
Abstract
‘Thermobaculum terrenum’ Botero et al. 2004 is the sole species within the proposed genus ‘Thermobaculum’. Strain YNP1T is the only cultivated member of an acid tolerant, extremely thermophilic species belonging to a phylogenetically isolated environmental clone group within the phylum Chloroflexi. At present, the name ‘Thermobaculum terrenum’ is not yet validly published as it contravenes Rule 30 (3a) of the Bacteriological Code. The bacterium was isolated from a slightly acidic extreme thermal soil in Yellowstone National Park, Wyoming (USA). Depending on its final taxonomic allocation, this is likely to be the third completed genome sequence of a member of the class Thermomicrobia and the seventh type strain genome from the phylum Chloroflexi. The 3,101,581 bp long genome with its 2,872 protein-coding and 58 RNA genes is a part of the Genomic Encyclopedia of Bacteria and Archaea project.
Similar content being viewed by others

Introduction
Strain YNP1T (= ATCC BAA-798 = CCMEE 7001) is the proposed type strain of the not yet validly published species ‘Thermobaculum terrenum’, which represents the type species of the not yet validly published genus name ‘Thermobaculum’ [1]. The strain was cultivated from a moderately acidic (pH 3.9) extreme thermal soil in Yellowstone National Park (YNP), Wyoming (USA) for which a thorough chemotaxonomic characterization was published by Botero et al. in 2004 [1]. Although the biological characteristics of the novel strain fulfill all criteria required for the type strain of a novel genus, the proposed name ‘Thermobaculum terrenum’ (= hot small rod belonging to earth/soil) has not yet been validly published (= included in one of the updates of the Validation List that is regularly published in Int J Syst Evol Bacteriol), because rule 30 (3a) of the Bacteriological Code (1990 Revision), which requires that as of 1st January 2001 the description of a new species [...] must include the designation of a type strain, and a viable culture of that strain must be deposited in at least two publicly accessible service collections in different countries from which subcultures must be available [2]. Strain YNP1T is currently deposited only in two US culture collections. Here we present a summary classification and a set of features for ‘T. terrenum’ strain YNP1T, together with the description of the complete genomic sequencing and annotation.
Classification and features
Based on analyses of 16S rRNA gene sequences, strain YNPT is the sole cultured representative of the genus ‘Thermobaculum’. It has no close relatives among the validly described species within the Chloroflexi. The type strain of Sphaerobacter thermophilus [3] shares the highest pairwise similarity (84.9%), followed by Thermoleophilum album and T. minutum [4–6], the two sole members of the actinobacterial order Thermoleophilales [7] with 83.6% sequence identity, and three type strains from the clostridial genus Thermaerobacter (83.2-83.5%) [8], that are currently not placed within a named family. Only four uncultured bacterial clones in GenBank share a higher degree of sequence similarity with strain YNPT than the type strain of the ‘closest’ related species, S. thermophilus. These are clone DRV-SSB031 from rock varnish in the Whipple Mountains, California (92.1%) [9], and clones AY6_14 (FJ891044), AY6_27 (FJ891057) and AY6_18 (FJ891048) from quartz substrates in the hyperarid core of the Atacama Desert (86.9–87.9%). No phylotypes from environmental screening or metagenomic surveys could be linked to ‘T. terrenum’, indicating a rather rare occurrence in the habitats screened thus far (as of September 2010). A representative genomic 16S rRNA sequence of ‘T. terrenum’ YNPT was compared using BLAST with the most recent release of the Greengenes database [10] and the relative frequencies of taxa and keywords, weighted by BLAST scores, were determined. The three most frequent genera were Thermobaculum (81.2%), Sphaerobacter (10.3%) and Conexibacter (8.4%). The five most frequent keywords within the labels of environmental samples which yielded hits were ‘microbial’ (3.6%), ‘waste’ (3.3%), ‘soil’ (3.3%), ‘simulated’ (3.2%) and ‘level’ (3.1%). The five most frequent keywords within the labels of environmental samples which yielded hits of a higher score than the highest scoring species were ‘soil’ (4.5%), ‘structure’ (3.3%), ‘simulated’ (3.2%), ‘level/site/waste’ (2.9%) and ‘core’ (2.1%).
