
Abstract
Thermomonospora curvata Henssen 1957 is the type species of the genus Thermomonospora. This genus is of interest because members of this clade are sources of new antibiotics, enzymes, and products with pharmacological activity. In addition, members of this genus participate in the active degradation of cellulose. This is the first complete genome sequence of a member of the family Thermomonosporaceae. Here we describe the features of this organism, together with the complete genome sequence and annotation. The 5,639,016 bp long genome with its 4,985 protein-coding and 76 RNA genes is a part of the Genomic Encyclopedia of Bacteria and Archaea project.
Similar content being viewed by others


Introduction
Strain B9T (= DSM 43183 = ATCC 19995 = JCM 3096) is the type strain of Thermomonospora curvata, which in turn is the type species of the genus Thermomonospora [1]. T. curvata was effectively published in 1957 [1]. When the original strains R30 and R71 were no longer cultivable, strain B9 was proposed as the neotype in 1967 [2]. Currently, there are two species in the genus Thermomonospora, which in turn is one of the six genera in the family Thermomonosporaceae [3]. The generic name Thermomonospora was proposed by Henssen [1] for thermophilic actinomycetes isolated from composted stable manure [4]. Strain B9T was isolated from municipal refuse compost samples [1]. Other (rubber degrading) strains of T. curvata have been isolated from food residues used in animal husbandry in Egypt (strain E4), from compost in Germany (strain E5) [5], and also from municipal solid waste compost (probably USA) [6–9]. Cellulase biosynthesis has been studied in a catabolite repression-resistant mutant of T. curvata [10]. Here we present a summary classification and a set of features for T. curvata strain B9T, together with the description of the complete genomic sequencing and annotation.
Classification and features
The 16S rRNA gene sequence of the strain B9T (AF002262) shows 98.1% identity with the 16S rRNA gene sequence of T. curvata strain E5 (AY525766) [5].The distance of strain B9T to other members of this family ranged between 5% and 7%. Further analysis shows 94% 16S rRNA gene sequence identity with an uncultured bacterium, clone BG079 (HM362496) and 92% similarity to compost metagenome contig00434 (ADGO01000428) [11] from metagenomic libraries (env_nt) (status October 2010). A representative genomic 16S rRNA sequence of T. curvata was compared using NCBI BLAST under default settings (e.g., considering only the high-scoring segment pairs (HSPs) from the best 250 hits) with the most recent release of the Greengenes database [12] and the relative frequencies, weighted by BLAST scores, of taxa and keywords (reduced to their stem [13]) were determined. The five most frequent genera were Actinomadura (54.3%), Nocardiopsis (12.5%), Actinocorallia (8.8%), Jiangella (5.8%) and Actinoallomurus (5.0%) (208 hits in total). Regarding the two hits to sequences from members of the species, the average identity within HSPs was 99.9%, whereas the average coverage by HSPs was 96.2%. Regarding the single hit to sequences from other members of the genus, the average identity within HSPs was 95.2%, whereas the average coverage by HSPs was 58.4%. Among all other species, the one yielding the highest score was Actinomadura cremea, which corresponded to an identity of 96.3% and a HSP coverage of 85.3%. The highest-scoring environmental sequence was HM362496 (‘microbial naturally composting sugarcane piles decomposting bagasse clone BG079’), which showed an identity of 94.5% and a HSP coverage of 96.3%. Within the labels of environmental samples which yielded hits, the five most frequent keywords were ‘soil’ (4.7%), ‘compost’ (3.1%), ‘microbi’ (2.4%), ‘skin’ (2.0%) and ‘acid’ (2.0%) (41 hits in total). These keywords partially fit to the ecology of compost and food residues, from which the known strains have been isolated [1,5,6]. Environmental samples which yielded hits of a higher score than the highest scoring species were not found.
Figure 1 shows the phylogenetic neighborhood of T. curvata B9T in a 16S rRNA based tree. The sequences of the four 16S rRNA gene copies in the genome differ from each other by up to one nucleotide, and differ by up to five nucleotides from the previously published 16S rRNA sequence (D86945), which contains one ambiguous base call.

Phylogenetic tree highlighting the position of L. byssophila relative to the type strains of the other genera within the family Cytophagaceae. The tree was inferred from 1,340 aligned characters [15–16] of the 16S rRNA gene sequence under the maximum likelihood criterion [17] and rooted with the type strain of the closely related family Sphingobacteriaceae. The branches are scaled in terms of the expected number of substitutions per site. Numbers above branches are support values from 900 bootstrap replicates [18] if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [19] are shown in blue, published genomes in bold [4,20,21].
