
Abstract
Strain T5T is the type strain of the species Phaeobacter inhibens Martens et al. 2006, a secondary metabolite producing bacterium affiliated to the Roseobacter clade. Strain T5T was isolated from a water sample taken at the German Wadden Sea, southern North Sea. Here we describe the complete genome sequence and annotation of this bacterium with a special focus on the secondary metabolism and compare it with the genomes of the Phaeobacter inhibens strains DSM 17395 and DSM 24588 (2.10), selected because of the close phylogenetic relationship based on the 16S rRNA gene sequences of these three strains. The genome of strain T5T comprises 4,130,897 bp with 3.923 protein-coding genes and shows high similarities in genetic and genomic characteristics compared to P. inhibens DSM 17395 and DSM 24588 (2.10). Besides the chromosome, strain T5T possesses four plasmids, three of which show a high similarity to the plasmids of the strains DSM 17395 and DSM 24588 (2.10). Analysis of the fourth plasmid suggested horizontal gene transfer. Most of the genes on this plasmid are not present in the strains DSM 17395 and DSM 24588 (2.10) including a nitrous oxide reductase, which allows strain T5T a facultative anaerobic lifestyle. The G+C content was calculated from the genome sequence and differs significantly from the previously published value, thus warranting an emendation of the species description.
Similar content being viewed by others


Introduction
Strain T5T was isolated from a water sample taken on 25th of October 1999 above an intertidal mud flat of the German Wadden Sea (53°42′20″N, 07°43′11″E) and found to be closely related to the type strain of Roseobacter gallaeciensis [1]. Two years later Martens et al. (2006) reclassified Roseobacter gallaeciensis as Phaeobacter gallaeciensis and described strain T5T as type strain of the species Phaeobacter inhibens. As found for various Phaeobacter strains [2–7], P. inhibens strain T5T (= DSM 16374T = LMG 22475T = CIP 109289T) is able to produce the antibiotic tropodithietic acid (TDA) [8]. Furthermore, strains of P. gallaeciensis and P. inhibens, including strain T5T, are able to produce a brownish pigment, which is the basis of the genus name (phaeos = dark, brown) [1]. The epithet of the species name points to the strong inhibitory activity of P. inhibens against different taxa of marine bacteria and algae [1]. The genus Phaeobacter is known to have a high potential for secondary metabolite production, as indicated by biosynthesis of TDA and N-acyl homoserine lactones (AHLs), as well as presence of genes coding for polyketide synthases (PKS) and nonribosomal peptide synthetases (NRPS) [2,7–10]. Biosynthesis of many different bioactive natural products is mediated by PKSs or NRPSs, including antibiotics, toxins and siderophores. Moreover, production of volatile compounds is widespread over the Roseobacter clade. It displays a particularly high proportion of volatile sulfur-containing compounds and thus seems to play an important role in the sulfur cycle of the ocean [11]. The sulfur-containing TDA, for which the sulfur precursor has not yet been determined, plays an important role in the mutualistic symbioses of P. inhibens and marine algae [12]. p-Coumaric acid causes the organism to switch from a state of mutualistic symbiosis to a pathogenic lifestyle in which toxicity is mediated via the production of the algicidal roseobacticides, which, like p-coumaric, is also a sulfur-containing metabolite [13,14].
Here we present the genome of P. inhibens strain T5T with particular emphasis on the genes involved in secondary metabolism and comparison with the recently published genomes of the P. inhibens strains DSM 17395 and DSM 24588 (2.10) [3]. DSM 17395 and DSM 24588, originally deposited as P. gallaeciensis strains, were recently reclassified as P. inhibens [15].
Classification and features
16S rRNA gene analysis
Figure 1 shows the phylogenetic neighborhood of P. inhibens DSM 16374T in a tree based on 16S rRNA genes. The sequences of the three identical 16S rRNA gene copies differ by one nucleotide from the previously published 16S rRNA sequence (NCBI Accession No. AY177712).

Phylogenetic tree highlighting the position of P. inhibens relative to the type strains of the other species within the genus Phaeobacter and the neighboring genera Leisingera and Ruegeria [1,20–33]. The tree was inferred from 1,385 aligned characters [34,35] of the 16S rRNA gene sequence under the maximum likelihood (ML) criterion [36]. Rooting was done initially using the midpoint method [37] and then checked for its agreement with the current classification (Table 1). The branches are scaled in terms of the expected number of substitutions per site. Numbers adjacent to the branches are support values from 1,000 ML bootstrap replicates [38] (left) and from 1,000 maximum-parsimony bootstrap replicates [39] (right) if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [40] are labeled with one asterisk, those also listed as ‘Complete and Published’ with two asterisks [21]. The genomes of six more Leisingera and Phaeobacter species are published in the current issue of Standard in Genomic Science [41–46]. The 16S rRNA sequences of P. inhibens strain DSM 24588 and P. inhibens strain DSM 17395 are virtually identical to those of P. inhibens DSM 16374T (data not shown).