Figure 1 shows the phylogenetic neighborhood of ‘T. terrenum’ strain YNPT in a 16S rRNA based tree. The sequences of the two identical 16S rRNA gene copies in the genome do not differ from the previously published 1,333 nt long partial sequence generated from ATCC BAA-798 (AF391972).

Phylogenetic tree highlighting the position of ‘T. terrenum’ strain YNPT relative to the type strains of the other species within the phylum Chloroflexi. The trees were inferred from 1,316 aligned characters [11,12] of the 16S rRNA gene sequence under the maximum likelihood criterion [13] and rooted in accordance with the current taxonomy. The branches are scaled in terms of the expected number of substitutions per site. Numbers above the branches are support values from 1,000 bootstrap replicates [14] if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [15] are shown in blue, published genomes [16] and GenBank records [CP000804, CP000875, CP000909, CP001337] in bold, e.g. the GEBA genome S. thermophilus [17].
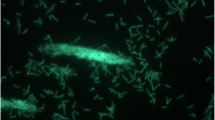
The cells of strain YNP1T are 1–1.5 × 2–3 µm long, non-motile rods (Figure 2 and Table 1), enveloped by a thick cell wall external to a cytoplasmic membrane [1]. YNP1T cells occur singly or in pairs, stain Gram-positive in the exponential growth-phase, are obligately aerobic, and non-spore-forming [1]. Colonies are pink-colored and growth occurs best at pH 6–8 (pHopt 7) and 67°C, with a possible temperature range of 41–75°C [1]. Culture doubling time at Topt was 4 hours and increases sharply above 70°C, whereas growth at the temperature extremes was relatively poor [1]. Cells grow best in complex media containing 0.5% NaCl and yeast extract (for growth factors) [1], but also on sucrose, fructose, glucose, ribose, xylose, sorbitol, and xylitol [1]. Strain YNP1T was positive for catalase, urease, and nitrate reduction, but tested negative for oxidases, and was also negative for fermentation of glucose or lactose [1]. No anaerobic growth was observed in the presence of sulfate, nitrate, ferric iron, or arsenate as possible electron acceptors [1]. No chemolithoautotrophic growth was observed in an experimental matrix that included the electron donors H2, H2S, or S0 with oxygen as the electron acceptor. Surprisingly, the in vitro pH optimum of strain YNP1T (pH 7) is much higher than that of the soil from which it was isolated (pH 4–5) [1]. In pure culture, strain YNP1T failed to grow at such low pH values, suggesting that the thermal soil habitat is not optimal for the strain [1].

Transmission electron micrograph of ‘T. terrenum’ strain YNP1T, scale bar 0.1 µm
Chemotaxonomy
Murein is present in large amounts, which is consistent with the observed thick (approximately 34 nm) cell walls with a muramic acid content similar to that of Bacillus subtilis [1]. The muramic acid content of strain YNP1T was roughly one quarter of that measured for B. subtilis) but almost 40-fold greater than in E. coli [1]. Lipopolysaccharide (LPS) was not detected [1]. Major fatty acids were dominated by straight and branched chain saturated acids: C18:0 (27.0%); iso-C17:0 (11.6%); iso-C19:0 (12.9%); anteiso-C18:0 (12.5%); C20:0 (16.5%) and C19:0 (6.6%). The pink pigment associated with strain YNP1T exhibited a significant absorption at wavelengths 267, 326, 399, 483, 511, and 549 nm [1].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position [26], and is part of the Genomic Encyclopedia of Bacteria and Archaea project [27]. The genome project is deposited in the Genome OnLine Database [15] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
T. terrenum strain YNP1T, ATCC BAA-798, was grown in ATCC medium 1981 (M-R2A medium) [28] at 60°C. The culture used to prepare genomic DNA (gDNA) for sequencing was only two transfers from the original deposit. The purity of the culture was determined by growth on general maintenance media under both aerobic and anaerobic conditions. Cells where harvested after 24 hours by centrifugation and gDNA was extracted from lysozyme-treated cells using CTAB and phenol-chloroform. The purity, quality and size of the bulk gDNA preparation was assessed according to DOE-JGI guidelines. Amplification and partial sequencing of the 16S rRNA gene confirmed the isolate as ‘T. terrenum’. The quantity of the DNA was determined on a 1% agarose gel using mass markers of known concentration supplied by JGI. The average fragment size of the purified gDNA determined to be ∼43kb by pulsed-field gel electrophoresis.
