
Abstract
Desulfobulbus propionicus Widdel 1981 is the type species of the genus Desulfobulbus, which belongs to the family Desulfobulbaceae. The species is of interest because of its great implication in the sulfur cycle in aquatic sediments, its large substrate spectrum and a broad versatility in using various fermentation pathways. The species was the first example of a pure culture known to disproportionate elemental sulfur to sulfate and sulfide. This is the first completed genome sequence of a member of the genus Desulfobulbus and the third published genome sequence from a member of the family Desulfobulbaceae. The 3,851,869 bp long genome with its 3,351 protein-coding and 57 RNA genes is a part of the Genomic Encyclopedia of Bacteria and Archaea project.
Similar content being viewed by others

Introduction
Strain 1pr3T “Lindhorst” (= DSM 2032 = ATCC 33891 = VKM B-1956) is the type strain of the species Desulfobulbus propionicus, which is the type species of the genus Desulfobulbus [1,2]. The genus currently consists of five validly published named species [3]. The genus name is derived from the Neo-Latin word ‘desulfo-’ meaning ‘desulfurizing’ and the Latin word ‘bulbus’ meaning ‘a bulb or an onion’, yielding the ‘onion-shaped sulfate reducer’ [2]. The species epithet is derived from the Neo-Latin word ‘acidum propionicum’ and the Latin suffix ‘-icus’ in the sense of ‘pertaining to’; ‘propionicus’ = ‘pertaining to propionic acid’ [2]. Strain 1pr3T “Lindhorst” was isolated by Fritz Widdel in 1982 from anaerobic mud of a village ditch in Lindhorst near Hannover [4]. Other strains have been isolated from anaerobic mud in a forest pond near Hannover and from a mud flat of the Jadebusen (North Sea) [4], from an anaerobic intertidal sediment in the Ems-Dollard estuary (Netherlands) [5], and from a sulfate-reducing fluidized bed reactor inoculated with mine sediments and granular sludge [6]. Several studies have been carried out on the metabolic pathways of the strain 1pr3T [4,7,8]. Here we present a summary classification and a set of features for D. propionicus strain 1pr3T, together with the description of the complete genomic sequencing and annotation.
Classification and features
A representative genomic 16S rRNA sequence of strain 1pr3T was compared using NCBI BLAST under default settings (e.g., considering only the high-scoring segment pairs (HSPs) from the best 250 hits) with the most recent release of the Greengenes database [9] and the relative frequencies, weighted by BLAST scores, of taxa and keywords (reduced to their stem [10]) were determined. The four most frequent genera were Desulfobulbus (76.1%), Desulfurivibrio (11.9%), Desulforhopalus (8.1%) and Desulfobacterium (3.9%) (19 hits in total). Regarding the eleven hits to sequences from members of the species, the average identity within HSPs was 95.1%, whereas the average coverage by HSPs was 94.7%. Regarding the nine hits to sequences from other members of the genus, the average identity within HSPs was 94.9%, whereas the average coverage by HSPs was 94.9%. Among all other species, the one yielding the highest score was Desulfobulbus elongatus, which corresponded to an identity of 96.9% and an HSP coverage of 93.8%. The highest-scoring environmental sequence was FJ517134 (“semiarid ‘Tablas de Daimiel National Park’ wetland (Central Spain) unraveled water clone TDNP Wbc97 92 1 234′), which showed an identity of 97.8% and a HSP coverage of 98.3%. The five most frequent keywords within the labels of environmental samples which yielded hits were ‘sediment’ (8.4%), ‘marin’ (2.9%), ‘microbi’ (2.5%), ‘sea’ (1.7%) and ‘seep’ (1.7%) (231 hits in total). These keywords are in line with habitats from which the cultivated strains of D. propionicus were isolated. Environmental samples which resulted in hits of a higher score than the highest scoring species were not found.
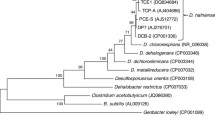
Figure 1 shows the phylogenetic neighborhood of D. propionicus in a 16S rRNA based tree. The sequences of the two 16S rRNA gene copies in the genome do not differ from each other, and differ by two nucleotides from the previously published 16S rRNA sequence (AY548789).