A representative genomic 16S rRNA gene sequence of P. inhibens DSM 16374T was compared using NCBI BLAST [16,17] under default settings (e.g., considering only the high-scoring segment pairs (HSPs) from the best 250 hits) with the most recent release of the Greengenes database [18] and the relative frequencies of taxa and keywords (reduced to their stem [19]) were determined, weighted by BLAST scores. The most frequently occurring genera were Ruegeria (32.5%), Phaeobacter (28.8%), Silicibacter (13.6%), Roseobacter (13.3%) and Nautella (3.5%) (141 hits in total). Regarding the single hit to sequences from the species, the average identity within HSPs was 99.8%, whereas the average coverage by HSPs was 99.3%. Regarding the nine hits to sequences from other species of the genus, the average identity within HSPs was 99.0%, whereas the average coverage by HSPs was 99.2%. Among all other species, the one yielding the highest score was P. gallaeciensis (NZ_ABIF01000004), which corresponded to an identity of 100.0% and an HSP coverage of 100.0%. (Note that the Greengenes database uses the INSDC (= EMBL/NCBI/DDBJ) annotation, which is not an authoritative source for nomenclature or classification). The highest-scoring environmental sequence was AJ296158 (Greengenes short name ‘Spain:Galicia isolate str. PP-154’), which showed an identity of 99.8% and an HSP coverage of 100.0%. The most frequently occurring keywords within the labels of all environmental samples which yielded hits were ‘microbi’ (3.1%), ‘marine’ (2.6%), ‘coral’ (2.3%), ‘biofilm’ (2.1%) and ‘membrane, structure, swro’ (1.8%) (100 hits in total). Environmental samples which yielded hits of a higher score than the highest scoring species were not found.
Morphology and physiology
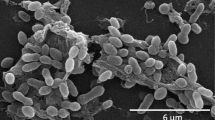
Cells of T5T are ovoid rods, 1.4–1.9 × 0.6–0.8 µm (Figure 2). Furthermore, T5T cells show the typical multicellular star-shaped structure described previously for P. gallaeciensis and other Roseobacter-clade organisms [2,4,47] (Figure 2). Cells of T5T are motile by means of a polar flagellum. T5T is a Gram-negative, marine, facultatively anaerobic, mesophilic bacterium with an optimal growth temperature between 27 and 29 °C and an optimal salinity between 0.51 and 0.68 M. The pH range for growth is 6.0–9.5, with an optimum at 7.5. On marine agar T5T forms smooth and convex colonies with regular edges and brownish pigmentation on ferric citrate containing media. T5T utilizes pentoses, hexoses, disaccharides and most amino acids as carbon and energy sources. No vitamin requirements were observed [1].

Scanning electron microscope pictures of P. inhibens strain DSM 16374T showing (a) the typical cell ovoid shape of strain T5T and (b) the multicellular, star-shaped structure as described previously for Phaeobacter and further Roseobacter-clade organisms.
Chemotaxonomy
There are no significant differences between the fatty-acid profile of strain T5T and other representatives of the Roseobacter clade [1]. Strain T5T has the highest profile similarity to P. gallaeciensis CIP 105210T [1]. The principal cellular fatty acids of strain T5T are the following saturated branched-chain fatty acids: C18:1ω7c (73.77%), 11-methyl C18:1ω7c (7.45%), C16:0 (3.83%), C18:0 (3.14%), 2-OH C16:0 (3.10%), C14:1 (2.19%), 3-OH C10:0 (1.71%), 3-OH C12:0 (1.59%), 3-OH C14:1/3 oxo-C14:0 (0.87%), C18.1ω9c (0.76%) and an unambiguously identified fatty acid (1.59%) [1]. The major polar lipids of strain T5T comprise phosphatidylglycerol, phosphatidylethanolamine, phosphatidylcholine, an aminolipid and two unidentified lipids [1].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of the DOE Joint Genome Institute Community Sequencing Program (CSP) 2010, CSP 441 “Whole genome type strain sequences of the genera Phaeobacter and Leisingera - a monophyletic group of physiological highly diverse organisms”. The genome project is deposited in the Genomes On Line Database [40] and the complete genome sequence is deposited in GenBank and the Integrated Microbial Genomes database (IMG) [56]. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI) using state of the art sequencing technology [57]. A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
A culture of DSM 16374T was grown aerobically in DSMZ medium 514 [58] at 25°C. Genomic DNA was isolated using the Jetflex Genomic DNA Purification Kit (GENOMED 600100) following the standard protocol provided by the manufacturer but modified by an incubation time of 40 min, the incubation on ice over night on a shaker, the use of additional 10 µl proteinase K, and the addition of 100 µl protein precipitation buffer. DNA is available from DSMZ through the DNA Bank Network [59].
Genome sequencing and assembly
For this genome, we constructed and sequenced an Illumina short-insert paired-end library with an average insert size of 225 bp, and an Illumina long-insert paired-end library with an average insert size of 9602 bp, which generated 18,471,132 reads and 11,906,846 reads, respectively, totaling 4,557 Mbp of Illumina data. All general aspects of library construction and sequencing performed can be found at the JGI website [60]. The initial draft assembly contained 13 contigs in 10 scaffold. The initial draft data was assembled with Allpaths [61] and the consensus was computationally shredded into 10 kbp overlapping fake reads (shreds). The Illumina draft data was also assembled with Velvet [62], and the consensus sequences were computationally shredded into 1.5 kbp overlapping fake reads (shreds). The Illumina draft data was assembled again with Velvet using the shreds from the first Velvet assembly to guide the next assembly. The consensus from the second Velvet assembly was shredded into 1.5 kbp overlapping fake reads. The fake reads from the Allpaths assembly and both Velvet assemblies and a subset of the Illumina CLIP paired-end reads were assembled using parallel phrap (High Performance Software, LLC) [63]. Possible mis-assemblies were corrected with manual editing in Consed [63]. Gap closure was accomplished using repeat resolution software (Wei Gu, unpublished), and sequencing of bridging PCR fragments with PacBio technologies. A total of 10 PCR PacBio consensus sequences were completed to close gaps and to raise the quality of the final sequence. The final assembly is based on 4,557 Mbp of Illumina draft data, which provides an average 1,111 × coverage of the genome.
Genome annotation
Genes were identified using Prodigal [64] as part of the DOE-JGI genome annotation pipeline [65], followed by a round of manual curation using the JGI GenePRIMP pipeline [66]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation were performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [56].