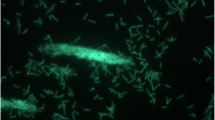
Strain B9T is facultatively aerobic, Gram-positive, non-acid-alcohol-fast, and chemoorganotrophic [1,4 Table 1]. Based on the original literature, the morphology of neotype B9T was the same as of the original strains [1–2]. Substrate mycelium was branched and bared aerial hyphae that differentiated into single or short chains of arthrospores [2–4] (Figure 2, arthrospores not visible). Spores were formed by the differentiation of the sporophores when they reached a given width [2]. Polymorphic and single spores in clusters appeared with a folded surface on branched and unbranched sporophores [2]. They had spindle, lemon or pear forms varying between 0.6–1.5 x 0.3–0.9 µm [2]. The optimal growth occurred at 50°C. However, weak growth was observed at 40°C and 65°C, but no growth at 28°C [2]. Colonies were white or yellow depending on culture medium [2]. On meat extract agar, the growth was moderate, aerial white mycelium formed and the colonies were yellow to brown [2]. On asparagine glucose agar, the growth was low and the aerial mycelium white [2]. On casein glucose agar, a few single colonies were observed [2]. The growth was good and the aerial mycelium white on cellulose agar medium [2]. On Czapek agar, a few spotty colonies were observed [2]. On Czapek peptone agar, the growth was good, almost no aerial mycelium formation [2]. When oatmeal agar was used as medium, the growth was good and the aerial mycelium white [2]. The growth on yeast agar was good, with thick aerial mycelium. In this case, colonies were partially yellow [2]. On yeast glucose agar, the growth was good, aerial mycelium developed later and was white while brownish colonies were formed [2]. On yeast starch agar, the growth was good, white aerial mycelium was formed and colonies were yellow to orange [2]. On potato agar I, spotty growth is observed, while no aerial mycelium was formed [2]. Few single colonies formed on potato agar II [2]. On starch agar medium, the growth was moderate and aerial mycelium was white [2]. Strain B9T showed endogluconase activity and attacks cellulose [4]. It was also active in the decomposition of municipal waste compost [6–9]. When grown on protein-extracted lucerne fiber compound, strain B9T released 16 times more β-glucosidases compared to growth on cellulose or purified cellulose [34]. Strain B9T grew well at pH 7.5 on any nutrient medium that contains some yeast extract. It showed significant growth even at pH 11 [35]. Tests of the nitrate reduction and phosphatase were positive [4]. The sole carbon sources (1%, w/v) were ribose and sucrose. L-arabinose, galactose, lactose and mannitol were not used [4]. Strain B9T was able to degrade agar, cellulose powder (MN300), carboxymethylcellulose, keratin, xylan, starch, Tween 20 and Tween 80 [4]. Growth was also observed in the presence of crystal violet (0.2 µg/ml), but it was inhibited by kanamycin and novobiocin (each 25µg/ml) [4]. The inability to utilize pectin is an important feature that differentiates strain B9T from other members of the genus Thermomonospora. Amylases of the strain B9T were extremely active and stable at 60–70°C and slightly acid to neutral pH [36–38]. Also, endoglucanase and exoglucanase were active in the strain [39]. Cellobiose was found to be a good cellulase inducer [40].

Scanning electron micrograph of L. byssophila 4M15T
Chemotaxonomy
Strain B9T possesses a cell wall type III with A1γ and with meso-diaminopimelic acid as major constituent [4,41,42]. The principal menaquinones are MK-9(H4), MK-9(H6) and MK-9(H8), with MK-9(H6) being the predominant one (the profile type sensu Kroppenstedt is termed 4B2 [43]) [4]. The fatty acid profile was described to be of type 3a [4]. Members of this type can synthesize terminally branched and 10-methyl-branched fatty acids [43]. T. curvata lacks madurose, a type C sugar and has polar lipids of type IV [4], represented by phosphatidylinositol (PI) and unknown phospholipids (PL), which according to Lechevalier [44–45] are, phosphatidyglycerol (PE), phosphatidylinositolmannosides(PIM) and diphosphatidylglycerol (DPG) [4].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position [46], and is part of the Genomic Encyclopedia of Bacteria and Archaea project [47]. The genome project is deposited in the Genomes OnLine Database [19] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
T. curvata B9T, DSM 43183, was grown in DSMZ medium 550 (CYC medium, modified following Cross and Attwell, 1973) [48] at 45°C. DNA was isolated from 0.5–1 g of cell paste using MasterPure Gram-positive NDA purification kit (Epicentre MGP04100) following the standard protocol as recommended by the manufacturer, with modification st/LALM for cell lysis as described in Wu et al. [47]. DNA is available through the DNA bank Network [49–50].