Phylogenetic tree highlighting the position of D. propionicus relative to the other type strains within the family Desulfobulbaceae. The tree was inferred from 1,425 aligned characters [11,12] of the 16S rRNA gene sequence under the maximum likelihood criterion [13] and rooted in accordance with the current taxonomy. The branches are scaled in terms of the expected number of substitutions per site. Numbers above branches are support values from 200 bootstrap replicates [14] if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [15] are shown in blue, published genomes [16] in bold.
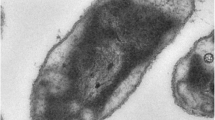
The cells of D. propionicus are ellipsoidal to lemon-shaped (1-1.3 by 1.8–2 µm) (Figure 2). D. propionicus is a Gram-negative and non-sporulating bacterium (Table 1) that produces fimbriae [4]. The temperature range for growth is between 10ºC and 43ºC, with an optimum at 39ºC [4]. The pH range for growth is between 6.0 and 8.6, with an optimum at pH 7.1–7.5 [4]. Strain 1pr3T is described to be nonmotile, with no flagellum detected by electron microscopy [4], although the genome sequence suggests it to be comprehensively equipped with the genes required for flagellar assembly (see below). The closely related strains 2pr4 and 3pr10 were motile by a single polar flagellum [4], suggesting either a recent mutational loss of flagellar motility in strain 1pr3T, or a failure to express the genes under the conditions of growth. D. propionicus was initially described to be a strictly anaerobic chemoorganotroph [4]. Further studies a decade later indicated that this organism was able to grow in the presence of oxygen while oxidizing sulfide, elemental sulfur, sulfite and polysulfide to sulfate [27], where mainly thiosulfate was formed from elemental sulfur [27,28]. D. propionicus is the first example of a pure culture known to disproportionate elemental sulfur to sulfate and sulfide [7]. But growth of D. propionicus with elemental sulfur as the electron donor and Fe(III) as a sulfide sink and/or electron acceptor was very slow [7]. It ferments three moles of pyruvate to two moles acetate and one mole of propionate stoichiometrically via the methylmalonyl-CoA pathway [8]. Strain 1pr3T was also found to reduce iron to sustain growth [7]. Fe(III) greatly stimulated sulfate production, and D. propionicus produced as much sulfate in the absence of Mn(IV) or Fe(III) as it did with Mn(IV) [7]. In the absence of sulfate, ethanol is fermented to propionate and acetate in a molar ratio of 2:1 [24], while i-propanol is produced during the fermentation of ethanol [24]. In the presence of H2 and CO2, ethanol is quantitatively converted to propionate [24]. H2-plus sulfate-grown cells of the strain 1pr3T were able to oxidize 1-propanol and 1-butanol to propionate and butyrate respectively with the concomitant reduction of acetate plus CO2 to propionate [24]. Growth on H2 required acetate as a carbon source in the presence of CO2 [4]. Strain 1pr3T is also able to grow mixotrophically on H2 in the presence of an organic compound [24]. When the amounts of sulfate and ethanol are limiting, D. propionicus competes successfully with Desulfobacter postgatei, another sulfate reducer [29]. Propionate, lactate, ethanol and propanol were used as electron donors and carbon sources [4]. Together with pyruvate, they are oxidized to acetate as an end-product [4]. Butyrate may be used in a few cases [4]. Sulfide oxidation in D. propionicus is biphasic, proceeding via oxidation to elemental sulfur, followed by sulfur disproportionation to sulfide and sulfate [7,27,30]. However, the uncoupler tetrachlorosalicylanilide (TCS) and the electron transport inhibitor myxothiazol inhibited sulfide oxidation to sulfate and caused accumulation of sulfur [30]. But in the presence of the electron transport inhibitor 2-n-heptyl-4-hydroxyquinoline-N-oxide (HQNO), sulfite and thiosulfate were formed [30]. When grown on lactate or pyruvate, the strain 1pr3T is able to grow without an external electron acceptor and formed propionate and acetate as fermentation products [4,31]. For this purpose, the substrates are fermented via the methylmalonyl-CoA pathway [31]. In the cells of D. propionicus, the activities of methylmalonyl-CoA: pyruvate transcarboxylase, a key enzyme of methylmalonyl-CoA pathway, as well as the other enzymes (pyruvate dehydrogenase, succinate dehydrogenase and malate dehydrogenase) involved in the pathway were detected [31]. D. propionicus can convert not only pyruvate but also alcohols via methylmalonyl-CoA pathway in the absence of sulfate [24,32,33]. Inorganic pyrophosphatase was present in strain 1pr3T at high levels of activity, but the enzyme was Mg2+-dependent and stimulated by Na2S2O4 [34]. However, isocitrate lyase and pyrophosphate-dependent acetate kinase were not detected [34]. Sulfate, sulfite and thiosulfate serve as electron acceptors and are reduced to H2S, but not elemental sulfur, malate, fumarate [4]. Nitrate also served as electron acceptor and was reduced to ammonia [4,27]. Acetate, valerate, higher fatty acids, succinate, fumarate, malate, sugars are not utilized [4]. Strain 1pr3T requires 4-aminobenzoic acid as growth factor [4,6]. Cell membrane and cytoplasmic fraction contain b- and c-type cytochromes [4].

