of EXT1/ EXT 2 genes [25.26]
Our study found both mutations in EXT1. The first was one-base deletion and caused early termination at codon 156 (p.Ser149fsX156), in which neither domain can be translated. Another appeared at c.2034T>G, causing truncated proteins (p.Tyr678X) with glycosyltransferase domain incomplete. Both mutated proteins were in premature form, their structures unstable and soon degraded in the cytoplasm.
EXT1/EXT2 germline mutations have been detected in most MHE cases; EXT1 mutates more often than EXT2, with variable prevalence among populations [17, 27]. Most are frame-shift, nonsense, and splice-site mutations, responsible for premature termination of translation, inducing rapid inactivation and degradation with nearly complete loss of their function [17].

Schematic profile of mutations on EXT1 and EXT2 genes in Taiwanese patients with multiple hereditary exostoses. GenBank accession numbers of mRNA sequences: EXT1: NM_000127; EXT2-v1 (isoform 1): NM_000401, EXT2-v2 (isoform 2-Reference): NM_207122, EXT2-v3 (isoform 3): NM_001178083.
We collected Taiwanese MHE genetic analysis results from literature and were summary in Figure 3 [28-31]. Total thirteen cases were included, nine were familial cases, and four were sporadic cases (31%), higher than previous study estimated (10%) [17]. Interestingly, in these results, six cases had mutant EXT1, seven mutant EXT2 (54%). This result implies that the incidence of EXT2 in Taiwanese MHE is equal or slightly high than EXT1, which differs from Western populations [12, 17]. According to previous literature, mutations observed in coding region of EXT1 and EXT2 that generate frame-shift or nonsense change are dominant [17, 32]. Consider mutation type found in Taiwanese MHE cases: nonsense mutation is major (7/12, 58%), frame-shift type had three (25%), splicing site and missense mutation had one each (8.3%). These proportions concurred with other population studies [12, 13, 15, 17].
To date, we had completed nine MHE genetic studies in Taiwan; all found mutant EXT1 or EXT2. Yet if mutation type is large fragment deletion/duplication, translocation/inversions, or epigenetic variants, it would not be detected by PCR-directed DNA sequencing [33]. According to previous literature, if MHE case could not detect point mutation, the second most probable type is large fragment deletion [12-17]. To augment detection rate, real-time quantitative PCR, multiplex ligation-dependent probe amplification (MLPA), fluorescence in situ hybridization (FISH) and DNA microarray can serve for large fragment deletion/duplication mutation analysis [12,13]. If other candidate genes (EXTL-1-3) were considered with MHE, linkage analysis should be done first to narrow down the possible gene location.
In conclusion, we identified two novel mutations in EXT1 from two MHE probands of unrelated Taiwanese families and extended the mutation EXT1 spectrum. Our patients showed similar incidence of EXT1 and EXT2, lower EXT1 than among Westerners. Collection mutation data in both genes could help diagnosis and genetic counseling for MHE patients and their families
Acknowledgements
Study was funded by grant DMR101-095 from China Medical University Hospital.
References
Schmale GA, Conrad EU 3rd, Raskind WH. The natural history of hereditary multiple exostoses. J Bone Joint Surg Am 1994;76:986–92.
Wicklund CL, Pauli RM, Johnston D, Hecht JT. Natural history study of hereditary multiple exostoses. Am J Med Genet 1995;55: 43–6.
Hennekam RC. Hereditary multiple exostoses. J Med Genet 1991;28:262–6.
Bovée JV. Multiple osteochondromas. Orphanet J Rare Dis 2008;13:3.
Akita S, Murase T, Yonenobu K, Shimada K, Masada K, Yoshikawa H. Long-term results of surgery for forearm deformities in patients with multiple cartilaginous exostoses. J Bone Joint Surg Am 2007;89:1993–9.
Ahn J, Lüdecke HJ, Lindow S, Horton WA, Lee B, Wagner MJ. Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1). Nat Genet 1995;11:137–43.
Lüdecke HJ, Ahn J, Lin X, Hill A, Wagner MJ, Schomburg L. Genomic organization and promoter structure of the human EXT1 gene. Genomics 1997;40:351–4.
Stickens D, Clines G, Burbee D, Ramos P, Thomas S, Hogue D, et al. The EXT2 multiple exostoses gene defines a family of putative tumour suppressor genes. Nat Genet 1996;14:25–32
Wuyts W, Van Hul W, Wauters J, Nemtsova M, Reyniers E, Van Hul EV, et al. Positional cloning of a gene involved in hereditary multiple exostoses. Hum Mol Genet 1996;5:1547–57.
Clines GA, Ashley JA, Shah S, Lovett M. The structure of the human multiple exostoses 2 gene and characterization of homologs in mouse and Caenorhabditis elegans. Genome Res 1997;7:359–67.
Lind T, Tufaro F, McCormick C, Lindahl U, Lidholt K. The putative tumor suppressors EXT1 and EXT2 are glycosyltransferases required for the biosynthesis of heparan sulfate. J Biol Chem 1998;273:26265–8.
Ciavarella M, Coco M, Baorda F, Stanziale P, Chetta M, Bisceglia L, et al. 20 novel point mutations and one large deletion in EXT1 and EXT2 genes: report of diagnostic screening in a large Italian cohort of patients affected by hereditary multiple exostosis. Gene 2013;515:339-48.
Sarrión P, Sangorrin A, Urreizti R, Delgado A, Artuch R, Martorell L, et al. Mutations in the EXT1 and EXT2 genes in Spanish patients with multiple osteochondromas. Sci Rep 2013;3:1346.