Genome sequencing and assembly
The genome of was sequenced using a combination of Sanger and 454 sequencing platforms. All general aspects of library construction and sequencing can be found at the JGI website [51]. Pyrosequencing reads were assembled using the Newbler assembler version 1.1.02.15 (Roche). Large Newbler contigs were broken into 6,203 overlapping fragments of 1,000 bp and entered into assembly as pseudo-reads. The sequences were assigned quality scores based on Newbler consensus q-scores with modifications to account for overlap redundancy and adjust inflated q-scores. A hybrid 454/Sanger assembly was made using the parallel phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with Dupfinisher [52] or transposon bombing of bridging clones (Epicentre Biotechnologies, Madison, WI). A total of 2,673 Sanger finishing reads were produced to close gaps, to resolve repetitive regions, and to raise the quality of the finished sequence. Illumina reads that were used to correct potential base errors and increase consensus quality using a software Polisher developed at JGI [53]. The error rate of the completed genome sequence is less than 1 in 100,000. Together, the combination of the Sanger and 454 sequencing platforms provided 36.3 × coverage of the genome. The final assembly contains 73,067 Sanger reads and 602,893 pyrosequencing reads.
Genome annotation
Genes were identified using Prodigal [54] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [55]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [56].
Genome properties
The genome consists of a 5,639,016 bp long chromosome with a 71.6% GC content (Table 3 and Figure 3). Of the 5,061 genes predicted, 4,985 were protein-coding genes, and 76 RNAs; ninety five pseudogenes were also identified. The majority of the protein-coding genes (64.7%) were assigned with a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.

Graphical circular map of the genome. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
References
Henssen A. Beiträge zur Morphologie und Systematik der thermophilen Actinomyceten. Arch Mikrobiol 1957; 26:373–414. PubMed doi:10.1007/BF00407588
Henssen A, Schnepf E. Zur Kenntnis thermophiler Actinomyceten. Arch Microbiol 1967; 57:214–231. doi:10.1007/BF00405948
Garrity G. NamesforLife. BrowserTool takes expertise out of the database and puts it right in the browser. Microbiol Today 2010; 37:9.
Kroppenstedt RM, Goodfelow M. 2006. The family Thermomonosporaceae: Actinocorallia, Actinomadura, Spirillospora and Thermomonospora. In: M Dworkin, S Falkow, E Rosenberg, KH Schleifer E Stackebrandt (eds), The Prokaryotes, 3. ed, vol. 7. Springer, New York, p. 682–724.
Ibrahim EM, Arenskotter M, Luftmann H, Steinbuchel A. Identification of poly (cis-1,4-isoprene) degradation intermediates during growth of moderately thermophilic actinomycetes on rubber and cloning of a functonal Icp homologue from Nocardia farcinaca strain E1. Appl Environ Microbiol 2006; 72:3375–3382. PubMed doi:10.1128/AEM.72.5.3375-3382.2006
Stutzenberger FJ. Cellulase production by Thermomonospora curvata isolated from municipal solid waste compost. Appl Microbiol 1971; 22:147–152. PubMed
Stutzenberger FJ. Cellulolytic activity of Thermomonospora curvata. 1: Nutritional requirements for cellulase production. Appl Microbiol 1972; 24:77–82. PubMed
Stutzenberger FJ. Cellulolytic activity of Thermomonospora curvata. 2: Optimal conditions, partial purification and product of the cellulase. Appl Microbiol 1972; 24:83–90. PubMed
Stutzenberger FJ, Kaufman AJ, Lossin RD. Cellulolytic activity in municipal solid waste compost. Can J Microbiol 1970; 16:553–560. PubMed doi:10.1139/m70-093
Fennington G, Neubauer D, Stutzenberger FJ. Cellulase biosynthesis in a catabolite repression-resistant mutant of Thermomonospora curvata. Appl Environ Microbiol 1984; 47:201–204. PubMed
Allgaier M, Reddy A, Park JI, Ivanova N, D’haeseleer P, Lowry S, Sapra R, Hazen TC, Simmons BA, VanderGheynst JS, et al. Targeted discovery of glycoside hydrolases from a switchgrass-adapted compost community. PLoS ONE 2010; 5:e8812. PubMed doi:10.1371/journal.pone.0008812
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie E, Keller K, Huber T, Dalevi D, Hu P, Andersen G. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069–5072. PubMed doi:10.1128/AEM.03006-05
Porter MF. An algorithm for suffix stripping. Program: electronic library and information systems 1980; 14:130–137.