Scanning electron micrograph of D. propionicus 1pr3T
Chemotaxonomy
Odd-chain fatty acids predominated in the fatty acid profile of the strain 1pr3T (77% of the total fatty acids vs. 23% for the even-chain fatty acids) [35,36], reflecting the use of propionate as a chain initiator for fatty acid biosynthesis [35]. The major fatty acids, when grown on propionate, were found to be C17:1ω6 (51.5%), C15:0 (28.3%), C16:0 (6.9%), C14:0 (5.2%), C18:0 (3.1%), C15:1 ω6 and C16:1 ω5, (2.4% each) and C18:1 ω7 (2.1%). The minor fatty acids were C17:0 (0.6% of the total fatty acids), C16:1 ω7 (0.9%), C18:1 ω9 and C15:1Δ7 (1.0% each), C12:0 (1.3%), C17:1 ω8 (1.6%) and C13:0 (1.7%) [36].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position [37], and is part of the Genomic Encyclopedia of Bacteria and Archaea project [38]. The genome project is deposited in the Genomes OnLine Database [15] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
D. propionicus 1pr3T, DSM 2032, was grown anaerobically in DSMZ medium 194 (Desulfobulbus medium) [39] at 37°C. DNA was isolated from 0.5–1 g of cell paste using MasterPure Gram-positive DNA purification kit (Epicentre MGP04100) following the standard protocol as recommended by the manufacturer, with modification st/LALM for cell lysis as described in Wu et al. [38]. DNA is available through the DNA Bank Network [40,41].
Genome sequencing and assembly
The genome was sequenced using a combination of Illumina and 454 sequencing platforms. All general aspects of library construction and sequencing can be found at the JGI website [42]. Pyrosequencing reads were assembled using the Newbler assembler version 2.0.00.20-PostRelease-11-05-2008-gcc-3.4.6 (Roche). The initial Newbler assembly consisting of 35 contigs in two scaffolds was converted into a phrap [43] assembly by making fake reads from the consensus, to collect the read pairs in the 454 paired end library. Illumina GAii sequencing data (327Mb) was assembled with Velvet [44] and the consensus sequences were shredded into 1.5 kb overlapped fake reads and assembled together with the 454 data. The 454 draft assembly was based on 145.0 Mb 454 draft data and all of the 454 paired end data. Newbler parameters are -consed -a 50 -l 350 -g -m -ml 20. The Phred/Phrap/Consed software package [43] was used for sequence assembly and quality assessment in the subsequent finishing process. After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with gapResolution [42], Dupfinisher [45], or sequencing cloned bridging PCR fragments with subcloning or transposon bombing (Epicentre Biotechnologies, Madison, WI). Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR primer walks (J.-F.Chang, unpublished). A total of 563 additional reactions and five shatter libraries were necessary to close gaps and to raise the quality of the finished sequence. Illumina reads were also used to correct potential base errors and increase consensus quality using a software Polisher developed at JGI [46]. The error rate of the completed genome sequence is less than 1 in 100,000. Together, the combination of the Illumina and 454 sequencing platforms provided 147.6 × coverage of the genome. The final assembly contained 475,513 pyrosequence and 11,740,513 Illumina reads.
Genome annotation
Genes were identified using Prodigal [47] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [48]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [49].
Genome properties
The genome consists of a 3,851,869 bp long chromosome with a GC content of 58.9% (Table 3 and Figure 3). Of the 3,408 genes predicted, 3,351 were protein-coding genes, and 57 RNAs; 68 pseudogenes were also identified. The majority of the protein-coding genes (70.5%) were assigned with a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.