Wu Y, Xing X, Xu S, Ma H, Cao L, Wang S, et al. Novel and recurrent mutations in the EXT1 and EXT2 genes in Chinese kindreds with multiple osteochondromas. J Orthop Res 2013;31:1492–9.
Kang QL, Xu J, Zhang Z, He JW, Fu WZ, Zhang ZL. Mutation screening for the EXT1 and EXT2 genes in Chinese patients with multiple osteochondromas. Arch Med Res 2013;44:542–8.
Cao L, Liu F, Kong M, Fang Y, Gu H, Chen Y, et al. Novel EXT1 mutation identified in a pedigree with hereditary multiple exostoses. Oncol Rep 2014;31:713–8.
Jennes I, Pedrini E, Zuntini M, Mordenti M, Balkassmi S, Asteggiano CG, et al. Multiple osteochondromas: mutation update and description of the multiple osteochondromas mutation database (MOdb). Hum Mutat 2009;30:1620–7.
Philippe C, Porter DE, Emerton ME, Wells DE, Simpson AH, Monaco AP. Mutation screening of the EXT1 and EXT2 genes in patients with hereditary multiple exostoses. Am J Hum Genet 1997;61:520–8.
Wuyts W, Van Hul W, De Boulle K, Hendrickx J, Bakker E, Vanhoenacker F, et al. Mutations in the EXT1 and EXT2 genes in hereditary multiple exostoses. Am J Hum Genet 1998;62:346–54.
Wise CA, Clines GA, Massa H, Trask BJ, Lovett M. Identification and localization of the gene for EXTL, a third member of the multiple exostoses gene family. Genome Res 1997;7:10–6.
Wuyts W, Van Hul W, Hendrickx J, Speleman F, Wauters J, De Boulle K, et al. Identification and characterization of a novel member of the EXT gene family, EXTL2. Eur J Hum Genet 1997;5:382–9.
McCormick C1, Leduc Y, Martindale D, Mattison K, Esford LE, Dyer AP, et al. The putative tumour suppressor EXT1 alters the expression of cell-surface heparan sulfate. Nat Genet 1998;19:158–61.
Van Hul W, Wuyts W, Hendrickx J, Speleman F, Wauters J, De Boulle K, et al. Identification of a third EXT-like gene (EXTL3) belonging to the EXT gene family. Genomics 1998;47:230-7.
Bellaiche, Y., The, I. & Perrimon, N. Tout-velu is a Drosophila homologue of the putative tumour suppressor EXT-1 and is needed for Hh diffusion. Nature 1998;394,85–8.
Raskind WH, Conrad EU, Chansky H, Matsushita M. Loss of heterozygosity in chondrosarcomas for markers linked to hereditary multiple exostoses loci on chromosomes 8 and 11. Am J Hum Genet 1995;56:1132–9.
Hecht JT, Hogue D, Strong LC, Hansen MF, Blanton SH, Wagner M. Hereditary multiple exostosis and chondrosarcoma: linkage to chromosome II and loss of heterozygosity for EXT-linked markers on chromosomes II and 8. Am J Hum Genet 1995;56:1125–31.
Wuyts W, Van Hul W. Molecular basis of multiple exostoses: mutations in the EXT1 and EXT2 genes. Hum Mutat 2000;15:220–7
Shi YR, Wu JY, Hsu YA, Lee CC, Tsai CH, Tsai FJ. Mutation screening of the EXT genes in patients with hereditary multiple exostoses in Taiwan. Genet Test 2002;6:237–43.
Chen WC, Chi CH, Chuang CC, Jou IM. Three novel EXT1 and EXT2 gene mutations in Taiwanese patients 27 with multiple exostoses. J Formos Med Assoc 2006;105:434–7.
Lin WD, Tsai CH, Chen CP, Tsai FJ. Human gene mutations. Gene symbol: EXT2. Disease: exostoses (multiple) 2. Hum Genet 2007;122:211.
Lin WD, Lin SP, Chen CP, Tsai FJ. Human gene mutations. Gene symbol: EXT1. Disease: exostoses (multiple) 1. Hum Genet 2007;122:212.
Jennes I, Entius MM, Van Hul E, Parra A, Sangiorgi L, Wuyts W. Mutation screening of EXT1 and EXT2 by denaturing high-performance liquid chromatography, direct sequencing analysis, fluorescence in situ hybridization, and a new multiplex ligation-dependent probe amplification probe set in patients with multiple osteochondromas. J Mol Diagn 2008;10:85–92.
Lonie L, Porter DE, Fraser M, Cole T, Wise C, Yates L, et al. Determination of the mutation spectrum of the EXT1/EXT2 genes in British Caucasian patients with multiple osteochondromas, and exclusion of six candidate genes in EXT negative cases. Hum Mutat. 2006;27:1160.
Author information
Authors and Affiliations
Corresponding author
Additional information
Declaration of Interest: Authors declare no conflicts of interest for this work.
Open Access. This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
*Corresponding author. Department of Pediatrics and Medical Genetics, China Medical University Hospital, No. 2, Yuh Der Road, 404, Taichung, Taiwan E-mail: d0704@mail.cmuh.org.tw (F.-J. Tsai).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, WD., Hwu, WL., Wang, CH. et al. Mutant EXT1 in Taiwanese Patients with Multiple Hereditary Exostoses. BioMed 4, 11 (2014). https://doi.org/10.7603/s40681-014-0011-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.7603/s40681-014-0011-4