Genome properties
The genome statistics are provided in Table 3 and Figure 3. The genome consists of six scaffolds with a total length of 4,130,897 bp and a G+C content of 60.0%. The scaffolds correspond to a chromosome 3,669,861 bp in length and four extrachromosomal elements as identified by their replication systems (see below). Of the 3,986 genes predicted, 3,923 were protein-coding genes, and 63 RNAs; 39 pseudogenes were also identified. The majority of the protein-coding genes (81.0%) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.

Graphical representation of the genome of P. inhibens T5T. From outside to the center: (1) sequence of P. inhibens T5T, (2) results of a blastn comparison from P. inhibens DSM 24588 (2.10) against P. inhibens T5T, (3) results of a blastn comparison of P. inhibens DSM 17395 against P. inhibens T5T, (4) G+C content. Comparisons and visualization are done with BRIG [67].
Insights into the genome
Genome sequencing of P. inhibens DSM 16374T revealed the presence of four extrachromosomal elements with sizes of 227 kb, 88 kb, 78 kb, and 69 kb (Figure 3; Table 5) and DnaA-like I, RepABC-8, RepB-I and RepA-I as replication systems, respectively [68]. The different replicases that mediate the initiation of replication are designated according to the established plasmid classification scheme [69]. With the exception of the 88 kb replicon, these extrachromosomal elements are highly syntenic to specific replicons in the genomes of P. inhibens strains DSM 17395 and DSM 24588 (Figure 3).
The locus tags of all replicases, plasmid stability modules and the large virB4 gene of a type IV secretion system are presented in Table 6. The plasmids pInhi_A227 and pInhi_B88 contain postsegregational killing systems (PSK) consisting of a typical operon with two small genes encoding a stable toxin and an unstable antitoxin [70]. Moreover, plasmid pInhi_B88 also contains a complete virB gene cluster of type IV secretion system, required for the formation of a transmembrane channel. However, the absence of the relaxase VirD2, which is necessary for the strand-specific DNA nicking at the origin of transfer (oriT), and the coupling protein VirD4 indicates that this plasmid is non-conjugative [71,72]. The RepA-I type replicon pInhi_D69 contains a complete rhamnose operon [73] and is dominated by genes required for polysaccharide biosynthesis.
As already indicated by the strong inhibitory activity of P. inhibens T5T [8] all 26 described genes involved in the production of TDA are present in the genome of this strain. As found for the P. inhibens strains DSM 17395 and DSM 24588, the key genes for TDA production tdaABCDEF (Inhi_3684 – _3688, Inhi_3701), paaZ2 (Inhi_3702) and a gene coding for a putative Na-dependent transporter (Inhi_3697) [3,74] are located on the 227 kb plasmid of T5T (Figure 3). The remaining 19 genes, containing genes of the phenylacetyl-CoA and assimilatory sulfate reduction pathways, are scattered over the chromosome as in the strains DSM 17395 and DSM 24588 [3]. Beside the tdaA gene, present on the 227 kb plasmid, we also found other genes involved in the regulation of TDA synthesis located on the chromosome, what is in agreement with Thole et al. (2012) and Berger et al. (2012) [3,75]. This includes the genes encoding transcriptional activator proteins (Inhi_2121; _2059; _0396) comparable with pgaR, iorR a transcriptional regulator (PGA1_c20730), a putative serine-protein kinase (Inhi_2265) and a putative signal peptide peptidase (Inhi_2227).
Two complete prophages and an additional cluster coding for the production of gene transfer agents (GTA) were found in the genome of strain T5T. The GTA gene cluster is equal in length and comprises the same genes (Inhi_0654 – Inhi_0670) as the GTA clusters of the strains DSM 17395 and DSM 24588. The two prophages of strain T5T consist of 52 ORFs (prophage 1; ∼37kb) and 63 ORFs (prophage 2; ∼48kb), respectively. Strain DSM 17395 possesses two prophages, but for DSM 24588 no prophages were detected [3]. Prophage 1 of strain T5T is similar to prophage 1 of strain DSM 17395, with the exception that a few ORFs are different (PGA1_c18280 – _c18310, PGA1_c18480 – _c18530 and PGA1_c18570 – _c18680; Inhi_1777, Inhi_1785 – _1788, Inhi_1803 – _1812 and Inhi_1816 – 1829). Prophage 2 of strain T5T is a Mu-like bacteriophage, not present in strain DSM 17395.
It was previously shown that strain T5T produces two different AHLs, i.e. C18-en-HSL and N-3-hydroxydecanoyl-homoserine lactone (3OHC10-HSL) [76]. In P. inhibens strain DSM 17395 TDA and pigment production are regulated via a pgaR-pgaI QS system [47]. The AHL synthase encoding gene pgaI in DSM 17395 is responsible for the production of 3OHC10-HSL. In the genome of strain T5T we found a homologous system probably coding for the 3OHC10-HSL producing AHL synthase (Inhi_2120, homolog to pgaI) and the respective regulator (Inhi_2121, homologous to pgaR) (Figure 3, QS system I). Thus TDA production in strain T5T might also be regulated by a QS system. In addition, two further QS systems (QS system II and III; Figure 3) were found on the chromosome of T5T. System II is formed by the genes Inhi_0506 and _0507 and is located in the prophage region 2. Orthologs for these QS system genes are also present in P. inhibens strain DSM 24588 (PGA2_c18960 and PGA2_c18970) but absent in strain DSM 17395. QS system III consists of the genes Inhi_1819 and _1820 and is unique for strain T5T compared to P. inhibens DSM 17395 and DSM 24588. It is also located in the potential prophage 1 region (Fig. 3). A homologous system was found in the genome of Phaeobacter caeruleus DSM 24564T and the neighboring genes show a high synteny. The location in the prophage region and the high synteny to the system of P. caeruleus suggest a possible gene transfer of this QS system via a bacteriophage. The functions of QS system II and III are currently unknown, but it is likely that the compound C18-en-HSL is produced by one of those systems.