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540–552. PubMed
Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452–464. PubMed doi:10.1093/bioinformatics/18.3.452
Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 2008; 57:758–771. PubMed doi:10.1080/10635150802429642
Yarza P, Richter M, Peplies J, Euzeby J, Amann R, Schleifer KH, Ludwig W, Glöckner FO, Rosselló-Móra R. The All-Species Living Tree project: A 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst Appl Microbiol 2008; 31:241–250. PubMed doi:10.1016/j.syapm.2008.07.001
Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? Lect Notes Comput Sci 2009; 5541:184–200. doi:10.1007/978-3-642-02008-7_13
Liolios K, Mavromatis K, Tavernarakis N, Kyrpides NC. The Genomes On Line Database (GOLD) in 2007: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2008; 36:D475–D479. PubMed doi:10.1093/nar/gkm884
Liolios K, Sikorski J, Jando M, Lapidus A, Copeland A, Rio TGD, Nolan M, Lucas S, Tice H, Cheng JF, et al. Complete genome sequence of Thermobispora bispora type strain (R51T). Stand Genomic Sci 2010; 2:318–326. doi:10.4056/sigs.962171
Nolan M, Sikorski J, Jando M, Lucas S, Lapidus A, Rio TGD, Chen F, Tice H, Pitluck S, Cheng JF, et al. Complete genome sequence of Streptosporangium roseum type strain (NI 9100T). Stand Genomic Sci 2010; 2:29–37. doi:10.4056/sigs.631049
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed doi:10.1038/nbt1360
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed doi:10.1073/pnas.87.12.4576
Garrity GM, Holt JG. The Road Map to the Manual. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 119–169.
Stackebrandt E, Rainey FA, Ward-Rainey NL. Proposal for a new hierarchic classification system, Actinobacteria classis nov. Int J Syst Bacteriol 1997; 47:479–491. doi:10.1099/002 07713-47-2-479
Buchanan RE. Studies in the nomenclature and classification of bacteria. II. The primary subdivisions of the Schizomycetes. J Bacteriol 1917; 2:155–164. PubMed
Skerman VBD, McGowan V, Sneath PHA. Approved lists of bacterial names. Int J Syst Bacteriol 1980; 30:225–420. doi:10.1099/00207713-30-1-225
Zhi XY, Li WJ, Stackebrandt E. An update of the structure and 16S rRNA gene sequence-based definition of higher ranks of the class Actinobacteria, with the proposal of two new suborders and four new families and emended descriptions of the existing higher taxa. Int J Syst Evol Microbiol 2009; 59:589–608. PubMed doi:10.1099/ijs.0.65780-0
Zhang Z, Kudo T, Nakajima Y, Wang Y. Clarification of the relationship between the members of the family Thermomonosporaceae on the basis of 16S rDNA, 16S–23S rRNA internal transcribed spacer and 23S rDNA sequences and chemotaxonomic analyses. Int J Syst Evol Microbiol 2001; 51:373–383. PubMed
Küster E. Genus IV. Thermomonospora Henssen 1957, 398. In: Buchanan RE, Gibbons NE (eds), Bergey’s Manual of Determinative Bacteriology, Eighth Edition, The Williams and Wilkins Co., Baltimore, 1974, p. 858–859.
Zhang Z, Wang Y, Ruan J. Reclassification of Thermomonospora and Microtetraspora. Int J Syst Bacteriol 1998; 48:411–422. PubMed
Classification of bacteria and archaea in risk groups. http://www.baua.de TRBA 466.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet 2000; 25:25–29. PubMed doi:10.1038/75556
Bernier R, Stutzenberger F. Extracellular and cell-associated forms of beta-glucosidase in Thermomonospora curvata. Lett Appl Microbiol 1988; 7:103–107. doi:10.1111/j.1472-765X.1988.tb01263.x
Kempf A. 1995. Untersuchungen über thermophile Actinomyceten: Taxonomie, Ökologie, und Abbau von Biopolymeren [PhD dissertation]. University of Darmstadt. Darmstadt, Germany.