Genome sequencing and assembly
The genome was sequenced using a combination of Sanger and 454 sequencing platforms. All general aspects of library construction and sequencing can be found at the JGI website (http://www.jgi.doe.gov/). Pyrosequencing reads were assembled using the Newbler assembler version 1.1.02.15 (Roche). Large Newbler contigs were broken into 3,926 overlapping fragments of 1,000 bp and entered into assembly as pseudo-reads. The sequences were assigned quality scores based on Newbler consensus q-scores with modifications to account for overlap redundancy and adjust inflated q-scores. A hybrid 454/Sanger assembly was made using the parallel phrap assembler (High Performance Software, LLC). Possible misassemblies were corrected with Dupfinisher or transposon bombing of bridging clones [29]. A total of 432 Sanger finishing reads were produced to close gaps, to resolve repetitive regions, and to raise the quality of the finished sequence. Illumina reads were used to improve the final consensus quality using an in-house developed tool (the Polisher [30]). The error rate of the completed genome sequence is less than 1 in 100,000. Together, the combination of the Sanger and 454 sequencing platforms provided 10.0× coverage of the genome. The final assembly contains 32,920 Sanger reads.
Genome annotation
Genes were identified using Prodigal [31] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [32]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [33].
Genome properties
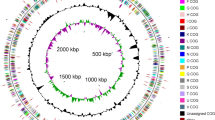
The genome consists of two chromosomes: the low G+C (48%) 2,026,947 bp long chromosome 1, and the high G+C (64%) 1,074,634 bp long chromosome 2 (Table 3, Figure 3, Figure 4). Of the 2,930 genes predicted (1,935 on chromosome 1 and 995 on chromosome 2), 2,872 were protein-coding genes, and 58 RNAs; forty one pseudogenes were also identified. The majority of the protein-coding genes (73.4%) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.

Graphical circular map of the 2Mb low G+C chromosome 1. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.

Graphical circular map of the 1 Mb high-G+C chromosome 2. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
References
Botero LM, Brown KB, Brumefield S, Burr M, Castenholz RW, Young M, McDermott TR. Thermobaculum terrenum gen. nov., sp. nov.: a non-phototrophic gram-positive thermophile representing an environmental clone group related to the Chloroflexi (green non-sulfur bacteria) and Thermomicrobia. Arch Microbiol 2004; 181:269–277. PubMed doi:10.1007/s00203-004-0647-7
Lapage SP, Sneath PHA, Lessel EF, Skerman VBD, Seeliger HPR, Clark WA. International Code of Nomenclature of Bacteria (1990) Revision. American Society for Microbiology, Washington, DC, 1992.
Demharter W, Hensel R, Smida J, Stackebrandt E. Sphaerobacter thermophilus gen. nov., sp. nov. A deeply rooting member of the actinomycetes subdivision isolated from thermophilically treated sewage sludge. Syst Appl Microbiol 1989; 11:261–266.
Zarilla KA, Perry JJ. Thermoleophilum album gen. nov. and sp. now., a bacterium obligately for thermophily and n-alkane substrates. Arch Microbiol 1984; 137:286–290. doi:10.1007/BF00410723
Zarilla KA, Perry JJ. Deoxyribonucleic acid homology and other comparisons among obligately thermophilic hydrocarbonoclastic bacteria, with a proposal for Thermoleophilum minutum sp. nov. Int J Syst Bacteriol 1986; 36:13–16. doi:10.1099/00207713-36-1-13
List editor. Validation list No. 20. Int J Syst Evol Microbiol 1986; 36:354–356.