Two functions were suggested that can possibly be used as unique chemotaxonomic markers for the species P. inhibens within the Roseobacter clade [3]. The genes coding for the first of these functions are located on the chromosome and are involved in cell wall development and surface attachment [dltA encoding a D-alanine-poly(phosphoribitol) ligase involved in biosynthesis of D-alanyl-lipoteichoic acid]. The second unique function is the biosynthesis and transport of iron-chelating siderophores, and the encoding genes are located on the plasmid pPGA1_78 and pPGA2_95, respectively. These two clusters are also present in the genome of strain T5T. The siderophore gene cluster (Inhi_3924 – Inhi_3928) is located on the 78 kb plasmid (Fig. 3) and the dltA gene cluster (Inhi_1065 – Inhi_1086) is located on the chromosome (Fig. 3). Screenings in the newly available Roseobacter genomes showed that Leisingera methylohalidivorans DSM 14336 [42] and Leisingera aquimarina DSM 24565 [41] also harbor the genes for siderophore synthesis. The uniqueness of the dltA gene cluster within the species P. inhibens, however, remains and can be used as chemotaxonomic marker.
The existence of genes coding for a polyketide synthase (Inhi_1972) and three non-ribosomal peptide synthetases (Inhi_1072, _1974 and _3983) confirm the results of Martens et al. (2007) [7]. These genes are present in the genomes of strains DSM 17395 and DSM 24588, too (PGA1_c04930 and PGA1_c05350, _c13760, _c28490; PGA2_c05370 and PGA2_c04910, _c13660, _71p110). The genes Inhi_3983 of P. inhibens strain T5T and PGA2_71p110 of P. inhibens strain DSM 24588 are located on the 69 kb plasmid (Fig. 3) and 71 kb plasmid, respectively. In contrast, the homologous gene (PGA1_c28490) of P. inhibens strain DSM 17395 is located on the chromosome.
For the P. inhibens strains DSM 17395 and DSM 24588 a surface-attached lifestyle was inferred from the genome analysis [3]. Even though strain T5T was isolated from a water sample, it exhibits the same genes associated with the biosynthesis and transport of polysaccharides as strains DSM 17395 and DSM 24588. This includes genes described as unique for the strains DSM 17395 and DSM 24588, i.e. a gene coding for a glycosyltransferase-like protein (Inhi_3961) and two ORFs (Inhi_3954 and Inhi_3955) related to a type I secretion system and used for the transport of exopolysaccharides. Production of extracellular polysaccharides is a major factor contributing to surface attachment [77,78]. Thus it appears likely that T5T is also well-adapted to a surface attached lifestyle.
P. inhibens was described as a strictly aerobic bacterium [1]. However, we found genes involved in the dissimilatory nitrate reduction pathway to nitrogen, including the gene coding for a copper containing nitrite reductase (Inhi_3645) and a nitric oxide reductase cluster (Inhi_3648 – Inhi_3654), both located on the replicon pInhi_A227. These genes are also present and located on the largest plasmids of P. inhibens DSM 17395 (PGA1_262p) and P. inhibens DSM 24588 (PGA2_239p) (Figure 3). In addition, P. inhibens strain T5T possesses a gene cluster coding for a nitrous oxide reductase (Inhi_3786 – Inhi_3792) located on the replicon pInhi_B88, which is absent in the strains DSM 17395 and DSM 24588 (Figure 3). Neither strain T5T nor DSM 17395 and DSM 24588 have genes coding for a nitrate reductase. The findings suggest that P. inhibens T5T has a complete dissimilatory nitrite reduction pathway, but is not able to reduce nitrate, as previously described by Martens et al. (2006) [1]. To confirm the results we tested strain T5T for its capability to grow anaerobically with nitrite. Anaerobic marine basal medium was prepared according to Cypionka and Pfennig (1986) [79] and supplemented with nitrite and glucose, both in a final concentration of 5 mM. After two weeks a decrease of nitrite was determined by photometric analysis at 545 nm by using the Griess reaction [80] and an increase of the turbidity was detected (results not shown). Thus it became clear that P. inhibens T5T is able to grow anaerobically with nitrite, suggesting an emended description of this organism as a facultatively anaerobic bacterium.
Phylogenetic analysis shows that P. inhibens and P. gallaeciensis form a cluster together with Phaeobacter arcticus (Figure 1). The cluster is set apart from the cluster comprising Leisingera aquimarina, Leisnigera nanhaiensis, Leisingera methylohalidivorans, Phaeobacter caeruleus and Phaeobacter daeponensis, but the backbone of the 16S rRNA gene tree shown in Figure 1 is rather unresolved. Using the online analysis tool “Genome-to-Genome Distance Calculator” 2.0 (GGDC) [81,82], we performed a preliminary phylogenetic analysis of the draft genomes of the type strains of the genera Leisingera and Phaeobacter and the finished genomes of P. inhibens strains DSM 17395 and DSM 24588. Table 7 shows the results of the in silico calculated DNA-DNA hybridization (DDH) similarities of P. inhibens to other Phaeobacter and Leisingera species. In the following analysis, we will refer only to the results of formula 2, as this formula is robust against the use of draft genomes such as AOQA01000000 (CIP 105210T) [83]. The use of GGDC revealed a high similarity of T5T (78%) to the strains P. inhibens DSM 17395 and DSM 24588, but a low similarity to P. gallaeciensis strain CIP 105210T (36%). DSM 17395 and CIP 105210T were previously supposed to be type-strain deposits for P. gallaeciensis [33] and we cross-compared them using GGDC. Formula 2 yielded a similarity of only 38.30% ± 2.50 between these two strains, thus indicating not only that they are not the same strain, but also do not even belong to the same species. The results are in agreement with the study of Buddruhs et al. (2013) [15] showing that strain DSM 17395 is the false deposit and belongs together with DSM 24588 to P. inhibens, whereas CIP 105210T is the correct type-strain deposit for P. gallaeciensis.