Graphical circular map of the chromosome. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
References
Validation List no. 7. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. Int J Syst Bacteriol 1981; 31:382–383. doi:10.1099/00207713-31-3-382
Widdel F. 1980. Anaerober Abbau von Fettsäuren und Benzoesäure durch neu isolierte Arten Sulfatreduzierender Bakterien. Dissertation. Georg August-Universität zu Göttingen. Lindhorst/Schaumburg-Lippe, Göttingen, Germany, 443 p.
Garrity G. NamesforLife. BrowserTool takes expertise out of the database and puts it right in the browser. Microbiol Today 2010; 37:9.
Widdel F, Pfennig N. Studies on dissimilatory sul-fate-reducing bacteria that decompose fatty acids II. Incomplete oxidation of propionate by Desul-fobulbus propionicus gen. nov., sp. nov. Arch Microbiol 1982; 131:360–365. doi:10.1007/BF00411187
Laanbroek HJ, Pfennig N. Oxidation of short-chain fatty acids by sulfate-reducing bacteria in freshwater and in marine sediments. Arch Microbiol 1981; 128:330–335. PubMed doi:10.1007/BF00422540
Kaksonen AH, Plumb JJ, Robertson WJ, Franzmann PD, Gibson JAE, Puhakka JA. Culturable diversity and community fatty acid profiling of sulfate-reducing fluidized-bed reactors treating acidic, metal-containing wastewater. Geomicrobiol J 2004; 21:469–480. doi:10.1080/01490450490505455
Lovley DR, Phillips EJP. Novel processes for anaerobic sulfate production from elemental sulfur by sulfate-reducing bacteria. Appl Environ Microbiol 1994; 60:2394–2399. PubMed
Tasaki M, Kamagata Y, Nakamura K, Okamura K, Minami K. Acetogenesis from pyruvate by Desul-fotomaculum thermobenzoicum and differences in pyruvate metabolism among three sulfate-reducing bacteria in the absence of sulfate. FEMS Microbiol Lett 1993; 106:259–263. doi:10.1111/j.1574-6968.1993.tb05973.x
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie E, Keller K, Huber T, Dalevi D, Hu P, Andersen G. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069–5072. PubMed doi:10.1128/AEM.03006-05
Porter MF. An algorithm for suffix stripping. Program: electronic library and information systems 1980; 14:130–137. doi:10.1108/eb046814
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540–552. PubMed
Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452–464. PubMed doi:10.1093/bioinformatics/18.3.452
Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 2008; 57:758–771. PubMed doi:10.1080/10635150802429642
Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? Lect Notes Comput Sci 2009; 5541:184–200. doi:10.1007/978-3-642-02008-7_13
Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Hugenholtz P, Markowitz VM, Kyrpides NC. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2010; 38:D346–D354. PubMed doi:10.1093/nar/gkp848
Rabus R, Ruepp A, Frickey T, Rattei T, Fartmann B, Stark M, Bauser M, Zibat A, Lombardot T, Becker I, et al. The genome of Desulfotalea psychrophila, a sulfate-reducing bacterium from permanently cold Arctic sediments. Environ Microbiol 2004; 6:887–902. PubMed doi:10.1111/j.1462-2920.2004.00665.x
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed doi:10.1038/nbt1360
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed doi:10.1073/pnas.87.12.4576
Garrity GM, Holt JG. The Road Map to the Manual. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 1. Springer, New York 2001:119–169.
Validation List No. 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 2006; 56:1–6. PubMed doi:10.1099/ijs.0.64188-0
Kuever J, Rainey FA, Widdel F. Class IV. Deltaproteobacteria class. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 922.
Kuever J, Rainey FA, Widdel F. Order III. Desul-fobacterales ord. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 959.
Kuever J, Rainey FA, Widdel F. Family II. Desul-fobulbaceae fam. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part C, Springer, New York, 2005, p. 988.