Reddy GSN, Garcia-Pichel F. Description of Patulibacter americanus sp. nov., isolated from biological soil crusts, emended description of the genus Patulibacter Takahashi et al. 2006 and proposal of Solirubrobacterales ord. nov. and Thermoleophilales ord. nov. Int J Syst Evol Microbiol 2009; 59:87–94. PubMed doi:10.1099/ijs.0.64185-0
Chun J, Lee JH, Jung Y, Kim M, Kim S, Kim BK, Lim YW. EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol 2007; 57:2259–2261. PubMed doi:10.1099/ijs.0.64915-0
Kuhlman KR, Fusco WG, La Duc MT, Allenbach LB, Ball CL, Kuhlman GM, Anderson RC, Erickson IK, Stuecker T, Benardini J, et al. Diversity of microorganisms within rock varnish in the Whipple Mountains, California. Appl Environ Microbiol 2006; 72:1708–1715. PubMed doi:10.1128/AEM.72.2.1708-1715.2006
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069–5072. PubMed doi:10.1128/AEM.03006-05
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540–552. PubMed
Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452–464. PubMed doi:10.1093/bioinformatics/18.3.452
Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 2008; 57:758–771. PubMed doi:10.1080/10635150802429642
Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? Lect Notes Comput Sci 2009; 5541:184–200. doi:10.1007/978-3-642-02008-713
Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Hugenholtz P, Markowitz VM, Kyrpides NC. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2010; 38:D346–D354. PubMed doi:10.1093/nar/gkp848
Wu D, Raymond J, Wu M, Chatterji S, Ren Q, Graham JE, Bryant DA, Robb F, Colman A, Tallon LJ, et al. Complete genome sequence of the aerobic CO-oxidizing thermophile Thermomicrobium roseum. PLoS ONE 2009; 4:e4207. PubMed doi:10.1371/journal.pone.0004207
Pati A, LaButti K, Pukall R, Nolan M, Glavina del Rio T, Tice H, Cheng JF, Lucas S, Chen F, Lucas S, et al. Complete genome sequence of Sphaerobacter thermophilus type strain (S 6022T). Stand Genomic Sci 2010; 2:49–56. doi:10.4056/sigs.601105
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed doi:10.1038/nbt1360
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed doi:10.1073/pnas.87.12.4576
Garrity GM, Holt JG. Phylum BVI. Chloroflexi phy. nov. In: DR Boone, RW Castenholz, GM Garrity (eds): Bergey’s Manual of Systematic Bacteriology, second edition, vol. 1 (The Archaea and the deeply branching and phototrophic Bacteria), Springer-Verlag, New York, 2001, p. 427–446.
List Editor. Validation List no. 85. Validation of publication of new names and new combinations previously effectively published outside the IJ-SEM. Int J Syst Evol Microbiol 2002; 52:685–690. PubMed doi:10.1099/ijs.0.02358-0
Garrity GM, Holt JG. The Road Map to the Manual. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 119–169.
Hugenholtz P, Stackebrandt E. Reclassification of Sphaerobacter thermophilus from the subclass Sphaerobacteridae in the phylum Actinobacteria to the class Thermomicrobia (emended description) in the phylum Chloroflexi (emended description). Int J Syst Evol Microbiol 2004; 54:2049–2051. PubMed doi:10.1099/ijs.0.03028-0
LGC Advanced Catalogue Search.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet 2000; 25:25–29. PubMed doi:10.1038/75556
Klenk HP, Göker M. En route to a genome-based classification of Archaea and Bacteria? Syst Appl Microbiol 2010; 33:175–182. PubMed doi:10.1016/j.syapm.2010.03.003
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056–1060. PubMed doi:10.1038/nature08656
Sims D, Brettin T, Detter JC, Han C, Lapidus A, Copeland A, Glavina Del Rio T, Nolan M, Chen F, Lucas S, et al. Complete genome sequence of Kytococcus sedentarius type strain (541T). Stand Genomic Sci 2009; 1:12–20. doi:10.4056/sigs.761
Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008.
Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119. PubMed doi:10.1186/1471-2105-11-119
Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455–457. PubMed doi:10.1038/nmeth.1457
Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271–2278. PubMed doi:10.1093/bioinformatics/btp393
Acknowledgements
This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396, UT-Battelle and Oak Ridge National Laboratory under contract DE-AC05-00OR22725.
Author information
Authors and Affiliations
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Kiss, H., Cleland, D., Lapidus, A. et al. Complete genome sequence of ‘Thermobaculum terrenum’ type strain (YNP1T). Stand in Genomic Sci 3, 153–162 (2010). https://doi.org/10.4056/sigs.1153107
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.1153107