The differences in the G+C content (55.7%) published earlier [1] and the value calculated directly from the genome (Table 3) warrants an update of the taxonomic description on P. inhibens [84]. Moreover, genomic and experimental evidence indicates that P. inhibens is not strictly aerobic but facultatively anaerobic.
Conclusion
Emended description of the species Phaeobacter inhibens Martens et al. 2006
The description of the species Phaeobacter inhibens is the one given by Martens et al. 2006 [1], with the following modification. The G+C content, rounded to zero decimal places, is 60%. Phaeobacter inhibens is a facultative anaerobic bacterium by using nitrite reduction.
References
Martens T, Heidorn T, Pukall R, Simon M, Tindall BJ, Brinkhoff T. Reclassification of Roseobacter gallaeciensis Ruiz-Ponte et al. 1998 as Phaeobacter gallaeciensis gen. nov., comb. nov., description of Phaeobacter inhibens sp. nov., reclassification of Ruegeria algicola (Lafay et al. 1995) Uchino et al. 1999 as Marinovum algicola gen. nov., comb. nov., and emended descriptions of the genera Roseobacter, Ruegeria and Leisingera. Int J Syst Evol Microbiol 2006; 56:1293–1304. PubMed http://dx.doi.org/10.1099/ijs.0.63724-0
Bruhn JB, Nielsen KF, Hjelm M, Hansen M, Bresciani J, Schulz S, Gram L. Ecology, inhibitory activity and morphogenesis of a potential marine fish larvae probiotic bacteria, Roseobacter strain 27-4. Appl Environ Microbiol 2005; 71:7263–7270. PubMed http://dx.doi.org/10.1128/AEM.71.11.7263-7270.2005
Thole S, Kalhoefer D, Voget S, Berger M, Engelhardt T, Liesegang H, Wollherr A, Kjelleberg S, Daniel R, Simon M, et al. Phaeobacter gallaeciensis genomes from globally opposite locations reveal high similarity of adaptation to surface life. ISME J 2012; 6:2229–2244. PubMed http://dx.doi.org/10.1038/ismej.2012.62
Bruhn JB, Gram L, Belas R. Production of antibacterial compounds and biofilm by Roseobacter species are influenced by culture conditions. Appl Environ Microbiol 2007; 73:442–450. PubMed http://dx.doi.org/10.1128/AEM.02238-06
D’Alvise PW, Melchiorsen J, Porsby CH, Nielsen KF, Gram L. Inactivation of Vibrio anguillarum by attached and planktonic Roseobacter Cells. Appl Environ Microbiol 2010; 76:2366–2370. PubMed http://dx.doi.org/10.1128/AEM.02717-09
Prado S, Montes J, Romalde JL, Barja JL. Inhibitory activity of Phaeobacter strains against aquaculture pathogenic bacteria. Int Microbiol 2009; 12:107–114. PubMed
Martens T, Gram L, Grossart HP, Kessler D, Müller R, Simon M, Wenzel SC, Brinkhoff T. Bacteria of the Roseobacter clade show potential for secondary metabolite production. Microb Ecol 2007; 54:31–42. PubMed http://dx.doi.org/10.1007/s00248-006-9165-2
Brinkhoff T, Bach G, Heidorn T, Liang L, Schlingloff A, Simon M. Antibiotic production by a Roseobacter clade-affiliated species from the German Wadden Sea and its antagonistic effects on indigenous isolates. Appl Environ Microbiol 2004; 70:2560–2565. PubMed http://dx.doi.org/10.1128/AEM.70.4.2560-2565.2003
Rao D, Webb JS, Kjelleberg S. Competitive interactions in mixed-species biofilms containing the marine bacterium Pseudoalteromonas tunicata. Appl Environ Microbiol 2005; 71:1729–1736. PubMed http://dx.doi.org/10.1128/AEM.71.4.1729-1736.2005
Ruiz-Ponte C, Samain JF, Sanchez JL, Nicolas JL. The benefit of a Roseobacter species on the survival of scallop larvae. Mar Biotechnol (NY) 1999; 1:52–59. PubMed http://dx.doi.org/10.1007/PL00011751
Thiel V, Brinkhoff T, Dickschat JS, Wickel S, Grunenberg J, Wagner-Döbler I, Simon M, Schulz S. Identification and biosythesis of tropone derivatives and sulfur volatiles produced by bacteria of the marine Roseobacter clade. Org Biomol Chem 2010; 8:234–246. PubMed http://dx.doi.org/10.1039/b909133e
Riclea R, Gleitzmann J, Bruns H, Junker C, Schulz B, Dichschat JS. Algicidal lactones from the marine Roseobacter clade bacterium Ruegeria pomeroyi. Beilstein J Org Chem 2012; 8:941–950. PubMed http://dx.doi.org/10.3762/bjoc8.106
Seyedsayamdost MR, Case RJ, Kolter R, Clardy J. The Jekyll-and-Hyde chemistry of Phaeobacter gallaeciensis. Nat Chem 2011; 3:331–335. PubMed http://dx.doi.org/10.1038/nchem.1002
Seyedsayamdost MR, Carr R, Kolter R, Clardy J. Roseobacticides: small molecule modulators of an algal-bacterial symbiosis. J Am Chem Soc 2011; 133:18343–18349. PubMed http://dx.doi.org/10.1021/ja207172s
Buddruhs N, Pradella S, Göker M, Päuker O, Pukall R, Spröer C, Schumann P, Petersen J, Brinkhoff T. Molecular und phenotypic analyses reveal the non-identity of Phaeobacter gallaeciensis type strain deposits CIP 105210T and DSM 17395. Int J Syst Evol Microbiol 2013; (accepted). PubMed http://dx.doi.org/10.1099/ijs.0.053900-0
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215:403–410. PubMed
Korf I, Yandell M, Bedell J. BLAST, O’Reilly, Sebastopol, 2003.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069–5072. PubMed http://dx.doi.org/10.1128/AEM.03006-05
Porter MF. An algorithm for suffix stripping. Program: electronic library and information systems 1980; 14:130–137.