Laanbroek HJ, Abee T, Voogd IL. Alcohol conversions by Desulfobulbus propionicus Lindhorst in the presence and absence of sulfate and hydrogen. Arch Microbiol 1982; 133:178–184. doi:10.1007/BF00414998
Classification of bacteria and archaea in risk groups. http://www.baua.de TRBA 466.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet 2000; 25:25–29. PubMed doi:10.1038/75556
Dannenberg S, Kroder M, Dilling W, Cypionka H. Oxidation of H2, organic compounds and inorganic sulfur compounds coupled to reduction of O2 or nitrate by sulfate-reducing bacteria. Arch Microbiol 1992; 158:93–99. doi:10.1007/BF00245211
Cypionka H, Widdel F, Pfennig N. Survival of sulfate-reducing bacteria after oxygen stress, and growth in sulfate-free oxygen-sulfide gradients. FEMS Microbiol Ecol 1985; 31:39–45. doi:10.1111/j.1574-6968.1985.tb01129.x
Laanbroek HJ, Geerligs HJ, Sijtsma L, Veldkamp H. Competition for sulfate and ethanol among Desulfobacter, Desulfobulbus, and Desulfovibrio species isolated from intertidal sediments. Appl Environ Microbiol 1984; 47:329–334. PubMed
Fuseler K, Cypionka H. Elemental sulfur as an intermediate of sulfide oxidation with oxygen by Desulfobulbus propionicus. Arch Microbiol 1995; 164:104–109. doi:10.1007/BF02525315
Tasaki M, Kamagata Y, Nakamura K, Okamura K, Minami K. Acetogenesis from pyruvate by Desul-fotomaculum thermobenzoicum and differences in pyruvate metabolism among three sulfate-reducing bacteria in the absence of sulfate. FEMS Microbiol Lett 1993; 106:259–263. doi:10.1111/j.1574-6968.1993.tb05973.x
Stams AJM, Kremer DR, Nicolay K, Weenk GH, Hansen TA. Pathway of propionate formation in Desulfobulbus propionicus. FEMS Microbiol Lett 1988; 49:273–277.
Tasaki M, Kamagata Y, Nakamura K, Mikami E. Propionate formation from alcohols or aldehydes by Desulfobulbus propionicus in the absence of sulfate. J Ferment Bioeng 1992; 73:329–331. doi:10.1016/0922-338X(92)90195-Z
Kremer DR, Hansen TA. Pathway of propionate degradation in Desulfobulbus propionicus. FEMS Microbiol Lett 1988; 49:273–277. doi:10.1111/j.1574-6968.1988.tb02729.x
Taylor J, Parkes RJ. The cellular fatty acids of the sulphate-reducing bacteria, Desulfobacter sp., Desulfobulbus sp. and Desulfovibvio desulfuvicans. J Gen Microbiol 1983; 129:3303–3309.
Parkes RJ, Calder AG. The cellular fatty acids of three strains of Desulfobulbus, a propionateutilising sulphate-reducing bacterium. FEMS Microbiol Ecol 1985; 31:361–363. doi:10.1111/j.1574-6968.1985.tb01172.x
Klenk HP, Göker M. En route to a genome-based classification of Archaea and Bacteria? Syst Appl Microbiol 2010; 33:175–182. PubMed doi:10.1016/j.syapm.2010.03.003
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056–1060. PubMed doi:10.1038/nature08656
List of growth media used at DSMZ: http://www.dsmz.de/microorganisms/media_list.php.
Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch A, Berendsohn WG, Wägele JW. The DNA Bank Network: the start from a German initiative. Biopreservation and Biobanking (In press).
DNA Bank Network. http://www.dnabank-network.org
DOE Joint Genome Institute. http://www.jgi.doe.gov
Phrap and Phred for Windows. MacOS, Linux, and Unix. http://www.phrap.com
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821–829. PubMed doi:10.1101/gr.074492.107
Han C, Chain P. 2006. Finishing repeat regions automatically with Dupfinisher. in Proceeding of the 2006 international conference on bioinformatics & computational biology. Edited by Hamid R. Arabnia & Homayoun Valafar, CSREA Press. June 26–29, 2006: 141–146.
Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008.
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119. PubMed doi:10.1186/1471-2105-11-119
Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455–457. PubMed doi:10.1038/nmeth.1457
Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271–2278. PubMed doi:10.1093/bioinformatics/btp393
Acknowledgements
We would like to gratefully acknowledge the help of Katja Steenblock (DSMZ) for growing D. propionicus cultures. This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396, UT-Battelle and Oak Ridge National Laboratory under contract DE-AC05-00OR22725, as well as German Research Foundation (DFG) INST 599/1-2.
Author information
Authors and Affiliations
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Pagani, I., Lapidus, A., Nolan, M. et al. Complete genome sequence of Desulfobulbus propionicus type strain (1pr3T). Stand in Genomic Sci 4, 100–110 (2011). https://doi.org/10.4056/sigs.1613929
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.1613929