Kuo MJ, Hartman PA. Purification and partial characterization of Thermomonospora vulgaris amylases. Can J Microbiol 1967; 13:1157–1163. PubMed doi:10.1139/m67-160
Lupo D, Stutzenberger F. Changes in endoglucanase patterns during growth of Thermomonospora curvata on cellulose. Appl Environ Microbiol 1988; 54:588–589. PubMed
Stutzenberger F, Carnell R. Amylase production by Thermomonospora curvata. Appl Environ Microbiol 1977; 34:234–236. PubMed
Stutzenberger FJ, Lupo D. pH-dependent thermal activation of endo-1,4-β-glucanase in Thermomonospora curvata. Enzyme Microb Technol 1986; 8:205–208. doi:10.1016/0141-0229(86)90088-8
Stutzenberger FJ, Kahler G. Cellulase biosynthesis during degradation of cellulose derivatives by Thermomonospora curvata. J Appl Bacteriol 1986; 61:225–233.
Lechevalier MP, Lechevalier HA. 1970. Composition of whole-cell hydrolysates as a criterion in the classification of aerobic Actinomycetes. In: H. Prauser (Ed.) The Actinomycetales. Gustav Fischer-Verlag. Jena, Germany. p 311–316.
Schleifer KH, Kandler O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev 1972; 36:407–477. PubMed
Kroppenstedt RM. 1985. Fatty acid and menaquinone analysis of actinomycetes and related organisms. In: M. Goodfellow and D. E. Minnikin (Eds.) Chemical Methods in Bacterial Systematics. Academic Press. London, UK. 173–199.
Lechevalier MP. Biévre Cd, Lechevalier HA. Chemotaxonomy of aerobic actinomycetes: Phospholipid composition. Biochem Ecol Syst 1977; 5:249–260. doi:10.1016/0305-1978(77)90021-7
Lechevalier MP, Stern AE, Lechevalier HA. 1981. Phospholipids in the taxonomy of Actinomycetes. In: K.P. Schaal and G. Pulverer (Eds.) Actinomycetes. Gustav Fischer-Verlag. Jena, Germany.
Klenk HP, Göker M. En route to a genome-based classification of Archaea and Bacteria? Syst Appl Microbiol 2010; 33:175–182. PubMed doi:10.1016/j.syapm.2010.03.003
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056–1060. PubMed doi:10.1038/nature08656
List of growth media used at DSMZ: http://www.dsmz.de/microorganisms/media_list.php.
DNA bank Network. http://www.dnabank-network.org.
Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch A, Berendsohn WG, Wägele JW. The DNA Bank Network: the start from a German initiative. Biopreservation and Biobanking. (In press).
DOE Joint Genome Institute. http://www.jgi.doe.gov
Han CS, Chain P. Finishing repeat regions automatically with Dupfinisher. in Proceeding of the 2006 international conference on bioinformatics & computational biology. Edited by Hamid R. Arabnia & Homayoun Valafar, CSREA Press. June 26–29, 2006: 141–146.
Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008.
Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119. PubMed doi:10.1186/1471-2105-11-119
Pati A, Ivanova N, Mikhailova N, Ovchinikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: A gene prediction improvement pipeline for microbial genomes. Nat Methods 2010; 7:455–457. PubMed doi:10.1038/nmeth.1457
Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271–2278. PubMed doi:10.1093/bioinformatics/btp393
Acknowledgements
We would like to gratefully acknowledge the help of Marlen Jando for growing T. curvata cultures and Susanne Schneider for DNA extraction and quality analysis (both at DSMZ). This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396, UT-Battelle and Oak Ridge National Laboratory under contract DE-AC05-00OR22725, as well as German Research Foundation (DFG) INST 599/1-1.
Author information
Authors and Affiliations
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Chertkov, O., Sikorski, J., Nolan, M. et al. Complete genome sequence of Thermomonospora curvata type strain (B9T). Stand in Genomic Sci 4, 13–22 (2011). https://doi.org/10.4056/sigs.1453580
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.1453580