Huo YY, Xu XW, Li X, Liu C, Cui HL, Wang CS, Wu M. Ruegeria marina sp. nov., isolated from marine sediment. Int J Syst Evol Microbiol 2011; 61:347–350. PubMed http://dx.doi.org/10.1099/ijs.0.022400-0
Moran MA, Buchan A, González JM, Heidelberg JF, Whitman WB, Kiene RP, Henriksen JR, King GM, Belas R, Fuqua C, et al. Genome sequence of Silicibacter pomeroyi reveals adaptation to the marine environment. Nature 2004; 432:910–913. PubMed http://dx.doi.org/10.1038/nature03170
Kim YO, Park S, Nam BH, Kang SJ, Hur YB, Lee SJ, Oh TK, Yoon JH. Ruegeria halocynthiae sp. nov., isolated from the sea squirt Halocynthia roretzi. Int J Syst Evol Microbiol 2012; 62:925–930. PubMed http://dx.doi.org/10.1099/ijs.0.031609-0
Yi H, Lim YW, Chun J. Taxonomic evaluation of the genera Ruegeria and Silicibacter: a proposal to transfer the genus Silicibacter Petursdottir and Kristjansson 1999 to the genus Ruegeria Uchino et al. 1999. Int J Syst Evol Microbiol 2007; 57:815–819. PubMed http://dx.doi.org/10.1099/ijs.0.64568-0
Muramatsu Y, Uchino Y, Kasai H, Suzuki K, Nakagawa Y. Ruegeria mobilis sp. nov., a member of the Alphaproteobacteria isolated in Japan and Palau. Int J Syst Evol Microbiol 2007; 57:1304–1309. PubMed http://dx.doi.org/10.1099/ijs.0.64572-0
Vandecandelaere I, Nercessian O, Segaert E, Achouak W, Faimali M, Vandamme P. Ruegeria scottomollicae sp. nov., isolated from a marine electroactive biofilm. Int J Syst Evol Microbiol 2008; 58:2726–2733. PubMed http://dx.doi.org/10.1099/ijs.0.65843-0
Oh KH, Jung YT, Oh TK, Yoon JH. Ruegeria faecimaris sp. nov., isolated from a tidal flat sediment. Int J Syst Evol Microbiol 2011; 61:1182–1188. PubMed http://dx.doi.org/10.1099/ijs.0.025999-0
Schaefer JK, Goodwin KD, McDonald IR, Murrell JC, Oremland RS. Leisingera methylohalidivorans gen. nov., sp. nov., a marine methylotroph that grows on methyl bromide. Int J Syst Evol Microbiol 2002; 52:851–859. PubMed http://dx.doi.org/10.1099/ijs.0.01960-0
Vandecandelaere I, Segaert E, Mollica A, Faimali M, Vandamme P. Leisingera aquimarina sp. nov., isolated from a marine electroactive biofilm, and emended descriptions of Leisingera methylohalidivorans Schaefer et al. 2002, Phaeobacter daeponensis Yoon et al. 2007 and Phaeobacter inhibens Martens et al. 2006. Int J Syst Evol Microbiol 2008; 58:2788–2793. PubMed http://dx.doi.org/10.1099/ijs.0.65844-0
Sun F, Wang B, Liu X, Lai Q, Du Y, Li G, Luo J, Shao Z. Leisingera nanhaiensis sp. nov., isolated from marine sediment. Int J Syst Evol Microbiol 2010; 60:275–280. PubMed http://dx.doi.org/10.1099/ijs.0.010439-0
Vandecandelaere I, Segaert E, Mollica A, Faimali M, Vandamme P. Phaeobacter caeruleus sp. nov., a blue-coloured, colony-forming bacterium isolated from a marine electroactive biofilm. Int J Syst E vol Microbiol 2009; 59:1209–1214. PubMed http://dx.doi.org/10.1099/ijs.0.002642-0
Yoon JH, Kang SJ, Lee SY, Oh TK. Phaeobacter daeponensis sp. nov., isolated from a tidal flat of the Yellow Sea in Korea. Int J Syst Evol Microbiol 2007; 57:856–861. PubMed http://dx.doi.org/10.1099/ijs.0.64779-0
Zhang DC, Li HR, Xin YH, Liu HC, Chi ZM, Zhou PJ, Yu Y. Phaeobacter arcticus sp. nov., a psychrophilic bacterium isolated from the Arctic. Int J Syst Evol Microbiol 2008; 58:1384–1387. PubMed http://dx.doi.org/10.1099/ijs.0.65708-0
Ruiz-Ponte C, Cilia V, Lambert C, Nicolas JL. Roseobacter gallaeciensis sp. nov., a new marine bacterium isolated from rearings and collectors of the scallop Pecten maximus. Int J Syst Bacteriol 1998; 48:537–542. PubMed http://dx.doi.org/10.1099/00207713-48-2-537
Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452–464. PubMed http://dx.doi.org/10.1093/bioinformatics/18.3.452
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540–552. PubMed http://dx.doi.org/10.1093/oxfordjournals.molbev.a026334
Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web-servers. Syst Biol 2008; 57:758–771. PubMed http://dx.doi.org/10.1080/10635150802429642
Hess PN, De Moraes Russo CA. An empirical test of the midpoint rooting method. Biol J Linn Soc Lond 2007; 92:669–674. http://dx.doi.org/10.1111/j.1095-8312.2007.00864.x
Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? Lect Notes Comput Sci 2009; 5541:184–200. http://dx.doi.org/10.1007/978-3-642-02008-7_13
Swofford DL. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4.0 b10. Sinauer Associates, Sunderland, 2002.
Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Hugenholtz P, Markowitz VM, Kyrpides NC. The Genomes OnLine Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2010; 38: D346–D354. PubMed http://dx.doi.org/10.1093/nar/gkp848
Riedel T, Teshima H, Petersen J, Fiebig A, Davenport K, Dalingault H, Erkkila T, Gu W, Munk C, Xu Y, et al. Genome sequence of the Leisingera aquimarina type strain (DSM 24565T), a member of the Roseobacter clade rich in extrachromosomal elements. Stand Genomic Sci 2013; 8:389–402. http://dx.doi.org/10.4056/sigs.3858183
Buddruhs N, Chertkov O, Fiebig A, Petersen J, Chen A, Pati A, Ivanova N, Lapidus A, Goodwin LA, Chain P, et al. Complete genome sequence of the marine methyl-halide oxidizing Leisingera methylohalidivorans type strain (DSM 14336T), a member of the Roseobacter clade. Stand Genomic Sci 2013; 9:128–141. http://dx.doi.org/10.4056/sigs.4297965
Dogs M, Teshima H, Petersen J, Fiebig A, Chertkov O, Dalingault H, Chen A, Pati A, Goodwin LA, Chain P, et al. Genome sequence of Phaeobacter daeponensis type strain (DSM 23529T), a facultatively anaerobic bacterium isolated from marine sediment, and emendation of Phaeobacter daeponensis. Stand Genomic Sci 2013; 9:142–159. http://dx.doi.org/10.4056/sigs.4287962
Beyersmann PG, Chertkov O, Petersen J, Fiebig A, Chen A, Pati A, Ivanova N, Lapidus A, Goodwin LA, Chain P, et al. Genome sequence of Phaeobacter caeruleus type strain (DSM 24564T), a surface-associated member of the marine Roseobacter clade. Stand Genomic Sci 2013; 8:403–419. http://dx.doi.org/10.4056/sigs.3927626
Freese H, Dalingault H, Petersen J, Pradella S, Fiebig A, Davenport K, Teshima H, Chen A, Pati A, Ivanova N, et al. Genome sequence of the plasmid and phage-gene rich marine Phaeobacter arcticus type strain (DSM 23566T). Stand Genomic Sci 2013; 8:450–464. http://dx.doi.org/10.4056/sigs.383362
Breider S, Teshima H, Petersen J, Fiebig A, Chertkov O, Dalingault H, Chen A, Pati A, Ivanova N, Lapidus A, et al. Genome sequence of Leisingera nanhaiensis strain DSM 23252T isolated from marine sediment. Stand Genomic Sci 2013; 8:128–141.
Berger M, Neumann A, Schulz S, Simon M, Brinkhoff T. Tropodithietic acid production in Phaeobacter gallaeciensis is regulated by N-acyl homoserine lactone-mediated quorum sensing. J Bacteriol 2011; 193:6576–6585. PubMed http://dx.doi.org/10.1128/JB.05818-11
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed http://dx.doi.org/10.1038/nbt1360
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms. Proposal for the domains Archaea, Bacteria and Eucarya. Proc Natl Acad Sci USA 1990; 87.4576–4579.
Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: DJ Brenner, NR Krieg, JT Staley, GM Garrity (eds), Bergey’s Manual of Systematic Bacteriology, second edition, vol. 2 (The Proteobacteria), part B (The Gammaproteobacteria), Springer, New York, 2005, p. 1.
Garrity GM, Bell JA, Lilburn T. Class I. Alphaproteobacteria class. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 1.
Validation List No. 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 2006; 56:1–6. PubMed http://dx.doi.org/10.1099/ijs.0.64188-0
Garrity GM, Bell JA, Lilburn T. Family I. Rhodobacteraceae fam. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 161.
BAuA. Classification of Bacteria and Archaea in risk groups. TRBA 2010; 466:93.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25–29. PubMed http://dx.doi.org/10.1038/75556
Markowitz VM, Mavromatis K, Ivanova NN, Chen IA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. 2009; 25:2271–2278.
Mavromatis K, Land ML, Brettin TS, Quest DJ, Copeland A, Clum A, Goodwin L, Woyke T, Lapidus A, Klenk HP, et al. The fast changing landscape of sequencing technologies and their impact on microbial genome assemblies and annotation. PLoS ONE 2012; 7:e48837. PubMed http://dx.doi.org/10.1371/journal.pone.0048837
List of growth media used at the DSMZ: http://www.dmsz.de/catalogues/cataloque-microorganisms/culture-technology/list-of-media-for-microorganisms.html.
Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch A, Berendsohn WGWJ. The DNA Bank Network: the start from a German initiative. Biopres Biobanking 2011; 9:51–55. http://dx.doi.org/10.1089/bio.2010.0029
The DOE Joint Genome Institute. http://www.jgi.doe.gov.
Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, Lander ES, Nusbaum CJD. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res 2008; 18:810–820. PubMed http://dx.doi.org/10.1101/gr.7337908
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821–829. PubMed http://dx.doi.org/10.1101/gr.074492.107
Phrap and Phred for Windows. MacOS, Linux, and Unix. http://www.phrap.com.
Hyatt D, Chen G, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119. PubMed http://dx.doi.org/10.1186/1471-2105-11-119
Mavromatis K, Ivanova NN, Chen IM, Szeto E, Markowitz VM, Kyrpides NC. The DOE-JGI Standard operating procedure for the annotations of microbial genomes. Stand Genomic Sci 2009; 1:63–67. PubMed http://dx.doi.org/10.4056/sigs.632
Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455–457. PubMed http://dx.doi.org/10.1038/nmeth.1457
Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 2011; 12:402. PubMed http://dx.doi.org/10.1186/1471-2164-12-402
Petersen J. Phylogeny and compatibility: plasmid classification in the genomics era. Arch Microbiol 2011; 193:313–321. PubMed
Petersen J, Brinkmann H, Berger M, Brinkhoff T, Päuker O, Pradella S. Origin and evolution of a novel DnaA-like plasmid replication type in Rhodobacterales. Mol Biol Evol 2011; 28:1229–1240. PubMed http://dx.doi.org/10.1093/molbev/msq310
Zielenkiewicz U, Ceglowski P. Mechanisms of plasmid stable maintenance with special focus on plasmid addiction systems. Acta Biochim Pol 2001; 48:1003–1023. PubMed
Cascales E, Christie PJ. The versatile bacterial type IV secretion systems. Nat Rev Microbiol 2003; 1:137–149. PubMed http://dx.doi.org/10.1038/nrmicro753
Petersen J, Frank O, Göker M, Pradella S. Extrachromosomal, extraordinary and essential-the plasmids of the Roseobacter clade. [Epub ahead of print]. Appl Microbiol Biotechnol 2013. PubMed http://dx.doi.org/10.1007/s00253-013-4746-8
Giraud MF, Naismith JH. The rhamnose pathway. Curr Op in Struct Biol 2000; 10:687–696. PubMed http://dx.doi.org/10.1016/S0959-440X(00)00145-7
Geng H, Bruhn JB, Nielsen KF, Gram L, Belas R. Genetic dissection of tropdithietic acid biosynthesis by marine roseobacters. Appl Environ Microbiol 2008; 74:1535–1545. PubMed http://dx.doi.org/10.1128/AEM.02339-07
Berger M, Brock NL, Liesegang H, Dogs M, Preuth I, Simon M, Dickschat JS, Brinkhoff T. Genetic analysis of the upper phenylacetate catabolic pathway in the production of tropodithietic acid by Phaeobacter gallaeciensis. Appl Environ Microbiol 2012; 78:3539–3551. PubMed http://dx.doi.org/10.1128/AEM.07657-11
Wagner Döbler I, Thiel V, Eberl L, Allgaier M, Bodor A, Meyer S, Ebner S, Henning A, Pukall R, Schulz S. Discovery of complex mixtures of novel long-chain quorum sensing signals in free-living and host-associated marine Alphaproteobacteria. ChemBioChem 2005; 6:2195–2206. PubMed http://dx.doi.org/10.1002/cbic.200500189
Danhorn T, Fuqua C. Biofilm formation by plant-associated bacteria. Annu Rev Microbiol 2007; 61:401–422. PubMed http://dx.doi.org/10.1146/annurev.micro.61.080706.093316
Vu B, Chen M, Crawford RJ, Ivanova EP. Bacterial extracellular polysaccharides involved in biofilm formation. Molecules 2009; 14:2535–2554. PubMed http://dx.doi.org/10.3390/molecules14072535
Cypionka H, Pfennig N. Growth yields of Desulfotomaculum orientes with hydrogen in chemostat culture. Arch Microbiol 1986; 143:396–399. http://dx.doi.org/10.1007/BF00412808
Griess JE. Ber. Dtsch. Chem. Ges. 1879; 12:426. http://dx.doi.org/10.1002/cber.187901201117
Auch AF, Klenk HP, Göker M. Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand Genomic Sci 2010; 2:142–148. PubMed http://dx.doi.org/10.4056/sigs.541628
Auch AF, Von Jan M, Klenk HP, Göker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci 2010; 2:117–134. PubMed http://dx.doi.org/10.4056/sigs.531120
Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics (In press). PubMed
Meier-Kolthoff JP, Klenk HP, Göker M. Taxonomic use of DNA G+C content and DNA-DNA hybridization in the genomic age. Int J Syst Evol Microbiol 2014; (In press).
Acknowledgements
The authors gratefully acknowledge the assistance of Iljana Schröder for growing P. inhibens cultures and Evelyne-Marie Brambilla for DNA extraction and quality control (both at the DSMZ). The work conducted by the U.S. Department of Energy Joint Genome Institute was supported by the Office of Science of the U.S. Department of Energy under contract No. DE-AC02-05CH11231; the work conducted by members of the Roseobacter consortium was supported by the German Research Foundation (DFG) Transregio-SFB 51. We also thank the European Commission which supported phenotyping via the Microme project 222886 within the Framework 7 program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Dogs, M., Voget, S., Teshima, H. et al. Genome sequence of Phaeobacter inhibens type strain (T5T), a secondary metabolite producing representative of the marine Roseobacter clade, and emendation of the species description of Phaeobacter inhibens. Stand in Genomic Sci 9, 334–350 (2013). https://doi.org/10.4056/sigs.4448212
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.4448212